

Abstract
Objective:
To characterize changes in myeloid and lymphoid innate immune cells in patients with relapsing-remitting multiple sclerosis (MS) during a 6-month follow-up after alemtuzumab treatment.
Methods:
Circulating innate immune cells including myeloid cells and innate lymphoid cells (ILCs) were analyzed before and 6 and 12 months after onset of alemtuzumab treatment. Furthermore, a potential effect on granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)–23 production by myeloid cells and natural killer (NK) cell cytolytic activity was determined.
Results:
In comparison to CD4+ T lymphocytes, myeloid and lymphoid innate cell subsets of patients with MS expressed significantly lower amounts of CD52 on their cell surface. Six months after CD52 depletion, numbers of circulating plasmacytoid dendritic cells (DCs) and conventional DCs were reduced compared to baseline. GM-CSF and IL-23 production in DCs remained unchanged. Within the ILC compartment, the subset of CD56bright NK cells specifically expanded under alemtuzumab treatment, but their cytolytic activity did not change.
Conclusions:
Our findings demonstrate that 6 months after alemtuzumab treatment, specific DC subsets are reduced, while CD56bright NK cells expanded in patients with MS. Thus, alemtuzumab specifically restricts the DC compartment and expands the CD56bright NK cell subset with potential immunoregulatory properties in MS. We suggest that remodeling of the innate immune compartment may promote long-term efficacy of alemtuzumab and preserve immunocompetence in patients with MS.
Alemtuzumab (NCT00548405; Lemtrada; Genzyme, Cambridge, MA) is a humanized monoclonal antibody specific for the membrane glycoprotein CD52. Alemtuzumab provides long-lasting suppression of disease activity in relapsing-remitting multiple sclerosis (RRMS). Through in vivo targeting of CD52 on the cell surface, alemtuzumab induces various biological effects such as complement-dependent cell lysis, antibody-dependent cellular cytotoxicity, and apoptosis resulting in the elimination of circulating T lymphocytes.1–4 However, the effect of alemtuzumab on the innate immune compartment has not been comprehensively analyzed in RRMS.
Innate immune cells mediate the first line of defense against pathogens and play essential roles in regulating tissue homeostasis and inflammation.5,6 This heterogeneous population comprises myeloid cells such as dendritic cells (DCs) and macrophages and the family of innate lymphoid cells (ILCs). As orchestrators of immunity and tolerance induction, plasmacytoid DCs (pDCs) have been shown to modulate pathogenic T-cell responses, thus affecting autoimmune neuroinflammation.7–9 ILCs consist of 4 major subsets, including cytotoxic natural killer cells (NK cells) and 3 tissue-resident non-cytotoxic subsets, namely ILC1, ILC2, and group 3 ILC (ILC3 and lymphoid tissue inducer cells [LTi]).6 Frequencies of circulating LTis, and ILC subsets implicated in chronic inflammation, are increased in patients with multiple sclerosis (MS).10 Furthermore, NK-mediated control of T-cell activity11 has been shown to be impaired in MS,12,13 but can be restored by treatment with daclizumab.13
In this study, we investigated the phenotype and responses of innate immune cells in a 6-month follow-up study of alemtuzumab treatment to gain a better understanding of alemtuzumab-mediated effects on the innate immune response.
METHODS
Patients and biomaterial.
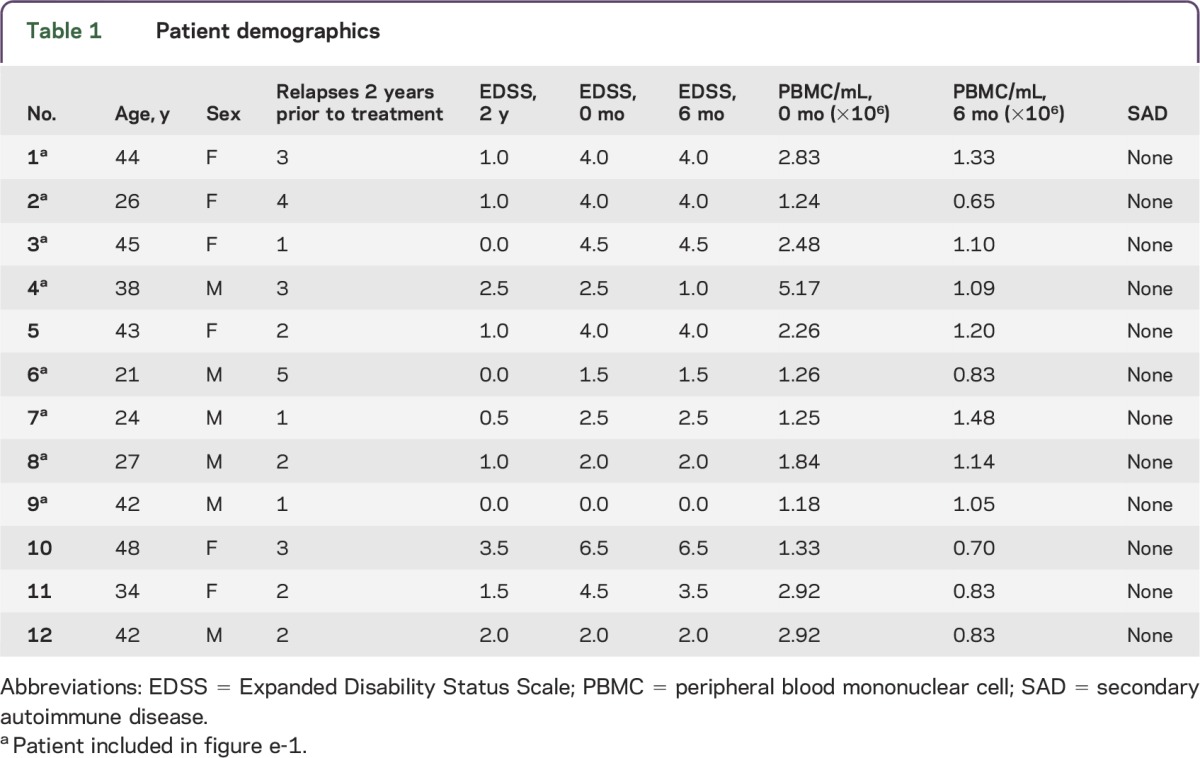
All patients were recruited at the Department of Neurology at the University Hospital Münster, Germany. Twelve patients with RRMS prior to and on alemtuzumab (Lemtrada) treatment (table 1, age 21–48 years, mean age 36.2 years, 6 female, 6 male) were included in the current study. Mean number of relapses was 2.4 ± 1.2 and mean Expanded Disability Status Scale (EDSS) progression was 1.2 ± 1.1 2 years prior to alemtuzumab initiation (table 1). Three patients were therapy-naive and the other patients received pretreatments including azathioprine, β-interferons, glatiramer acetate, teriflunomide, fingolimod, natalizumab, mitoxantrone, and siponimod (within a clinical trial). So far none of the patients included in the study exhibited a secondary autoimmune disease. PBMCs were isolated from ethylenediaminetetraacetic acid blood derived from these patients at baseline (n = 12) and 6 (n = 12) and 12 months (n = 8) after standard treatment regimen of alemtuzumab (table 1) and cryopreserved as previously described.14
Table 1.
Patient demographics
Standard protocol approvals, registrations, and patient consents.
This study was performed according to the Declaration of Helsinki and approved by the local ethics committee (2014-398-f-S). All patients gave written informed consent.
Stimulation of DCs.
For the identification of cytokine production in myeloid cells, freshly thawed PBMCs were stimulated with 200 ng/mL lipopolysaccharide (Sigma-Aldrich, St. Louis, MO) in X-Vivo 15 (Lonza Group, Basel, Switzerland) supplemented with Brefeldin A (5 μg/mL) and Monensin (2 μM) (BioLegend, San Diego, CA) at a concentration of 1 × 107 cells/mL for 10 hours at 37°C, 5% CO2. Subsequently, cells were stained for flow cytometry as described below.
Flow cytometry.
Flow cytometry of thawed PBMCs was performed as previously described14 using the respective fluorochrome-conjugated antibodies at the indicated working concentrations (table e-1 at Neurology.org/nn). Staining for chemokine receptors was done at 37°C. Intracellular staining for cytokines was performed using the intracellular staining kit (eBioscience, San Diego, CA) following the manufacturer's instructions. Samples were acquired on a 10-color Navios (Beckman Coulter, Sharon Hill, PA) or FACSCanto II (BD Biosciences, East Rutherford, NJ) flow cytometer and analyzed by FlowJo v10 and Kaluza 1.3. The gating strategies to determine innate myeloid and lymphoid subsets are described in the respective figures. T-regulatory cells were phenotypically characterized by FoxP3 expression among gated CD3+CD4+ T cells.
NK-cell degranulation assay.
Degranulation of interleukin (IL)–15-activated NK cells in response to K562, 721.221, and allogeneic CD4+ T cells was determined as previously described.13,15 Staphylococcal enterotoxin B (SEB; Sigma-Aldrich)–activated CD4+ T cells were generated by stimulation of thawed PBMCs in RPMI 1640 medium supplemented with 10% heat-inactivated FCS, 200 IU/mL recombinant human IL-2 (eBioscience), and 100 μg/mL SEB at cell densities of 2 × 106 cells/mL for 4 days at 37°C. CD4+ T cells were isolated using the CD4+ T cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions.
Statistical analysis.
Data were analyzed using GraphPad Prism v5.04 (La Jolla, CA). Depending on the outcome of D'Agostino and Pearson omnibus normality test, groups of 2 datasets were compared by paired or unpaired Student t test and Wilcoxon matched-pairs signed rank test or Mann-Whitney test, respectively. Datasets of 3 or more groups were analyzed by 1-way analysis of variance (ANOVA) or repeated-measures ANOVA with Bonferroni posttest or for non-Gauss distributed data Kruskal-Wallis test or Friedman test with Dunn post-test, respectively. Datasets with too few data points to perform testing for Gaussian distribution were analyzed as Gaussian distributed. Results were regarded as statistically significant with the following p values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
RESULTS
In accordance with previous studies, proportions and numbers of CD4+ T cells markedly declined 6 months after alemtuzumab treatment (figure e-1A). In comparison to CD4+ T cells, DCs (figure 1A), monocytes (figure 1B), and ILCs (figure 1C) derived from patients with RRMS expressed significantly lower amounts of CD52 on their cell surface. Interestingly, despite low cell surface expression of CD52, alemtuzumab treatment altered proportions of pDCs (figure e-1B), monocytes (figure e-1C), and ILCs (figure e-1D), with most of the changes occurring 6 months after treatment onset. Thus, the effect of alemtuzumab treatment on these innate myeloid and lymphoid cell subsets as well as their phenotype and function were further investigated using PBMCs derived from 12 patients with RRMS (table 1) before and 6 months after treatment onset. Patients with RRMS remained clinically stable during the 6-month follow-up (table 1, EDSS 3.2 ± 1.7 at baseline vs 3.0 ± 1.8 at 6 months) and none of the investigated patients experienced any further relapse. In accordance with the alemtuzumab-induced lymphopenia, total numbers of PBMCs were strongly reduced in patients with RRMS following 6 months of alemtuzumab treatment (table 1, 2.22 ± 1.17 × 106 PBMC/mL at baseline vs 1.02 ± 0.26 × 106 PBMC/mL at 6 months).
Figure 1. CD52 expression on innate myeloid and lymphoid cell subsets.

Peripheral blood mononuclear cells derived from treatment-naive patients with relapsing-remitting multiple sclerosis (n = 5, down triangles) were stained with fluorochrome-conjugated antibodies specific for (A) dendritic cell subsets, (B) monocyte subsets, and (C) innate lymphoid cell subsets as well as for CD52. CD52 expression on CD4+ T cells served as a positive control. Left: histograms of one representative donor show CD52 expression of the respective cell subset (dark gray) in comparison to the isotype control (light gray). Right: graphs display the median fluorescence intensities (MFI) of the respective cell subsets. Error bars indicate the SD. p Values were calculated by unpaired Student t test, ***p < 0.001, ****p < 0.0001. ILC = innate lymphoid cell; LTi = lymphoid tissue inducer cell.
Within the innate myeloid cell compartment (figure 2A), decreased numbers of circulating conventional CD141+ DCs and CD1c+ DCs as well as pDCs were observed in alemtuzumab-treated patients (figures 2B and e-2A), resulting in a relative increase of monocytes after 6 months (figures 3, A and B, and e-2B).
Figure 2. Alemtuzumab-induced changes in the dendritic cell compartment.

(A) Gating strategy for dendritic cells. Peripheral blood mononuclear cells (PBMCs) were gated by forward scatter vs side scatter characteristics. Lineage (CD3, CD14, CD19) negative cells expressing human leukocyte antigen–DR (HLA-DR) were further categorized into CD141+ conventional DCs, CD1c+ conventional DCs, and CD303+ plasmacytoid dendritic cells (pDCs). (B) Graphs display proportions of Lin−HLA-DR+ cells within PBMCs or the respective dendritic cell (DC) subsets within Lin−HLA-DR+ (upper row) and total cell numbers (lower row) of DC subsets derived from alemtuzumab-treated patients with relapsing-remitting multiple sclerosis (n = 12) at baseline (filled triangles) and 6-month follow-up (open triangles). p Values were calculated by paired Student t test or Wilcoxon matched-pairs signed rank test, respectively, *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 3. Alemtuzumab-induced changes in monocytes.

(A) Gating strategy for monocytes. Monocytes and their subsets were separated from the total lineage (CD3, CD19) negative compartment based on the expression of CD14, CD16, and human leukocyte antigen–DR (HLA-DR), as previously described.32 (B) Graphs display proportions of monocytes within peripheral blood mononuclear cells or the respective subsets within monocytes (upper row) and total cell numbers (lower row) of monocyte subsets derived from alemtuzumab-treated patients with relapsing-remitting multiple sclerosis (n = 12) at baseline (filled triangles) and 6-month follow-up (open triangles). p Values were calculated by paired Student t test or Wilcoxon matched-pairs signed rank test, respectively, *p < 0.05, **p < 0.01, ***p < 0.001.
While lower numbers of circulating DC subsets were observed in alemtuzumab-treated patients, IL-23 and granulocyte-macrophage colony-stimulating factor (GM-CSF) production by DCs remained unaltered in patients with MS and healthy donors (figure 4A and data not shown). GM-CSF production by monocytes also remained unaltered (figure 4, B and C), whereas proportions of IL-23-producing monocytes (figure 4B) including the CD14+CD16− classical monocyte subset (figure 4C and data not shown) were mildly enhanced in alemtuzumab-treated patients and healthy donors compared to baseline.
Figure 4. Alemtuzumab-mediated effects on interleukin (IL)–23 and granulocyte-macrophage colony-stimulating factor (GM-CSF) production in innate myeloid cells.
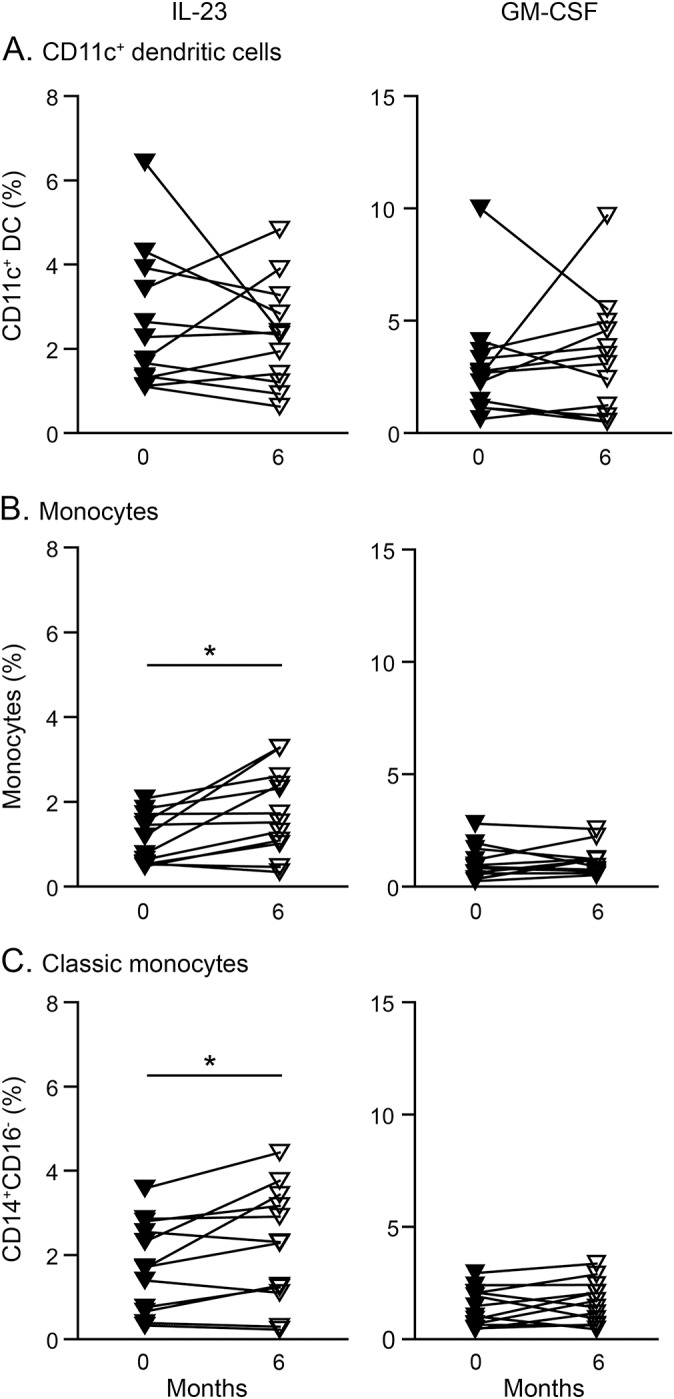
Graphs display proportions of IL-23 and GM-CSF producing (A) CD11c+ dendritic cells (DCs), (B) monocytes, and (C) CD14+CD16− classical monocytes derived from alemtuzumab-treated patients with relapsing-remitting multiple sclerosis (n = 12) at baseline (filled triangles) and 6-month follow-up. p Values were calculated by paired Student t test or Wilcoxon matched-pairs signed rank test, respectively, *p < 0.05.
With regard to CD4+ T-cell subsets, a relative increase of proinflammatory T-helper (Th) 17 as well as immune-regulatory CD4+ T cells within the CD4+ subset was observed, while proportions within PBMCs and total numbers of these subsets were decreased 6 months after alemtuzumab treatment (figure e-3, A and B). Interestingly, alemtuzumab-induced decrease of circulating CD4+ Th cells was accompanied by significantly increased proportions of ILC subsets (figure 5, A and B), the innate counterparts of the respective Th cell subset. Within the ILC compartment, reduced proportions and numbers of proinflammatory LTi were observed, whereas frequencies and numbers of immune-regulatory CD56bright NK cells were significantly increased (figure 5B). In accordance with the relative increase of ILCs, CD56dim NK cells the major ILC subset, were also enhanced within the PBMCs (figure e-4). However, this increase was relative, because proportions of CD56dim NK cells within the ILC subpopulation as well as their cell numbers remained unaltered (figure 5B). Finally, cytolytic activity of NK cells toward major histocompatibility complex–I-deficient target cell lines as well as antigen-activated CD4+ T cells was sustained in patients treated with alemtuzumab (figure 5C).
Figure 5. Alemtuzumab-induced changes in the innate lymphoid cell (ILC) compartment.

(A) Gating strategy for ILCs. ILCs were defined as lineage (CD3, CD14, CD19, CD20)–negative peripheral blood mononuclear cells (PBMCs) devoid for dendritic cell (DC) markers CD123 and CD11c. CD56brightCD16dim/− natural killer (NK) cells were gated from CD56+NKp46+ ILC, whereas CD117+CRTH2− lymphoid tissue inducer cells (LTis) were gated from CD56−NKp46− ILCs. (B) Graphs display proportions of ILCs within PBMCs or ILC subsets within ILCs (upper row) and total cell numbers (lower row) of ILC subsets derived from alemtuzumab-treated patients with relapsing-remitting multiple sclerosis (n = 12) at baseline (filled triangles) and 6-month follow-up (open triangles). (C) Release of cytolytic granules by NK cells at baseline and 6 months after alemtuzumab therapy (n = 7) in response to K562 (left), 721.221 (middle), or antigen-activated allogenic CD4+ T cells. p Values were calculated by paired Student t test or Wilcoxon matched-pairs signed rank test, respectively, *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
We investigated phenotype and effector function of innate immune cells in patients with RRMS in a 6-month follow-up study after a first cycle of alemtuzumab treatment. We found that alemtuzumab reduced numbers of specific DC subsets capable of eliciting detrimental immune responses in MS, but did not affect their IL-23 and GM-CSF production. Alemtuzumab-induced decrease of CD4+ T cells was accompanied by an increase in ILC frequencies, mainly caused by expansion of the CD56bright NK cells subset known to harbor potent immunoregulatory properties.11,13,16 However, the cytolytic function of NK cells was not altered 6 months after alemtuzumab treatment. These data indicate that alemtuzumab treatment in patients with RRMS restricts DC responses, while NK-cell activity is preserved. We suggest that these immunomodulatory properties of alemtuzumab might contribute to its long-term beneficial effects and preserved immunocompetence in patients with RRMS.
Despite their low surface expression of CD52, DCs were effectively depleted by alemtuzumab treatment,17,18 and rapid reconstitution of DCs has been observed in recipients of allogeneic stem cell transplantation with prior immune conditioning.19,20 Thus, it was an unexpected finding of our study that numbers of peripheral DCs including conventional DCs as well as pDCs were reduced 6 months after alemtuzumab treatment. Both DC subsets have been shown to exhibit specialized functions pivotal in the pathogenesis of MS.21,22 Conventional DCs induce Th1- and Th2-associated cytokines in RRMS and are cellular producers of IL-23.23,24 pDCs instead display a limited capacity to induce T-cell responses under steady-state conditions, but undergo functional specialization in MS with the capacity to prime IL-17-secreting T cells.25 Our results suggest that alemtuzumab targeting of pDCs might provide an additional beneficial mechanism in RRMS by downregulating detrimental adaptive immune responses.
Alemtuzumab did not affect surface expression levels and activation-dependent upregulation of the costimulatory molecules CD40 and CD80 on DCs in a low number of patients analyzed. In addition, human leukocyte antigen–DR expression was equally inducible after 6 months in CD1c+ DCs as compared to stimulated cells from untreated patients (figure e-5A). Additional pilot data from this study point to an equivalent production of tumor necrosis factor, IL-6, and IL-12 by myeloid cells 6 months after alemtuzumab treatment (figure e-5, B and C). We showed before that specifically IL-23 production by DCs is essential for the maintenance of Th17 lymphocytes and has previously been suggested as a therapeutic target in MS.9,26 Alemtuzumab did not alter GM-CSF and IL-23 production in DCs and mildly increased IL-23-producing monocytes 6 months after treatment. Additional study data from 8 treated patients pointed towards a reduction of IL-23-producing monocyte counts to baseline levels after 12 months (figure e-5D). The relevance of transient enhanced IL-23 production in this monocyte subset in alemtuzumab patients is unclear but may point towards compensatory effects.
NK cells have been suggested as additional players of the innate compartment in regulating T-cell responses in MS.11,13,27 Beneficial effects of daclizumab, a humanized monoclonal antibody directed against the IL-2 receptor chain,28 are associated with increased numbers and function of the CD56bright NK-cell subset in patients with RRMS.11,13 Strikingly, our data revealed that alemtuzumab treatment also expanded this immune-regulatory NK-cell subset, emphasizing a potential role for the CD56bright NK-cell subset in alemtuzumab-treated patients with RRMS. However, in contrast to daclizumab, alemtuzumab did not change the cytolytic activity of NK cells in response to antigen-activated CD4+ T cells. Of note, increased numbers of CD56bright NK cells have also been observed in several autoimmune settings such as Hashimoto thyroiditis.29 Secondary autoimmune diseases including Hashimoto thyroiditis are common adverse reactions following alemtuzumab therapy.30,31 Although the underlying mechanisms of autoimmunity following alemtuzumab therapy have not yet been resolved, one can speculate on the functional implication of CD56bright NK cells in this process.
Altogether, this study elucidates the repopulation dynamics and immune responses of innate immune cells in alemtuzumab-treated patients with RRMS. A limitation of our approach is the low number of patients included and the lack of functional assays addressing antigen uptake, processing, and presentation capacities of DCs that preclude final conclusions from our study.
Future controlled studies may expand our observations by addressing the role of innate immunity in alemtuzumab-treated patients with RRMS. Additional studies are required to understand the effect of alemtuzumab on the innate immune system in patients with RRMS. Our study suggests that alemtuzumab exerts disease-modulating activity by reducing DC numbers and preserving NK-cell responses in RRMS. Thus, remodeling of the innate immune compartment might add a beneficial treatment effect by alemtuzumab in RRMS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Daniela Roosterman, Verena Schütte, Kerstin Gottschalk, Schumina Säuberlich, Karla Musiolik, and Sarah Spielmann for technical assistance and all participants who donated blood in order to realize this study.
GLOSSARY
- ANOVA
analysis of variance
- DC
dendritic cell
- EDSS
Expanded Disability Status Scale
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IL
interleukin
- ILC
innate lymphoid cell
- LTi
lymphoid tissue inducer cell
- MS
multiple sclerosis
- NK cell
natural killer cell
- PBMC
peripheral blood mononuclear cell
- pDC
plasmacytoid dendritic cell
- RRMS
relapsing-remitting multiple sclerosis
- SEB
Staphylococcal enterotoxin B
- Th
T helper
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
J.A. and C.C.G. conceptualized the study, designed research, analyzed and interpreted the data, and wrote the manuscript. D.A. and A.S.M. performed research, collected and analyzed data, and helped with writing the manuscript. S.G.M. and T.R. organized patient recruitment, took care of patients, provided clinical information, and critically edited the manuscript. K.S. performed research and collected data. S.S. designed research and critically edited the manuscript. L.K., N.M., S.W., and B.G. were responsible for patient care, provided clinical information, and critically edited the manuscript. H.W., V.A., and S.J. helped to conceptualize the project and critically edited the manuscript.
STUDY FUNDING
This study was supported by the Krankheitsbezogene Kompetenznetz Multiple Sklerose (Disease-Related Competence Network for Multiple Sclerosis) funded by the Federal Ministry of Education and Research (FKZ 01FI1603A to H.W., L.K., and C.C.G.; 01GI0907B1 to H.W.; 01GI1603D to S.G.M., T.R., and H.W.) and the Collaborative Research Centre CRC TR128 “Initiating/effector vs regulatory mechanisms in multiple sclerosis: progress towards tackling the disease” (Projects A9 to H.W. and C.C.G. and Z2 to H.W.). The work of C.C.G. has been funded by the individual research grant “The role of natural killer cells in the immunoregulation of multiple sclerosis” by the German Research Foundation (DFG, GR3946/2-1). Also supported by DFG EXC 1003, Grant FF-2014-01, Cells in Motion–Cluster of Excellence, Münster, Germany, to J.A.; DFG SCHE692/3-1 to S.S.; and Strategic Research Fund of the Heinrich Heine University Düsseldorf (SFF-F2012/79-5-Scheu) to S.S. This study has also been supported by Genzyme Therapeutics Ltd., United Kingdom (“Alemtuzumab in autoimmune inflammatory neurodegeneration: mechanisms of action and neuroprotective potential” [ALAIN] to H.W., S.G.M., and T.R.).
DISCLOSURE
J.A., D.A., A.S.M., K.S., S.J., S.S., and B.G. have no financial disclosures. C.C.G. received speaker honoraria and travel expenses for attending meetings from Genzyme, Novartis Pharma GmbH, and Bayer Health Care. T.R. has received travel expenses and financial research support from Genzyme and Novartis and has received honoraria for lecturing from Genzyme, Biogen, and Teva. S.W. received speaker honoraria from Bayer Vital, Biogen, Merck-Serono, Novartis Pharma, and TEVA Pharma. N.M. received honoraria for lecturing and travel expenses for attending meetings from Biogen Idec, GlaxoSmithKline, Teva, Novartis Pharma, Bayer Healthcare, and Fresenius Medical Care and received financial research support from Fresenius Medical Care. L.K. received compensation for serving on Scientific Advisory Boards for Genzyme. She received speaker honoraria and travel support from Novartis, Merck Serono, and CSL Behring. She receives research support from Novartis. V.A. is a member of advisory boards and/or gave presentations for the following companies: Astra-Zeneca, Eli Lilly, Janssen-Organon, Lundbeck, Otsuka, Servier, and Trommsdorff. H.W. received compensation for serving on Scientific Advisory Boards/Steering Committees for Bayer Healthcare, Biogen, Genzyme, Merck Serono, Novartis, and Sanofi Aventis. He also received speaker honoraria and travel support from Bayer Vital GmbH, Bayer Schering AG, Biogen, CSL Behring, Fresenius Medical Care, Genzyme, Glaxo Smith Kline, GW Pharmaceuticals, Lundbeck, Merck Serono, Omniamed, Novartis, and Sanofi-Aventis. He received compensation as a consultant from Biogen, Merck Serono, Novartis, and Sanofi-Aventis. H.W. received research support from Bayer Vital, Biogen, Genzyme Merck Serono, Novartis, Sanofi Aventis Germany, and Sanofi US. S.G.M. has received honoraria for lecturing and travel expenses for attending meetings and has received financial research support from Bayer, Bayer Schering, Biogen Idec, Genzyme, Merck Serono, Merck Sharp & Dohme, Novartis, Novo Nordisk, Sanofi-Aventis, and Teva. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Hale G, Xia MQ, Tighe HP, Dyer MJ, Waldmann H. The CAMPATH-1 antigen (CDw52). Tissue Antigens 1990;35:118–127. [DOI] [PubMed] [Google Scholar]
- 2.Rao SP, Sancho J, Campos-Rivera J, et al. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS One 2012;7:e39416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Y, Turner MJ, Shields J, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 2009;128:260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mone AP, Cheney C, Banks AL, et al. Alemtuzumab induces caspase-independent cell death in human chronic lymphocytic leukemia cells through a lipid raft-dependent mechanism. Leukemia 2006;20:272–279. [DOI] [PubMed] [Google Scholar]
- 5.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science 2010;327:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 2011;12:21–27. [DOI] [PubMed] [Google Scholar]
- 7.Gross CC, Wiendl H. Dendritic cell vaccination in autoimmune disease. Curr Opin Rheumatol 2013;25:268–274. [DOI] [PubMed] [Google Scholar]
- 8.Zozulya AL, Clarkson BD, Ortler S, Fabry Z, Wiendl H. The role of dendritic cells in CNS autoimmunity. J Mol Med 2010;88:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poppensieker K, Otte DM, Schurmann B, et al. CC chemokine receptor 4 is required for experimental autoimmune encephalomyelitis by regulating GM-CSF and IL-23 production in dendritic cells. Proc Natl Acad Sci USA 2012;109:3897–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry JS, Han S, Xu Q, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med 2012;4:145ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci USA 2006;103:5941–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laroni A, Armentani E, Kerlero de Rosbo N, et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J Autoimmun 2016;72:8–18. [DOI] [PubMed] [Google Scholar]
- 13.Gross CC, Schulte-Mecklenbeck A, Rünzi A, et al. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc Natl Acad Sci USA 2016;113:E2973–E2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Posevitz-Fejfar A, Posevitz V, Gross CC, et al. Effects of blood transportation on human peripheral mononuclear cell yield, phenotype and function: implications for immune cell biobanking. PLoS One 2014;9:e115920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulte-Mecklenbeck A, Bittner S, Ehling P, et al. The two-pore domain K2 P channel TASK2 drives human NK-cell proliferation and cytolytic function. Eur J Immunol 2015;45:2602–2614. [DOI] [PubMed] [Google Scholar]
- 16.Cooper MA, Fehniger TA, Turner SC, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001;97:3146–3151. [DOI] [PubMed] [Google Scholar]
- 17.Buggins AG, Mufti GJ, Salisbury J, et al. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood 2002;100:1715–1720. [PubMed] [Google Scholar]
- 18.Klangsinsirikul P, Carter GI, Byrne JL, Hale G, Russell NH. Campath-1G causes rapid depletion of circulating host dendritic cells (DCs) before allogeneic transplantation but does not delay donor DC reconstitution. Blood 2002;99:2586–2591. [DOI] [PubMed] [Google Scholar]
- 19.Auffermann-Gretzinger S, Lossos IS, Vayntrub TA, et al. Rapid establishment of dendritic cell chimerism in allogeneic hematopoietic cell transplant recipients. Blood 2002;99:1442–1448. [DOI] [PubMed] [Google Scholar]
- 20.Chklovskaia E, Nowbakht P, Nissen C, Gratwohl A, Bargetzi M, Wodnar-Filipowicz A. Reconstitution of dendritic and natural killer-cell subsets after allogeneic stem cell transplantation: effects of endogenous flt3 ligand. Blood 2004;103:3860–3868. [DOI] [PubMed] [Google Scholar]
- 21.Gross CC, Jonuleit H, Wiendl H. Fulfilling the dream: tolerogenic dendritic cells to treat multiple sclerosis. Eur J Immunol 2012;42:569–572. [DOI] [PubMed] [Google Scholar]
- 22.Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol 2013;13:566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karni A, Abraham M, Monsonego A, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol 2006;177:4196–4202. [DOI] [PubMed] [Google Scholar]
- 24.Vaknin-Dembinsky A, Murugaiyan G, Hafler DA, Astier AL, Weiner HL. Increased IL-23 secretion and altered chemokine production by dendritic cells upon CD46 activation in patients with multiple sclerosis. J Neuroimmunol 2008;195:140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwab N, Zozulya AL, Kieseier BC, Toyka KV, Wiendl H. An imbalance of two functionally and phenotypically different subsets of plasmacytoid dendritic cells characterizes the dysfunctional immune regulation in multiple sclerosis. J Immunol 2010;184:5368–5374. [DOI] [PubMed] [Google Scholar]
- 26.Klotz L, Wiendl H. Monoclonal antibodies in neuroinflammatory diseases. Expert Opin Biol Ther 2013;13:831–846. [DOI] [PubMed] [Google Scholar]
- 27.Wiendl H, Gross CC. Modulation of IL-2Ralpha with daclizumab for treatment of multiple sclerosis. Nat Rev Neurol 2013;9:394–404. [DOI] [PubMed] [Google Scholar]
- 28.Kappos L, Wiendl H, Selmaj K, et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2015;373:1418–1428. [DOI] [PubMed] [Google Scholar]
- 29.Rizzo R, Zatelli MC, Rotola A, et al. Increase in peripheral CD3(-)CD56(bright)CD16(-) natural killer cells in Hashimoto's thyroiditis associated with HHV-6 infection. Adv Exp Med Biol 2016;897:113–120. [DOI] [PubMed] [Google Scholar]
- 30.Ruck T, Bittner S, Wiendl H, Meuth SG. Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int J Mol Sci 2015;16:16414–16439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obermann M, Ruck T, Pfeuffer S, Baum J, Wiendl H, Meuth SG. Simultaneous early-onset immune thrombocytopenia and autoimmune thyroid disease following alemtuzumab treatment in relapsing-remitting multiple sclerosis. Mult Scler 2016;22:1235–1241. [DOI] [PubMed] [Google Scholar]
- 32.Abeles RD, McPhail MJ, Sowter D, et al. CD14, CD16 and HLA-DR reliably identifies human monocytes and their subsets in the context of pathologically reduced HLA-DR expression by CD14(hi)/CD16(neg) monocytes: expansion of CD14(hi)/CD16(pos) and contraction of CD14(lo)/CD16(pos) monocytes in acute liver failure. Cytometry A 2012;81:823–834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.