

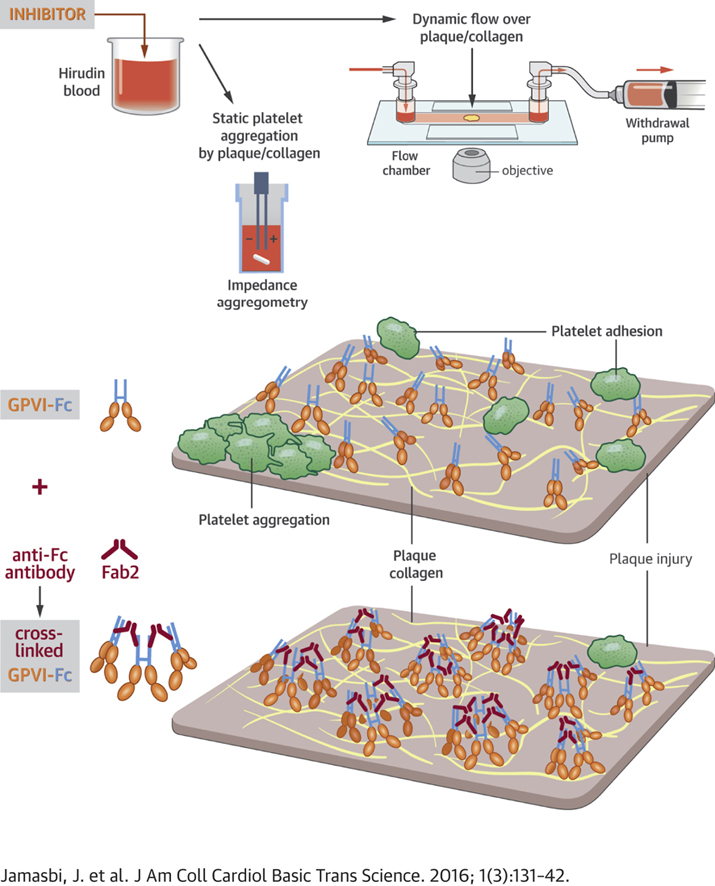
Summary
To enhance the antithrombotic properties of recombinant glycoprotein VI fragment crystallizable (GPVI-Fc), the authors incubated GPVI-Fc with anti-human Fc antibodies to cross-link the Fc tails of GPVI-Fc. Cross-linking potentiated the inhibition of human plaque- and collagen-induced platelet aggregation by GPVI-Fc under static and flow conditions without increasing bleeding time in vitro. Cross-linking with anti-human-Fc Fab2 was even superior to anti-human-Fc immunoglobulin G (IgG). Advanced optical imaging revealed a continuous sheath-like coverage of collagen fibers by cross-linked GPVI-Fc complexes. Cross-linking of GPVI into oligomeric complexes provides a new, highly effective, and probably safe antithrombotic treatment as it suppresses platelet GPVI-plaque interaction selectively at the site of acute atherothrombosis.
Key Words: antithrombotic, atherothrombosis, glycoprotein VI, plaque rupture
Abbreviations and Acronyms: Fc, fragment crystallizable; GPVI, glycoprotein VI; IgG, immunoglobulin G; PE, phycoerythrin; SIM, structured illumination microscopy; STED, stimulated emission depletion; XL, cross-linked
Visual Abstract
Highlights
-
•
The GPVI collagen receptor mediates platelet activation to collagen exposed after rupture or injury of atherosclerotic plaques. Revacept®, a GPVI-Fc fusion protein binding to plaque collagen and concealing binding sites for platelet GPVI, acts as a lesion-focused antiplatelet drug. To optimize its specific inhibition of plaque-induced platelet activation, we cross-linked the Fc tails of GPVI-Fc with anti-human-Fc IgG or Fab2 antibodies.
-
•
Cross-linking yielded oligomeric GPVI-Fc complexes, which inhibited atherosclerotic plaque- induced platelet aggregation in static and flow assays as efficiently as antibodies blocking GPVI receptors on platelets. Under arterial flow, GPVI-Fc cross-linking with anti-Fc-Fab2 was superior to cross-linking with anti-Fc IgG.
-
•
Advanced optical imaging revealed a rapid and stable sheath-like coverage of collagen fibers by cross-linked GPVI-Fc complexes preventing platelet attachment to collagen.
-
•
Cross-linked GPVI-Fc did not increase bleeding time in vitro.
-
•
Transformation of GPVI into multivalent GPVI complexes may provide a new superior strategy to suppress atherothrombosis without increasing systemic bleeding risk.
Erosion or rupture of atherosclerotic plaques exposes material to circulating blood that triggers thrombosis and can precipitate myocardial infarction and stroke, which are leading causes of death worldwide 1, 2. Decisive thrombogenic plaque components include collagen type I and III fibers, which trigger platelet deposition under static and flow conditions by activating the platelet glycoprotein VI (GPVI) receptor 3, 4, 5, 6. The alternative collagen receptor, α2β1-integrin, is not involved in plaque-induced platelet aggregation 4, 6. Therefore, targeting GPVI might preferentially inhibit atherosclerotic plaque–induced thrombosis and should not affect other cells because GPVI expression is restricted to platelets and megakaryocytes (7). Only the dimeric form of GPVI binds to collagen with high affinity 8, 9, recognizing mostly tandem glycine-proline-hydroxyproline motifs but also other peptide sequences of collagen fibers 10, 11.
The GPVI–collagen interaction can be inhibited either by occupation of GPVI-binding sites on collagen using a dimeric glycoprotein VI–fragment crystallizable (GPVI-Fc) fusion protein (Revacept; advanceCOR, Munich, Germany) or by antibodies directed against platelet GPVI 7, 12, 13, 14, 15. Anti-GPVI antibodies might increase bleeding, as observed in some patients with anti-GPVI autoantibodies 14, 15, whereas GPVI-Fc might be safer because it did not increase bleeding time in a Phase I study (13). Anti-GPVI antibodies are systemic and potent inhibitors of plaque- and collagen-induced platelet aggregation under static and flow conditions, whereas GPVI-Fc acts locally at the site of plaque rupture and is in vitro most effective under high shear flow (16). However, GPVI-Fc inhibition of plaque-induced platelet aggregation in static assays and at low arterial shear flow is inferior to anti-GPVI antibodies (16). GPVI-Fc, even at very high concentrations, apparently cannot saturate all binding sites on plaque collagen. The goal of the present study, therefore, was to improve the efficacy of GPVI-Fc binding to exposed plaque collagen to combine its lesion selectivity with the full potential of GPVI antagonism as demonstrated by the anti-GPVI antibodies.
Methods
Carotid atherosclerotic plaque tissue donated by patients undergoing endarterectomy for high-grade carotid stenosis was processed, homogenized, and preserved as described elsewhere 5, 16, 17. Plaque homogenates from 5 patients were pooled, and aliquots were used to stimulate platelet aggregation under static conditions or were coated onto glass coverslips for flow studies 16, 18, 19. In addition to plaque, which contains mainly type I and III collagens 4, 6, blood was exposed to Horm collagen fibers also consisting of type I and III collagens (16).
Nanoscopic imaging of GPVI-Fc binding and platelet adhesion to collagen under flow was performed by structured illumination microscopy (SIM) (20) and stimulated emission depletion (STED) microscopy. Additional details are presented in the Supplemental Appendix.
Statistical analysis
The means of 2 parallel experimental conditions were compared by using the Student t test or, if normality was not assured, by using the Mann-Whitney U test as indicated in the figure legends. More than 2 concurrent experimental conditions using the same samples were tested by using repeated measures analysis of variance. If normality was not assured by repeated measures, analysis of variance on ranks was used followed by pair-wise comparisons with p values based on a Tukey adjustment for multiple testing.
Results
GPVI-Fc, even at very high concentrations, apparently cannot saturate all binding sites on plaque collagen and incompletely inhibits platelet aggregation (16). To gain further insights into the underlying mechanism, we studied the kinetics and spacial distribution at low shear flow (600/s) of how fluorescent GPVI-Fc added to blood accretes on collagen fibers and how this affects platelet adhesion, which precedes platelet aggregation. We noted that GPVI-Fc binding to collagen was very fast (Video 1), and dense GPVI-Fc dots could repel arrested platelets from stably adhering to collagen (Figure 1A). However, the platelets settled preferentially on segments of collagen fibers carrying little GPVI-Fc (Figures 1A and 1B), and they were even able to displace neighboring collagen-bound GPVI-Fc to firmly attach to these collagen segments. Use of SIM confirmed that platelets stably adhered to the segments of collagen fibers, which carried little GPVI-Fc (Figure 1C, Video 2).
Figure 1.
Dynamics of GPVI-Fc Binding and Platelet Adhesion to Collagen Under Flow
Glycoprotein VI–fragment crystallizable (GPVI-Fc) (50 μg/ml final concentration; 333 nM) labeled with phycoerythrin (PE)-conjugated anti–human-Fc antibody (red) was added to blood containing abciximab (to inhibit platelet aggregation and allow only platelet adhesion) before perfusion over collagen (550/s). Differential interference contrast (DIC) and fluorescence images were taken by video microscopy (1 frame/2 s) using a 100× NA1.4 oil objective. Images are representative for 6 individual experiments. Upper rows: overlay of DIC (collagen/platelets) and fluorescence (PE-labeled GPVI-Fc) images. Bottom rows: fluorescence images of PE-labeled GPVI-Fc. See also Video 1. (A) A single platelet (thick arrow) rolls over PE-labeled GPVI-Fc (thin arrows in row below) bound to collagen. Two platelets (asterisks) adhere to segments of collagen fibers that contain less fluorescent GPVI-Fc. (B) A single platelet (arrow, top row) attaching downstream of the fiber and moving back against the blood flow displaces GPVI-Fc-PE from collagen (arrows in bottom row). (C) Structured illumination microscopy imaging. Platelets adhere to collagen segments carrying little GPVI-Fc. Collagen coated onto glass coverslips was stained with anti–collagen type I and type III antibody (Ab) and AlexaFluor405-conjugated secondary Ab (blue). PE-labeled GPVI-Fc (50 μg/ml) was added to blood containing Arg-Gly-Asp-Ser (to inhibit platelet aggregation and allow only platelet adhesion) before the start of perfusion (shear rate 550/s). After 4 min of flow, platelets were fixed and stained with anti-CD41 Ab and DyLight 488-conjugated secondary Ab (green). The fluorescence micrograph shows a maximum intensity projection of 0.15 μm z sections (total z 2.5 μm) of a structured illumination microscopy image (ELYRA PS.1; Carl Zeiss MicroImaging GmbH, Jena, Germany). i and ii, Identical pictures. i, Binding of GPVI-Fc (red) onto collagen fibers (blue). Green channel (platelets) was omitted. ii, Picture as in i but with platelets (green). Image is representative of 5 others from different experiments. Video 2 presents a 3-dimensional representation.

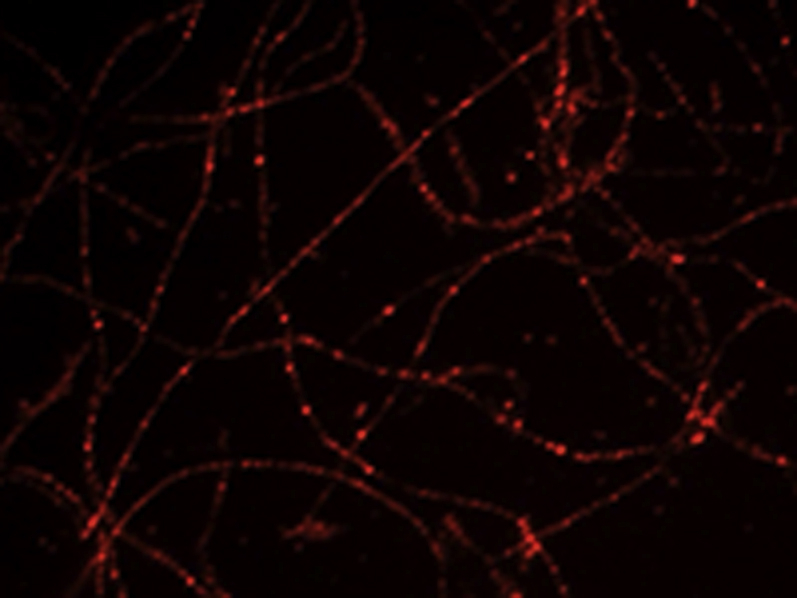
GPVI-Fc binding to collagen
GPVI-Fc (50μg/ml final concentration; 333nM) labeled with PE-conjugated anti-human-Fc antibody (red) was added to blood containing abciximab before perfusion over collagen (600/s). PE-labeled GPVI-Fc was detected by fluorescence microscopy (excitation 560nm, emission 605nm) using a 100x NA 1.4 oil objective. Flow direction: from right to left.
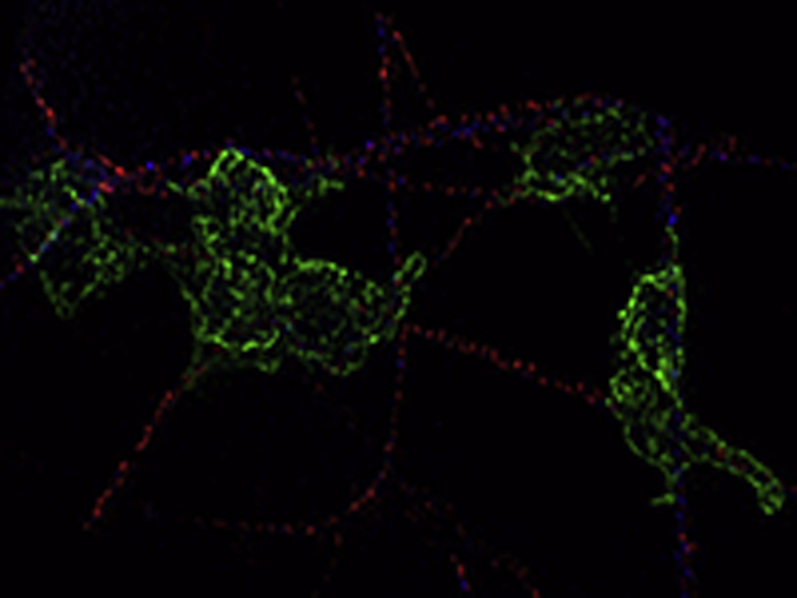
Platelets adhere mainly to collagen segments carrying little GPVI-Fc
Three-dimensional animation of the SIM image in Fig. 1 C processed with Huygens software. Platelet attachment (green) to collagen (blue) in the presence of GPVI-Fc (red).
We therefore aimed to modify GPVI-Fc in such a way that it would assure a more continuous binding to collagen and resist displacement by platelets. We hypothesized that anti–Fc-mediated cross-linking of the Fc tails would more closely align GPVI-Fc molecules to their corresponding binding motifs on collagen. We thus pre-incubated anti–human-Fc immunoglobulin G (IgG) and Fab2 antibodies at equimolar concentrations with GPVI-Fc to allow the formation of cross-linked (XL) complexes (GPVI-Fc*IgG-XL and GPVI-Fc*Fab2-XL). Pre-incubation of blood with GPVI-Fc*IgG-XL or GPVI-Fc*Fab2-XL inhibited static platelet aggregation stimulated by plaque or collagen more effectively than GPVI-Fc alone (Figure 2A). For plaque-stimulated samples, inhibition by GPVI-Fc*IgG-XL was 72 ± 12% compared with 35 ± 13% by GPVI-Fc, and inhibition by GPVI-Fc*Fab2-XL was 84 ± 10% compared with 40 ± 13% by GPVI-Fc. For collagen-stimulated samples, inhibition by GPVI-Fc*IgG-XL was 60 ± 17% compared with 20 ± 17% by GPVI-Fc, and inhibition by GPVI-Fc*Fab2-XL was 67 ± 21% compared with 18 ± 9% by GPVI-Fc. Bare Fc protein cross-linked with anti–human-Fc IgG or anti–human-Fc Fab2 (Fc*IgG-XL; Fc*Fab2-XL) or IgG or Fab2 alone did not inhibit platelet aggregation. Concentration–response curves of GPVI-Fc*Fab2-XL showed superior inhibition by GPVI-Fc*Fab2-XL at all concentrations starting with 33 nM for plaque- and collagen-stimulated samples (Figure 2B). These concentrations are well below the maximal plasma GPVI-Fc concentrations (50 μg/ml = 333 nM) that were reached after intravenous application in a previous Phase I clinical study (13).
Figure 2.

Cross-Linking of GPVI-Fc With Anti–Human-Fc IgG and Anti–Human-Fc Fab2 Increases Inhibition of Static Platelet Aggregation Induced by Collagen or Plaque Compared With GPVI-Fc
(A) Bar diagrams show the effects of glycoprotein VI–fragment crystallizable (GPVI-Fc) (20 μg/ml; 133 nM), GPVI-Fc, and Fc cross-linked (XL) with anti–human-Fc immunoglobulin G (IgG) (GPVI-Fc*IgG-XL, Fc*IgG-XL; 133 nM) (left panels) or with anti–human-Fc Fab2 (GPVI-Fc*Fab2-XL, Fc*Fab2-XL; 133 nM) (right panels) on plaque- or collagen-induced platelet aggregation. Further controls were anti–human-Fc IgG (133 nM) (left panels) and anti–human-Fc Fab2 (133 nM) (right panels). Cumulative platelet aggregation (AU*min) was measured by using multiple electrode aggregometry. Mean ± SD; n = 4. Repeated measures analysis of variance (overall p < 0.001) with secondary pair-wise comparisons (as indicated by bars) by Tukey correction. (B) Concentration dependency of the effects of GPVI-Fc*Fab2-XL on plaque- and collagen-induced platelet aggregation compared with GPVI-Fc. Mean ± SD; n = 4 to 6. Comparisons were made by using the Student t test or, if inappropriate, by using the Mann-Whitney U test (at 333 nM with plaque and at 33 nM with collagen). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We then explored the effects of GPVI-Fc antibody cross-linking on plaque-induced platelet aggregation under flow. Coronary thrombosis mostly arises from rupture of thin-capped (<65 μm) fibroatheroma, exposing the plaque lipid core containing collagenous structures to circulating blood 6, 21, 22. This action creates a new thrombogenic and rough luminal surface that will influence platelet adhesion and aggregation dynamics probably similar to our plaque model. In our model, plaque fragments of different sizes and consisting of lipids and collagenous structures are exposed to arterially flowing blood 4, 16. The subtle (<8 μm) roughness of the plaque surface in our model created local differences in dynamics and extent of plaque-induced platelet aggregate formation at flow. For comparison, flow experiments using collagen-coated surfaces were also performed.
As depicted in the micrographs and diagrams in Figures 3A and 3B, GPVI-Fc*IgG-XL inhibited plaque-stimulated aggregate formation at any time after the start of flow much more efficiently than GPVI-Fc. With GPVI-Fc*Fab2-XL, the inhibition was even more pronounced, and GPVI-Fc*Fab2-XL virtually abolished platelet accretion on plaque material. Interestingly, inhibition of plaque-induced platelet aggregate formation by GPVI-Fc*IgG-XL and GPVI-Fc*Fab2-XL at the end of flow was more pronounced than after collagen stimulation.
Figure 3.
Cross-Linking of GPVI-Fc With Anti–Human-Fc IgG or Anti–Human-Fc Fab2 Increases Its Inhibition of Platelet Aggregation Stimulated by Collagen or Plaque Under Flow
Buffer (control), GPVI-Fc, or GPVI-Fc (50 μg/ml; 333 nM) premixed with equimolar anti–human-Fc IgG or anti–human-Fc Fab2 antibodies for crosslinking was added to blood containing DiOC6 for platelet visualization and perfused over plaque homogenate at a shear rate of 600/s. (A) Representative micrographs display platelet coverage of plaque at 2, 5, and 9 min after start of blood flow. (B) Effect of buffer, GPVI-Fc, GPVI-Fc*IgG-XL, or GPVI-Fc*Fab2-XL on the kinetics of platelet deposition from flowing blood onto plaque and collagen. Measurements are each second. Mean (solid line) ± SD (shaded area); n = 4 to 6. Comparison at 2, 5, and 9 min by using repeated measures analysis of variance or if inappropriate by repeated measures analysis of variance on ranks (only 2 min). Significance of secondary pair-wise comparisons by Tukey correction is indicated by bars. ∗p < 0.05. (C) Kinetics of not cross-linked and cross-linked GPVI-Fc binding to collagen. (Videos 3 and 4 present additional details.) GPVI-Fc pre-incubated with PE-labeled anti–human-Fc Fab2 (20:1 mol/mol) (GPVI-Fc*Fab2) or with an equimolar mixture of PE-labeled and unlabeled anti–human-Fc Fab2 (1:10) for cross-linking (GPVI-Fc*Fab2-XL), was added to blood (333 nM GPVI-Fc final concentration) containing abciximab and perfused over collagen at a shear rate of 600/s. Binding of PE-labeled GPVI-Fc (cross-linked or not cross-linked) to collagen was quantified every second by fluorescence microscopy using a 10× objective. Top: Time course of GPVI-Fc*Fab2 and GPVI-Fc*Fab2-XL binding to collagen (area coverage). Mean (solid line) ± SD (shaded area); n = 4. Bottom: Fluorescence intensity surface plots of GPVI-Fc*Fab2 and GPVI-Fc*Fab2-XL bound to collagen at 3 min after start of blood flow. Color gradients indicate increase of fluorescence intensity. Abbreviations as in Figures 1 and 2.


Kinetics of not cross-linked GPVI-Fc binding to collagen under flow
GPVI-Fc was incubated with PE-labeled anti-human-Fc Fab2 antibodies in a 20:1 molar ratio. The mixtures were added to blood (333nM GPVI-Fc f.c.) containing abciximab before perfusion over collagen at a shear rate of 600/sec for 3min. Binding of PE-labeled GPVI-Fc*Fab2 to collagen was recorded by fluorescence video microscopy (1frame/sec) using a 10x objective.
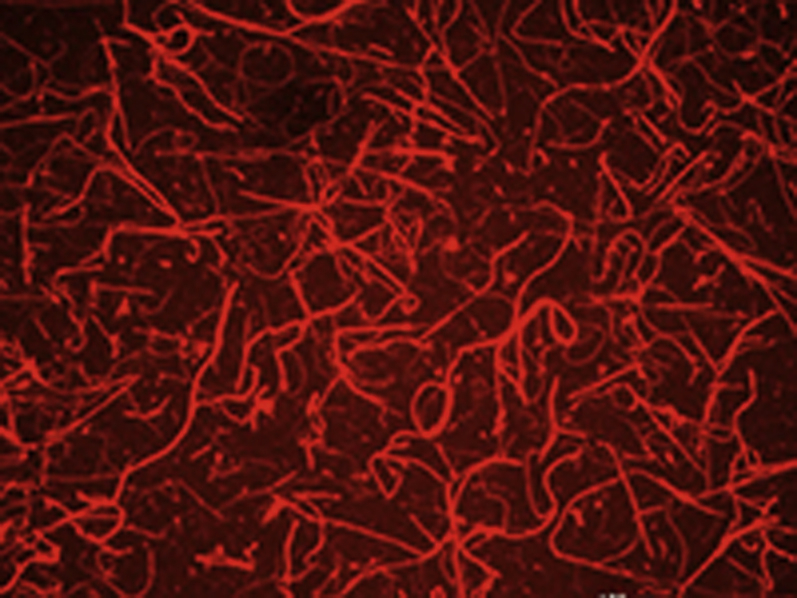
Kinetics of cross-linked GPVI-Fc binding to collagen under flow
GPVI-Fc was incubated with 10% PE-labeled and 90% unlabeled anti-human-Fc Fab2 antibodies in a 1:1 molar ratio for GPVI-Fc cross-linking. The mixtures were added to blood (333nM GPVI-Fc f.c.) containing abciximab before perfusion over collagen at a shear rate of 600/sec for 3min. Binding of PE-labeled GPVI-Fc*Fab2-XL to collagen was recorded by fluorescence video microscopy (1frame/sec) using a 10x objective.
We then compared the binding kinetics of labeled GPVI-Fc and cross-linked GPVI-Fc to collagen to explore whether a faster binding of cross-linked GPVI-Fc might explain its better inhibitory effect. Perfusion of collagen-coated coverslips with blood containing phycoerythrin (PE)-labeled cross-linked GPVI-Fc (GPVI-Fc*Fab2-XL) or PE-labeled GPVI-Fc (GPVI-Fc*Fab2) revealed that GPVI-Fc*Fab2-XL bound only slightly faster than GPVI-Fc*Fab2 (Figure 3C, Videos 3 and 4). Similar observations were made with GPVI-Fc*IgG and GPVI-Fc*IgG-XL (data not shown). As demonstrated by the fluorescence intensity surface plots, the final GPVI-Fc*Fab2-XL binding to collagen was much higher than that of GPVI-Fc*Fab2. The very rapid binding of cross-linked GPVI-Fc to collagen delayed and reduced the formation of collagen-induced platelet aggregate formation at 2 and 5 min after start of flow (Figures 3B and 3C).
As in the static assays, bare Fc protein cross-linked with anti–human-Fc IgG or anti–human-Fc Fab2 did not inhibit plaque- and collagen-stimulated platelet aggregation under flow (Supplemental Figure 1). We found that the ratio of GPVI-Fc to antibody was crucial for platelet inhibition. A 1:1 ratio of GPVI-Fc:anti–human-Fc Fab2 (presumably generating longer linear complexes) was more effective in inhibiting plaque-induced platelet aggregation in flowing blood than a ratio of 1:0.5 (Supplemental Figure 2). GPVI-Fc*Fab2-XL seemed to inhibit plaque-stimulated platelet aggregation under flow even better than anti-GPVI antibodies (Supplemental Figure 3).
The protein complexes formed by the interaction of GPVI-Fc with anti–human-Fc IgG or anti–human-Fc Fab2 antibodies were characterized by analytical ultracentrifugation (Supplemental Figure 4). This method provides hydrodynamic and thermodynamic information concerning the size, shape, and interactions of macromolecules. For not cross-linked and cross-linked complexes of GPVI-Fc with anti–human-Fc IgG, we found 3 peaks with similar Svedberg values but a different distribution of the protein’s abundance. When anti–human-Fc Fab2 was used, a shift to higher Svedberg values was found, indicating the formation of protein complexes of higher molecular weight and possibly different configurations of Fab2 cross-linked complexes. Tentative structures of GPVI-Fc*IgG-XL and GPVI-Fc*Fab2-XL complexes based on the approximate molecular weight of the main peaks (approximately 400 to 800 kDa) are depicted in Supplemental Figure 4B. They show the predominant formation of oligomeric ([GPVI-Fc]2 and [GPVI-Fc]3) complexes.
The accretion of fluorescent-labeled GPVI-Fc and cross-linked GPVI-Fc from flowing blood onto collagen fibers and the relation to platelet adhesion was studied by using nanoscopy. Conditions and GPVI-Fc/antibody ratios were similar to that shown in Supplemental Figure 4, in which the samples had been characterized by analytical ultracentrifugation. Imaging using structured illumination microscopy (SIM) of samples fixed after blood perfusion showed that not cross-linked GPVI-Fc*IgG and GPVI-Fc*Fab2 was bound to collagen in a dotty and inhomogeneous manner, leaving segments on collagen free which, in fact, platelets used for surface membrane anchoring (Figure 4). In contrast, GPVI-Fc*IgG-XL and GPVI-Fc*Fab2-XL binding to collagen was abundant, continuous, and rapid enough to virtually prevent platelet attachment. The qualitative observations were confirmed by quantitative measurements. The superior potency of GPVI-Fc*IgG-XL and GPVI-Fc*Fab2-XL to prevent platelet adhesion to collagen fibers compared with GPVI-Fc was clearly visible by SIM (Figure 5). Stable platelet adhesion measured in the same samples was drastically reduced by cross-linking of GPVI-Fc (–76%) with anti–human-Fc Fab2.
Figure 4.

SIM Imaging of Binding of GPVI-Fc (Not Cross-Linked and Cross-Linked) and Platelets to Collagen
GPVI-Fc was incubated with anti–human-Fc antibodies in a 20:1 molar ratio (GPVI-Fc*IgG, GPVI-Fc*Fab2) or in a 1:1 molar ratio for crosslinking (GPVI-Fc*IgG-XL, GPVI-Fc*Fab2-XL). The antibodies contained 10% PE-conjugated anti–human-Fc IgG or anti–human-Fc Fab2. The mixtures were added to blood (333 nM GPVI-Fc) containing abciximab before perfusion over collagen at a shear rate of 600/s. After 4 min of flow, platelets were fixed and stained with anti-CD41 antibody and DyLight 488-conjugated secondary Ab. (Left) Structured illumination microscopy (SIM) images showing binding of PE-labeled GPVI-Fc (not cross-linked and cross-linked) (red) and platelets (green) to collagen with enlarged sections (×800) on the right. Maximum intensity projections of 0.15 μm z sections (total z 2.5 μm) are shown. (Right) Line intensity profiles (maximum intensity 100%) drawn along comparable, platelet-free segments of collagen fibers in the designated images (left). Images are representative of 5 different experiments. Binding of not cross-linked GPVI-Fc to collagen is discontinuous, showing fiber segments that are not occupied by GPVI-Fc (zero intensity in line profiles). Abbreviations as in Figures 1 and 2.
Figure 5.

SIM and STED Imaging of Collagen Stained With Not Cross-Linked GPVI-Fc (GPVI-Fc*IgG, GPVI-Fc*Fab2) and Cross-Linked GPVI-Fc (GPVI-Fc*IgG-XL, GPVI-Fc*Fab2-XL)
GPVI-Fc was incubated with Alexa Fluor 488-conjugated anti–human-Fc IgG or anti–human-Fc Fab2 antibodies in either a 10:1 molar ratio (not cross-linked GPVI-Fc) or a 1:1 molar ratio (cross-linked GPVI-Fc). The mixtures were then incubated with collagen coated glass coverslips. (A) SIM imaging. The SIM micrographs are maximum intensity projections of 0.15 μm z sections (total z 1.5 μm). The image is representative of 5 others from different experiments. (B) Stimulated emission depletion (STED) microscopy imaging. Collagen coated glass coverslips were stained simultaneously with AlexaFluor594-labeled anti-collagen type I and type III antibodies (red) in addition to GPVI-Fc, which was either labeled or cross-linked with anti–human-Fc antibodies. Yellow indicates co-localization of cross-linked GPVI-Fc with anti-collagen antibodies. Abbreviations as in Figures 1, 2, and 4.
Optical nanoscopy using SIM and STED microscopy (for even higher resolution) of collagen fibers incubated with fluorescent-labeled GPVI-Fc and GPVI-Fc cross-linked with anti–human-Fc IgG or anti–human-Fc Fab2 revealed a continuous and homogeneous binding compared with the dotted pattern of not cross-linked GPVI-Fc (Figures 5A and 5B). In the STED micrographs, in which collagen was simultaneously stained with AlexaFluor594-conjugated anti-collagen type I and III antibodies, binding of cross-linked GPVI-Fc to collagen showed complete co-localization with the anti-collagen antibodies.
To confirm our hypothesis that it is in fact cross-linking that potentiates the inhibitory effect of GPVI-Fc, inhibition of platelet aggregation on plaque and collagen fibers by GPVI-Fc*Fab2-XL were compared with that by GPVI-Fc*Fab. Fab antibodies bear only 1 antigen-binding site and are unable to cross-link antigens. Plaque- or collagen-coated surfaces were pre-incubated with either GPVI-Fc*Fab or GPVI-Fc*Fab2-XL. As expected, virtually no effect of GPVI-Fc*Fab was observed in contrast to a complete inhibition of plaque- and collagen-induced platelet aggregation by GPVI-Fc*Fab2-XL, indicating that indeed cross-linking causes the potent inhibition by GPVI-Fc*Fab2-XL (Figure 6A). Analysis by using SIM imaging demonstrated the absence of platelet adhesion to collagen fibers in the GPVI-Fc*Fab2-XL samples and a reticulated, dense binding pattern of GPVI-Fc*Fab2-XL binding in contrast to the staggered pattern of GPVI-Fc*Fab binding; this approach left fiber segments free for platelet membrane contacts. The qualitative observations were confirmed by quantitative measurements (Figure 6B).
Figure 6.

GPVI-Fc*Fab in Contrast to GPVI-Fc*Fab2-XL Does Not Inhibit Platelet Aggregation Induced by Plaque and Collagen Under Flow: Functional and SIM Imaging Studies
GPVI-Fc (100 μg/ml; 666 nM) was incubated with 3-fold molar excess of anti–human-Fc Fab-AlexaFluor594 (GPVI-Fc*Fab) or equimolar concentrations of anti–human-Fc Fab2 or anti-human-Fc Fab2–PE (GPVI-Fc*Fab2-XL). (A) Effect of GPVI-Fc*Fab and GPVI-Fc*Fab2-XL on the kinetic of platelet deposition onto plaque and collagen in flowing blood. Plaque homogenate and collagen coated onto coverslips were incubated with GPVI-Fc*Fab and GPVI-Fc*Fab2-XL complexes before perfusion with blood at a shear rate of 600/s. Fluorescence images of platelet deposition were continuously recorded (1 frame/5 s) and analyzed as detailed in the Methods section. Mean ± SD; n = 4. Comparison by using analysis of variance for repeated measures at full 2 to 5 min and secondary pair-wise comparisons by using Tukey correction. Significance of pair-wise comparison of GPVI-Fc*Fab2-XL to control and to GPVI-Fc*Fab was equal at all time points and is only indicated by a single line with asterisks. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. (B) SIM imaging of GPVI-Fc*Fab and GPVI-Fc*Fab2-XL binding and platelet adhesion to collagen fibers. Collagen-coated coverslips were incubated with GPVI-Fc*Fab-AlexaFluor594 or GPVI-Fc*Fab2-PE-XL (red) before perfusion with blood containing abciximab at a shear rate of 600/s. After 4 min of flow, platelets were fixed and stained with anti-CD41 antibody and DyLight 488-conjugated second Ab (green). (Left) SIM micrographs (maximum intensity projections of 0.15 μm z sections (total z 2.5 μm) with enlarged sections (×800) showing the different pattern of GPVI-Fc*Fab and GPVI-Fc*Fab2-XL binding (red) to collagen. Platelet adhesion (green) to collagen is observed in GPVI-Fc*Fab but not GPVI-Fc*Fab2-XL treated samples (left). Images are representative of 5 different experiments. (Right) Line intensity profiles (maximum intensity 100%) along comparable, platelet-free sections of collagen fibers in the designated images (left). Binding of GPVI-Fc*Fab to collagen is discontinuous with segments of fibers lacking significant binding (<10% in line profiles). Abbreviations as in Figures 1, 2, and 4.
To test whether cross-linked GPVI-Fc would increase bleeding time, measurements were conducted by using the platelet function analyzer PFA-200 (Siemens Healthcare, Germany). This device simulates primary hemostasis in vitro and is used for routine screening of patients with potential hemorrhagic risk 23, 24. GPVI-Fc*Fab2-XL did not significantly increase the closure times measured with the PFA-200 collagen/adenosine diphosphate or collagen/epinephrine cartridges compared with buffer or GPVI-Fc (Figure 7). As expected, aspirin produced a prolonged closure time with the collagen/epinephrine cartridge.
Figure 7.

GPVI-Fc*Fab2-XL Does Not Increase In Vitro Bleeding
Blood was either pre-incubated with no supplement, buffer, aspirin (ASA) (300 μg/ml), GPVI-Fc (333 nM), or GPVI-Fc*Fab2-XL (333 nM) before transfer to collagen/adenosine diphosphate (ADP) or collagen/epinephrine cartridges and determination of the in vitro closure time with the platelet function analyzer PFA-200. Mean ± SD; n = 6. ***p < 0.001 by repeated measures analysis of variance and secondary pair-wise comparison of ASA versus all other conditions. Abbreviations as in Figure 2.
Discussion
We found that cross-linking of GPVI-Fc with Fc-directed antibodies drastically increased its potency to inhibit atherosclerotic plaque- and collagen-induced platelet aggregation under static and flow conditions. The increase in efficacy was obviously due to cross-linking per se and not to saturation of other Fc-binding sites with anti–human-Fc antibodies. First, analytical ultracentrifugation confirmed the formation of cross-linked GPVI-Fc complexes after incubating GPVI-Fc with equimolar amounts of anti–human-Fc IgG or Fab2 antibodies. Second, functional platelet experiments under flow and nanoscopic imaging studies comparing the effect of GPVI-Fc incubated with anti–human-Fc Fab2 or Fab′ antibodies (which cannot cross-link) unequivocally showed that cross-linking of the Fc domains of GPVI-Fc is the underlying mechanism.
In blood, the addition of GPVI-Fc cross-linked with anti–human-Fc Fab2 led to an even stronger inhibition of plaque- and collagen-induced platelet aggregation than the addition of GPVI-Fc cross-linked with anti–human-Fc IgG, particularly under flow conditions (Figure 3). A possible explanation for the stronger inhibition with Fab2-containing complexes could be that GPVI-Fc*Fab2-XL complexes cannot again be stripped from plaque collagen by blood cells carrying the Fc receptor what might happen to GPVI-Fc*IgG-XL complexes. Particularly after rapid binding of cross-linked GPVI-Fc*IgG-XL to plaque collagen in the flow studies, the goat Fc tails might be well accessible for the Fc receptors of nearby flowing blood cells. This might explain the different kinetics and degree of inhibition by anti-Fc Fab2- and IgG cross-linking of GPVI-Fc in the flow studies (Figure 3B).
Advanced microscopic imaging studies provided insights into the mechanism of improved platelet inhibition by GPVI-Fc cross-linking. Notably, collagen fibers bound labeled GPVI-Fc from flowing blood very rapidly and stably; there was only a small difference in the binding kinetics between cross-linked and not cross-linked GPVI-Fc but a higher plateau level of cross-linked GPVI-Fc binding. Nanoscopy using SIM and STED microscopy showed that GPVI-Fc cross-linked with anti–human-Fc IgG or anti–human-Fc Fab2 bound to collagen in a more continuous and homogeneous manner compared with the dotted binding pattern of not cross-linked GPVI-Fc. Cross-linked GPVI-Fc covered collagen similar to a sheath, thereby impeding platelets to contact binding motifs on collagen. Cross-linking obviously generates multiple linear oligomeric GPVI-Fc complexes providing high-density alignment of GPVI domains with their corresponding collagen-binding motifs (Figure 8).
Figure 8.

Model of GPVI-Fc and Cross-Linked GPVI-Fc Binding to Plaque Collagen
Cross-linking of dimeric glycoprotein VI–fragment crystallizable (GPVI-Fc) fusion protein (GPVI, brown; Fc, blue) by anti–Fc Fab2 creates oligomeric (n = 2 to 4) GPVI-Fc molecules, which bind to their corresponding motifs on collagen fibers more densely (bottom) than GPVI-Fc (top).
The increase in efficacy by GPVI-Fc cross-linking could lead to several advantages for future antiplatelet treatments of cardiovascular patients. First, cross-linked GPVI-Fc offers effective platelet inhibition on sites of plaque rupture independent of the actual flow rate. Second, the inhibitory effect of cross-linked GPVI-Fc on plaque-induced platelet aggregation is at least as potent as that of anti-GPVI antibodies, but it is focused on the injured lesion and not burdened with systemic antiplatelet effects (unlike that of anti-GPVI antibodies). In fact, anti-GPVI antibodies may increase bleeding. Although the tail bleeding time of GPVI-deficient mice is only moderately increased (14), a variable bleeding diathesis is observed in patients with rare genetic or acquired GPVI defects (25).
Finally, due to the high efficacy, cross-linked GPVI-Fc can be used in much lower doses than GPVI-Fc. Moreover, under arterial flow inhibition by cross-linked GPVI-Fc, complexes appeared to be even more pronounced if stimulated by human plaque material than by collagen fibers alone (Figure 3). This further focuses its action to the injured plaque and may translate to a reduced risk of systemic bleeding. Indeed, by measuring closure time with the PFA-200 device, we found no evidence that cross-linked GPVI-Fc increases bleeding time in vitro.
When atherothrombosis causes acute myocardial infarction or ischemic stroke, a highly efficient antiplatelet therapy is needed. We demonstrated that the new principle of generating multimeric GPVI complexes (e.g., by antibody cross-linking of GPVI-Fc) exploits the full local potential of GPVI antagonism in human plaque–induced platelet adhesion and aggregation while leaving systemic platelet function intact. Cross-linked GPVI-Fc complexes in the bloodstream very rapidly accrete onto denuded plaque collagen fibers and cover them continuously and stably. This approach efficiently prevents platelet arrest and aggregate formation. The rapid deployment to the triggering plaque lesion, its high local potency, and the lack of systemic platelet impairment combine to make an attractive pharmacodynamic profile that should inspire the development of new poly-GPVI domain–based drugs targeting plaque collagen–platelet interaction. We envision that GPVI oligomers could be further tailored for clinical application. Antibody cross-linked GPVI-Fc complexes with persistent coating of collagen exposed at the lesion but limited stability in the circulation due to dissociation of the antibody from GPVI-Fc may be preferable; an example is for percutaneous coronary interventions in which a short-term, but highly effective, lesion-focused antithrombotic action is desired at the moment of dilation of a stenosing plaque. In fact, human monoclonal antibodies against Fc selected from a human combinatorial antibody library are already available and could, in principal, be further developed for GPVI-Fc complexing in clinical studies. Other strategies might be preferred, including transforming GPVI domains by chemical linking into a multivalent format with long-term stability in the circulation and avoiding potential immunogenic problems of GPVI-Fc antibody complexes. These approaches might be used if a prolonged protection from thrombogenic de novo plaque collagen exposure is desired, such as in patients with recurrent embolism from plaque erosion (26).
Conclusions
Cross-linking GPVI-Fc into oligomeric complexes aligned GPVI domains densely with their corresponding binding motifs on collagen and strikingly enhanced GPVI-Fc suppression of platelet adhesion and plaque- and collagen-induced aggregation under static and flow conditions without increasing bleeding time in vitro. Transformation of GPVI into multivalent GPVI complexes may provide a new strategy for a highly effective and safe GPVI targeting treatment against acute atherothrombosis.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: GPVI mediates platelet activation by injured plaques exposing collagen. This scenario can be blocked by GPVI antibodies possibly burdened with systemic platelet dysfunction or, less potently, by a GPVI-Fc fusion protein that conceals GPVI-binding motifs on collagen of lesioned plaques from platelets but leaves systemic platelet function intact. We demonstrate here that cross-linking the Fc tails of GPVI-Fc to form oligomeric GPVI-Fc complexes results in a rapid continuous coverage of collagen fibers and complete prevention of platelet aggregation on human plaque in static and flow models without increasing bleeding time. Thus, the full potential of blocking the collagen–GPVI axis of platelet activation can be provided at the culprit lesion by linking GPVI-Fc to oligomeric complexes while systemic platelet function is preserved.
TRANSLATIONAL OUTLOOK: Cross-linked GPVI-Fc, which rapidly binds to the site of plaque injury, might prevent atherothrombosis with no increased risk of systemic bleeding. This approach may hold special promise in intervention-associated plaque trauma, in which suppression of initiating platelet activation is desired to attenuate ensuing steps to restenosis.
Acknowledgments
The authors thank K. von Oheimb for expert technical assistance. The results are part of the doctoral thesis of Ms. Jamasbi at the University of Munich.
Footnotes
The study was supported by grants from advanceCOR GmbH (to Ms. Jamasbi), the August-Lenz foundation, the Deutsche Forschungsgemeinschaft (SFB1123/Z01, INST 409/150-1 FUGG, STED 3X), and the European Research Council (ERC AdG °249949). Ms. Jamasbi received a scholarship from advanceCOR GmbH. Drs. Münch and Ungerer are managing directors of advanceCOR GmbH and own shares of the company. Dr. Uhland is an employee of advanceCOR GmbH. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Appendix
References
- 1.Fuster V., Moreno P.R., Fayad Z.A., Corti R., Badimon J.J. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. 2005;46:937–954. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- 2.Badimon L., Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276:618–632. doi: 10.1111/joim.12296. [DOI] [PubMed] [Google Scholar]
- 3.van Zanten G.H., de Graaf S., Slootweg P.J. Increased platelet deposition on atherosclerotic coronary arteries. J Clin Invest. 1994;93:615–632. doi: 10.1172/JCI117014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Penz S., Reininger A.J., Brandl R. Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. FASEB J. 2005;19:898–909. doi: 10.1096/fj.04-2748com. [DOI] [PubMed] [Google Scholar]
- 5.Reininger A.J., Bernlochner I., Penz S.M. A 2-step mechanism of arterial thrombus formation induced by human atherosclerotic plaques. J Am Coll Cardiol. 2010;55:1147–1158. doi: 10.1016/j.jacc.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 6.Schulz C., Penz S., Hoffmann C. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res Cardiol. 2008;103:356–367. doi: 10.1007/s00395-008-0722-3. [DOI] [PubMed] [Google Scholar]
- 7.Zahid M., Mangin P., Loyau S. The future of glycoprotein VI as an antithrombotic target. J Thromb Haemost. 2012;10:2418–2427. doi: 10.1111/jth.12009. [DOI] [PubMed] [Google Scholar]
- 8.Jung S.M., Moroi M., Soejima K. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J Biol Chem. 2012;287:30000–30013. doi: 10.1074/jbc.M112.359125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miura Y., Takahashi T., Jung S.M., Moroi M. Analysis of the interaction of platelet collagen receptor glycoprotein VI (GPVI) with collagen. A dimeric form of GPVI, but not the monomeric form, shows affinity to fibrous collagen. J Biol Chem. 2002;277:46197–46204. doi: 10.1074/jbc.M204029200. [DOI] [PubMed] [Google Scholar]
- 10.Herr A.B., Farndale R.W. Structural insights into the interactions between platelet receptors and fibrillar collagen. J Biol Chem. 2009;284:19781–19785. doi: 10.1074/jbc.R109.013219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smethurst P.A., Joutsi-Korhonen L., O'Connor M.N. Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood. 2004;103:903–911. doi: 10.1182/blood-2003-01-0308. [DOI] [PubMed] [Google Scholar]
- 12.Massberg S., Konrad I., Bultmann A. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004;18:397–399. doi: 10.1096/fj.03-0464fje. [DOI] [PubMed] [Google Scholar]
- 13.Ungerer M., Rosport K., Bultmann A. Novel antiplatelet drug revacept (dimeric glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 2011;123:1891–1899. doi: 10.1161/CIRCULATIONAHA.110.980623. [DOI] [PubMed] [Google Scholar]
- 14.Dutting S., Bender M., Nieswandt B. Platelet GPVI: a target for antithrombotic therapy?! Trends Pharmacol Sci. 2012;33:583–590. doi: 10.1016/j.tips.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Deckmyn H., De Meyer S.F., Broos K., Vanhoorelbeke K. Inhibitors of the interactions between collagen and its receptors on platelets. Handb Exp Pharmacol. 2012;(210):311–337. doi: 10.1007/978-3-642-29423-5_13. [DOI] [PubMed] [Google Scholar]
- 16.Jamasbi J., Megens R.T., Bianchini M. Differential inhibition of human atherosclerotic plaque-induced platelet activation by dimeric GPVI-Fc and anti-GPVI antibodies: Functional and imaging studies. J Am Coll Cardiol. 2015;65:2404–2415. doi: 10.1016/j.jacc.2015.03.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandl R., Richter T., Haug K., Wilhelm M.G., Maurer P.C., Nathrath W. Topographic analysis of proliferative activity in carotid endarterectomy specimens by immunocytochemical detection of the cell cycle-related antigen Ki-67. Circulation. 1997;96:3360–3368. doi: 10.1161/01.cir.96.10.3360. [DOI] [PubMed] [Google Scholar]
- 18.Bampalis V.G., Brantl S.A., Siess W. Why and how to eliminate spontaneous platelet aggregation in blood measured by multiple electrode aggregometry. J Thromb Haemost. 2012;10:1710–1714. doi: 10.1111/j.1538-7836.2012.04819.x. [DOI] [PubMed] [Google Scholar]
- 19.Dwivedi S., Pandey D., Khandoga A.L., Brandl R., Siess W. Rac1-mediated signaling plays a central role in secretion-dependent platelet aggregation in human blood stimulated by atherosclerotic plaque. J Transl Med. 2010;8:128. doi: 10.1186/1479-5876-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elia N., Ott C., Lippincott-Schwartz J. Incisive imaging and computation for cellular mysteries: lessons from abscission. Cell. 2013;155:1220–1231. doi: 10.1016/j.cell.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheruvu P.K., Finn A.V., Gardner C. Frequency and distribution of thin-cap fibroatheroma and ruptured plaques in human coronary arteries: a pathologic study. J Am Coll Cardiol. 2007;50:940–949. doi: 10.1016/j.jacc.2007.04.086. [DOI] [PubMed] [Google Scholar]
- 22.Megens R.T., oude Egbrink M.G., Merkx M., Slaaf D.W., van Zandvoort M.A. Two-photon microscopy on vital carotid arteries: imaging the relationship between collagen and inflammatory cells in atherosclerotic plaques. J Biomed Opt. 2008;13:044022. doi: 10.1117/1.2965542. [DOI] [PubMed] [Google Scholar]
- 23.Kundu S.K., Heilmann E.J., Sio R., Garcia C., Davidson R.M., Ostgaard R.A. Description of an in vitro platelet function analyzer—PFA-100. Semin Thromb Hemost. 1995;21(Suppl 2):106–112. doi: 10.1055/s-0032-1313612. [DOI] [PubMed] [Google Scholar]
- 24.Paniccia R., Priora R., Liotta A.A., Abbate R. Platelet function tests: a comparative review. Vasc Health Risk Manag. 2015;11:133–148. doi: 10.2147/VHRM.S44469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arthur J.F., Dunkley S., Andrews R.K. Platelet glycoprotein VI-related clinical defects. Br J Haematol. 2007;139:363–372. doi: 10.1111/j.1365-2141.2007.06799.x. [DOI] [PubMed] [Google Scholar]
- 26.Deyev S.M., Lebedenko E.N. Multivalency: the hallmark of antibodies used for optimization of tumor targeting by design. Bioessays. 2008;30:904–918. doi: 10.1002/bies.20805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.