

Abstract
In this Data in Brief article we provide a data package of GROMACS input files for atomistic molecular dynamics simulations of multicomponent, asymmetric lipid bilayers using the OPLS-AA force field. These data include 14 model bilayers composed of 8 different lipid molecules. The lipids present in these models are: cholesterol (CHOL), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE), 1-stearoyl-2-oleoyl-sn-glycero-3-phosphatidyl-ethanolamine (SOPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS), 1-stearoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (SOPS), N-palmitoyl-D-erythro-sphingosyl-phosphatidylcholine (SM16), and N-lignoceroyl-D-erythro-sphingosyl-phosphatidylcholine (SM24). The bilayers׳ compositions are based on lipidomic studies of PC-3 prostate cancer cells and exosomes discussed in Llorente et al. (2013) [1], showing an increase in the section of long-tail lipid species (SOPS, SOPE, and SM24) in the exosomes. Former knowledge about lipid asymmetry in cell membranes was accounted for in the models, meaning that the model of the inner leaflet is composed of a mixture of PC, PS, PE, and cholesterol, while the extracellular leaflet is composed of SM, PC and cholesterol discussed in Van Meer et al. (2008) [2]. The provided data include lipids׳ topologies, equilibrated structures of asymmetric bilayers, all force field parameters, and input files with parameters describing simulation conditions (md.mdp). The data is associated with the research article “Interdigitation of Long-Chain Sphingomyelin Induces Coupling of Membrane Leaflets in a Cholesterol Dependent Manner” (Róg et al., 2016) [3].
Keywords: Molecular dynamics simulations, Force field, Topology, Lipidomics, Lipid, GROMACS
Graphical abstract
Specifications Table
| Subject area | Chemistry, Biophysics |
| More specific subject area | Molecular dynamics simulations |
| Type of data | Text files |
| How data was acquired | Classical and quantum mechanics calculations |
| Data format | Consistent ordered package |
| Experimental factors | Software used: GROMACS 4 for MD, GAUSSIAN-03/09 for QM |
| Experimental features | OPLS-AA force field suit |
| Data source location | Not applicable |
| Data accessibility | Data are supplied with this article |
Value of the data
-
•
New parameters for MD simulations of PS and PE are provided.
-
•
New parameters for MD simulations of sphingomyelin are provided.
-
•
The lipid models given here are compatible with the OPLS-AA force field in studies of membrane proteins and drugs interacting with lipids.
-
•
The force field parameters and lipid topologies provided here may be used to construct lipid bilayers whose composition is closely related to that of biological membranes.
-
•
Equilibrated structures of asymmetric bilayer models may be used in MD studies of cell membranes.
1. Data
Lipidomics studies are becoming more and more popular [1] to identify in detail the lipid compositions found in cell membranes [2]. The emerging lipidomics data provide a very valuable source of information for atom-scale molecular dynamics (MD) simulations [3] that can clarify nanoscale dynamical phenomena in complex biomembrane systems.
To perform MD simulations, one needs detailed descriptions of the molecules that one aims to study [4]. First, one needs the topology, which includes information about all bonds, valence angles, and torsion angles in the molecule. Second, one needs a set of parameters, called the force field, to describe interactions related to all the interactions in the studied system. In this Data in Brief article, we provide a data package for the force field parameters and topologies for a number of lipids, in particular phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and sphingomyelin (SM).
2. Experimental design, materials and methods
In this article, we provide the topologies and force field parameters for 6 lipids compatible with the OPLS-AA force field [5], [6] to be used with the GROMACS simulation package [7]. The models described in this article are based on a combination of specific parameters for lipids derived in our previous studies [8], [9], [10], original OPLS-AA parameters, and a few specifically derived parameters for sphingomyelin. The description of cholesterol is entirely based on OPLS-AA parameters.
In our prior studies, we reparameterized (1) the torsion angles in the glycerol backbone and in the phosphocholine group, (2) the torsion angles and Lennard-Jones parameters in long saturated hydrocarbons [8], and (3) the torsion angles in unsaturated hydrocarbons [9]. All these components are necessary to describe a PC molecule. For PE and PS, we used the same parameters as for PC to describe the glycerol backbone, the phosphate group, and acyl chains, which are identical in these lipid classes. The parameters for the ethanolamine and serine groups were taken from the OPLS-AA force field derived for the amino acids׳ sidechain of lysine and serine, respectively.
The structure of SM is based on the amino alcohol sphingosine (Fig. 1) that is attached to a phosphatidylcholine head group and an acyl chain. To parameterize SM, we used previously derived parameters for the phosphatidylcholine group and long hydrocarbons [8], while the backbone part of sphingosine was reparameterized. First, partial charges were derived using a procedure described in our previous work [8], and the torsion angle potentials missing in the OPLS-AA set were calculated using a protocol described elsewhere [8]. Energy evaluation in torsion angles׳ potential parameterization was performed with the Hartree–Fock method by employing a polarized basis set 6−31G* augmented with diffuse functions (6−31+G**) on phosphorus and non-ester oxygen atoms. The effects of polar solvent were included implicitly by employing the Polarizable Continuum Model [11]. All electronic structure calculations were carried out using the GAUSSIAN-03/09 [12]. Fig. 1 shows the structure of SM16 with fragments of the molecule marked according to the source of parameters (Fig. 2).
Fig. 1.

Structure of SM16. The colors highlight the origin of force field parameterization for partial charges and torsional interactions. The color of the circle (red or blue) indicates the source of partial charges, and the color of the lines (red or black) connecting the circles indicates the source of torsional parameters. Red stands for parameterization based on [8], blue stands for parameterization based on new calculations reported in this work, and black corresponds to the original OPLS-AA parameters. There is reason to stress that all parameters for bonds and valence angle description are taken from the original OPLS-AA force field. Lennard-Jones parameters originate from OPLS-AA except for long hydrocarbons, which were derived in [8] (see text).
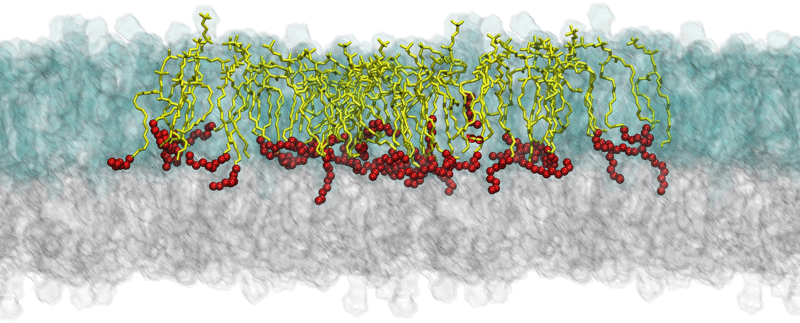
Fig. 2.

Example of the simulation results: snapshots of equilibrated structures of a multicomponent bilayer (model M3 in Ref. [3]) where SM24 molecules are shown as yellow sticks with the last eight carbon atoms depicted as red balls. The figure illustrates interdigitation of long SM24 tails into the opposite leaflet of the bilayer. Lipids in the outer leaflet are shown as transparent blue glass, while lipids in the inner leaflet are described as transparent gray glass. For clarity, the system is surrounded by its periodic image on the left and right sides (SM24 molecules are highlighted only in the central image).
Acknowledgments
We thank Arkadiusz Maciejewski for performing QM calculations of rotational energy profiles and fitting the torsional potentials. We also thank CSC – IT Center for Science (Espoo, Finland) for computing resources. We further acknowledge the Academy of Finland (Center of Excellence program), the European Research Council (Advanced Grant CROWDED-PRO-LIPIDS), the Research Council of Norway through its Centers of Excellence funding scheme (Project no. 179571), the Norwegian Cancer Society, and the South-Eastern Norway Regional Health Authority for financial support.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.dib.2016.03.067.
Appendix A. Supplementary material
Supplementary material
Supplementary material
References
- 1.Llorente A., Skotland T., Sylvänne T., Róg T., Kauhanen D., Orłowski A., Vattulainen I., Ekroos K., Sandvig K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta. 2013;1831:1302–1309. doi: 10.1016/j.bbalip.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 2.van Meer G., Voelker D.R., Feigenson G.W. Membranes lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Róg T., Orłowski A., Llorente A., Skotland T., Sylvänne T., Kauhanen D., Ekroos K., Sandvig K., Vattulainen I. Interdigitation of long-chain sphingomyelin induces coupling of membrane leaflets in a cholesterol dependent manner. Biochim. Biophys. Acta. 2016;1858:281–288. doi: 10.1016/j.bbamem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Vattulainen I., Róg T. Lipid simulations: a perspective on lipids in action. Cold Spring Harb. Perspect. Biol. 2011;3:a004655. doi: 10.1101/cshperspect.a004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jorgensen W.L., Maxwell D.S., TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 6.Robertson M.J., Tirado-Rives J., Jorgensen W.L. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 2015;11:3499–3509. doi: 10.1021/acs.jctc.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hess B., Kutzner C., van der Spoel D., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 8.Maciejewski A., Pasenkiewicz-Gierula M., Cramariuc O., Vattulainen I., Róg T. Refined OPLS-AA force field for saturated phosphatidylcholine bilayers at full hydration. J. Phys. Chem. B. 2014;118:4571–4581. doi: 10.1021/jp5016627. [DOI] [PubMed] [Google Scholar]
- 9.Kulig W., Pasenkiewicz-Gierula M., Róg T. Cis and trans unsaturated phosphatidylcholine bilayers: a molecular dynamics simulation study. Chem. Phys. Lipids. 2016;195:12–20. doi: 10.1016/j.chemphyslip.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Kulig W., Pasenkiewicz-Gierula M., Róg T. Topologies, structures and parameter files for lipid simulations in GROMACS with the OPLS-aa force field: DPPC, POPC, DOPC, PEPC, and cholesterol. Data Brief. 2015;5:333–336. doi: 10.1016/j.dib.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cossi M., Scalmani G., Rega N., Barone V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002;117:43–54. [Google Scholar]
- 12.M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery, T. Vreven, Jr., K.N. Kudin, J.C. Burant, et al., Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material