

Abstract
Proteins are constantly synthesized and degraded in living cells during their growth and division, often in response to metabolic and environmental conditions. The synthesis and breakdown of proteins under different conditions reveal information about their mechanism of function. The metabolic incorporation of non-natural amino acid azidohomoalanine (AHA) and subsequent labeling via click chemistry emerged as a non-radioactive strategy useful in the determination of protein kinetics and turnover. We used the method to monitor the degradation of two proteins involved in the multidrug efflux in E. coli, the inner membrane transporter AcrB and its functional partner membrane fusion protein AcrA. Together they form a functional complex with an outer membrane channel TolC to actively transport various small molecule compounds out of E. coli cells. We found that both AcrA and AcrB lasted for approximately six days in live E. coli cells, and the stability of AcrB depended on the presence of AcrA but not on active efflux. These results lead to new insight into the multidrug resistance in Gram-negative bacteria conferred by efflux.
Keywords: Pulse chase, protein lifetime, integral membrane protein, azidohomoalanine, click chemistry, multidrug efflux pump
Introduction
Proteins are a major component of all cells and play many essential roles as structural materials, building blocks, catalytic elements, transporters, channels, and signaling molecules. The stability and life time of specific proteins are closely correlated with their structure, function, and the metabolic status of the cell. Half-lives of most proteins in cells range from minutes to days [1–3]. The half-life of proteins in living cells is usually monitored using pulse chase experiments involving isotope labeling, in which cells are cultured in the presence of a specific isotope (for example, H3-Leu, C14-Leu, or S35-Met) for a short period of time (pulse) before switched into a normal culture (chase) [4]. The degradation of the isotope-labeled protein is then monitored over time. While the radioactive isotope labeling experiment has the advantage of being highly sensitive, convenient, and generally applicable to all proteins, it requires hazardous material with limited shelf-life and high costs in waste management. A non-radioactive method, which replaced isotope labeling with a non-natural amino acid azidohomoalanine (AHA), has been developed and applied to label proteins in several studies [5–9 10–13]. Due to their structural similarity, AHA can be incorporated into protein sequences by methionyl-tRNA synthetase. Subsequently, the AHA-containing protein can be detected either directly using mass spectroscopy or after purification/immune-precipitation and labeling with probe molecules containing an alkyne group via the well-established click chemistry [9]. For example, this approach has been used to track newly synthesized proteins in environmental microbes [7], whole mice [11], and rat hippocampal neurons [12]. It has also been used to monitor the origin, redistribution, and turnover of proteins in animal tissue cells [8] and track the kinetics of nucleosome turnover [13].
In this study we used the AHA-based pulse chase method to monitor the degradation of two proteins involved in the multidrug efflux in E. coli, a large integral membrane protein AcrB, and its functional partner AcrA, a periplasmic membrane fusion protein. As a constitutively expressed efflux pump system, the Resistance-Nodulation-Cell Division (RND) family transporter AcrA-AcrB-TolC actively transports a variety of compounds including antimicrobial agents, dyes, and detergent out of bacterial cells [14]. To span the two layers of cell membrane in Gram-negative bacteria, the efflux system contains three components: an inner membrane protein AcrB, a periplasmic membrane fusion protein (MFP) AcrA, and an outer membrane protein TolC. While the interaction between AcrA and AcrB is specific, TolC also functions in several other RND efflux systems such as AcrEF-TolC, EmrAB-TolC, and MacAB-TolC [15]. These systems have similar structural organization and are also involved in the efflux of antibiotics and other toxic compounds from the cell. During active efflux, the proteins assemble into a large complex that contains an AcrB trimer, a TolC trimer, and three or six subunits of AcrA. A proton translocation pathway exists in the transmembrane domain of AcrB, thus the inward proton gradient across the inner membrane is exploited to drive the active efflux of toxic compounds. Study of the degradation of AcrB and AcrA will help us gain valuable insight into the mechanism of multidrug resistance conferred by these proteins.
Materials and methods
Creation of gene knockout strains
Strain DL41 was obtained from the Coli Genetic Stock Center (Yale University). Gene knockout strains DL41ΔacrB and DL41ΔacrAB were created using Quick & Easy E. coli gene deletion kit (Gene Bridges GmbH, Heidelberg, Germany) following the manufacturer’s protocol. The target genes in the knockout strains were replaced with a kanamycin resistance cassette. Colony PCR was used to confirm that target genes had been replaced.
EtBr accumulation assay
The EtBr accumulation assay was performed following a published protocol [16]. E. coli strains (DL41 or DL41ΔacrB) containing different plasmids (pQE70 or pQE70-AcrB) as indicated were cultured overnight at 37°C in LB broth and diluted 100-fold into fresh LB broth the next morning. After growth to the late exponential phase (OD600 is ~1.0), cells were harvested at 4,500 rpm for 10 min at room temperature, washed once with 50 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl and 0.1% (vol/vol) glycerol, and resuspended in the same buffer to OD600 of 0.1. The accumulation of EtBr by E coli cells was monitored with a PerkinElmer LS-55 fluorescence spectrometer (PerkinElmer, Waltham, MA) at room temperature. The final concentration of EtBr used was 5 μM. The excitation and emission wavelengths were 520 and 590 nm, respectively, with slit widths at 5 nm for excitation and 5 nm for emission.
Growth of DL41 in different media
A single colony of DL41 was used to inoculate 3 mL LB media, which was cultured overnight. The next morning, the cell culture was diluted to a final OD600 of 0.01 into the M9 media supplemented with 20 essential amino acids (each at 4 mg/mL) and cultured to saturation. The culture was diluted again to a final OD600 of 0.01 into the M9 media with different supplement: 19 essential amino acids (each at 4 mg/mL) without Met, all 20 essential amino acids (each at 4 mg/mL), or 19 essential amino acids (each at 4 mg/mL) plus AHA (5 mg/mL). The OD600 of the cultures were recorded every 2 h for 16 h.
S35 Met pulse-chase experiment
E. coli cells (DL41ΔacrB) containing the respective plasmids were cultured overnight at 37°C in M9 medium, which was used to inoculate fresh M9 medium the next morning. When the OD reached 1.2, cells were harvested at 4,500 rpm for 10 min at room temperature, resuspended in 5 mL M9 medium with 19 amino acids (no methionine). S35-Met (1000 Ci/mmol) was added to a final concentration of 50 μCi/ml for 2 min, and then a 400-fold excess of non-radioactive Met (20 mM) was added. Immediately after the addition of the non-radioactive methionine (chase), 632 μL (1/8 of the total volume) of the culture was collected and centrifuged. The pellet was labeled as day 0 sample and kept at −80°C until use. The rest of the culture was also centrifuged (4000 rpm, 10 min) and resuspended in 5 mL of fresh media and cultured at 37°C overnight. After 24 h, 714 μL (1/7 of the total volume) of the culture was withdraw, centrifuged, and stored frozen as the day 1 sample. The rest of the culture was centrifuged and resuspended in 5 mL of fresh media, and cultured overnight. This processed was repeated each morning. For the day 2 to 6 samples, 833, 1000, 1250, 1670, and 2500 μL of culture were taken, which correspond to 1/6, 1/5, ¼, 1/3, and ½ of the volume of the culture, and the left over culture was always centrifuged and resuspended in 5 mL of fresh media. For the day 7 sample, the entire 5 mL culture was centrifuged and used in the analysis. With this method, decedents from the same amount of starting pulse-labelled cells were collected each day.
After all samples were collected, cell pellets were thaw on ice and resuspended in 200 μL buffer containing 20 mM sodium phosphate (pH 7.5), 0.2 M NaCl, 10% glycerol, 1 mM PMSF, and 0.4% lysozyme. Cells were lysed via 6 freezing/thawing (F/T) cycles: resuspended cells were incubated in ethanol/dry ice bath (−72 °C) for 3 min, then transferred to room temperature water bath for 5 min (20 °C). Between each F/T cycle, samples were vortexed for 1 min. The lysed cells were centrifuged to collect the pellet, which contained the membrane fraction. A sodium phosphate buffer (20 mM, pH 7.4) containing 1% (w/v) n-dodecyl-β-D-maltoside (DDM) was used to solubilize the membrane fractions and then the supernatant was collected, and 20 μL of buffer-equilibrated Ni-nitrilotriacetic acid (NTA) superflow resin was added. The samples were incubated for 1 hour with shaking on ice, and then briefly centrifuged and the supernatant removed. Bound proteins were eluted using the same buffer supplemented with 500 mM imidazole and analyzed using SDS-PAGE. Radioisotope-labeled protein was detected using a Typhoon FLA 9500 laser scanner.
AHA incorporation and labeling
Plasmids pQE70-AcrB and pQE70-AcrB-D407A were constructed in previous studies [17, 18]. The construct contains a vector derived C-terminal his-tag to facilitate convenient purification. They were transformed into DL41ΔacrB or DL41ΔacrAB as indicated for protein expression under basal condition without induction. Specifically, DL41ΔacrB containing pQE70-AcrB were cultured to collect samples that were used to analyze the degradation of AcrB and AcrA. DL41ΔacrAB containing pQE70-AcrB were used to analyze the degradation of AcrB in the absence of AcrA. DL41ΔacrB containing pQE70-AcrB-D407A were used to analyze the degradation of AcrB-D407A. E. coli cells was cultured overnight at 37°C in LB broth. On the next day, the overnight culture was diluted 250-fold into the M9 minimum medium and cultured at 37 °C overnight. The overnight culture was then diluted 20-fold into fresh M9 medium containing 19 amino acids (40 μg/ml) and AHA (50 μg/ml). For an experiment lasting for 7 days, eight 5 mL cultures were set up except for DL41ΔacrB-pQE70-AcrB, for which 10 mL cultures were set up. After incubated overnight, the cultures were centrifuged gently (4000 rpm, 10 min) to collect the cell pellet. One pellet was labeled at the day 0 sample and stored frozen at −80°C. The rest pellets were gently resuspended into 5 mL of fresh M9 medium containing 20 amino acids and returned to the incubator. After 24 h, all samples were centrifuged again, and one was frozen (the day 1 sample) while the rest was resuspended into fresh media and returned to the incubator. All samples were collected similarly. After the collection of the last sample, all samples were process together for labeling. Samples collected from the DL41ΔacrB-pQE70-AcrB culture were divided in half and used to analyze the degradation of AcrB and AcrA, respectively.
AcrB extraction and purification were conducted as described using Ni-NTA superflow resin except for the following modifications [17]. Briefly, E. coli cells (DL41ΔacrB or DL41ΔacrAB containing plasmid pQE70-AcrB, or DL41ΔacrB containing plasmid pQE70-AcrB-D407A) were resuspended and lysed in 20 mM sodium phosphate (pH 7.5), 0.2 M NaCl, 1% TritonX-100, and 1 mM phenyl methane-sulfonyl fluoride (PMSF), washed using the same buffer, and eluted using 10 mM sodium acetate, 100 mM NaCl, 3% SDS (pH 4.0). After elution, the pH of the fraction was adjusted back to pH 7.5 using an aliquot of 1 M phosphate buffer for the labeling reaction.
In the AcrA degradation experiment, E. coli cells (DL41ΔacrB containing pQE70-AcrB) were collected as described above. The cell pellets were resuspended in a buffer containing 20 mM sodium phosphate (pH 7.5), 0.2 M NaCl, 10% glycerol, and 1 mM PMSF. After cells were disrupted using sonication, the membrane fraction was collected by centrifugation and then solubilized in the same buffer supplemented with 1% Triton X-100 for 2 h. After centrifugation, the supernatant was incubated with 5 μl anti-AcrA antibody at 4 °C with shaking for 2 h. PMSF and protease inhibitor cocktail was added to inhibit protein degradation. Protein A beads were washed with sodium phosphate buffer and incubated with 5% BSA in sodium phosphate buffer at 4 °C for 4 h to decrease non-specific binding. After incubating protein extraction solution with the BSA saturated protein A beads at 4 °C for 1 h, beads were washed with 0.1% Tween-20 sodium phosphate buffer for 4 times. AcrA was eluted with 3% SDS sodium phosphate buffer (pH 7.5) by incubation for 10 min at 70 °C.
The click reaction was performed following a published protocol [19]. Breifly, purified AHA-containing protein was incubated with 200 μM Tris(benzyltriazolylmethyl) amine (TBTA), 500 μM ascorbic acid, 50 μM alkyne-biotin (PEG4 carboxamide-Propargyl Biotin), and 200 uM CuSO4. The reaction was allowed to proceed overnight at room temperature and analyzed directly using anti-biotin Western blot.
Results and Discussion
The incorporation of AHA did not affect protein activity
Before the AHA-incorporation method could be used to determine a protein’s degradation, it is important to confirm that the replacement of Met with AHA does not have a significant impact on the structure and function of the subject protein. While the incorporation of AHA into the sequence of proteins has been demonstrated in several cases, it is not always clear if the replacement of Met by AHA would affect the structure and function of the protein to be studied. We used a well-established EtBr efflux assay to investigate if the replacement of Met by AHA would affect the structure and function of the AcrAB-TolC system [16]. EtBr is actively transported by the AcrAB-TolC system. In the absence of AcrB, EtBr diffuses across the cell membrane into the bacteria, upon which its fluorescence increases drastically due to intercalation into nucleic acid. In the presence of active pumping, the outward efflux counter-acts the effect of the inward diffusion and EtBr is effectively removed from the cell. Therefore, the fluorescence does not increase as significantly.
We first created an acrB gene knockout E. coli strain of DL41 (DL41ΔacrB), and then transformed a plasmid encoding AcrB into the strain (DL41ΔacrB-pQE70-AcrB). As shown in Figure 1A, DL41ΔacrB rapidly accumulated EtBr, while the wild type DL41 did not. The introduction of pQE70-AcrB reduced the EtBr fluorescence of DL41ΔacrB to a level close to that of the parent strain, indicating that AcrB encoded in the plasmid is fully functional. Next, we used the acrB knockout strain to examine the effect of AHA incorporation on AcrB activity. In addition, since the incorporation of AHA is not protein-specific, it is necessary to confirm that the replacement of Met by AHA in general does not affect the growth and EtBr accumulation of the bacteria. DL41ΔacrB transformed with pQE70-AcrB or the empty vector pQE70 were cultured in a minimum media supplemented with 19 essential amino acids plus Met or AHA, and their EtBr accumulation plots were collected (Figure 1B). The fluorescence of DL41ΔacrB-pQE70-AcrB remained low in both Met- and AHA-containing media, indicating that the activity of the efflux system was not affected by replacing Met in the proteins with AHA. In the absence of the acrB gene, the fluorescence of DL41ΔacrB-pQE70 cultured in both Met-containing and AHA-containing media increased rapidly, revealing the accumulation of EtBr into the bacterial cells. These results indicate that the replacement of Met by AHA in AcrB does not affect the function, and thus the structure of the protein.
Figure 1.

A. EtBr accumulation assay of strains DL41 (black open circles), DL41ΔacrB-pQE70 (black filled circles), and DL41ΔacrB -pQE70-AcrB (red circles). The plasmid-encoded AcrB is functional in the knockout strain and effectively eliminates the accumulation of EtBr into the cytosol. B. EtBr accumulation assay of strains DL41ΔacrB-pQE70 (top two traces) and DL41ΔacrB -pQE70-AcrB (bottom two traces) cultured in the presence of Met (red) or AHA (black). The replacement of Met by AHA does not have a dramatic effect on the efflux of EtBr by AcrB. C. Growth of DL41 in the M9 media containing 19 essential amino acid without Met (squares), supplemented with Met (diamond), or supplemented with AHA (triangle).
To evaluate the effect of replacing Met by AHA on the growth of the cell, we cultured DL41 in the M9 media supplemented with 19 essential amino acids (no Met), 19 essential amino acids with AHA or all 20 essential amino acids. As shown in Figure 1C, DL41 barely grew in the absence of Met. In culture containing all 20 essential amino acids, the strain grew to saturation in ~10 hours. When Met was replaced by AHA, DL41 grew slower at the beginning and took 14 hours to reach saturation, but the saturation density of the culture was similar as the density of the culture containing Met.
The life-time of AcrB determined using the S35-Met pulse-chase experiment
To establish the benchmark values, we first used the conventional S35-Met pulse-chase experiment to estimate the lifetime of AcrB. DL41ΔacrB- pQE70-AcrB was used in this study. The bacteria were cultured in the presence of 50 μCi/ml (1000 Ci/mmol) S35-Met for 2 min, and then a 400-fold excess of non-radioactive Met (20 mM) was added. The cell culture was returned to the shaker, and aliquots of samples were withdrawn at the indicated time and analyzed. In a previous study, we found that the intensity of the radioactively labeled AcrB did not decrease over 24 hours [20]. Here we extended the experiment to 7 days (Figure 2A and 2B). The radioactive signals grew weaker overtime and reduced to the background level around day 7.
Figure 2.

A. Representative image of the result of S35-Met pulse-chase experiment over the period of seven days. Samples were collected at the indicated time after the chase. The band intensity reduced to less than 5% on day seven. B. Band intensities were measured and normalized for seven-day studies against the pre-chasing signal (time point 0). The average value and standard deviation of measurements from three independent measurements were shown. The blot image of two other experiments were shown in Electronic Supplementary Material (ESM, Fig. S1)
Monitoring the in vivo degradation of AcrB through the incorporation of AHA
Optimization of labeling condition
To detect AHA-containing proteins, a labeling step need to be performed in which an alkyne probe (containing a fluorophore or biotin) is used to react with the azido group in the side chain of AHA. A major challenge of applying the AHA-based tracing method to integral membrane proteins such as AcrB is the difficulty of reacting AHA with the alkyne probe. Our initial attempts following protocol from literature for soluble proteins yielded very low levels of labeling [5–9, 21]. In AcrB, the AHA side chains (originally Met side chains) seemed to be poorly accessible for reaction. We found that the following modifications were essential for the application of the method to AcrB:
The most important factor affecting the outcome of labeling is the unfolding of the target protein. We found that the protein needed to be fully unfolded to yield reproducible and high level of labeling. We experimented with unfolding AcrB using 8 M urea and 3% SDS before proceeding to the click reaction. Without the presence of the denaturing reagent, little labeling could be observed. When 8 M urea was used, a weak labeling signal could be observed, indicating certain AHA residues became accessible to reaction. Significant labeling could only be achieved after the protein was completely unfolded using 3% (w/v) SDS before labeling. In addition, we found that ascorbate worked best as the reducing reagent in our experiment [22]. Finally, we eluted our his-tagged protein using a change of pH as described in Materials and Method, since we found imidazole, even at a very low concentration, had an inhibitory effect on the subsequent labeling.
Monitoring the degradation of AcrB
To monitor the degradation of AcrB, we first cultured E. coli cells in the presence of AHA, and then chased using Met for 7 days (Figure 3). The upper panel is the coomassie blue stain to reflect the overall level of AcrB, while the lower panel is an image of the anti-biotin Western blot of the same set of samples. The biotinylated AHA-AcrB band is clearly visible for the first 5 days of incubation, and becomes much smaller in day 6 and 7, consistent with the result from the S35 Met pulse chase experiment.
Figure 3.

Tracing the disappearance of AHA-containing AcrB via biotinylation. A. Representative images of commassie blue stain (top, CB) and anti-biotin western blot (bottom, WB) of AcrB samples collected after the indicated time of chase using Met. From top to bottom, the molecular weight of the marker bands are 180, 130, 100, and 70 kD, respectively. B. Band intensities of the anti-biotin blot were measured and normalized against the pre-chasing signal (time point 0). The average value and standard deviation of measurements from three independent experiments were shown. The coomassie blue stained gel and blot image of two other experiments were shown in ESM (Fig. S2)
Degradation of a functionless AcrB mutant
AcrB is constitutively expressed. The level of AcrB in E. coli does not alter drastically with the concentration of potential inducers in the environment. For example, Rosenberg et al. have shown that the presence of various cholates induced the production of AcrB by 1.1 to 1.7 folds [23]. Therefore, we speculate that the long lifetime of AcrB is not correlated with its function, since the production of the protein is not very sensitive to the presence of substrate in the environment (and thus the need for active efflux). To examine if this is the case, we examined the degradation of a functionless mutant, AcrB-D407A. D407 is an essential residue on the proton translocation pathway of AcrB. The D407 to Ala mutation does not affect the overall structure of the protein, and yet the function is completely abolished [24]. We monitored the degradation of AHA-labeled AcrB-D407A as described above, and found that it also lasted for 6 days before the AHA signal reduced to less than 5%, indicating that active efflux is not a prerequisite for the long lifetime of the protein (Figure 4).
Figure 4.
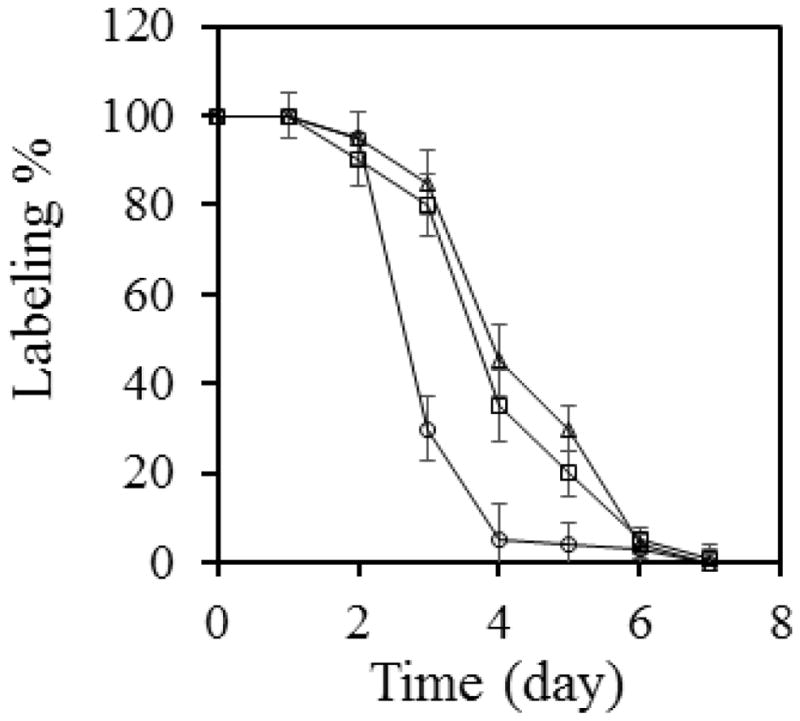
The lifetime of AcrB does not depend on function, but is affected by the presence of AcrA. The disappearance of AHA-containing AcrB was followed via biotinylation over the period of 7 days for AcrB (squares) or AcrB-D407A (triangles), or AcrB in an acrA-deficient strain (circles). Band intensities of the anti-biotin blot were measured and normalized against the before-chasing signal (time point 0). The average value and standard deviation of measurements from three independent experiments were shown.
Effect of AcrA on AcrB stability
AcrA is a periplasmic protein partner of AcrB. Both proteins, as well as the outer membrane channel TolC, are critical for drug efflux. TolC is not likely to form a constant complex with AcrA and AcrB, as it also interacts and functions with several other proteins. It is less clear if AcrA and AcrB interact with each other in the absence of active efflux. If the presence of AcrA influences the degradation of AcrB, then they are more likely to form a complex even without active efflux. To examine the effect of AcrA on AcrB degradation, we created an acrA and acrB double knockout strain, DL41ΔacrAB, and then examined the degradation of AcrB (Figure 4). We found that in the absence of AcrA, the AHA-AcrB signal reduced to ~5% in 4 days. This observation suggests that AcrA interacts with AcrB even in the absence of active efflux. And such interaction helps to stabilize the structure of AcrB in the cell membrane and increase its stability in E. coli.
In vivo degradation of AcrA
The genes of AcrA and AcrB are very close to each other in E. coli chromosome, with the starting codon of acrB only 23 bases downstream of the stop codon of acrA. Therefore, they are likely to share the same mRNA during protein production. Since the functions of AcrA and AcrB depend closely on each other, we speculate that the stability of AcrA is likely to be comparable to that of AcrB. Using the same method, we examined the degradation of AcrA (Figure 5). We found that AHA-AcrA also lasted for 6 days in E. coli cells under the same experimental condition, consistent with our expectation.
Figure 5.
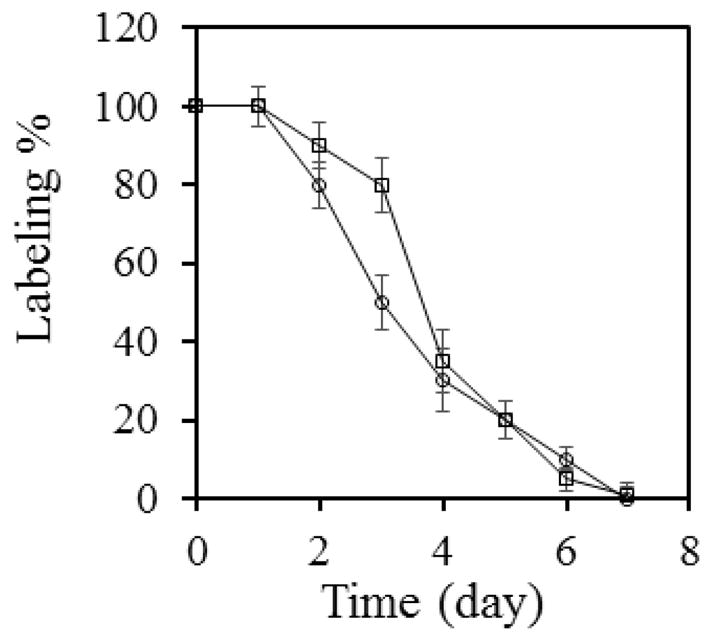
The disappearance of AHA-containing AcrA over the period of 7 days (circles). Data for AcrB were also shown in the same graph (squares) for comparison. Band intensities of the anti-biotin blot were measured and normalized against the pre-chasing signal (time point 0). The average value and standard deviation of measurements from three independent experiments were shown. The coomassie blue stained gel and blot image of these experiments were shown in ESM (Fig. S3)
Conclusion
In summary, here we used a non-radioactive pulse chase method to monitor the in vivo degradation of AcrA and AcrB and found they lasted 6–7 days in E. coli cells, which is reasonable as AcrA and AcrB are likely co-translated and their cellular functions are correlated. We also found that the stability of AcrB does not correlate with active efflux, but depends on the presence of AcrA.
The average lifetimes of proteins in different organisms have been the focus of several studies. Belle et al. measured the half-life of 3,751 proteins in the yeast proteome and found the distribution of half-lives to be approximately log-normal, with a mean and median half-life of ~43 min [25]. Systematic studies of mammalian cell lines cultured in vitro reveal that the average half-life of proteins ranges from hours to days, while the degradation rates of different protein families seemed to be conserved between mouse and human [26, 27]. Similar studies on E. coli were performed several decades ago. The half-life for bacterial proteins has been reported to be ~20 h [28–31]. In a more recent study, Larrabee et al. monitored the turnover of ~250 soluble proteins in E. coli growing at the log phase and found that the majority of them were not degraded to any appreciable extent in 70 min [32]. These studies indicate that although the doubling time and protein synthesis rate of bacteria cells are many fold faster than that of the eukaryotic cells, the protein degradation rate in general is not significantly faster. Different proteins in the same cell could have drastically different rates of degradation. AcrA and AcrB are undoubtedly among the longer-lived ones. This is consistent with their preventative protection role in the cell.
Supplementary Material
Fig. S1 Results from two additional repeats of the S35-Met pulse chase experiment result (see Fig. 2)
Fig. S2 Results from two additional repeats of the AcrB degradation experiments detected using the AHA labeling method (see Fig. 3)
Fig. S3 Results from three repeats of the AcrA degradation experiments detected using the AHA labeling method (see Fig. 5)
Acknowledgments
This work was supported by the National Science Foundation (MCB 1158036, YW) and National Institute of Allergy and Infectious Diseases (1R21AI103717, YW; R01 AI051517, RED; and F31 fellowship AI120653-01, SRW). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Compliance with Ethical Standards
The authors declare no potential conflicts of interest.
References
- 1.Goldberg A, ST John A. Annu Rev Biochem. 1976;45:747–803. doi: 10.1146/annurev.bi.45.070176.003531. [DOI] [PubMed] [Google Scholar]
- 2.Mayer R, Doherty F. FEBS Lett. 1986;198:181–93. doi: 10.1016/0014-5793(86)80403-3. [DOI] [PubMed] [Google Scholar]
- 3.Dice JF. FASEB J. 1987;1:349–57. doi: 10.1096/fasebj.1.5.2824267. [DOI] [PubMed] [Google Scholar]
- 4.Simon E, Kornitzer D. Methods Enzymol. 2014;536:65–75. doi: 10.1016/B978-0-12-420070-8.00006-4. [DOI] [PubMed] [Google Scholar]
- 5.Martell J, Weerapana E. Molecules. 2014;19:1378–93. doi: 10.3390/molecules19021378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avti PK, Maysinger D, Kakkar A. Molecules. 2013;18:9531–49. doi: 10.3390/molecules18089531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hatzenpichler R, Scheller S, Tavormina PL, Babin BM, Tirrell DA, Orphan VJ. Environ Microbiol. 2014;16:2568–90. doi: 10.1111/1462-2920.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dieck ST, Kochen L, Hanus C, Heumüller M, Bartnik I, Nassim-Assir B, Merk K, Mosler T, Garg S, Bunse S, Tirrell DA, Schuman EM. Nat methods. 2015;12:411–4. doi: 10.1038/nmeth.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Link AJ, Tirrell DA. J Am Chem Soc. 2003;125:11164–5. doi: 10.1021/ja036765z. [DOI] [PubMed] [Google Scholar]
- 10.Simon M, Stefan N, Borsig L, Pluckthun A, Zangemeister-Wittke U. Mol Cancer Ther. 2014;13:375–85. doi: 10.1158/1535-7163.MCT-13-0523. [DOI] [PubMed] [Google Scholar]
- 11.McClatchy DB, Ma YH, Liu C, Stein BD, Martinez-Bartolome S, Vasquez D, Hellberg K, Shaw RJ, Yates JR. J Proteome Res. 2015;14:4815–22. doi: 10.1021/acs.jproteome.5b00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dieterich DC, Hodas JJL, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deal RB, Henikoff JG, Henikoff S. Science. 2010;328:1161–4. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiroshi N, Yumiko R. Biochim Biophys Acta Proteins Proteomics. 2009;1794:769–81. [Google Scholar]
- 15.Koronakis V, Eswaran J, Hughes C. Annu Rev Biochem. 2004;73:467–89. doi: 10.1146/annurev.biochem.73.011303.074104. [DOI] [PubMed] [Google Scholar]
- 16.Li XZ, Poole K, Nikaido H. Antimicrob Agents Chemother. 2003;47:27–33. doi: 10.1128/AAC.47.1.27-33.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu W, Zhong M, Wei Y. Protein Pept Lett. 2011;18:863–71. doi: 10.2174/092986611796011446. [DOI] [PubMed] [Google Scholar]
- 18.Lu W, Chai Q, Zhong M, Yu L, Fang J, Wang T, Li H, Zhu H, Wei Y. J Mol Biol. 2012;423:123–34. doi: 10.1016/j.jmb.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Link AJ, Vink MK, Tirrell DA. J Am Chem Soc. 2004;126:10598–602. doi: 10.1021/ja047629c. [DOI] [PubMed] [Google Scholar]
- 20.Chai Q, Wang Z, Webb S, Dutch RE, Wei Y. Biochemistry. 2016;5:2301–4. doi: 10.1021/acs.biochem.6b00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinz FI, Dieterich DC, Tirrell DA, Schuman EM. ACS Chem Neurosci. 2012;3:40–9. doi: 10.1021/cn2000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596–9. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg EY, Bertenthal D, Nilles ML, Bertrand KP, Nikaido H. Mol Microbiol. 2003;48:1609–19. doi: 10.1046/j.1365-2958.2003.03531.x. [DOI] [PubMed] [Google Scholar]
- 24.Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. J Bacteriol. 2006;188:7290–6. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Proc Natl Acad Sci USA. 2006;103:13004–9. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cambridge SB, Gnad F, Nguyen C, Bermejo JL, Krüger M, Mann M. J Proteome Res. 2011;10:5275–84. doi: 10.1021/pr101183k. [DOI] [PubMed] [Google Scholar]
- 27.Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Science. 2008;322:918–23. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- 28.Willetts NS. Biochem J. 1967;103:453–61. doi: 10.1042/bj1030453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch AL, Levy HR. J Biol Chem. 1955;217:947–57. [PubMed] [Google Scholar]
- 30.Luscombe M, Phelps CF. Biochem J. 1967;102:110–9. doi: 10.1042/bj1020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borek E, Ponticorvo L, Rittenberg D. Proc Natl Acad Sci USA. 1958;44:369–74. doi: 10.1073/pnas.44.5.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larrabee KL, Phillips JO, Williams GJ, Larrabee AR. J Biol Chem. 1980;255:4125–30. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Results from two additional repeats of the S35-Met pulse chase experiment result (see Fig. 2)
Fig. S2 Results from two additional repeats of the AcrB degradation experiments detected using the AHA labeling method (see Fig. 3)
Fig. S3 Results from three repeats of the AcrA degradation experiments detected using the AHA labeling method (see Fig. 5)