

Abstract
Hydrogen sulfide (H2S), the gas with the odor of rotten eggs, was formally discovered in 1777, over 239 years ago. For many years, it was considered an environmental pollutant and a health concern only in occupational settings. Recently, however, it was discovered that H2S is produced endogenously and plays critical physiological roles as a gasotransmitter. Although at low physiological concentrations it is physiologically beneficial, exposure to high concentrations of H2S is known to cause brain damage, leading to neurodegeneration and long‐term neurological sequelae or death. Neurological sequelae include motor, behavioral, and cognitive deficits, which are incapacitating. Currently, there are concerns about accidental or malicious acute mass civilian exposure to H2S. There is a major unmet need for an ideal neuroprotective treatment, for use in the field, in the event of mass civilian exposure to high H2S concentrations. This review focuses on the neuropathology of high acute H2S exposure, knowledge gaps, and the challenges associated with development of effective neuroprotective therapy to counteract H2S‐induced neurodegeneration.
Keywords: brain, hydrogen sulfide, neuropathology, neurodegeneration, neuroprotection
Introduction
Hydrogen sulfide (H2S) exemplifies compounds that are beneficial in low doses and toxic in high doses. It is both a vital physiological endogenous signaling molecule and a highly reactive and toxic xenobiotic gas. H2S was discovered 239 years ago in 1777 by a Swedish scientist, Carl Wilhelm Scheel.1, 2 It is a colorless irritant gas, has a higher density than air, and has a characteristic rotten‐egg odor. It is at least twice as soluble in organic solvents as it is in water.3 Because of its higher density, H2S tends to spread laterally near the ground and is particularly dangerous in confined spaces. The brain is the primary target organ of toxicity, but the respiratory and cardiovascular systems are additional targets of acute H2S poisoning.1, 4, 5, 6, 7
Sources
There are many sources of H2S, both endogenous and environmental. Endogenously, its presence in the gut has been recognized for a long time. A decade or so ago, it was also discovered that H2S is produced in other tissues of the human body, including the brain.8, 9 It is now known that H2S is a gasotransmitter that, along with nitric oxide and carbon monoxide, participates in cell signaling. The control of endogenous production of H2S has therefore become a target of multiple potential therapeutic applications.10, 11, 12, 13
H2S is also produced by the endogenous metabolism of carbonyl sulfide (COS), which occurs in the environment, in food, and as a pollutant. COS is metabolized in vivo by the enzyme carbonic anhydrase to H2S, the presumed proximate toxicant responsible for causing neurotoxicity and neurodegeneration.14 COS is also used as an industrial chemical and grain fumigant and is highly neurotoxic.
H2S is both an environmental pollutant and a hazard in more than 70 occupational settings.5, 6, 15, 16 In the environment, well‐recognized sources of H2S include release from volcanic eruptions, marshes, and bogs, and geological formations associated with natural gas and other fossil fuels.6, 15, 16, 17 In this regard, H2S is a well‐known hazard to petroleum and natural gas extraction personnel.18 Currently, there are major concerns about the potential for hydraulic fracking serving as a source of H2S exposure to workers and the community.19, 20 H2S is also produced in abundance by rotting organic matter.21 H2S gas produced from organic decomposition is a hazard in the food processing and sewage industries and to farmers and workers engaged in intensive food animal–production facilities, such as intensive swine confinement operations.6, 22 Almost every year in rural Middle America, farm workers die of acute H2S exposure in the hog industry.23 H2S is also an industrial chemical used in bulk quantities.15, 24 Because of this, there are additional concerns about potential mass civilian exposure following catastrophic industrial accidents leading to release of H2S in highly populated areas. Malfunction of crude oil or natural gas pipelines have also led to mass civilian population exposures to H2S. A short list of the major sources or occupational settings associated with H2S is given in Table 1.9
Table 1.
A short list of occupational settings and other sources of hydrogen sulfide
| Petrochemical refineries of crude oils with sulfur | Livestock farmers |
| Natural gas plants | Sewerage plants and sewer workers |
| Coke production from coal with sulfur | Animal manure disposal |
| Kraft wood pulp production | Pipeline maintenance |
| Tanneries | Food processing factories |
| Sulfur production | Production of deuterated water |
| Hydrogen sulfide gas production and storage | Construction industry (e.g., manhole workers, asphalt use and storage) |
| Viscose rayon production | Carbon disulfide production |
| Hydraulic fracking | Mixing household chemicals for suicide |
| Gypsum drywall | Carbonyl sulfide by in vivo metabolism |
Note: Partially modified from Ref. 9.
In the past 10 years, a new form of acute H2S poisoning has emerged: suicide by H2S inhalation has increasingly been reported.25, 26 Intentional H2S poisoning is accomplished by mixing acid with common household chemicals readily available in neighborhood stores. Victims take their lives in confined spaces, such as in cars or apartments. H2S was previously used in World War I as a chemical weapon.27 However, the ready accessibility to H2S‐generating chemicals or H2S gas in industrial quantities, combined with its highly toxic nature, emphasizes the potential for weaponization and misuse to harm the public.
Acute hydrogen sulfide–induced neurotoxicity and neurodegeneration in human beings
In humans, the most important route of H2S exposure is by inhalation. The brain is the primary target organ,24 with the respiratory and cardiovascular systems also affected.6, 16, 22 The acute toxicodrome of H2S poisoning in humans is well known and is characterized by “knockdown” (sudden collapse), pulmonary edema, conjunctivitis, and olfactory paralysis. The acute toxicity of H2S is more dependent on concentration than time. In other words, the higher the concentration of H2S, the more severe the acute, toxic outcome. The dose‐related effects following acute H2S exposure are summarized in Table 2.17 As an example from Table 2, inhalation of air containing 1000 ppm H2S can cause death after only a few breaths. On the lower end of the spectrum, conjunctivitis is the major clinical presentation in humans.
Table 2.
Dose‐related effects of hydrogen sulfide inhalation exposure
| 3–5 ppm | Offensive “rotten egg” smell |
| 10–20 ppm | Eye irritation |
| 50–100 ppm | Conjunctivitis, severe eye injury |
| 100–150 ppm | Olfactory fatigue; can no longer smell hydrogen sulfide odor |
| 150–250 ppm | Irritation of nose and lungs, nausea, headache, vomiting, dizziness |
| 250–500 ppm | Severe respiratory irritation, pulmonary edema |
| 500–1000 ppm | CNS stimulation, seizures, hyperpnoea, apnea, coma, death |
| 1000 ppm and above | Immediate knockdown, death from respiratory paralysis |
Central nervous system–related symptoms of acute H2S poisoning include a wide range of neurological effects, such as interference with olfactory sensations, persistent headache, fainting, ataxia, anxiety, depression, insomnia, knockdown, seizures, coma, and suppression of the respiratory center, leading to acute death.16, 28 Sudden loss of consciousness and collapse, colloquially known as “knockdown,” is a distinct and incapacitating characteristic of H2S poisoning.29, 30 About 8% of petroleum industry workers experience knockdown.31 Once unconscious, victims of acute H2S exposure are usually unable to escape further exposure to this highly toxic gas. With additional exposure, victims often slip into a coma and die. First responders, including family members, are especially susceptible to becoming secondary victims30 because of the immediate toxicity of H2S. Acute H2S exposure does not, however, always result in a fatality. Workers in the oil industry are documented to have recovered, apparently completely, from one or several short‐term knockdown episodes.6
It is common for victims of acute H2S intoxication to develop chronic neurological sequelae. These neurologic effects include recurrent seizures, persistent headache, nausea, vomiting, fatigue, hearing impairment, movement disorders (e.g., spasticity, ataxia), altered psychological states, memory impairment, vision impairment (blindness and color discrimination errors), anosmia, amnesia, psychosis, prolonged coma, persistent vegetative state, anxiety, depression, and sleeping disorders.6, 7, 30, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45
Typically, neurological sequelae have been reported in human victims of H2S poisoning who experience coma for periods ranging from 5 to 30 minutes.34, 35 In some reported cases, victims treated for H2S exposure recover from the acute effects of H2S poisoning and are discharged, only for postexposure neurologic complications to manifest days after apparent recovery.34, 35 In the worst‐case scenarios, some survivors of acute H2S exposure later descend into permanent vegetative states.34, 35 Apparently, there is a wide variability in susceptibility to neurologic damage among individuals exposed to single, acute exposures. Factors that most likely account for the wide variability in toxic outcome include genetic predisposition, health status, and the amount of time that victims spend in coma, among others.
The underlying mechanisms leading to the development of neurological sequelae are not currently known. Ischemic hypoxia and hypotension are widely reported triggers, but the downstream molecular pathways leading from hypoxia to H2S‐mediated neurotoxicity remain undefined. It is also probable that H2S itself is a direct trigger of molecular toxic pathways in neurons, astrocytes, or microglia, leading to neurotoxicity and neurodegeneration. Recently acquired knowledge indicating that H2S is a gasotransmitter with direct cellular effects strongly supports this possibility. Thus, hypoxia, hypotension, and direct cellular effects of H2S could all be responsible for the pathogenesis of neurotoxicity and neurodegeneration.
Reflecting the heterogeneous nature of the brain, different brain regions have varying sensitivity to H2S‐induced neurotoxicity. Medical imaging techniques and neurohistopathology of the brains of victims of acute H2S exposure have revealed pathology in the basal ganglia, thalamus, cortex, and brain stem.22, 29, 30, 34, 35, 36, 37, 38 The underlying mechanisms for the increased susceptibility of these regions to H2S‐induced neurotoxicity are not clear.
Lessons from veterinary medicine
H2S in high, acute exposures is toxic to most forms of life, including pets, farm animals, and wildlife, with farm animals commonly affected.46 In veterinary medicine, H2S exposure in animals results both from inhalation of toxic gases and an oral route through ingestion of feeds and water with high sulfur content. Acute exposure by inhalation is a frequent cause of massive deaths in confinement‐raised pigs, owing to agitation of manure pits resulting in sudden release of high environmental concentrations of H2S.47 As in human beings, inhalation of high levels of H2S gas concentrations causes death in pigs after a few breaths. To date, there has been no retrospective tracking of porcine survivors of acute H2S poisoning to determine whether surviving pigs develop neurological sequelae.
A rather common but widely unknown H2S‐induced disease of ruminants is polioencephalomalacia, a neurodegenerative disease.33, 46, 48 This disease, commonly seen in cattle, sheep, and goats, is associated with ingestion of water and/or feeds with high sulfur content.47 Rumen microbial metabolism transforms the water‐ or food‐borne sulfur into H2S gas that is absorbed and circulated to the brain, where it causes laminar necrosis of gray matter. The mechanism of H2S absorption in affected ruminants is still debated. Given the solubility of H2S in lipids3 and movement across mucosal epithelial surfaces, it is possible that H2S produced in the rumen is absorbed directly across the ruminal wall and circulated to the brain, where it causes neurotoxicity and neurodegeneration. An alternative hypothesis is that the rumen H2S is inhaled following eructation, causing H2S poisoning in ruminants. This is based on a publication indicating that a substantial amount of eructed gases are inhaled.49 Inhalation of eructed rumen gases is an important component of normal ruminant physiology. When present in inhaled eructed gases, H2S molecules are absorbed easily across the pulmonary alveolar wall. It is likely that both absorption across the ruminal wall and inhalation of eructed gases contribute to the pathogenesis of neurodegeneration.
There are striking similarities in the distribution of brain lesions in H2S‐induced polioencephalomalacia and those in the brains of human or nonhuman primate survivors of acute H2S poisoning.4, 32, 34, 35, 46 In polioencephalomalacia of ruminants, neurodegeneration is observed in the cortex, thalamus, and the inferior colliculi areas.33, 46 Generally, lesions are bilateral, but they are sometimes unilateral. At necropsy, the classical lesion of polioencephalomalacia is autofluorescence of regions of cerebral cortical necrosis under ultraviolet illumination. The lesions appear to start in the inferior colliculus region and spread to other sensitive brain regions including the thalamus and caudate nuclei.46 Pseudolaminar cortical necrosis is characteristic of ruminants dying of H2S‐induced polioencephalomalacia. Grossly, brains of animals with H2S‐induced polioencephalomalacia are swollen because of edema, to the extent that gyri are flattened as a result of the enlarged brain pressing on the calvarium. Often, this leads to herniation of the medulla and cerebellum47 (Fig. 1). As in ruminant H2S poisoning, cerebral edema and increased intracranial pressure is reported in human victims and is one of the causes of death by acute H2S poisoning.50 In ruminants, clinically affected animals are typically blind, lack appetite, exhibit muscle tremors of the head, have intermittent clonic convulsions, and exhibit head pressing.46, 48 It has been suggested that the blindness exhibited is of cortical origin.46 Several of these clinical signs are recognized neurological sequelae of H2S‐induced neurotoxicity in humans as well.35 Brain edema is attributed to H2S injury to endothelial cells of brain capillaries. The underlying mechanisms and the molecular pathways of neurodegeneration remain understudied both in the ruminant and the human condition.
Figure 1.
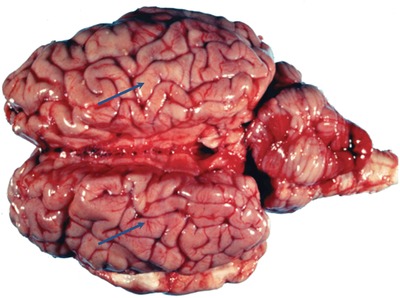
Brain of a cow that died of H2S‐induced polioencephalomalacia. Note the flattened gyri (arrow) and cerebellar coning and herniation caused by brain edema. Cerebral edema is a contributing factor in H2S‐induced mortality in humans. Photo courtesy of Dr. Steve Ensley.
Lessons from acute H2S exposure in experimental animal models
Extensive research has been conducted on the neurotoxicology of acute H2S poisoning using animal models, including rodents, rabbits, and nonhuman primates. However, a knowledge gap on the neuropathology of acute H2S poisoning still exists. In particular, our understanding of molecular pathways of H2S‐mediated neurotoxicity is incomplete.4, 6, 51, 52, 53, 54, 55, 56 There is an outstanding need to develop an animal model of acute H2S‐induced neurotoxicity that will improve our understanding of the pathogenesis of neurodegeneration and neurological sequelae.
An ideal animal model should recapitulate the human condition, using the inhalation route, a common route of human exposure. A rodent model of acute human H2S exposure by inhalation leading to neurodegeneration and attendant neurological sequelae has not yet been fully described. However, there have been challenges recapitulating the acute H2S‐induced neurotoxicity and neurodegeneration observed in humans using rodent models. These challenges were highlighted in the elegant study, which showed the difficulty of producing cerebral necrosis in rats injected with sodium hydrogen sulfide (NaHS), a H2S donor, to simulate single high‐dose exposures to H2S in humans.52 The study was characterized by high mortality, and only one of three surviving, unventilated rats injected with 120 mg/kg NaHS developed neuronal necrosis.52 Also, only one ventilated rat that survived the 200 mg/kg NaHS exhibited necrosis in the cerebral cortex.52 Pulmonary edema and hypotension were characteristic of rats that developed cerebral cortical necrosis, which led these investigators to conclude that hypotension and pulmonary edema are key factors in the pathogenesis of neuronal necrosis. Recently, a rat study by Sonobe et al., which delivered H2S by NaHS injection, was also characterized by high mortality, with only a few surviving rats exhibiting brain lesions.57 Two of the surviving rats exhibited neurodegeneration in the cortex and thalamus.57
Neurodegeneration, characterized by cerebrocortical necrosis, has been reported in rhesus monkeys exposed by inhalation to 500 ppm H2S for 22 minutes.4 The cerebellar cortex of these monkeys also showed reductions in Purkinje cell numbers. The basal ganglia, which includes the substantia nigra, is consistently affected by acute H2S poisoning in humans.32, 38 Basal ganglia lesions were also reported in monkeys,4 suggesting that the monkey recapitulated the human condition. What makes the monkey model even more attractive is that the inhalation route was used in these studies, reflecting the relevant route of exposure in human intoxications.52, 57 Unfortunately, primate models are more expensive than nonprimate models, limiting their broad application for translational research.
A novel animal model of H2S poisoning involves exposure to COS, a gas known to be metabolized to H2S in vivo. COS is metabolized to H2S by carbonic anhydrase enzyme in the brain.14 Using carbonic anhydrase inhibitors to prevent the metabolism of COS to H2S, Chengelis et al. showed significant attenuation of brain lesions in rats exposed to COS by inhalation.14 In this model, brain lesions were consistently observed in the colliculi of the brain stem, among other sites. Consistent with this report, our laboratory has produced lesions in the cortex, central inferior colliculus, and thalamus of mice exposed by inhalation to COS (Fig. 2). Lesions induced in our mouse model of COS inhalation are akin to that of acute H2S‐induced neuropathology in humans and have striking similarities with H2S‐induced polioencephalomalacia in ruminants. In both H2S‐ and COS‐exposed mice, we have observed histologic features of glial responses consistent with activation of the innate immune system, suggesting that inflammatory processes are involved in the pathogenesis of acute H2S‐induced brain injury (Fig. 3).
Figure 2.
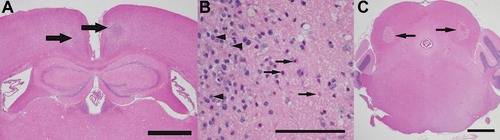
Bilateral foci of necrosis (arrows) efface the architecture of the retrosplenial cortex of a mouse (A) exposed to 476 ppm carbonyl sulfide for 4 h on day 0 and for 1 h on day 1, with euthanasia on day 8. A higher‐magnification photomicrograph (B) of the retrosplenial cortex in this mouse reveals degenerating (eosinophilic, “red, dead”) neurons (arrows), a glial scar with numerous reactive microglia (arrowheads), and vacuolar change (clear spaces) in the surrounding neuropil. In mice exposed to H2S by inhalation, similar foci of necrosis (arrows) were typically present in the inferior colliculus of the brain stem (C) and in the thalamus. Hematoxylin and eosin (H&E) stain; scale bar is 1000 μm in A and C, and 100 μm in B.
Figure 3.
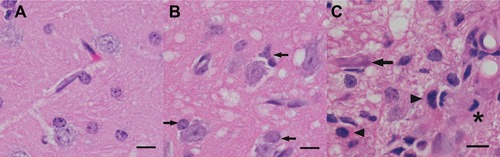
Inhalation of hydrogen sulfide or carbonyl sulfide results in damage to the thalamus (panels B and C, respectively), as well as other locations in the CNS (not shown). Vacuolation of the neuropil adjacent to neurons and capillaries is prominent in both, although the affected cell types are not identifiable in H&E‐stained sections. (B) The glial response is composed of increased numbers of glia (arrows) surrounding neurons (satellitosis). (C) Several gemistocytic astrocytes (arrowheads) are prominent in an area of gliosis that is almost devoid of neurons (a single degenerating neuron is identified in this field, arrow). A mitotic figure (to the left of the asterisk) likely represents microglial/histiocytic activation and proliferation. Note the rarity of perineural glia and homogeneity of the neuropil in the thalamus of a mouse not exposed to H2S or COS (A). H&E stain; scale bar is 10 μm.
Molecular mechanisms leading to H2S‐induced neurodegeneration
The cellular and molecular pathways leading to H2S‐induced neurodegeneration have yet to be defined. Our hypothesis of the broader pathophysiologic mechanisms of H2S‐induced neuronal cell death is summarized in Figure 4. A summary of our hypothesis of the potential cellular and molecular mechanisms involved in acute H2S‐induced neurotoxicity and neurodegeneration is presented in Figure 5. Much of the literature suggests that H2S‐induced neurodegeneration is a result of ischemic hypoxia arising from the combination of H2S‐induced hypotension, severe pulmonary edema impairing oxygen absorption, and inhibition of cytochrome c oxidase enzymatic activity in the mitochondria.22, 52, 58 Hypoxic ischemia is known to cause neurodegeneration triggered by cyanide, carbon monoxide, and stroke, among others mechanisms.59, 60, 61 Research linking H2S‐induced hypoxia to cell death is needed in order to identify potential molecular therapeutic targets for treating this disease. Currently, the literature, both in human and veterinary medicine, largely rests on descriptions of H2S‐induced neurological lesions and regional distribution of those lesions in the brain.
Figure 4.
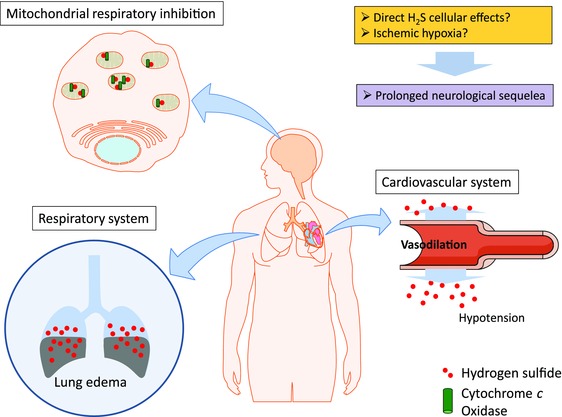
Proposed pathophysiology of H2S‐induced neurotoxicity. Acute exposure to high levels of H2S induces lung edema, leading to reduced oxygen absorption in the lungs. H2S also affects the cardiovascular system, inducing vasodilation leading to hypotension. Together, both lead to development of ischemic hypoxia in the brain. The function of cytochrome c oxidase in the mitochondrial electron transport chain is also inhibited by H2S, leading to reduced ATP production. Collectively, these pathophysiologic effects, coupled with direct cellular effects, account for the acute neurotoxic effects and the subsequent development of prolonged neurological sequelae.
Figure 5.
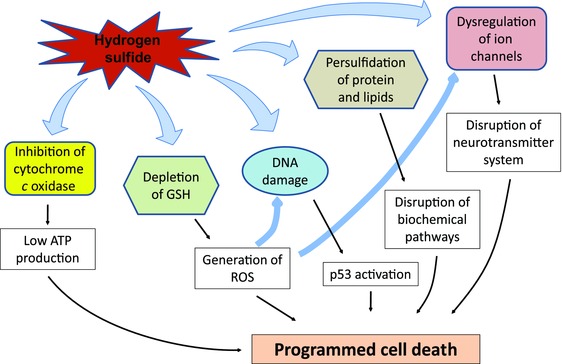
Potential mechanisms of H2S‐induced cytotoxicity. Hydrogen sulfide inhibits cytochrome c oxidase in mitochondria, leading to low ATP production. H2S also disrupts calcium homeostasis, leading to high intracellular calcium. Depletion of reduced glutathione leads to generation of reactive oxygen species (ROS). H2S induces DNA damage, persulfidation of protein and lipids, and dysregulation of ion channels, which are further aggravated by excessive intracellular levels of ROS. Collectively, these H2S‐induced effects may lead to programmed cell death in neurons and glia.
H2S binds and inhibits cytochrome c oxidase (complex IV), an enzyme in the electron transport chain.31, 62 This is the penultimate step before ATP synthesis in complex V. It is widely stated that the resulting ATP depletion leads to cellular energy deficiency.54, 55 Because the brain is a highly energy‐consuming organ, ATP depletion interferes with vital cellular functions of neurons, such as neurotransmission, that are highly dependent on levels of ATP. Cyanide and H2S have a similar mechanism of inhibiting cytochrome c oxidase. However, to what extent ATP depletion per se contributes to neurotoxicity and neurodegeneration is still debatable. In one cyanide study, Yamamoto showed that ATP depletion was present in the liver, but not in the brain, of cyanide‐treated rodents.63 He concluded that loss of consciousness in cyanide‐treated mice was due to hyperammonemia and increased brain aromatic amino acids but not due to ATP depletion. A recent in vitro study employing cultured neuronal cells did not find reduced ATP levels after H2S treatment.31
Oxidative stress is another pathophysiologic mechanism that is likely involved in H2S neurotoxicity. Acute exposure to high concentration of H2S can induce generation of ROS in hepatocytes.64 Treatment of hepatocytes with NaHS (up to 500 μM) results in depletion of glutathione (GSH), a molecule that plays a critical role in managing reactive oxygen species (ROS) in cells. Co‐administration with ROS scavengers rescued hepatocytes from the H2S‐induced cytotoxicity, indicating that the disruption of normal endogenous GSH balance resulted in sensitization of cellular defenses to ROS. Disruption of the normal ROS protective mechanisms is likely important in H2S‐mediated neurotoxicity, since H2S has recently been reported to induce more oxidative stress than cyanide and to induce apoptosis and inhibit DNA synthesis in vitro.31
Other homeostatic pathways worthy of consideration in H2S‐induced neurotoxicity and neurodegeneration include disruption of cell signaling, neuroinflammation, glutamate‐induced excitotoxicity, dysregulation of calcium homeostasis, and protein modification, among others.3, 65, 66, 67, 68 These are neurotoxic mechanisms that are commonly shared among other toxicants, including cyanide, 3‐nitropropionic acid, and nitrobenzene.69, 70, 71 H2S can induce DNA damage and apoptosis. Treatment with the H2S donor NaHS induced upregulation of p53 expression in fibroblasts, which is known to lead to upregulation of proapoptotic genes, such as p21 and Bax, and the release of cytochrome c from mitochondria, a step in programmed cell death. Similarly, treatment with NaHS upregulated expression of Bad and Bax and released cytochrome c into the cytosol of mouse cortical neurons.72
Disruption of cell signaling through the mitogen‐activated protein (MAP) kinase pathway has also been shown to be involved in H2S‐induced cytotoxicity. The MAPK pathway is a fundamental intracellular signaling mechanism that functions in a variety of homeostatic and responsive cell processes. Treatment of mouse cerebral cortical neurons with 1 mM NaHS resulted in ERK phosphorylation, leading to proapoptotic upregulation of Bad and Bax and release of cytochrome c from mitochondria. Co‐treatment with an MEK inhibitor with NaHS abolished ERK phosphorylation and protected cortical neurons.72 Human fibroblasts exposed to 1 mM NaHS have also been shown to phosphorylate JNK and ERK, supporting this theory.31
Excitotoxicity due to excessive levels of the neurotransmitter glutamate is theorized in H2S toxicity. Glutamate neurotoxicity was shown in acute H2S exposure to rat cerebellar granule neurons (CGN).73 Exposure to 200–300 μM NaHS induced cell death in rat CGN by activating the L‐type calcium channel, which was prevented by co‐treatment of L‐type calcium channel blocker. Blockage of N‐methyl‐d‐aspartate (NMDA) receptor binding by an antagonist prevented H2S‐induced neurotoxicity, supporting the premise that dysregulation of glutamate signaling at synapses contributes to H2S neurotoxicity.
H2S is an endogenously produced gasotransmitter that regulates synaptic activity, thereby affecting the function of both neurons and glia. This process has been shown to be mediated by protein modification of protein sulfhydration.31, 74, 75 Considering this, the direct effects of excessive concentrations of H2S on cells in the CNS warrant further consideration as potential mechanisms of neurotoxicity.9, 10
The roles of H2S in regulation of immune responses in the brain appear to be very complex. Toxic metabolites, such as H2S, are known to induce chronic neuroinflammation through mechanisms that activate glial cells and modulate cytokine production. Nontoxic levels of H2S are demonstrated to have anti‐inflammatory effects on microglia and astrocytes, possibly mediated through inducible nitric oxide synthase and MAPK signaling pathways.76, 77 Proinflammatory effects of H2S have been demonstrated in diverse diseases, such as pancreatitis, arthritis, and sepsis.78 The effects of supraphysiologic (toxic) levels of H2S on the immune system are not yet described. On the basis of other diseases caused by uncontrolled immune responses, we theorize that exogenous H2S has the potential to cause dysregulation of microglial function and induction of neuroinflammation in patients that survive initial acute H2S exposure.
Medical management of H2S poisoning and prevention of neurodegeneration
Currently, there is no suitable antidote for the prevention or treatment of acute H2S poisoning. The need is particularly acute for treatment of mass civilian casualties of H2S poisoning in the field. Difficulties hampering the development of effective therapies include an incomplete knowledge of the pathophysiologic mechanisms underlying toxicity, variability in toxic effects among individuals, a narrow range between no‐effect and toxic concentrations, and a lack of a suitable small animal model recapitulating the human condition. The fact that H2S is metabolized very rapidly in vivo provides additional challenges in development of suitable antidotes.
Treatment approaches to date have involved the use of sodium nitrite, hyperbaric oxygen, and 4‐dimethylaminophenol and administration of antioxidants, such as thiosulfates.21, 50, 79 Nitrite, which is given intravenously, converts iron in hemoglobin in circulating blood to methemoglobin, which then binds H2S. However, nitrite has disadvantages. It is a hypotensive agent, and, since H2S also causes vascular relaxation and hypotension, nitrite does not appear to be an ideal therapeutic approach. Nitrite has to be injected intravenously, a route not ideal for field treatment of mass civilian casualties. Furthermore, nitrite has a short half‐life because of its rapid excretion in urine.
The rationale for hyperbaric oxygen is to treat hypoxia, and this approach is efficacious,21, 79 but, unfortunately, hyperbaric oxygen chambers are not widely available. Both nitrite and hyperbaric oxygen therapy are hospital‐based treatments and cannot be used in the field for treatment of mass civilian casualties following acute H2S poisoning.
Field‐based, prehospital emergency care for patients exposed to H2S is needed, particularly because H2S is a potent toxicant that can cause peracute death. Currently, there is no medication that can be used in the field for treatment of mass civilian victims of H2S poisoning in the event of a catastrophic industrial accident or following use of H2S by terrorists. An ideal antidote for field applications should be available for use by intramuscular route in the form of an autoinjector. Needle‐free therapeutic agents formulated for intranasal administration, similar to transnasal Narcan™, are also appealing for treatment of mass casualties in the field. Developing an ideal antidote to bind H2S for use in field applications remains an area of great need.80
Summary
H2S is a strong example of dose‐dependent toxicity. It is a gasotransmitter critical to homeostatic mechanisms, including synaptic transmission, cardiovascular tone, and many other cellular responses. However, acute exposure to toxic levels of H2S or its precursors, NaHS and COS, results in a range of pathologies from mild to severe and acute to chronic. Neurological sequelae of H2S intoxication include neurotoxicity, cognition and memory deficits, persistent headaches, and motor and sensory deficits, among others. H2S‐induced neurodegenerative processes are believed to be mediated, in part, by hypoxia, hypotension, and ischemia; by perturbation of cellular metabolism; apoptosis and oxidative stress; and protein modification. Present knowledge of these mechanisms, however, only provides a partial explanation for observed clinical effects and pathology, suggesting that other mechanisms, such as induction of neuroinflammation, are likely involved. In particular, a knowledge gap exists linking molecular triggers with the downstream activation of a molecular cascade that ultimately leads to cell death or other phenotype changes. Through a more comprehensive understanding of the underlying pathophysiology, therapies targeted to specific neurotoxic pathways can be created. Currently, there is no drug available to effectively treat H2S toxicity or the prevention of H2S‐induced neurodegeneration and its attendant neurological sequelae. Reproducible, cost‐effective animal models that recapitulate the human condition following acute exposure to H2S by inhalation are also required to dissect mechanisms and responses to candidate drugs. The potential for H2S weaponization and consequential public health effects, as well as existing occupational health concerns, dictate the development of comprehensive pathophysiologic information and targeted neuroprotective therapies for H2S.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
This study was partly funded by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), by the National Institute of Neurological Disorders and Stroke (NINDS) (Grant Number NS0849487), and by start‐up funds to Dr. Wilson Rumbeiha.
References
- 1. Smith, R.P. & Gosselin R.E.. 1979. Hydrogen sulfide poisoning. J. Occup. Med. 21: 93–97. [DOI] [PubMed] [Google Scholar]
- 2. From Alchemy to Chemistry: Five hundred years of rare and interesting books . University of Urbana‐Champaign Rare Book Room Exhibit. Accessed 06/22/16. http://www.scs.illinois.edu/~mainzv/exhibit/scheele.htm.
- 3. Cuevasanta, E. et al 2012. Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS One 7: e34562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lund, O.E. & Wieland H.. 1966. Pathologic–anatomic findings in experimental hydrogen sulfide poisoning (H2S). A study on rhesus monkeys. Int. Arch. Arbeitsmed. 22: 46–54. [PubMed] [Google Scholar]
- 5. Beauchamp, R.O., Jr. et al 1984. A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 13: 25–97. [DOI] [PubMed] [Google Scholar]
- 6. Woodall, G.M. , Smith R.L. & Granville G.C.. 2005. Proceedings of the Hydrogen Sulfide Health Research and Risk Assessment Symposium October 31–November 2, 2000. Inhal. Toxicol. 17: 593–639. [DOI] [PubMed] [Google Scholar]
- 7. EPA . 2003. Toxicological review of hydrogen sulfide. EPA, Washington, DC: pp. 1–66. [Google Scholar]
- 8. Flannigan, K.L. et al 2013. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: a site‐specific, pro‐resolution mechanism. PLoS One 8: e71962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu, Y‐C. 2013. Signaling mechanisms underlying the hydrogen sulfide effects: identification of hydrogen sulfide “receptors” In Hydrogen Sulfide and Its Therapeutic Applications. Kimura H., Ed.: 83–108. New York: Springer. [Google Scholar]
- 10. Zheng, Y. et al 2015. Hydrogen sulfide prodrugs—a review. Acta Pharm. Sin. B 5: 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li, L. , Mohamed M.S.B. & Moore P.K.. 2013. Multiple roles of H2S in inflammation: a new class of therapeutics? In Hydrogen Sulfide and its Therapeutic Applications. Kimura H., Ed.: 63–82. New York: Springer. [Google Scholar]
- 12. Ichinose, F. 2013. Biological effects of H2S inhalation and its therapeutic potential In Hydrogen Sulfide and its Therapeutic Applications. Kimura H., Ed.: 157–172. New York: Springer. [Google Scholar]
- 13. Wang, M. et al 2015. Hydrogen sulfide functions as a neuromodulator to regulate striatal neurotransmission in a mouse model of Parkinson's disease. J. Neurosci. Res. 93: 487–494. [DOI] [PubMed] [Google Scholar]
- 14. Chengelis, C.P. & Neal R.A.. 1980. Studies of carbonyl sulfide toxicity: metabolism by carbonic anhydrase. Toxicol. Appl. Pharmacol. 55: 198–202. [DOI] [PubMed] [Google Scholar]
- 15. Council N.R.. 1979. “Hydrogen sulfide” In Medical and Biological Effects of Environmental Pollutants. Baltimore, MD: University Park Press; pp. 1–83. [Google Scholar]
- 16. Reiffenstein, R.J. , Hulbert W.C. & Roth S.H.. 1992. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol Toxicol. 32: 109–134. [DOI] [PubMed] [Google Scholar]
- 17. WHO . 1981. “Hydrogen sulfide” In Environmental Health Criteria 19. Vol. 2015. Geneva: ICPS International Programme on Chemical Safety; Accessed June 10, 2016. http://www.inchem.org/documents/ehc/ehc/ehc019.htm. [Google Scholar]
- 18. Rischitelli, G.D. & Schaumburg H.H.. 2000. “Hydrogen sulfide” In Experimental and Clinical Neurotoxicology. Spencer P. & Schaumburg H.H., Eds.: 655–658. New York: Oxford University Press. [Google Scholar]
- 19. Evans, R.B. , Prezant D. & Huang Y.C.. 2015. Hydraulic fracturing (fracking) and the Clean Air Act. Chest 148: 298–300. [DOI] [PubMed] [Google Scholar]
- 20. Adgate, J.L. , Goldstein B.D. & McKenzie L.M.. 2014. Potential public health hazards, exposures and health effects from unconventional natural gas development. Environ. Sci. Technol. 48: 8307–8320. [DOI] [PubMed] [Google Scholar]
- 21. Lindenmann, J. et al 2010. Severe hydrogen sulphide poisoning treated with 4‐dimethylaminophenol and hyperbaric oxygen. Diving Hyperb. Med. 40: 213–217. [PubMed] [Google Scholar]
- 22. Guidotti, T.L. 2010. Hydrogen sulfide: advances in understanding human toxicity. Int. J. Toxicol. 29: 569–581. [DOI] [PubMed] [Google Scholar]
- 23. Farm Safety Association . 2002. Manure gas dangers fact sheet. Vol. 2015. Accessed May 24, 2016. www.farmsafety.ca. [Google Scholar]
- 24. OSHA . Hydrogen sulfide In OSHA Fact Sheet. Vol. 2015. Accessed May 24, 2016. https://www.osha.gov/SLTC/hydrogensulfide/hazards.html. [Google Scholar]
- 25. Maebashi, K. et al Toxicological analysis of 17 autopsy cases of hydrogen sulfide poisoning resulting from the inhalation of intentionally generated hydrogen sulfide gas. Forensic Sci. Int. 207: 91–95. [DOI] [PubMed] [Google Scholar]
- 26. Sams, R.N. et al Suicide with hydrogen sulfide Am. J. Forensic Med. Pathol. 34: 81–82. [DOI] [PubMed] [Google Scholar]
- 27. Foulkes, C.H. 2009. “Gas!” The Story of the Special Brigade. Uckfield: Naval and Military Press Ltd. [Google Scholar]
- 28. Guidotti, T.L. 1994. Occupational exposure to hydrogen sulfide in the sour gas industry: some unresolved issues. Int. Arch. Occup. Environ. Health. 66: 153–160. [DOI] [PubMed] [Google Scholar]
- 29. Nogue, S. et al 2011. Fatal hydrogen sulphide poisoning in unconfined spaces. Occup. Med. (Lond.) 61: 212–214. [DOI] [PubMed] [Google Scholar]
- 30. Snyder, J.W. et al 1995. Occupational fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide. Am. J. Emerg. Med. 13: 199–203. [DOI] [PubMed] [Google Scholar]
- 31. Jiang, J. et al 2016. Hydrogen sulfide—mechanisms of toxicity and development of an antidote. Sci. Rep. 6: 20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsuo, M. , Cummins J.W. & Anderson R.E.. 1979. Neurological sequelae of massive hydrogen sulfide inhalation. Arch. Neurol. 36: 451–452. [DOI] [PubMed] [Google Scholar]
- 33. McAllister, M.M. , Gould D.H. & Hamar D.W.. 1992. Sulphide‐induced polioencephalomalacia in lambs. J. Comp. Pathol. 106: 267–278. [DOI] [PubMed] [Google Scholar]
- 34. Tvedt, B. et al 1991. Brain damage caused by hydrogen sulfide: a follow‐up study of six patients. Am. J. Ind. Med. 20: 91–101. [DOI] [PubMed] [Google Scholar]
- 35. Tvedt, B. et al 1991. Delayed neuropsychiatric sequelae after acute hydrogen sulfide poisoning: affection of motor function, memory, vision and hearing. Acta Neurol. Scand. 84: 348–351. [DOI] [PubMed] [Google Scholar]
- 36. Parra, O. et al 1991. Inhalation of hydrogen sulphide: a case of subacute manifestations and long term sequelae. Br. J. Ind. Med. 48: 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kilburn, K.H. 2003. Effects of hydrogen sulfide on neurobehavioral function. South Med. J. 96: 639–646. [DOI] [PubMed] [Google Scholar]
- 38. Gaitonde, U.B. , Sellar R.J. & O'Hare A.E.. 1987. Long term exposure to hydrogen sulphide producing subacute encephalopathy in a child. Br. Med. J. (Clin. Res. Ed.) 294: 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arnold, I.M. et al 1985. Health implication of occupational exposures to hydrogen sulfide. J. Occup. Med. 27: 373–376. [DOI] [PubMed] [Google Scholar]
- 40. Hoidal, C.R. et al 1986. Hydrogen sulfide poisoning from toxic inhalations of roofing asphalt fumes. Ann. Emerg. Med. 15: 826–830. [DOI] [PubMed] [Google Scholar]
- 41. Hurwitz, L. & Taylor G.. 1954. Poisoning by sewer gas with unusual sequelae. Lancet 266: 1110–1112. [DOI] [PubMed] [Google Scholar]
- 42. Wasch, H.H. et al 1989. Prolongation of the P‐300 latency associated with hydrogen sulfide exposure. Arch. Neurol. 46: 902–904. [DOI] [PubMed] [Google Scholar]
- 43. Sanz‐Gallen, P. et al 1994. [Acute poisoning caused by hydrogen sulphide: clinical features of 3 cases]. An. Med. Interna 11: 392–394. [PubMed] [Google Scholar]
- 44. Fenga, C. , Cacciola A. & Micali E.. 2002. Cognitive sequelae of acute hydrogen sulphide poisoning. A case report. Med. Lav. 93: 322–328. [PubMed] [Google Scholar]
- 45. Shivanthan, M.C. et al 2013. Hydrogen sulphide inhalational toxicity at a petroleum refinery in Sri Lanka: a case series of seven survivors following an industrial accident and a brief review of medical literature. J. Occup. Med. Toxicol. 8: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jubb, K.V.F. , Kennedy P.C. & Palmer N.. 1992. Pathology of Domestic Animals. San Diego: Academic Press Inc., Harcourt Brace Jovanovich. [Google Scholar]
- 47. Borst, G.H. 2001. [Acute poisoning of pigs with hydrogen sulfide as a result of acidification of slurry on a pig farm]. Tijdschr. Diergeneeskd. 126: 104–105. [PubMed] [Google Scholar]
- 48. Gould, D.H. 2000. Update on sulfur‐related polioencephalomalacia. Vet. Clin. North Am. Food Anim. Pract. 16: 481–496, vi–vii. [DOI] [PubMed] [Google Scholar]
- 49. Dougherty, R. & Cook H.. 1962. Routes of eructed gas expulsion in cattle—a quantitative study. Am. J. Vet. Res. 23: 997–100. [PubMed] [Google Scholar]
- 50. Lindenmann, J. et al 2010. Hyperbaric oxygen in the treatment of hydrogen sulphide intoxication. Acta Anaesthesiol. Scand. 54: 784–785. [DOI] [PubMed] [Google Scholar]
- 51. Luo, Y. et al 2014. Aggravation of seizure‐like events by hydrogen sulfide: involvement of multiple targets that control neuronal excitability. CNS Neurosci. Ther. 20: 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baldelli, R.J. , Green F.H. & Auer R.N.. 1993. Sulfide toxicity: mechanical ventilation and hypotension determine survival rate and brain necrosis. J. Appl. Physiol. (1985) 75: 1348–1353. [DOI] [PubMed] [Google Scholar]
- 53. Solnyshkova, T.G. 2003. Demyelination of nerve fibers in the central nervous system caused by chronic exposure to natural hydrogen sulfide‐containing gas. Bull. Exp. Biol. Med. 136: 328–332. [DOI] [PubMed] [Google Scholar]
- 54. Roth, S.H. et al 1997. Neurotoxicity of hydrogen sulfide may result from inhibition of respiratory enzymes. Proc. West Pharmacol. Soc. 40: 41–43. [PubMed] [Google Scholar]
- 55. Savolainen, H. et al 1980. Cumulative biochemical effects of repeated subclinical hydrogen sulfide intoxication in mouse brain. Int. Arch. Occup. Environ. Health. 46: 87–92. [DOI] [PubMed] [Google Scholar]
- 56. Greer, J.J. et al 1995. Sulfide‐induced perturbations of the neuronal mechanisms controlling breathing in rats. J. Appl. Physiol. (1985) 78: 433–440.7759410 [Google Scholar]
- 57. Sonobe, T. et al 2015. Immediate and long‐term outcome of acute H2S intoxication induced coma in unanesthetized rats: effects of methylene blue. PLoS One 10: e0131340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Milby, T.H. & Baselt R.C.. 1999. Hydrogen sulfide poisoning: clarification of some controversial issues. Am. J. Ind. Med. 35: 192–195. [DOI] [PubMed] [Google Scholar]
- 59. Riudavets, M.A. , Aronica‐Pollak P. & Troncoso J.C.. 2005. Pseudolaminar necrosis in cyanide intoxication: a neuropathology case report. Am. J. Forensic Med. Pathol. 26: 189–191. [PubMed] [Google Scholar]
- 60. Dirnagl, U. , Iadecola C. & Moskowitz M.A.. 1999. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22: 391–397. [DOI] [PubMed] [Google Scholar]
- 61. Graham, D.I. et al 1990. Neuropathologic consequences of internal carotid artery occlusion and hemorrhagic hypotension in baboons. Stroke 21: 428–434. [DOI] [PubMed] [Google Scholar]
- 62. Nicholls, P. et al 2013. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 41: 1312–1316. [DOI] [PubMed] [Google Scholar]
- 63. Yamamoto, H. 1989. Hyperammonemia, increased brain neutral and aromatic amino acid levels, and encephalopathy induced by cyanide in mice. Toxicol. Appl. Pharmacol. 99: 415–420. [DOI] [PubMed] [Google Scholar]
- 64. Truong, D.H. et al 2006. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab. Rev. 38: 733–744. [DOI] [PubMed] [Google Scholar]
- 65. Eghbal, M.A. , Pennefather P.S. & O'Brien P.J.. 2004. H2S cytotoxicity mechanism involves reactive oxygen species formation and mitochondrial depolarisation. Toxicology 203: 69–76. [DOI] [PubMed] [Google Scholar]
- 66. Baskar, R. , Li L. & Moore P.K.. 2007. Hydrogen sulfide‐induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. FASEB J. 21: 247–255. [DOI] [PubMed] [Google Scholar]
- 67. Yong, Q.C. et al 2010. Effect of hydrogen sulfide on intracellular calcium homeostasis in neuronal cells. Neurochem. Int. 56: 508–515. [DOI] [PubMed] [Google Scholar]
- 68. Cheung, N.S. et al 2007. Hydrogen sulfide induced neuronal death occurs via glutamate receptor and is associated with calpain activation and lysosomal rupture in mouse primary cortical neurons. Neuropharmacology 53: 505–514. [DOI] [PubMed] [Google Scholar]
- 69. O'Donoghue, J.L. 2000. Nitrobenzene In Experimental and Clinical Neurotoxicology. Spencer P. & Schaumburg H.H., Eds.: 864–866. New York: Oxford University Press. [Google Scholar]
- 70. Tor‐Agbidye, J. & Spencer P.. 2000. Cyanides and related compounds In Experimental and Clinical Neurotoxicology. Spencer P. & Schaumburg H.H., Eds.: 428–435. New York: Oxford University Press. [Google Scholar]
- 71. He, F. & Ludolf A.C.. 2000. 3‐Nitropropionic acid and related compounds In Experimental and Clinical Neurotoxicology. Spencer P. & Schaumburg H.H., Eds.: 870–875. New York: Oxford University Press. [Google Scholar]
- 72. Kurokawa, Y. et al 2011. Involvement of ERK in NMDA receptor‐independent cortical neurotoxicity of hydrogen sulfide. Biochem. Biophys. Res. Commun. 414: 727–732. [DOI] [PubMed] [Google Scholar]
- 73. Garcia‐Bereguiain, M.A. et al 2008. Hydrogen sulfide raises cytosolic calcium in neurons through activation of L‐type Ca2+ channels. Antioxid. Redox Signal. 10: 31–42. [DOI] [PubMed] [Google Scholar]
- 74. Kimura, H. et al 2005. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid. Redox Signal. 7: 795–803. [DOI] [PubMed] [Google Scholar]
- 75. Shibuya, N. et al 2009. 3‐Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11: 703–714. [DOI] [PubMed] [Google Scholar]
- 76. Lee, M. , McGeer E.G. & McGeer P.L.. 2016. Sodium thiosulfate attenuates glial‐mediated neuroinflammation in degenerative neurological diseases. J. Neuroinflammation 13: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hu, L.F. et al 2007. Hydrogen sulfide attenuates lipopolysaccharide‐induced inflammation by inhibition of p38 mitogen‐activated protein kinase in microglia. J. Neurochem. 100: 1121–1128. [DOI] [PubMed] [Google Scholar]
- 78. Bhatia, M. 2015. H2S and substance P in inflammation. Methods Enzymol. 555: 195–205. [DOI] [PubMed] [Google Scholar]
- 79. Herbert, A.F. 1989. Treatment of hydrogen sulphide intoxication In Proceedings of International Conference on Hydrogen Sulphide Toxicity. Prior M.G. et al, Eds.: 211–215, Banff, AB. [Google Scholar]
- 80. U.S. Department of Health and Human Services . 2007. NIH strategic plan and research agenda for medical countermeasures against chemical threats. National Institute of Health, Bethesda, MD. Accessed 06/20/16. https://www.niaid.nih.gov/topics/BiodefenseRelated/ChemicalCountermeasures/Documents/nihstrategicplanchem.pdf.