

Summary
Down syndrome (DS) is the leading genetic cause of mental retardation and is caused by a third copy of human chromosome 21. The different pathologies of DS involve many tissues with a distinct array of neural phenotypes. Here we characterize embryonic stem cell lines with DS (DS-ESCs), and focus on the neural aspects of the disease. Our results show that neural progenitor cells (NPCs) differentiated from five independent DS-ESC lines display increased apoptosis and downregulation of forehead developmental genes. Analysis of differentially expressed genes suggested RUNX1 as a key transcription regulator in DS-NPCs. Using genome editing we were able to disrupt all three copies of RUNX1 in DS-ESCs, leading to downregulation of several RUNX1 target developmental genes accompanied by reduced apoptosis and neuron migration. Our work sheds light on the role of RUNX1 and the importance of dosage balance in the development of neural phenotypes in DS.
Graphical Abstract
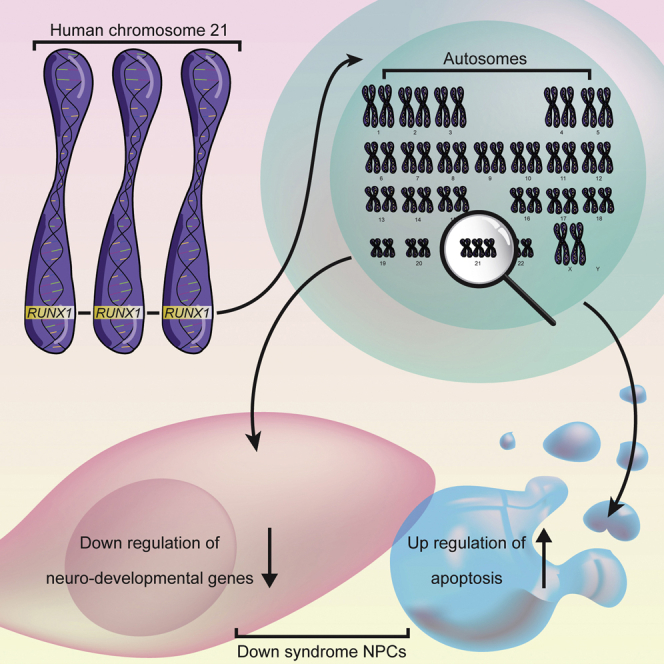
Highlights
-
•
Down syndrome NPCs exhibit increased apoptosis and downregulation of neural genes
-
•
RUNX1 regulates many of the downregulated genes in Down syndrome NPCs
-
•
All three copies of RUNX1 were deleted by genome editing in Down syndrome ESCs
-
•
Deletion of RUNX1 leads to changes in neural migration genes and apoptotic levels
In their work, Benvenisty and colleagues utilize neural progenitor cells (NPCs) derived from five embryonic stem cell lines with trisomy 21 to show the involvement of RUNX1 in the development of the neural phenotype of Down syndrome (DS). Ablation of all three copies of RUNX1 using genome editing led to reduced apoptosis and changes to neuron migration-related genes in DS-NPCs.
Introduction
Down syndrome (DS) is the leading genetic cause of mental impairment (Pulsifer, 1996), resulting from an extra copy of human chromosome 21. Individuals with DS display various phenotypes that affect multiple tissues (Korenberg et al., 1994), the most prevalent of which include cognitive defects, premature Alzheimer's disease, aging, and distinct dysmorphic facial features (Briggs et al., 2013, Galdzicki et al., 2001, Roizen and Patterson, 2003). It is thought that the pathologies of DS result from dosage sensitivity of several genes that play a role in the development of different tissues, and from inter- and intra-chromosomal regulatory interactions (Briggs et al., 2013).
Although chromosome 21 harbors about 350 genes, only a minimal region of about 50 genes within the chromosome is responsible for most of the phenotypes associated with DS. This region, which localizes to the long arm of chromosome 21, is considered the “DS-critical region”, and a third copy of this region is sufficient to cause most of the phenotypes of DS (Briggs et al., 2013, Delabar et al., 1993, Dierssen, 2012, Korenberg et al., 1994, McCormick et al., 1989, Mégarbané et al., 2009, Rahmani et al., 1989). Genes within the DS-critical region also play an important transcriptional regulatory role in different developmental processes. Thus, the effect of the dosage imbalance is not limited to genes on chromosome 21 alone, but also extends to target genes found on other chromosomes.
Mouse models for DS have been the primary tool for studying this disorder in past years. The most complex mouse models developed to study DS are either mice containing a third copy of three chromosomal regions orthologous to human chromosome 21, or mice carrying the complete human chromosome 21 as an extra copy (O'Doherty et al., 2005, Yu et al., 2010). These and other mouse models have proved to be very useful in understanding different aspects of the disorder. However, several DS phenotypes are not recapitulated due to limitations of genetic engineering or inter-species differences (Dierssen, 2012, Olson et al., 2004).
The use of embryonic stem cells (ESCs) for disease modeling has enabled the study of numerous human disorders that could not have been modeled in animals due to a lack of relevant phenotypes, appearance of different phenotypes, or even embryonic lethality (Avior et al., 2016, Halevy and Urbach, 2014). In contrast to induced pluripotent stem cells (iPSCs), which are reprogrammed from adult cells, ESC models for human disorders are derived from early embryos that were found to carry a mutation or a chromosomal aberration by preimplantation genetic diagnosis (PGD) or preimplantation genetic screening (PGS), respectively. This difference is important in modeling syndromes such as DS, as only a small fraction of trisomy-21 embryos survive to term (Morris et al., 1999, Spencer, 2001). By analyzing ESCs derived from early-stage embryos, we can study the molecular pathways altered by the presence of a third copy of chromosome 21 more faithfully, as well as the ways in which this chromosomal aberration may affect embryonic development.
We have previously isolated three PGS-derived ESC lines with trisomy 21, and suggested that ESCs carrying a third copy of chromosome 21 can be used as an in vitro model for DS (Biancotti et al., 2010). We have further demonstrated by global gene-expression analysis that the third copy of chromosome 21 is actively transcribed in DS-ESCs (Biancotti et al., 2010). In this study, we analyzed neural differentiation of five individual DS-ESC lines to identify molecular and cellular pathways involved in the development of this disease. Our data point to RUNX1, a gene that resides within the DS-critical region, as a key transcriptional regulator in DS neural progenitor cells (DS-NPCs). The contribution of this gene to the molecular phenotype of DS was further validated by its disruption via gene editing.
Results
To investigate the molecular and cellular phenotypes perturbed in DS, we compared DS and normal ESCs and their neural derivatives. In the past, we have isolated three DS-ESC lines, namely CSES13, CSES20, and CSES21, from PGS-derived embryos with trisomy of chromosome 21 (Figure 1A) (Biancotti et al., 2010). To extend the number of analyzed DS-ESC lines, we have established two additional DS-ESC lines, CSES32 and CSES44, which also carry trisomy 21 (Figure 1A). These DS-ESC lines were characterized in terms of morphology, alkaline phosphatase staining (Figure 1B), and expression of OCT4 (Figure 1C). All five DS-ESC lines showed expression of characteristic markers of pluripotent stem cells with average expression levels similar to those of wild-type (WT) cells (Figure 1D). Finally, CSES32 and CSES44 DS-ESC lines were differentiated into cells from the three embryonic germ layers upon induction of teratomas in vivo, showing structures of endodermal, mesodermal, and ectodermal tissues (Figure 1E).
Figure 1.

Cellular Characterization of DS-ESCs
Karyotype analysis was performed on all five DS cell lines.
(A) Metaphases of the five cell lines CSES13, 20, 21, 32, and 44 show a third copy of chromosome 21 in all lines (47,XX,+21 or 47,XY,+21). CSES32 and CSES44 cell lines were characterized for markers of pluripotent stem cells.
(B) CSES32 and CSES44 colonies stained positive for alkaline phosphatase.
(C) CSES32 and CSES44 colonies show positive staining for OCT4.
(D) All DS-ESC lines show expression of pluripotent genes such as NANOG, SOX2, and OCT4 similar to WT cells. The WT column represents the average of three different WT cell lines, and the DS column represents the average expression level of five different DS cell lines. Error bars represent SEM.
(E) CSES32 and CSES44 cell lines were differentiated in vivo by injecting them into immunodeficient mice to create teratomas. Teratoma sectioning and staining with H&E show differentiation into the three germ layers: EC marks ectoderm, ME marks mesoderm, and EN marks endoderm.
To better understand the neural phenotype of DS cells compared with normal cells, we differentiated all five DS-ESC lines into NPCs. Gene-expression analysis shows that in DS-ESCs, embryoid bodies (EBs), and NPCs, the relative expression of genes on chromosome 21 is about 1.5-fold higher than that of genes on chromosomes 20 or 22 (Figure 2A). These data suggest that in both undifferentiated and differentiated DS cells, all three copies of chromosome 21 are actively transcribed. This upregulation, however, accounts for only a minority of the differences observed in the global gene-expression profile between normal and DS-ESCs. Notably, the majority of the differentially expressed genes between the two cell types were located on autosomal chromosomes other than chromosome 21. Because DS patients have a striking developmental phenotype related to the CNS, we focused on the neural phenotype of DS-NPCs. To study the neural phenotype, we compared data of expression arrays of NPCs of three different WT cell lines with those of five different DS cell lines. The genes were then sorted according to their expression levels, whereas genes expressed more than 2-fold in DS-NPCs compared with WT-NPCs were considered to be upregulated in DS, while genes expressed less than 0.5-fold in DS-NPCs were considered to be downregulated. Functional annotation analysis of differentially expressed genes between DS- and control NPCs using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang et al., 2009a, Huang et al., 2009b) showed downregulation of genes related to forebrain development and upregulation of genes related to apoptosis (Figure 2B). The forebrain developmental genes downregulated in DS-NPCs include key neuronal genes such as POU3F2 (also known as BRN2) and ASCL1 (Figure 2C). To verify the predicted changes in apoptosis, we performed flow cytometry analysis to quantify the levels of programmed cell death in DS-NPCs derived from the five DS cell lines and compared them with control NPCs derived from three WT cell lines. The results showed an increase in the tendency of DS-NPCs to activate apoptosis when compared with control NPCs, assessed from the populations of both annexin V+/propidium iodide (PI)− and annexin V+/PI+ cells (Figure 2D).
Figure 2.

Molecular Characterization of DS-NPCs
All five DS cell lines were differentiated into EBs and NPCs.
(A) Gene-expression analysis of undifferentiated ESCs, EBs, and NPCs of all five DS cell lines show a 1.5-fold higher expression of chromosome 21 compared with chromosomes 20 and 22 as seen by the moving average plot.
(B) Functional annotation clustering, based on three arrays of WT cells and five arrays of DS cells, shows downregulation of forebrain development genes and upregulation of apoptosis-related genes in DS-NPCs; only genes that were up- or downregulated by 2-fold were analyzed.
(C) DS-NPCs show downregulation of several key neuro-developmental genes. For WT, three microarrays of three different cell lines were used, for DS, five microarrays of the five independent DS cell lines were used. Error bars represent SEM.
(D) Flow cytometry analysis performed on five DS-NPC lines and three WT-NPC lines exhibits more apoptotic cells in DS cells by the summation of annexin V+(FITC+)/PI− and annexin V+(FITC+)/PI+ cell populations. Shown are representative results from DS-NPCs (middle panel) and WT-NPCs (left panel). Bar graph represents the average summation of annexin V+/PI− and annexin V+/PI+ populations as percent apoptosis in DS-NPCs and WT-NPCs of all lines used. Error bars represent SEM.
We next analyzed whether the differential expression of genes we observed in DS cells results, at least partly, from an extra copy of a transcription factor residing on chromosome 21. For this purpose, we analyzed all upregulated genes with at least 2-fold change of expression in DS-NPCs, the majority of which reside on the autosomes other than chromosome 21 (Figure 3A), using the Promoter Integration in Microarray Analysis (PRIMA) software that searches for binding site enrichments on a given promoter set (Elkon et al., 2003). The analysis found the binding site of the nuclear protein Runt-related transcription factor 1 (RUNX1) to be significantly and highly enriched in the upregulated genes (p < 0.05 with 3.5-fold enrichment) (Figure 3B). RUNX1 is a transcription factor that localizes to the critical region of chromosome 21 (Figure 3B). To better understand the involvement of RUNX1 in the molecular pathology of DS, we used the CRISPR/Cas9 gene-editing system and designed a guide RNA to specifically target all isoforms of RUNX1 in DS-ESCs and create DS-CRISPR-deleted RUNX1 (DSCR) ESCs to observe the maximal effect of RUNX1 dosage differences (Figure 3C). Among the various clones isolated, we identified two clones, DSCR8 and DSCR75, with complete ablation of the RUNX1 protein by western blot analysis (Figure 3D), indicating disruption of all three RUNX1 alleles. Next, we differentiated the two DSCR clones into NPCs and performed global gene-expression analysis by DNA microarrays. We found that 162 genes were downregulated in our DSCR-NPCs compared with their isogenic DS-NPCs. We then analyzed these downregulated genes using the DAVID software and the USCS transcription factor binding site search. The analysis revealed that nearly 70% of the downregulated genes in the DSCR-NPCs (111 genes) were putative targets of RUNX1 with a Benjamini-corrected p value of 0.026. Among the downregulated targets of RUNX1 are several key developmental genes (Figure 4A), with some genes such as IGFBP5, CCL2, LGR5, FBLN5, and TLR4 showing a RUNX1-dosage-dependent expression (Figure 4B). One of these genes, CCL2, showed a much stronger downregulation when analyzed by qRT-PCR (using the primers listed in Table 1) compared with the expression array data, probably due to a less stringent probe set in the expression array. Functional annotation analysis revealed that the downregulated genes in DSCR-NPCs were enriched for neuron/cell migration and regulation of cell growth (Figure 4C). Finally, we analyzed whether the ablation of RUNX1 allowed the correction of the cellular phenotype of apoptosis. Indeed, flow cytometry analysis demonstrated that the DSCR clones had a reduced level of apoptosis when compared with their parental DS lines (Figure 4D).
Figure 3.

Identification of RUNX1 and Genetic Editing of DS Cells
(A) Analysis of differentially expressed genes shows many of the upregulated genes of more than 2-fold expression to be located on different chromosomes. Each dot represents a gene and red lines represent average fold change in expression for each chromosome between DS- and WT-NPCs.
(B) RUNX1 was found to be a common transcription factor regulating many of the upregulated genes in DS-NPCs. Height of bars represents -log(p value) while the number inside the bar represents fold enrichment of the motif in the promoter set of the upregulated genes in DS-NPCs. RUNX1 resides at chromosome 21q22.12, within the critical region responsible for DS.
(C) Guide RNA was designed to target the exon common to all RUNX1 gene isoforms. Blue letters represent the guide sequence and red letters represent the PAM sequence of the guide RNA.
(D) Western blot analysis showing a complete ablation of RUNX1 in edited DS-ESCs (DSCR), clones DSCR8 and DSCR75 compared with their parental line (DS) and with WT-ESCs.
Figure 4.

Molecular and Cellular Analysis of Genomically Edited DS-NPCs
(A) DSCR-NPCs show downregulation of several key developmental genes, all putative targets of RUNX1. Microarray expression data of two DSCR cell lines were compared with microarray expression data of their isogeneic parental DS line. DSCR bars represent the average levels of expression of two DSCR microarrays.
(B) Several of these genes were further analyzed for their expression by qRT-PCR and showed a RUNX1 dosage-dependent expression pattern in DS-, WT-, and DSCR-NPCs. For DS, the parental cell line was used in three independent experiments, for DSCR, the two isogenic cell lines were used in three independent experiments, and for WT, three different cell lines were used. Error bars represent SEM.
(C) Functional annotation analysis of all downregulated genes in DSCR-NPCs shows significant enrichment for neuron migration, cell migration, and regulation of cell growth.
(D) Comparison of the apoptotic levels of the isogenic DS- and DSCR-NPCs were analyzed and show downregulation of apoptosis in the edited cells as seen by the summation of annexin V+(FITC+)/PI− and annexin V+(FITC+)/PI+ cell populations in DSCR-NPCs (middle panel, representing one cell line) and DS-NPCs (left panel, representing one cell line). Bar graph represents the average summation of annexin V+/PI− and annexin V+/PI+ populations as percentage of apoptotic cells. For DS the parental cell line was used in three independent experiments, and for DSCR the two isogenic cell lines were used in three independent experiments. Error bars represent SEM. ∗p < 0.05 using Student's t test.
Table 1.
List of Primers
| Gene Symbol | Forward Primer | Reverse Primer |
|---|---|---|
| FEZF2 | TATCCACACCCAGGAAAAGC | GTGGGTCAGCTTGTGGTTCT |
| POU3F2 | GCGGATCAAACTGGGATTTA | AAAGGCTTCAGCTTGCACAT |
| LHX2 | TCTCGGACCGCTACTACCTG | GCTACCGTCCTTGCTGAAAC |
| ASCL1 | GTCTCCCGGGGATTTTGTAT | AGAGAACTTGGGTGCAGGAA |
| NR2F1 | TACGTGAGGAGCCAGTACCC | CCTACCAAACGGACGAAGAA |
| RFX4 | CATCACCAAGCAAACCCTTT | GACTCGATGGGAGACTGCTC |
| IGFBP5 | AAGGTGTGGCACTGAAAGTCCC | AAGCAGTGCAAACCTTCCCGT |
| CCL2 | TCTCGCCTCCAGCATGAAAGT | GCATTGATTGCATCTGGCTGA |
| LGR5 | ACTGCAAACCTGGAGAGTCTGA | GATACGCACAGCACTTGGAGAT |
| FBLN5 | TTGCTGCTGATGCTGTGTGTG | TGCGGATGTATGTAGGCTGGAG |
| TLR4 | TTTTATCACGGAGGTGGTTCCT | CAGGTCCAGGTTCTTGGTTGA |
| GAPDH | AGCCACATCGCTCAGACAC | GTACTCAGCGGCCAGCATCG |
Discussion
Much of our knowledge on DS comes from the documentation of symptoms in patients and analysis of mouse models. Although the contribution of both sources has been crucial in gaining a better understanding of the syndrome, the molecular pathways leading to the development of DS are still largely unknown. The derivation of ESCs with trisomy 21 enabled us to study the molecular processes that underlie DS in human cells and address questions that could not be addressed in other models. In recent years several studies have used both ESCs and iPSCs with trisomy 21 to study different aspects of DS such as hematopoiesis, heart development, and neural differentiation (Bosman et al., 2015, Chang et al., 2015, Chou et al., 2012, Maclean et al., 2012, Murray et al., 2015). Our analysis of DS-NPCs showed aberrant expression of key neuronal genes. In fact, two of the downregulated genes in DS-NPCs, POU3F2 and ASCL1, have been used together with MYT1L for direct conversion of fibroblasts into functional neurons, thus highlighting the developmental perturbation of DS cells (Vierbuchen et al., 2010). In recent years, the role of RUNX1 in neural development has been studied in different models. These studies suggested that RUNX1 plays an important role in the proliferation and differentiation of NPCs, the control of neurite outgrowth, and the impact on axonal pathfinding (Inoue et al., 2008, Theriault et al., 2005, Yoshikawa et al., 2015, Yoshikawa et al., 2016). Moreover, RUNX1 has been suggested to play a role in the peripheral and CNS development, in defining different brain compartments and in consolidation of specific neuronal identity in the developing mouse nervous system (Levanon et al., 2001, Simeone et al., 1995, Stifani et al., 2008, Zagami and Stifani, 2010). However, the role of RUNX1 in human neural development is still obscure. RUNX1 has been associated with DS in terms of its contribution to the increased risk of leukemia as seen in DS patients (De Vita et al., 2010). However, the involvement of RUNX1 in the neural phenotype of the syndrome has not been fully addressed. Based on our experimental data, we suggest that the extra copy of RUNX1 in DS-NPCs may disrupt different molecular pathways during neural development. This in turn could lead to perturbation in forebrain development and increased apoptosis as indicated by our data. In this study, we disrupted the expression of RUNX1 to demonstrate the importance of this gene in the phenotypes of DS. Ablation of RUNX1 resulted in downregulation of key developmental genes and cellular pathways related to neuron migration and cell growth, with reduced apoptosis in gene-edited DS-NPCs. These results highlight the importance of dosage balance of RUNX1 in DS cells. Our results are supported by a study based on a meta-analysis of DS, suggesting that RUNX1 is a transcription regulator that has a global dosage effect on other chromosomes, affecting genes related to CNS development and neuron differentiation (Vilardell et al., 2011). One hallmark of the facial phenotypes of DS patients is a protruding tongue and speech impediment. It was previously shown that these phenotypes of DS patients are, at least partially, the result of abnormal neuromuscular junctions in tongue muscles (Yarom et al., 1986). Interestingly, a recent study of RUNX1 demonstrated its involvement in the axonal pathfinding to specific tongue muscles (Yoshikawa et al., 2015). Our work links the roles of RUNX1 in the development of the nervous system to the neural phenotype observed in DS patients and suggests that this gene carries out a key function in the development of several of the phenotypes seen in DS. The expression patterns and role of RUNX1 in the human developing peripheral and central nervous systems should be furthered explored. Understanding the molecular processes underlying DS will help in the search for targeted therapy and provide further insights into the genetic dosage imbalance associated with this syndrome.
Experimental Procedures
Derivation of Embryonic Stem Cells with Down Syndrome
Derivation and characterization of ESCs with trisomy 21 were performed as previously described (Biancotti et al., 2010).
Cell Culture
Cell lines used in this study are as follows. For controls, cell lines CSES7, CSES15, and H9 were used (Biancotti et al., 2010, Lavon et al., 2008, Narwani et al., 2010, Thomson, 1998). For DS cell lines we used CSES13, CSES20, CSES21, CSES32, and CSES44 cell lines. Cell lines CSES13, 20, and 21 were previously described (Biancotti et al., 2010). Cells were cultured in standard human ESC culture media containing KnockOut DMEM (Gibco-Invitrogen) supplemented with 15% Knockout serum replacement (Gibco-Invitrogen), 2 mM L-glutamine (Sigma-Aldrich), 1:100 dilution of non-essential amino acids (Gibco-Invitrogen), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 8 ng/mL basic fibroblast growth factor (PeproTech), penicillin (50 units/ml), and streptomycin (50 μg/mL) (Gibco-Invitrogen). Mouse embryonic fibroblasts (MEFs) were grown in DMEM (Gibco-Invitrogen) supplemented with 10% FCS (Biological Industries), penicillin (50 units/ml) and streptomycin (50 μg/mL). EB formation was described previously (Biancotti et al., 2010). NPC differentiation was carried out according to a neural differentiation protocol with dorsomorphin (Tocris Bioscience) and SB431542 (Cayman Chemical) as described by Kim et al. (2010). NPCs were then sorted by fluorescence-activated cell sorting for NCAM1-positive cells with NCAM1 antibody (R&D Systems). Acquisition and sorting were performed using the FACSAria Cell-Sorting System (Becton Dickinson).
RNA Isolation and Reverse Transcription
RNA was isolated using a PerfectPure RNA Cultured Cell Kit-50 (5 PRIME). One microgram of total RNA was used for reverse transcription reaction using ImProm-II reverse transcriptase (Promega). For sequencing and quantitative experiments, PCRs were performed with ReadyMix (Sigma); for overexpression experiments, PCR reactions used Herculase II Fusion DNA polymerase (Agilent Technologies). Real-time qPCR was performed with 1 μg of RNA reverse transcribed to cDNA and TaqMan Universal Master Mix or SYBR Green qPCR Supermix (see primer list in Table 1; Applied Biosystems) and analyzed with the 7300 real-time PCR system (Applied Biosystems).
DNA Microarray Analysis
Total RNA was extracted according to the manufacturer's protocol (Affymetrix). RNA was subjected to the HG-U133plus2 Affymetrix microarray platform, as previously described (Biancotti et al., 2010) or the Human Gene 1.0 ST microarray platform (Affymetrix); washing and scanning were performed according to the manufacturer's protocol. Arrays were analyzed using Robust Multichip Analysis in the Affymetrix Expression Console.
Functional Annotations and Motif Search
Functional annotations were done by subjecting differentially expressed genes to the DAVID functional annotation clustering tool (http://david.abcc.ncifcrf.gov/) (Huang et al., 2009a, Huang et al., 2009b). Motifs were searched using the integrated PRIMA and EXPANDER software (http://acgt.cs.tau.ac.il/prima/) and analyzed for promoter enrichment (Elkon et al., 2003).
Apoptosis Assay and Flow Cytometry Analysis
For quantification of apoptosis, an annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (eBioscience) was used according to the manufacturer's instructions. Cells were then analyzed using either the FACSCalibur system or the FACSAria Cell-Sorting system (Becton Dickinson).
Genome Editing by CRISPR/Cas9
Genome editing was performed according to the Ran et al. (2013) protocol with slight modifications. CRISPR/Cas9 pSpCas9(BB)-2A-GFP plasmid was obtained from the Addgene repository. CRISPR guide RNA was designed using the CRISPR Design Tool website (http://crispr.mit.edu/). Oligos for plasmid cloning, F: CACCGATGAGCGAGGCGTTGCCGC and R: AAACGCGGCAACGCCTCGCTCATC, were cloned into the CRISPR/Cas9 plasmid. The CRISPR/Cas9 plasmid was co-transfected with modified pEGFP-N1 without GFP by Clontech into DS-ESCs. Selection for positive clones was carried out with ESC medium containing G418.
Western Blot Analysis
Polyacrylamide gel (8%) was used for protein separation. The gel was transferred to a nitrocellulose membrane, and antibody hybridization and chemiluminescence were performed according to the standard procedures. The primary antibodies used in this analysis were mouse anti-RUNX1 (A-2) sc-365644 (Santa Cruz Biotechnology) and anti-GAPDH (14C10; Cell Signaling Technology). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Author Contributions
T.H., J.-C.B., and N.B. designed the research, interpreted the results, and prepared the manuscript. T.H., J.-C.B., and O.Y. performed the experiments. T.G.-L. performed the karyotype analysis. N.S. supervised the work.
Acknowledgments
We thank members of the Azrieli Center for Stem Cells and Genetic Research at the Hebrew University for their contribution, and especially Mordecai Peretz for assistance with the figures, and Ido Sagi for critical reading of the manuscript and assistance with the graphical design. N.B. is the Herbert Cohn Chair in Cancer Research. This work was partially supported by the Israel Science Foundation (grant number 269/12), by the Israel Science Foundation – Morasha Foundation (grant number 1252/12), by the Rosetrees Trust, and by the Azrieli Foundation.
Published: September 8, 2016
Accession Numbers
The accession number for the microarray data reported in this paper is GEO: GSE84887.
References
- Avior Y., Sagi I., Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- Biancotti J.-C., Narwani K., Buehler N., Mandefro B., Golan-Lev T., Yanuka O., Clark A., Hill D., Benvenisty N., Lavon N. Human embryonic stem cells as models for aneuploid chromosomal syndromes. Stem Cells. 2010;28:1530–1540. doi: 10.1002/stem.483. [DOI] [PubMed] [Google Scholar]
- Bosman A., Letourneau A., Sartiani L., Del Lungo M., Ronzoni F., Kuziakiv R., Tohonen V., Zucchelli M., Santoni F., Guipponi M. Perturbations of heart development and function in cardiomyocytes from human embryonic stem cells with trisomy 21. Stem Cells. 2015;33:1434–1446. doi: 10.1002/stem.1961. [DOI] [PubMed] [Google Scholar]
- Briggs J.A., Mason E.A., Ovchinnikov D.A., Wells C.A., Wolvetang E.J. Concise review: new paradigms for Down syndrome research using induced pluripotent stem cells: tackling complex human genetic disease. Stem Cells Transl. 2013;2:175–184. doi: 10.5966/sctm.2012-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-Y., Chen S.-M., Lu H.-E., Lai S.-M., Lai P.-S., Shen P.-W., Chen P.-Y., Shen C.-I., Harn H.-J., Lin S.-Z. N-butylidenephthalide attenuates Alzheimer’s disease-like cytopathy in Down syndrome induced pluripotent stem cell-derived neurons. Sci. Rep. 2015;5:8744. doi: 10.1038/srep08744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S.T., Byrska-Bishop M., Tober J.M., Yao Y., Vandorn D., Opalinska J.B., Mills J.A., Choi J.K., Speck N.A., Gadue P. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2012;109:17573–17578. doi: 10.1073/pnas.1211175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vita S., Canzonetta C., Mulligan C., Delom F., Groet J., Baldo C., Vanes L., Dagna-Bricarelli F., Hoischen A., Veltman J. Trisomic dose of several chromosome 21 genes perturbs haematopoietic stem and progenitor cell differentiation in Down’s syndrome. Oncogene. 2010;29:6102–6114. doi: 10.1038/onc.2010.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delabar J.M., Theophile D., Rahmani Z., Chettouh Z., Blouin J.L., Prieur M., Noel B., Sinet P.M. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur. J. Hum. Genet. 1993;1:114–124. doi: 10.1159/000472398. [DOI] [PubMed] [Google Scholar]
- Dierssen M. Down syndrome: the brain in trisomic mode. Nat. Rev. Neurosci. 2012;13:844–858. doi: 10.1038/nrn3314. [DOI] [PubMed] [Google Scholar]
- Elkon R., Linhart C., Sharan R., Shamir R., Shiloh Y. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 2003;13:773–780. doi: 10.1101/gr.947203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdzicki Z., Siarey R., Pearce R., Stoll J., Rapoport S.I. On the cause of mental retardation in Down syndrome: extrapolation from full and segmental trisomy 16 mouse models. Brain Res. Brain Res. Rev. 2001;35:115–145. doi: 10.1016/s0926-6410(00)00074-4. [DOI] [PubMed] [Google Scholar]
- Halevy T., Urbach A. Comparing ESC and iPSC-based models for human genetic disorders. J. Clin. Med. 2014;3:1146–1162. doi: 10.3390/jcm3041146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Lempicki R.A., Sherman B.T. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K., Shiga T., Ito Y. Runx transcription factors in neuronal development. Neural Dev. 2008;3:20. doi: 10.1186/1749-8104-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.-S., Lee J.S., Leem J.W., Huh Y.J., Kim J.Y., Kim H.-S., Park I.-H., Daley G.Q., Hwang D.-Y., Kim D.-W. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 2010;6:270–281. doi: 10.1007/s12015-010-9138-1. [DOI] [PubMed] [Google Scholar]
- Korenberg J.R., Chen X.N., Schipper R., Sun Z., Gonsky R., Gerwehr S., Carpenter N., Daumer C., Dignan P., Disteche C. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA. 1994;91:4997–5001. doi: 10.1073/pnas.91.11.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavon N., Narwani K., Golan-Lev T., Buehler N., Hill D., Benvenisty N. Derivation of euploid human embryonic stem cells from aneuploid embryos. Stem Cells. 2008;26:1874–1882. doi: 10.1634/stemcells.2008-0156. [DOI] [PubMed] [Google Scholar]
- Levanon D., Brenner O., Negreanu V., Bettoun D., Woolf E., Eilam R., Lotem J., Gat U., Otto F., Speck N. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis. Mech. Dev. 2001;109:413–417. doi: 10.1016/s0925-4773(01)00537-8. [DOI] [PubMed] [Google Scholar]
- Maclean G.A., Menne T.F., Guo G., Sanchez D.J., Park I.-H., Daley G.Q., Orkin S.H. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc. Natl. Acad. Sci. USA. 2012;109:17567–17572. doi: 10.1073/pnas.1215468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick M.K., Schinzel A., Petersen M.B., Stetten G., Driscoll D.J., Cantu E.S., Tranebjaerg L., Mikkelsen M., Watkins P.C., Antonarakis S.E. Molecular genetic approach to the characterization of the “Down syndrome region” of chromosome 21. Genomics. 1989;5:325–331. doi: 10.1016/0888-7543(89)90065-7. [DOI] [PubMed] [Google Scholar]
- Mégarbané A., Ravel A., Mircher C., Sturtz F., Grattau Y., Rethoré M.-O., Delabar J.-M., Mobley W.C. The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of Down syndrome. Genet. Med. 2009;11:611–616. doi: 10.1097/GIM.0b013e3181b2e34c. [DOI] [PubMed] [Google Scholar]
- Morris J.K., Wald N.J., Watt H.C. Fetal loss in Down syndrome pregnancies. Prenat. Diagn. 1999;19:142–145. [PubMed] [Google Scholar]
- Murray A., Letourneau A., Canzonetta C., Stathaki E., Gimelli S., Sloan-Bena F., Abrehart R., Goh P., Lim S., Baldo C. Brief report: isogenic induced pluripotent stem cell lines from an adult with mosaic down syndrome model accelerated neuronal ageing and neurodegeneration. Stem Cells. 2015;33:2077–2084. doi: 10.1002/stem.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narwani K., Biancotti J.-C., Golan-Lev T., Buehler N., Hill D., Shifman S., Benvenisty N., Lavon N. Human embryonic stem cells from aneuploid blastocysts identified by pre-implantation genetic screening. In Vitro Cell Dev. Biol. Anim. 2010;46:309–316. doi: 10.1007/s11626-010-9303-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty A., Ruf S., Mulligan C., Hildreth V., Errington M.L., Cooke S., Sesay A., Modino S., Vanes L., Hernandez D. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309:2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson L.E., Richtsmeier J.T., Leszl J., Reeves R.H. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science. 2004;306:687–690. doi: 10.1126/science.1098992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsifer M.B. The neuropsychology of mental retardation. J. Int. Neuropsychol. Soc. 1996;2:159–176. doi: 10.1017/s1355617700001016. [DOI] [PubMed] [Google Scholar]
- Rahmani Z., Blouin J.L., Creau-Goldberg N., Watkins P.C., Mattei J.F., Poissonnier M., Prieur M., Chettouh Z., Nicole A., Aurias A. Critical role of the D21S55 region on chromosome 21 in the pathogenesis of Down syndrome. Proc. Natl. Acad. Sci. USA. 1989;86:5958–5962. doi: 10.1073/pnas.86.15.5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizen N.J., Patterson D. Down’s syndrome. Lancet. 2003;361:1281–1289. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- Simeone A., Daga A., Calabi F. Expression of runt in the mouse embryo. Dev. Dyn. 1995;203:61–70. doi: 10.1002/aja.1002030107. [DOI] [PubMed] [Google Scholar]
- Spencer K. What is the true fetal loss rate in pregnancies affected by trisomy 21 and how does this influence whether first trimester detection rates are superior to those in the second trimester. Prenat. Diagn. 2001;21:788–789. doi: 10.1002/pd.134. [DOI] [PubMed] [Google Scholar]
- Stifani N., Freitas A.R.O., Liakhovitskaia A., Medvinsky A., Kania A., Stifani S. Suppression of interneuron programs and maintenance of selected spinal motor neuron fates by the transcription factor AML1/Runx1. Proc. Natl. Acad. Sci. USA. 2008;105:6451–6456. doi: 10.1073/pnas.0711299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault F.M., Nuthall H.N., Dong Z., Lo R., Barnabe-Heider F., Miller F.D., Stifani S. Role for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in the mammalian nervous system. J. Neurosci. 2005;25:2050–2061. doi: 10.1523/JNEUROSCI.5108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J.A. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Südhof T.C., Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardell M., Rasche A., Thormann A., Maschke-Dutz E., Pérez-Jurado L.A., Lehrach H., Herwig R. Meta-analysis of heterogeneous Down syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics. 2011;12:229. doi: 10.1186/1471-2164-12-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarom R., Sagher U., Havivi Y., Peled I.J., Wexler M.R. Myofibers in tongues of Down’s syndrome. J. Neurol. Sci. 1986;73:279–287. doi: 10.1016/0022-510x(86)90152-8. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M., Hirabayashi M., Ito R., Ozaki S., Aizawa S., Masuda T., Senzaki K., Shiga T. Contribution of the Runx1 transcription factor to axonal pathfinding and muscle innervation by hypoglossal motoneurons. Dev. Neurobiol. 2015;75:1295–1314. doi: 10.1002/dneu.22285. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M., Masuda T., Kobayashi A., Senzaki K., Ozaki S., Aizawa S., Shiga T. Runx1 contributes to the functional switching of bone morphogenetic protein 4 (BMP4) from neurite outgrowth promoting to suppressing in dorsal root ganglion. Mol. Cell. Neurosci. 2016;72:114–122. doi: 10.1016/j.mcn.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Yu T., Li Z., Jia Z., Clapcote S.J., Liu C., Li S., Asrar S., Pao A., Chen R., Fan N. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum. Mol. Genet. 2010;19:2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagami C.J., Stifani S. Molecular characterization of the mouse superior lateral parabrachial nucleus through expression of the transcription factor Runx1. PLoS One. 2010;5:e13944. doi: 10.1371/journal.pone.0013944. [DOI] [PMC free article] [PubMed] [Google Scholar]