

Abstract
Objectives
Our aim was to evaluate the safety and efficacy of ipilimumab combined with standard first-line chemotherapy for patients with extensive-stage SCLC.
Methods
Patients with chemotherapy-naive extensive-stage SCLC were treated with carboplatin and etoposide for up to six cycles. Ipilimumab, 10 mg/kg, was given on day 1 of cycles 3 to 6 and every 12 weeks. Response was assessed by the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.0, and immune-related response criteria. The primary end point was 1-year progression-free survival (PFS) according to RECIST. Secondary end points included PFS according to immune-related PFS and overall survival. Autoantibody serum levels were evaluated and correlated with clinical outcomes.
Results
A total of 42 patients were enrolled between September 2011 and April 2014; 39 were evaluable for safety and 38 for efficacy. Six of 38 patients (15.8% [95% confidence interval (CI): 7.4–30.4]) were alive and progression-free at 1-year by RECIST. Median PFS was 6.9 months (95% CI: 5.5–7.9). Median immune-related PFS was 7.3 months (95% CI: 5.5–8.8). Median overall survival was 17.0 months (95% CI: 7.9–24.3). Of the patients evaluable for response, 21 of 29 (72.4%) achieved an objective response by RECIST and 28 of 33 (84.8%) achieved an objective response by the immune-related response criteria. All patients experienced at least one adverse event; at least one grade 3 or higher toxicity developed in 35 of 39 patients (89.7%); in 27 patients (69.2%) this was related to ipilimumab. Five deaths were reported to be related to ipilimumab. Positivity of an autoimmune profile at baseline was associated with improved outcomes and severe neurological toxicity.
Conclusions
Ipilimumab in combination with carboplatin and etoposide might benefit a subgroup of patients with advanced SCLC. Autoantibody analysis correlates with treatment benefit and toxicity and warrants further investigation.
Keywords: Small cell lung cancer, Ipilimumab, Autoantibodies, Biomarker, CTLA-4 immunotherapy
Introduction
SCLC accounts for approximately 15% to 20% of all lung cancers. Despite the high percentage of initial responses to chemotherapy, overall prognosis remains dismal, with median survival times of 9.5 months for extensive-stage disease.1 No therapeutic strategy except for the addition of radiotherapy to chemotherapy has produced improvements in survival.2, 3, 4, 5, 6, 7
Harnessing the immune response to attack tumor cells with antibodies directed against checkpoint molecules has had dramatic impact in the treatment of melanoma8, 9 and other solid tumors.10, 11
Clinical evidence supports immune recognition of SCLC in the form of paraneoplastic immune-mediated syndromes (PNSs). PNSs are associated with the cross-reactivity of immune responses with self-antigens, which are frequently neuronal antigens physiologically expressed by the normal nervous system and ectopically by cancer cells,12 but the T-cell–based mechanisms for paraneoplastic events remain poorly understood.13 The presence of autoimmune disease seems to be associated with better outcomes.14, 15 These findings suggest that the effective antitumor immune responses are linked to autoimmune manifestations.16
Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is expressed by lymphocytes early in the adaptive immune response, and it binds to B7 expressed in antigen-presenting cells to down-regulate T-cell responses.17 Additionally, CTLA-4 is highly expressed on regulatory T-cells, and antibody binding to CTLA-4 leads to their removal by antibody-dependent cytotoxicity.18 Release of these “brakes” with anti–CTLA-4 antibodies has been successfully tested in several tumors.
Ipilimumab is an anti–CTLA-4 antibody approved for the treatment of metastatic melanoma.8, 19, 20 However, how effective ipilimumab is in rapidly progressing tumors such as SCLC is unclear. However, chemotherapy for SCLC is effective in killing tumor cells, and cell death will release tumor antigen.21 It is therefore possible that in the context of immune modulation with ipilimumab, recognition of these antigens might induce clinically useful antitumor immunity.
In 2013, a study assessing ipilimumab added to carboplatin and paclitaxel by randomizing patients with extensive-stage SCLC to chemotherapy only or chemotherapy with concurrent or phased ipilimumab was published.22 This study suggested that phased ipilimumab after two cycles of chemotherapy was a promising strategy.
The current study enrolled patients with extensive-stage SCLC treated with standard carboplatin and etoposide in the first-line setting and aimed to evaluate the safety and efficacy of ipilimumab added to this combination and explore predictive biomarkers (ClinicalTrials.gov identifier NCT01331525).
Patients and Methods
Patient Population
The patients were men and women aged 18 and older who had a histological or cytological diagnosis of SCLC; no previous systemic therapy for SCLC; an Eastern Cooperative Oncology Group performance status of 0 or 1; adequate baseline laboratory test results; and no active or chronic infection with human immunodeficiency virus, hepatitis B, or hepatitis C. Exclusion criteria included limited-stage SCLC appropriate for radical treatment with chemoirradiation, symptomatic central nervous system metastases, or autoimmune disease; a history of live vaccines (for up to 1 month before or after any dose of ipilimumab); and a history of prior treatment with a CD137 agonist or CTLA-4 inhibitor or agonist and concomitant therapy with any of the following: interleukin-2, interferon, or other immunotherapy regimens; immunosuppressive agents; other investigational therapies; and chronic use of systemic corticosteroids.
Study Design and Treatment Plan
This single-stage nonrandomized phase II study examined the efficacy and toxicity of ipilimumab (10 mg/kg) together with carboplatin (area under the curve = 6, intravenously [IV] on day 1) and etoposide (120 mg/m2 IV on day 1 and 100 mg twice a day orally on days 2 and 3 every 21 days) (ICE). Patients could enroll in the trial at any point until cycle 3. Patients received carboplatin and etoposide up to six cycles. Chemotherapy was discontinued in the event of progressive disease (PD) (according to the Response Evaluation Criteria in Solid Tumors [RECIST], version [v] 1.0) or excessive toxicity. Ipilimumab, 10 mg/kg, was given IV on day 1 of chemotherapy cycles 3 to 6 and then once every 12 weeks from week 30 until immune-related disease progression or excessive toxicity.
Patients could be offered prophylactic cranial irradiation (PCI) after completion of induction chemoimmunotherapy.
The trial was conducted in accordance with good clinical practice, and ethical approval was obtained (Multicenter Research Ethics Committee 10/H0502/95; International Standard Randomized Controlled Trials Number: 14095893); written informed consent was provided by all patients before enrollment.
Study Assessments
Tumor assessments were conducted by computerized tomography every 6 weeks for the first year (until week 54) and then every 12 weeks until disease progression by both RECIST, v 1.0, and immune response criteria (irRC).23 A baseline brain computerized tomography scan (not a magnetic resonance imaging scan) was performed for central nervous system disease evaluation if clinically indicated.
Patients who received at least one dose of ipilimumab were considered evaluable for safety and assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), v. 4.0 (http://ctep.cancer.gov). As no safety data were available for the combination, a planned interim safety monitoring assessment was performed. Once nine patients had been treated with the combination for at least 6 weeks, a first clinical safety assessment was performed to identify any early safety signals from ipilimumab given in combination with carboplatin and etoposide. In addition, a review of safety was triggered throughout the trial if a grade 3 (G3) or higher toxicity thought to be related to the study drugs developed in at least 40% of the patients treated, if at least 10% of patients experienced an unexpected ipilimumab-related G3 or higher toxicity that could not be alleviated or controlled by appropriate care and/or steroid and/or infliximab therapy within 14 days of the initiation of such therapy, or in response to any ipilimumab-related deaths unless attributed to disease progression.
Data on adverse events (AEs) and immune-related AEs (irAEs) were collected at each study visit and until 90 days after the last dose of ipilimumab; irAE was defined as an AE that was treatment related and considered to be immune mediated.
All irAEs were managed according to international guidelines and package inserts/product labels. No dose reductions were allowed for ipilimumab. Dose modifications for carboplatin and etoposide were made according to local practice.
Biomarker Assessment
Detection of autoantibodies was performed at baseline and during follow-up in cases where clinically indicated. Anti-VGCC and anti-VGKC antibodies were determined with radioimmunoprecipitation assays.24, 25 Antibodies against intracellular neuronal antigens were detected using indirect immunohistochemistry on primate cerebellum (NOVA Lite, Inova, Werfen, Warrginton, UK), immunoblotting (Ravo PNS Blot, Rravo Diagnostika, Freiberg, Germany), and a semiautomated enzyme-linked immunosorbent assay.26 Interpretation was done according to protocol instructions.
Statistical Analysis
The sample size was based on A’Hern’s single-stage phase II design, with a two-sided significance level of 0.05, 80% power, p0 (clinically uninteresting true progression-free survival [PFS] according to RECIST, v 1.0) = 10%, and p1 (sufficiently promising true PFS according to RECIST v 1.0) = 25%. The design required recruiting 40 evaluable patients, and the efficacy of the treatment was considered worth developing further if eight or more patients were alive and progression-free at 1 year.
The intention-to-treat population consisted of all registered patients. Toxicity was assessed using the safety population, which excluded patients who did not receive any ipilimumab. Baseline, treatment and efficacy information was performed on the efficacy analysis population, which consisted of all eligible patients included in the safety population.
Outcome Analysis
The primary end point was 1-year PFS according to RECIST v 1.0. PFS was defined as the time from day 1 of the first cycle of chemotherapy to the date of progression or death from any cause.
Secondary end points included PFS; PFS by irRC (irPFS); overall survival (OS) defined as the time from the date of day 1, cycle 1 of chemotherapy to the date of death from any cause; best overall response defined as the maximum response by RECIST v 1.0 compared with the baseline scan at study entry; duration of response defined as the time from first response by RECIST v 1.0 to disease progression or death from any cause; duration of response by irRC; and toxicity assessment according to the National Cancer Institute Common Terminology Criteria for Adverse Events, v 4.0.
Patients who had not died or progressed were censored for survival end points at the last documented clinical review. Survival analyses were performed using Kaplan-Meier estimates, and 95% confidence intervals (CIs) for proportions were calculated using the Wilson interval as recommended for small n by Brown et al.27 Summary statistics and plots were used to examine other secondary end points and characterize response rates.
Immunological data were recorded for each patient. Ad hoc exploratory analysis was carried out to assess associations between antibody positivity and clinical outcomes (irRC, irPFS, OS, and toxicity occurrence).
Results
Patients and Treatment
Forty-two patients with no previous systemic therapy for SCLC were registered into this study between September 2011 and April 2014 at six sites in the United Kingdom (Fig. 1). Three patients withdrew from the trial before receiving ipilimumab, and atypical carcinoid was retrospectively diagnosed in one patient. Baseline demographics and disease characteristics are shown in Table 1 for the evaluable population (n = 38). Most patients were male (66%), with a performance status (PS) of 1 (66%) and involvement of the lung, lymph nodes, and liver. The presence of autoantibodies was investigated at baseline in 38 patients (Table 2). Seventeen patients (45%) had at least one confirmed positive autoimmune antibody at baseline.
Figure 1.

CONSORT diagram showing the disposition of patients in the ICE (ipilimumab, carboplatin, and etoposide) study. ITT, intention to treat.
Table 1.
Baseline Patient Demographics and Disease Characteristics of the Efficacy Population (n = 38)
| Demographic or Disease Characteristics | Value |
|---|---|
| Age, y | |
| Median | 63 |
| Range | 44–84 |
| Sex, % (n) | |
| Female | 13 (34.2) |
| Male | 25 (65.8) |
| ECOG PS, n (%) | |
| 0 | 11 (34.4) |
| 1 | 21 (65.6) |
| Missing | 6 |
| Index and nonindex lesions, n (%) | |
| Lung | 27 (71.1) |
| Lymph node | 27 (71.1) |
| Liver | 15 (39.5) |
| Bone | 3 (7.9) |
| CNS | 1 (2.6) |
| Effusion | 2 (5.3) |
| Soft tissue | 7 (18.4) |
| Other | 13 (34.2) |
| LDH (IU/L) | |
| Median | 398 |
| Range | 186–1252 |
| Missing | 4 |
| IgG (g/L) | |
| Median | 8.10 |
| Range | 0–18.00 |
| Not performed/missing | 3 |
| IgA(g/L) | |
| Median | 2.20 |
| Range | 0.70–4.20 |
| Not performed/missing | 4 |
| IgM(g/L) | 0.75 |
| Median | 0.20–2.60 |
| Range | 4 |
Note: Denominator is nonmissing data for the analysis population for each test performed.
ECOG PS, Eastern Cooperative Oncology Group performance status; CNS, central nervous system; LDH, lactate dehydrogenase; IgG, immunoglobulin G; IgA, immunoglobulin A; IgM, immunoglobulin M.
Table 2.
Autoantibody Analysis at Baseline in the Efficacy Population (n = 38)
| Autoantibody Assays | n (%) |
|---|---|
| Anti-SOX2 | |
| Positive | 9 (23.7) |
| Negative | 29 (76.3) |
| Not performed/missing | 0 |
| Anti-Hu | |
| Positive | 6 (15.8) |
| Negative | 32 (84.2) |
| Not performed/missing | 0 |
| Anti-Yo | |
| Positive | 2 (6.5) |
| Negative | 29 (93.5) |
| Not performed/missing | 7 |
| Anti-VGCCA | |
| Positive | 0 |
| Negative | 24 (100) |
| Not performed/missing | 14 |
| Anti-VGPCA | |
| Positive | 2 (8.3) |
| Negative | 22 (91.7) |
| Not performed/missing | 14 |
| Thyroid peroxidase | |
| Positive | 4 (16.0) |
| Negative | 21 (84.0) |
| Not performed/missing | 13 |
| Rheumatoid factors | |
| Positive | 3 (12.5) |
| Negative | 21 (87.5) |
| Not performed/missing | 14 |
| Antimuscle antibodies | |
| Positive | 0 |
| Negative | 33 (100) |
| Not performed/missing | 5 |
| ANA | |
| Positive | 10 (28.6) |
| Negative | 25 (71.4) |
| Not performed/missing | 3 |
| ANCA | |
| Positive | 2 (8.3) |
| Negative | 22 (91.7) |
| Not performed/missing | 14 |
Note: Denominator is nonmissing data for the analysis population for each test performed.
SOX2, SRY-box 2; anti-Hu, anti-human; anti-Yo, purkinje cell cytoplasmic antibody type 1; VGCCA, voltage-gated calcium channel antibody; VGPCA, anti-voltage gated potassium channel antibody; ANA, antinuclear antibody; ANCA, antineutrophil cytoplasmic antibody.
At the final database lock (November 3, 2015) after a minimum follow-up of 6.8 months (median 8.5 months) no patients were still receiving treatment.
The main reason for discontinuation of treatment was toxicity (10 of 39 [26%]).
Thirty-seven of 38 patients started ipilimumab treatment on the third cycle of chemotherapy. The median number of cycles of the combination treatment for the efficacy analysis population (n = 38) was six (range 3–6). Of these patients, 24 (63%) completed the chemoimmunotherapy phase. Twenty-three patients (61%) had at least one chemotherapy dose delayed and 15 (40%) had dose modifications. Fifteen patients (40%) had at least one dose of ipilimumab delayed and 13 (34%) missed at least one dose during the combination phase. The number of patients who received at least one maintenance dose of ipilimumab was nine (24%), and one patient received treatment for 78 weeks.
Nine patients (24%) received PCI and eight (21%) received radiotherapy to the chest.
Efficacy
Of the 38 patients (the efficacy analysis population), six (15.8% [95% CI: 7.4%–30.4%]) were progression-free at 1 year by RECIST. Median PFS was 6.9 months (95% CI: 5.5–7.9) (Fig. 2). Median irPFS was 7.3 months (95% CI: 5.5–8.8) with an irPFS at 1 year of 12.6% (95% CI: 4.0–26.3). Median OS was 17.0 months (95% CI: 7.9–24.3) (Fig. 3). Response information by RECIST and irRC was available for 29 and 33 patients, respectively, 21 of whom (72.4%) achieved an objective response according to RECIST and 28 of whom (84.8%) achieved an objective response according to irRC (Table 3). Supplementary Table 1 compares both patterns of response.
Figure 2.

Kaplan-Meier plots for progression-free survival (PFS) according to the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.0, (A) and immune-related response criteria (B) and according to autoantibody status at baseline (C). CI, confidence interval; irRC, immune-related response criteria; NR, not reached.
Figure 3.

Kaplan-Meier plots for overall survival (A) and according to autoantibody status at baseline (B). OS, overall survival; CI, confidence interval.
Table 3.
Best Overall Tumor Response in the Efficacy Population (n = 38)
| Tumor response | Value |
|---|---|
| RECIST version 1.0 | |
| Complete response | 1 (3.4%) |
| Partial response | 20 (69.0%) |
| Stable disease | 3 (10.3%) |
| Progressive disease | 5 (17.2%) |
| Not assessed/missing | 9 |
| Immune-related response criteria | |
| Complete response | 2 (6.1%) |
| Partial response | 26 (78.8%) |
| Stable disease | 5 (15.2%) |
| Progressive disease | 0 |
| Not assessed/missing | 5 |
RECIST, Response Evaluation Criteria in Solid Tumors.
Patients receiving PCI had a numerically superior OS (median OS 18.5 versus 12.3 months, respectively), but this difference did not reach statistical significance (p = 0.447).
Safety
All toxicities are listed in Supplementary Table 2. Table 4 summarizes the incidence of treatment related G3 or higher AEs for the safety analysis population (n = 39). All patients experienced at least one AE. At least one G3 or higher toxicity developed in 35 (90%); in 27 (69%) this toxicity was thought to be related to ipilimumab. Neurological AEs were reported for 19 patients (49%), although only four patients (10%) experienced five high-grade AEs and three (8%) of these were related to ipilimumab. Central neuropathies (described as mild encephalopathy and cerebellar syndromes mimicking PNS) developed in two patients and one had severe headaches with deterioration of performance status. No association was observed between the occurrence of neurological toxicity and PCI treatment.
Table 4.
Summary of Grade 3 or Higher Toxicities in Patients Receiving at Least One Cycle of Ipilimumab (n = 39)
| Toxicity | Total | Ipilimumaba | Carboplatina | Etoposidea |
|---|---|---|---|---|
| Patients with at least one grade 3 or higher AE | 35 (89.7%) | 27 (69.2%) | 25 (64.1%) | 25(64.1%) |
| Neurological | ||||
| Generalized muscle weakness | 1 (2.6%) | 1 (2.6%) | 0 | 0 |
| Headache | 1 (2.6%) | 1 (2.6%) | 0 | 0 |
| Agitation | 1 (2.6%) | 1 (2.6%) | 0 | 0 |
| Nervous system disorder | 1 (2.6%) | 1 (2.6%) | 0 | 0 |
| Central neuropathy | 1 (2.6%) | 1 (2.6%) | 0 | 0 |
| Other immune related | ||||
| ALT increase/transaminitis | 3 (7.7%) | 3 (7.7%) | 0 | 0 |
| Alkaline phosphatase increase | 3 (7.7%) | 3 (7.7%) | 0 | 0 |
| Autoimmune disorder | 2 (5.1%) | 2 (5.1%) | 0 | 0 |
| Colitisb/diarrhea | 19 (48.7%) | 19 (48.7%) | 6 (15.4%) | 7 (18%) |
| Hyperglycemia | 2 (5.1%) | 1 (2.6%) | 1 (2.6%) | 1 (2.6%) |
| Lymphocyte count decrease | 2 (5.1%) | 0 | 1 (2.6%) | 1 (2.6%) |
| Neutrophil count decrease | 9 (23.1%) | 2 (5.1%) | 8 (20.5%) | 8 (20.5%) |
| Rash | 1 (2.6%) | 1 (2.6%) | 0 | 0 |
| Thrombocytopenia | 2 (5.1%) | 1 (2.6%) | 2 (5.1%) | 2 (5.1%) |
| Other | ||||
| Anemia | 6 (15.4%) | 0 | 6 (15.4%) | 6 (15.4%) |
| Dyspnea | 3 (7.7%) | 1 (2.6%) | 1 (2.6%) | 1 (2.6%) |
| Fatigue | 3 (7.7%) | 1 (2.6%) | 2 (5.1%) | 2 (5.1%) |
| Febrile neutropenia | 3 (7.7%) | 0 | 2 (5.1%) | 2 (5.1%) |
| Hyponatremia | 3 (7.7%) | 0 | 1 (2.6%) | 0 |
| Infection | 11 (28.2%) | 3 (7.7%) | 7 (18%) | 7 (18%) |
| Sepsis | 4 (10.3%) | 2 (5.1%) | 2 (5.1%) | 2 (5.1%) |
| Thromboembolic event | 2 (5.1%) | 2 (5.1%) | 2 (5.1%) | 2 (5.1%) |
AE, adverse event; ALT, alanine transaminase.
Toxicities assessed by site principal investigator to be definitely, probably, or possibly related to the study drug.
One case of ileitis is included.
Other frequent AEs (probably irAEs) were diarrhea in 28 patients (72%) and skin rash in 20 patients (51%), respectively. For 18 patients (46%), treatment delays were associated with ipilimumab-related toxicity. Five deaths (13%) were reported to be related to ipilimumab. Two of the deaths (due to cardiac arrest and neutropenic sepsis) happened while the patients where receiving treatment or shortly thereafter, but the remaining three (due to pneumonia, autoimmune encephalitis, and sepsis) happened 4 to 5 months after the last treatment.
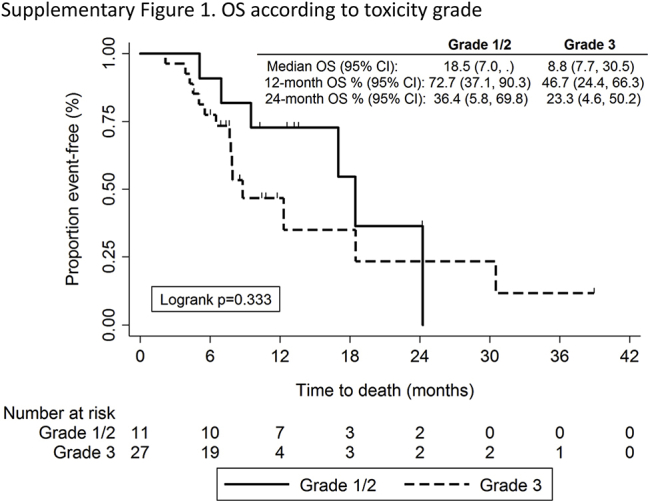
In an unplanned analysis, we evaluated whether severity of irAEs was associated with outcome. Patients who had more severe (≥G3) irAEs had numerically worse OS (Supplementary Fig. 1), but this was not statistically significant. Moreover, 73% of patients with grade 1 or 2 irAEs were alive at 1 year compared with 47% of patients with severe (≥G3) irAEs.
Autoantibodies as Predictive Biomarkers
In an ad hoc analysis, we explored the association between positivity of autoantibodies at baseline and clinical outcomes.
The most frequently detected antibodies were antinuclear antibodies in 10 patients and anti–SRY-box 2 (SOX2) in nine patients (Table 2). Twenty-three patients (60.5%) had at least one positive autoantibody detection. Antineuronal antibodies were more frequently positive (44.7%) than were the rest of the autoantibodies (31.6%). We assessed the association between autoantibodies and response (according to irRC). We found that none of 14 patients with positive antineuronal antibodies versus five of 19 patients with no positivity showed immune-related stable disease or immune-related PD (p = 0.049). Any autoantibody positivity showed a trend to association with response (p = 0.066).We then evaluated the association between autoantibody positivity and irPFS. We observed that patients with any positive autoantibody detected at baseline experienced a significantly longer median irPFS (8.8 months [95% CI: 5.1–10.7] versus 7.3 months [95% CI: 2.9–7.9, p = 0.036] (Fig. 2C). Antinuclear antibody positivity predicted for a significantly prolonged irPFS (10.2 months versus 6.9 months [p = 0.032]). Patients with any positive autoimmune antibody showed a trend to prolonged survival (18.5 months versus 17 months [p = 0.144]) (Fig. 3B).
We assessed the correlation between autoantibodies and toxicity. We found that three of 15 patients with positivity for SOX2 and/or anti-human antibodies presented with ipilimumab-related G3 or higher neurological toxicity compared with none of 23 patients with negativity for these antibodies (p = 0.054). One of these patients had more than one positive antineuronal autoantibody (anti-SOX2 and purkinje cell cytoplasmic antibody type 1).
Discussion
In our trial, we observed substantial excess toxicity from the combination ICE, which made the delivery of the chemoimmunotherapy and the maintenance ipilimumab challenging. Delays were frequent, as were interruptions of treatment due to toxicity.
Ipilimumab has a well-defined toxicity profile, and combination treatments have shown increased toxicity when compared with monotherapy. In the current study, the rate of G3 or higher toxicity is considerably higher (69%) (including five-related deaths). These figures are significantly higher than the toxicity reported in the randomized trial by Reck et al.,22 ranging from 43% to 50% (one toxic death in the concurrent arm). This increased toxicity might be explained by the better tolerance of the chemotherapy regimen used in that study and might also reflect excess toxicity from combining ipilimumab and etoposide. Combining a third drug (i.e., sunitinib or thalidomide) with the platinum and etoposide doublet in advanced SCLC has been challenging owing to increased toxic death rates,6, 7 and protocols have been amended to pursue a maintenance strategy.6 Moreover, the dose used in this study (10 mg/kg) was higher than the dose currently approved for melanoma (3 mg/kg), and data suggest increased toxicity with higher doses.28 Therefore, using ipilimumab at 3 mg/kg might be more appropriate in combination as well as in a sequential approach of immunotherapy after chemotherapy. Newer agents, such as anti–programmed cell death-1 (anti–PD-1)/anti–programmed death ligand-1 drugs, with a more favorable toxicity profile might be easier to combine with chemotherapy. Moreover, ipilimumab in combination with the anti–PD-1 agent nivolumab seems to have an acceptable toxicity profile and added clinical benefit in early-phase testing in patients with SCLC.29
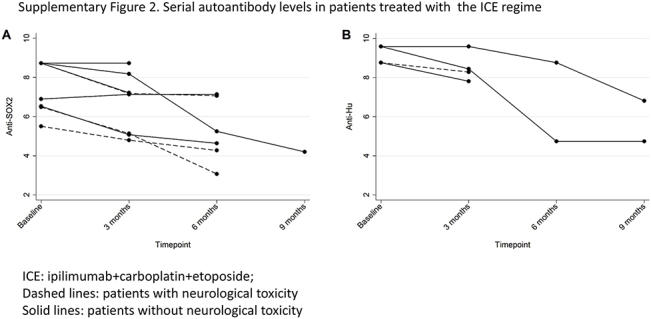
In our study, the rate of G3 or higher ipilimumab-related neurological toxicity was 7.6%. A comprehensive study of the prevalence of neurological PNS in a similar population of patients with SCLC observed that 9.1% had a PNS by clinical evaluation,30 with most of them (83%) having symptoms preceding the diagnosis of SCLC. Patients with clinical evidence of autoimmunity were however excluded from our study. As neurological toxicities developed after treatment initiation, they are most likely treatment related. Autoimmunity to the intracellular antigens SOX2 and Hu has been associated with PNS in several publications.30, 31, 32, 33 Our exploratory analysis revealed an association between anti-SOX2 and anti-Hu autoantibodies and severe neurological toxicities. Among patients with anti-SOX2 or anti-Hu antibodies at baseline we could not find differences (in antibody titers or subsequent antibody levels [Supplementary Fig. 2]) between those in whom neurological syndromes did and did not develop. The absence of antineuronal autoantibodies at diagnosis might therefore reflect a decreased likelihood of development of severe neurological toxicities triggered by ipilimumab. This suggests that careful monitoring of neurological symptoms in patients with antineuronal autoantibodies at baseline is important if immunotherapy is chosen as a strategy. We recognize that these findings need further validation and may additionally reflect the particular method of action of ipilimumab.
Our study is not randomized and therefore we cannot rule out that the neurological syndromes we clinically attributed to ipilimumab might have happened regardless of treatment with this drug. In two of the three patients with severe neurological toxicity mimicking PNS, the onset of the neurological syndrome preceded and perhaps therefore heralded disease progression. In the remaining case the progression was observed before the PNS. Thus, it remains possible that in spite of the absence of PNS at primary diagnosis, the neurological syndrome after treatment was caused by progression-related cross-reactive immune responses.
Markers of the function of regulatory T cells (Tregs) are lower in patients with autoantibodies and concomitant PNS as compared with in those with no neurological syndromes.34 Tregs express high levels of CTLA-4 and are down-regulated or removed by ipilimumab, and this is a desirable effect to enhance immune response against the tumor.35, 36 Our data are consistent with the hypothesis that downregulation of Tregs in patients with antineuronal autoantibodies by ipilimumab could promote development of autoimmune PNS.
The primary end point of the study was not met. Median PFS was 6.9 months. Interestingly, although irPFS seems to better reflect the efficacy of immunotherapeutic agents, in our study both parameters gave similar results (median irPFS 7.3 months). These results are consistent with the 6.4-month median irPFS observed in the phased ipilimumab arm in the study of Reck et al.22 Four patients with PD according to RECIST criteria were classified as being responders or having stable disease according to irRC. In cases involving other tumor types, patients with RECIST-defined PD but irRC-defined response or stable disease seem to have a better outcome than do those with PD according to both parameters.37 There is no previous assessment of this question in SCLC. Because of the low numbers, we were not able to compare survival of these patients with that of the RECIST responders.
A key secondary end point was OS. Although this study involved a relatively small cohort and it cannot be directly compared with other studies, the median OS of 17 months exceeds the OS reported in other recent trials in this setting6, 22 which is approximately 14 months. Interestingly, this happened despite the low rate of patients receiving PCI or thoracic radiotherapy (24%). Fifty-six percent of the patients were alive at 1 year, 29% were alive at 2 years, and almost 10% were alive at 3 years. This is consistent with findings in other studies in which improved OS is the key benefit from ipilimumab.19 More definitive data about the potential benefit of this combination will be available from the completed randomized trial (NCT 01450761).
To investigate potential biomarkers of benefit, we evaluated the association between autoimmunity and outcomes. We observed that a positive autoimmune profile at baseline predicted better response, irPFS with a trend to increased survival. The presence of autoantibodies at baseline has been linked to prognosis in this disease with conflicting results.31, 33, 38, 39, 40 Overall, there is evidence of patients benefiting from naturally occurring tumor immunity, with improved responses to tumor treatment or, in rare cases, complete eradication of tumor without tumor treatment.41, 42, 43
This would be consistent with our results suggesting that a preexisting immune response enhanced by ipilimumab could result in a beneficial effect from this drug. Although interesting, these results are hypothesis generating and need further validation. Moreover, the lack of a control chemotherapy-only arm precludes us from demonstrating a predictive versus a merely prognostic role.
In conclusion, ICE as first-line treatment for SCLC shows beneficial effects, particularly in patients with preexisting autoimmunity. However, toxicity was significant, suggesting that sequential immunotherapy after chemotherapy might be a more feasible approach, maybe in combination with other immune modulators such as PD-1 or programmed death ligand-1 inhibitors. More work is needed to demonstrate whether autoantibodies can serve as biomarkers for toxicity.
Acknowledgments
This study was funded by Bristol-Myers Squibb and Cancer Research UK (grant number C491/A12135). Dr. Arriola is supported by grant PI13/00140/FEDER. Drs. Ottensmeier, Wheater, and Nolan contributed to the study design. Dr. Ottensmeier, Dr. Galea, Dr. Woll, Mrs. Cross, Mr. Maishman, Mrs. Stanton, Mrs. Hamid, Dr. Cave, Dr. Geldart, Dr. Mulatero, Dr. Potter, Dr. Danson, Dr. Griffiths, and Dr. Nolan contributed to data collection. Dr. Arriola, Dr. Galea, Mr. Maishman, Mrs. Hamid, Mrs. Cross, Mrs. Stanton, Dr. Nolan, and Dr. Ottensmeier contributed to data analysis and interpretation. Dr. Arriola, Dr. Wheater, Dr. Galea, Mrs. Cross, Mr. Maishman, Mrs. Hamid, Mrs. Stanton, Dr. Cave, Dr. Geldart, Dr. Mulatero, Dr. Potter, Dr. Danson, Dr. Woll, Dr. Griffiths, Dr. Nolan, and Dr. Ottensmeier contributed to manuscript writing and review.
Footnotes
Disclosure: Dr. Arriola declares nonfinancial support from AstraZeneca and personal fees from Lilly outside the submitted work. Dr. Mulatero reports personal fees from Clovis Oncology, PRMA consulting, Novartis, Lilly, Pfizer, GlaxoSmithKline, Ernst and Young, Amgen, and Astra Zeneca; personal fees, nonfinancial support, and research funding from Boehringer Ingelheim; and research funding and nonfinancial support from Bristol-Myers Squibb, Pierre Fabre, Merck Sharp and Dome, and Roche outside the submitted work. Dr. Danson reports research funding from Astex Pharmaceuticals, Lilly, Plexicon, Bayer, Synta, Amgen, Boehringer Ingelheim, and GlaxoSmithKline; nonfinancial support and other from Bristol-Myers Squibb; and nonfinancial support from Merck Sharp and Dohme outside the submitted work. Dr. Geldart declares personal fees from Bristol-Myers Squibb and Pfizer outside the submitted work. Dr. Wheater declares personal fees and nonfinancial support from Bristol-Myers Squibb outside the submitted work. Dr. Griffiths declares personal fees from Bristol-Myers Squibb outside the submitted work. Dr. Cave reports nonfinancial support from Novartis, Boehringer Ingelheim, and Lilly outside the submitted work. Dr. Woll reports nonfinancial support from Bristol-Myers Squibb outside the submitted work. Dr. Nolan declares personal fees from Bristol-Myers Squibb and Merck outside the submitted work. Dr. Ottensmeier reports grants from Bristol Myers Squibb during the conduct of the study; he also reports personal fees from Transgene, Bristol Myers Squibb, Immatics, and Merck; research funding from Inovio; personal fees, nonfinancial support, and other from Bristol-Myers Squibb and Merck Sharp and Dohme; other from Verastem, Biontech AG, Serametrix, and Touchlight Genetics; and personal fees and nonfinancial support from Roche outside the submitted work. The remaining authors declare no conflict of interest.
Note: To access the supplementary material accompanying this article, visit the online version of the Journal of Thoracic Oncology at www.jto.org and at http://dx.doi.org/10.1016/j.jtho.2016.05.028.
Supplementary Data
Supplementary Figure 1.
Supplementary Figure 2.
References
- 1.Oze I., Hotta K., Kiura K. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: disappointing results. PloS One. 2009;4:e7835. doi: 10.1371/journal.pone.0007835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slotman B.J., van Tinteren H., Praag J.O. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet. 2015;385:36–42. doi: 10.1016/S0140-6736(14)61085-0. [DOI] [PubMed] [Google Scholar]
- 3.Ettinger D.S., Finkelstein D.M., Abeloff M.D. A randomized comparison of standard chemotherapy versus alternating chemotherapy and maintenance versus no maintenance therapy for extensive-stage small-cell lung cancer: a phase III study of the Eastern Cooperative Oncology Group. J Clin Oncol. 1990;8:230–240. doi: 10.1200/JCO.1990.8.2.230. [DOI] [PubMed] [Google Scholar]
- 4.Arnold A.M., Seymour L., Smylie M. Phase II study of vandetanib or placebo in small-cell lung cancer patients after complete or partial response to induction chemotherapy with or without radiation therapy: National Cancer Institute of Canada Clinical Trials Group Study BR.20. J Clin Oncol. 2007;25:4278–4284. doi: 10.1200/JCO.2007.12.3083. [DOI] [PubMed] [Google Scholar]
- 5.Pujol J.L., Breton J.L., Gervais R. Phase III double-blind, placebo-controlled study of thalidomide in extensive-disease small-cell lung cancer after response to chemotherapy: an intergroup study FNCLCC cleo04 IFCT 00-01. J Clin Oncol. 2007;25:3945–3951. doi: 10.1200/JCO.2007.11.8109. [DOI] [PubMed] [Google Scholar]
- 6.Ready N.E., Pang H.H., Gu L. Chemotherapy with or without maintenance sunitinib for untreated extensive-stage small-cell lung cancer: a randomized, double-blind, placebo-controlled phase II study-CALGB 30504 (Alliance) J Clin Oncol. 2015;33:1660–1665. doi: 10.1200/JCO.2014.57.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee S.M., Woll P.J., Rudd R. Anti-angiogenic therapy using thalidomide combined with chemotherapy in small cell lung cancer: a randomized, double-blind, placebo-controlled trial. J Natl Cancer Inst. 2009;101:1049–1057. doi: 10.1093/jnci/djp200. [DOI] [PubMed] [Google Scholar]
- 8.Schadendorf D., Hodi F.S., Robert C. Pooled analysis of long-term survival data from phase II and phase III trials of Ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Postow M.A., Chesney J., Pavlick A.C. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garon E.B., Rizvi N.A., Hui R. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 11.Borghaei H., Paz-Ares L., Horn L. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darnell R.B., Posner J.B. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543–1554. doi: 10.1056/NEJMra023009. [DOI] [PubMed] [Google Scholar]
- 13.Roberts W.K., Deluca I.J., Thomas A. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuD-specific type 2 CD8+ T cells. J Clin Invest. 2009;119:2042–2051. doi: 10.1172/JCI36131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maddison P., Newsom-Davis J., Mills K.R. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet. 1999;353:117–118. doi: 10.1016/S0140-6736(05)76153-5. [DOI] [PubMed] [Google Scholar]
- 15.Wirtz P.W., Lang B., Graus F. P/Q-type calcium channel antibodies, Lambert-Eaton myasthenic syndrome and survival in small cell lung cancer. J Neuroimmunol. 2005;164:161–165. doi: 10.1016/j.jneuroim.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Pignolet B.S., Gebauer C.M., Liblau R.S. Immunopathogenesis of paraneoplastic neurological syndromes associated with anti-Hu antibodies: a beneficial antitumor immune response going awry. Oncoimmunology. 2013;2:e27384. doi: 10.4161/onci.27384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson C.B., Allison J.P. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997;7:445–450. doi: 10.1016/s1074-7613(00)80366-0. [DOI] [PubMed] [Google Scholar]
- 18.Romano E., Kusio-Kobialka M., Foukas P.G. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodi F.S., O'Day S.J., McDermott D.F. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maio M., Grob J.J., Aamdal S. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol. 2015;33:1191–1196. doi: 10.1200/JCO.2014.56.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y., Kepp O., Ghiringhelli F. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol. 2010;22:113–124. doi: 10.1016/j.smim.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Reck M., Bondarenko I., Luft A. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24:75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 23.Wolchok J.D., Hoos A., O'Day S. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 24.Mason W.P., Graus F., Lang B. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120:1279–1300. doi: 10.1093/brain/120.8.1279. [DOI] [PubMed] [Google Scholar]
- 25.Shillito P., Molenaar P.C., Vincent A. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol. 1995;38:714–722. doi: 10.1002/ana.410380505. [DOI] [PubMed] [Google Scholar]
- 26.Chapman C.J., Thorpe A.J., Murray A. Immunobiomarkers in small cell lung cancer: potential early cancer signals. Clin Cancer Res. 2011;17:1474–1480. doi: 10.1158/1078-0432.CCR-10-1363. [DOI] [PubMed] [Google Scholar]
- 27.Brown L.D., Cai T.T., DasGupta A. Interval estimation for a binomial proportion. Statistical Science. 2001;16:101–117. [Google Scholar]
- 28.Wolchok J.D., Neyns B., Linette G. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 29.Antonia S.J., Bendel J.C., Taylor M.H. Phase I/II study of nivolumab with or without ipilimumab for treatment of recurrent small cell lung cancer (SCLC) J Clin Oncol. 2015;33(suppl):7503. [abstract] [Google Scholar]
- 30.Gozzard P., Woodhall M., Chapman C. Paraneoplastic neurologic disorders in small cell lung carcinoma: a prospective study. Neurology. 2015;85:235–239. doi: 10.1212/WNL.0000000000001721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Titulaer M.J., Klooster R., Potman M. SOX antibodies in small-cell lung cancer and Lambert-Eaton myasthenic syndrome: frequency and relation with survival. J Clin Oncol. 2009;27:4260–4267. doi: 10.1200/JCO.2008.20.6169. [DOI] [PubMed] [Google Scholar]
- 32.Graus F., Keime-Guibert F., Rene R. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138–1148. doi: 10.1093/brain/124.6.1138. [DOI] [PubMed] [Google Scholar]
- 33.Maddison P., Thorpe A., Silcocks P. Autoimmunity to SOX2, clinical phenotype and survival in patients with small-cell lung cancer. Lung Cancer. 2010;70:335–339. doi: 10.1016/j.lungcan.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Tani T., Tanaka K., Idezuka J. Regulatory T cells in paraneoplastic neurological syndromes. J Neuroimmunol. 2008;196:166–169. doi: 10.1016/j.jneuroim.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Read S., Greenwald R., Izcue A. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006;177:4376–4383. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selby M.J., Engelhardt J.J., Quigley M. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 37.Chiou V.L., Burotto M. Pseudoprogression and immune-related response in solid tumors. J Clin Oncol. 2015;33:3541–3543. doi: 10.1200/JCO.2015.61.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monstad S.E., Drivsholm L., Storstein A. Hu and voltage-gated calcium channel (VGCC) antibodies related to the prognosis of small-cell lung cancer. J Clin Oncol. 2004;22:795–800. doi: 10.1200/JCO.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 39.Gozzard P., Chapman C., Vincent A. Novel humoral prognostic markers in small-cell lung carcinoma: a prospective study. PloS One. 2015;10:e0143558. doi: 10.1371/journal.pone.0143558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vural B., Chen L.C., Saip P. Frequency of SOX Group B (SOX1, 2, 3) and ZIC2 antibodies in Turkish patients with small cell lung carcinoma and their correlation with clinical parameters. Cancer. 2005;103:2575–2583. doi: 10.1002/cncr.21088. [DOI] [PubMed] [Google Scholar]
- 41.Darnell R.B., DeAngelis L.M. Regression of small-cell lung carcinoma in patients with paraneoplastic neuronal antibodies. Lancet. 1993;341:21–22. doi: 10.1016/0140-6736(93)92485-c. [DOI] [PubMed] [Google Scholar]
- 42.Darnell R.B., Posner J.B. Observing the invisible: successful tumor immunity in humans. Nat Immunol. 2003;4:201. doi: 10.1038/ni0303-201. [DOI] [PubMed] [Google Scholar]
- 43.Graus F., Dalmou J., Rene R. Anti-Hu antibodies in patients with small-cell lung cancer: association with complete response to therapy and improved survival. J Clinical Oncol. 1997;15:2866–2872. doi: 10.1200/JCO.1997.15.8.2866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.