

Abstract
Key points
Blood pressure and flow exert mechanical forces on the walls of small arteries, which are detected by the endothelial and smooth muscle cells, and lead to regulation of the diameter (basal tone) of an artery.
CaV3.2 T‐type calcium channels are expressed in the wall of small arteries, although their function remains poorly understood because of the low specificity of T‐type blockers.
We used mice deficient in CaV3.2 channels to study their role in pressure‐ and flow‐dependent tone regulation and the possible impact of ageing on this role.
In young mice, CaV3.2 channels oppose pressure‐induced vasoconstriction and participate in endothelium‐dependent, flow‐mediated dilatation. These effects were not seen in mature adult mice.
The results of the present study demonstrate an age‐dependent impact of CaV3.2 T‐type calcium channel deletion in rodents and suggest that the loss of CaV3.2 channel function leads to more constricted arteries, which is a risk factor for cardiovascular disease.
Abstract
The myogenic response and flow‐mediated vasodilatation are important regulators of local blood perfusion and total peripheral resistance, and are known to entail a calcium influx into vascular smooth muscle cells (VSMCs) and endothelial cells (ECs), respectively. CaV3.2 T‐type calcium channels are expressed in both VSMCs and ECs of small arteries. The T‐type channels are important drug targets but, as a result of the lack of specific antagonists, our understanding of the role of CaV3.2 channels in vasomotor tone at various ages is scarce. We evaluated the myogenic response, flow‐mediated vasodilatation, structural remodelling and mRNA + protein expression in small mesenteric arteries from CaV3.2 knockout (CaV3.2KO) vs. wild‐type mice at a young vs. mature adult age. In young mice only, deletion of CaV3.2 led to an enhanced myogenic response and a ∼50% reduction of flow‐mediated vasodilatation. Ni2+ had both CaV3.2‐dependent and independent effects. No changes in mRNA expression of several important K+ and Ca2+ channel genes were induced by CaV3.2KO However, the expression of the other T‐type channel isoform (CaV3.1) was reduced at the mRNA and protein level in mature adult compared to young wild‐type arteries. The results of the present study demonstrate the important roles of the CaV3.2 T‐type calcium channels in myogenic tone and flow‐mediated vasodilatation that disappear with ageing. Because increased arterial tone is a risk factor for cardiovascular disease, we conclude that CaV3.2 channels, by modulating pressure‐ and flow‐mediated vasomotor responses to prevent excess arterial tone, protect against cardiovascular disease.
Key points
Blood pressure and flow exert mechanical forces on the walls of small arteries, which are detected by the endothelial and smooth muscle cells, and lead to regulation of the diameter (basal tone) of an artery.
CaV3.2 T‐type calcium channels are expressed in the wall of small arteries, although their function remains poorly understood because of the low specificity of T‐type blockers.
We used mice deficient in CaV3.2 channels to study their role in pressure‐ and flow‐dependent tone regulation and the possible impact of ageing on this role.
In young mice, CaV3.2 channels oppose pressure‐induced vasoconstriction and participate in endothelium‐dependent, flow‐mediated dilatation. These effects were not seen in mature adult mice.
The results of the present study demonstrate an age‐dependent impact of CaV3.2 T‐type calcium channel deletion in rodents and suggest that the loss of CaV3.2 channel function leads to more constricted arteries, which is a risk factor for cardiovascular disease.
Abbreviations
- CaV3.2KO
CaV3.2 knockout mice
- CSA
cross‐sectional area
- EC
endothelial cell
- EDH
endothelium‐dependent hyperpolarization
- FMVD
flow‐mediated vasodilatation
- IKCa
small conductance calcium‐activated potassium channel
- K9.5
Krebs buffer with 9.5 mm KCl
- K75
Krebs buffer with 75 mm KCl
- MR
myogenic response
- MT
myogenic tone
- NA
noradrenaline
- NO
nitric oxide
- PGI2
prostacyclin
- qPCR
quantitative real‐time PCR
- SKCa
intermediate conductance calcium‐activated potassium channels
- SMA
small mesenteric artery
- SNAP
S‐nitroso‐N‐acetyl‐dl‐penicillamine
- TRAM‐34
1‐[(2‐chlorophenyl)diphenylmethyl]‐1H‐pyrazole
- U46619
9,11‐dideoxy‐11α,9α‐epoxymethanoprostaglandin F2α
- VSMC
vascular smooth muscle cell
- WT
wild‐type
Introduction
Local regulation of blood perfusion in peripheral tissues relies in part on the mechanical forces exerted on the wall of small arteries and arterioles via changes in the intraluminal pressure (i.e. wall stress) or flow (i.e. shear stress). The myogenic response (MR) is a reflex by which an increase in pressure elicits vasoconstriction and a decrease in pressure elicits vasodilatation, such that blood flow is maintained as relatively constant throughout a broad range of pressures characteristic for each organ. Pressure is sensed by vascular smooth muscle cells (VSMCs) via mechano‐sensitive proteins in the extracellular matrix coupled to membrane‐spanning integrins (Hill & Meininger, 2012). Downstream myogenic depolarization, voltage‐dependent Ca2+ entry, Ca2+ sensitization and actin polymerization are commonly considered as key events leading to acute and sustained myogenic vasoconstriction (Hill et al., 2009; Walsh & Cole, 2013). Flow‐mediated vasodilatation (FMVD) is initiated by an increase in lumen shear stress sensed by the endothelial cells (ECs) via a mechanism that involves Ca2+ influx via TRPV4 channels (Kohler et al. 2006; Mendoza et al. 2010; Bubolz et al. 2012) and/or P2X4 receptors (Yamamoto et al. 2000; Ando & Yamamoto, 2013). Shear stress‐induced Ca2+ entry into ECs may evoke vasodilatation via the production of vasodilators such as nitrogen oxide (NO), prostacyclin (PGI2), epoxyeicosatrienoic acids and hydrogen peroxide (Bhagyalakshmi & Frangos, 1989; Fleming & Busse, 1999; Qiu et al. 2001; Huang et al. 2005; Liu et al. 2011). Several studies show that endothelium‐dependent vasodilatation is sensitive to inhibition of the small and intermediate‐conductance calcium‐activated potassium channels, SKCa and IKCa (Shimokawa et al. 1996; Takamura et al. 1999; Brandes et al. 2000; Dora et al. 2000; Parkington et al. 2002; Dora et al. 2008; Brahler et al. 2009; Potocnik et al. 2009). Thus, an increase in EC intracellular Ca2+ concentration ([Ca2+]i) to shear stress (Nilius & Droogmans, 2001) may activate SKCa and IKCa channels, leading to hyperpolarization of VSMCs (endothelium‐dependent hyperpolarization; EDH) and vasodilatation (Miura et al. 2001; Si et al. 2006; Brahler et al. 2009). Accordingly, it has been suggested that the EDH mechanism plays a more prominent role in small resistance vessels than the NO‐mediated pathway (Shimokawa et al. 1996; Brandes et al. 2000; Scotland et al. 2005; Brahler et al. 2009).
The family of voltage‐dependent Ca2+ channels consisting of 10 different genes is divided into the high voltage‐activated channels, comprising the L‐type, P/Q‐type, N‐type and R‐type, as well as the low voltage‐activated channels comprising the T‐type Ca2+ channels. The L‐type channel is the major Ca2+ entry channel coupling excitation with contraction in VSMC, and thus is of paramount importance for basal tone and blood pressure (Nelson et al. 1990; Smirnov & Aaronson, 1992; Moosmang et al. 2003). Mesenteric small arteries from rat and mouse abundantly express two of the three T‐type Ca2+ channel subtypes, namely the CaV3.1 and CaV3.2 channels (Gustafsson et al. 2001; Jensen et al. 2004; Ball et al. 2009; Braunstein et al. 2009; Björling et al. 2013); however, their function in the resistance vasculature remains poorly understood primarily as a result of the very low selectivity of pharmacological T‐type modulators. As noted in previous studies, the T‐type channels, with small unitary conductance, voltage‐dependent activation/inactivation at hyperpolarized potentials and fast inactivation, may not be well suited for operating under sustained activity of VSMCs in vivo where the membrane potential is between –45 and –35 mV under resting physiological pressure (Perez‐Reyes, 2003; Cribbs, 2006; Jensen & Holstein‐Rathlou, 2009; Smirnov et al. 2013). This has been explained by the existence of window currents by which T‐type channels can mediate basal Ca2+ entry through a small fraction of non‐inactivating channels (Bijlenga et al. 2000; Perez‐Reyes, 2003; Jensen & Holstein‐Rathlou, 2009). Furthermore, the existence of native T‐type splice variants with voltage‐dependent activation and inactivation curves shifted to more depolarized potentials than their recombinant counterparts may explain how Ca2+ entry through T‐type channels could have a function in arterioles (Chemin et al. 2001; Emerick et al. 2006; Jensen & Holstein‐Rathlou, 2009; Kuo et al. 2014). A recent study addressed the significance of molecular CaV3.1 channels in vascular responses to large depolarization using high‐KCl vs. small depolarization and stepwise increases from low to high arterial pressure. The CaV3.1 channels did not contribute to KCl‐ or noradrenaline (NA)‐induced Ca2+ entry or constrictions of mouse small mesenteric arteries (SMAs), whereas, at pressures of between 20 and 80 mmHg, CaV3.1 channels were important for myogenic tone (MT) (Björling et al. 2013). Moreover, inhibition of CaV3.2 channels using NiCl2 at micromolar concentrations did not inhibit KCl‐induced Ca2+ entry in CaV3.1 deficient mouse mesenteric arterioles in the presence of the L‐type inhibitor nifedipine (Björling et al. 2013). Thus, in certain physiological responses or vascular beds, where the membrane potential may be rather hyperpolarized and only small changes in membrane potential occur, the T‐type channels may have a distinct role compared to the L‐type channels. The CaV3.2 T‐type channel was shown to be expressed in VSMC and/or EC in several different resistance arteries (Blanks et al. 2007; Braunstein et al. 2009; Kuo et al. 2010; Poulsen et al. 2011), which makes the functional role of this channel particularly interesting. Because genetic ablation of the CaV3.2 T‐type channel isoform leads to permanently contracted coronary arterial smooth muscle, loss of NO‐mediated coronary vasodilator function and cardiac hypertrophy (with the latter being aggravated by increasing age), it appears that this isoform is involved in vasodilatation instead of vasoconstriction (Chen et al. 2003). The mechanism was explained to involve local Ca2+ entry through CaV3.2 channels activating co‐localized large conductance Ca2+‐activated K+ channels (BKCa), leading to hyperpolarization of VSMC, decreased cytosolic Ca2+ concentration and vasodilatation (Chen et al. 2003; Harraz et al. 2014 a). To clarify the specific role of CaV3.2 channel expression in mesenteric resistance vessels, we investigated the possibility that CaV3.2 channels oppose MT and contribute to a physiological endothelium‐dependent vasodilatation induced by increases in intraluminal flow. Our rigorous comparison of wild‐type (WT) vs. CaV3.2KO mouse arteries in all experiments enabled us to test the specificity of the putative CaV3.2 inhibitor NiCl2. Because there was an age‐dependent effect on cardiac fibrosis in CaV3.2 deficient mice (Chen et al. 2003) and because ageing is normally considered to be a risk factor for high blood pressure, we also tested the effect of ageing on the role of CaV3.2 channels in MT and FMVD responses. In addition, because the prolonged loss of vasodilator function and persistent increases in vascular tone might induce structural remodelling of the arterial wall, we evaluated the passive media‐to‐lumen ratio and media cross‐sectional area (CSA) in WT vs. CaV3.2KO mice in both age groups. Our results show a clear age‐dependency in the contribution of CaV3.2 channels to MT and FMVD. Furthermore, Ni2+ has both CaV3.2‐dependent and independent effects. The data are discussed in the context of CaV3.2 T‐type calcium channels having a specific vasodilator function in young individuals that is lost with advancing age, and we suggest that the loss of CaV3.2 channel function increases the risk for cardiovascular disease.
Methods
Animals and tissue preparations
Ethical approval for all animal procedures and transgenic mice were obtained from the Danish Animal Experiments Inspectorate and the Danish Working Environment Authority, respectively. CaV3.2−/− mice and age‐matched C57BL/J6 WT mice in two age groups were used (young: 8–17 weeks; adult: 28–56 weeks). Homozygous CaV3.2−/− mice, originally generated by Chen et al (2003) were bred by mating mice heterozygous for the deletion of amino acids 216−67 in the CaV3.2 protein, and backcrossing with C57BL/J6 WT mice for at least 10 generations. Genotyping was carried out as described previously (Chen et al. 2003). The mice were killed by cervical dislocation and the small intestine was transferred to a petri dish with aerated (5% CO2, 95% O2) Krebs buffer. Sprague–Dawley rats (7–12 weeks) were anaesthetized using CO2 followed by cervical dislocation. Second‐ or third‐order branches of SMAs from mice and rats were dissected using sharpened tweezers and ophthalmic scissors under a microscope (Leica M80; Leica Microsystems, Wetzlar, Germany) and used for pressure myography, immunofluorescence microscopy and quantitative real‐time PCR (qPCR).
Pressure myography
Mouse and rat mesenteric arteries were initially relaxed in Ca2+‐free Hepes buffered PSS (mounting buffer) and mounted on two cannulas with equal‐sized tips in a pressure myograph (model 120 CP; DMT A/S, Aarhus, Denmark) and then perfused with Krebs buffer to which 1% low‐endotoxin BSA had been added. After mounting the vessels, the myograph chamber was continuously superfused with Krebs buffer equilibrated with 5% CO2/95% O2 at 37°C at a rate of 2.3 mL min−1, thus exchanging one‐third of the bath volume per minute. The vessels were viewed through a 4× objective using an inverted fluorescence microscope (Olympus IX71; Olympus Denmark A/S, Ballerup, Denmark). Lumen and vessel (outer) diameters of the arteries were measured using a USB camera and MyoView II software (both DMT A/S) on a PC. For the experiments with rats, only the outer diameter was measured as a result of a change in the software.
Experimental protocol
Both rat and mouse mesenteric arteries were used for investigating FMVD, whereas only mouse mesenteric arteries were used for the MR. The initial mounting procedure was comparable for both types of experiment. Each experiment was divided into three trials: a control trial, an intervention and a trial using Ca2+‐free solution with EGTA to obtain the passive/maximal diameter of the vessel. For FMVD experiments, the vessels were equilibrated at 60 mmHg for 10–15 min until the bath was 37°C with flow enabled in the pressure myograph. Initially, the vitality of the vessel was tested using Krebs buffer with high [KCl] (75 mm; K75) and, subsequently, after relaxation in Krebs, U46619 (9,11‐dideoxy‐11α,9α‐epoxymethanoprostaglandin F2α; thromboxane A2 mimetic) was added directly to the bath (3.5–7 nm U46619) under no‐superfusion conditions until the vessel constricted 25–50% of the resting diameter. After stable pre‐constriction was obtained, flow was initiated in the same direction as the vessel had been exposed to in vivo, by changing the longitudinal pressure gradient (ΔP) along the vessel. A ΔP of 20 mmHg at a mean intraluminal pressure of 60 mmHg was controlled by changing the pressure head by 10 mmHg in both pipettes but in opposite directions, thus creating an equal pressure drop across both pipette tips with matched resistances. In this way there is no change in pressure in the middle of the vessel (Koller et al. 1993). The FMVD responses measured for 3 min at ΔP = 20 mmHg were reversibly induced three times in a row separated by 3 min at ΔP = 0 mmHg. Following the third FMVD response, when the vessel was still constricted at ΔP = 0 mmHg, the vitality of the endothelium was tested by directly adding 10 μm ACh to the bath. After resting in Krebs buffer, 30 μm NiCl2 was added directly to the bath under no‐superfusion conditions to inhibit the CaV3.2 Ca2+ channels. The same procedure as used in the control trial was carried out in the presence of U46619 to obtain the same level of pre‐constriction for each artery. At the end of each experiment, the vessel was superfused with Ca2+‐free Krebs buffer + EGTA to obtain the passive diameter for calculations of percentage dilatation and structural parameters. For MR experiments, the vessels were initially stretched at the maximal pressure of 120 mmHg to prevent bending, at the same time as the myograph bath was slowly heated to 37°C. In MR experiments, the luminal perfusate flow was blocked at the downstream end. The arteries were equilibrated for 15 min at 40 mmHg before initiation of the experiment. K75 was then added to constrict the vessel followed by washout with Krebs buffer, and again followed by constriction to 1 μm NA. After washout of NA, the vessels were equilibrated at 80 mmHg for 30–40 min, during which spontaneous myogenic constriction was usually observed. Next, the control pressure curve was constructed as described previously (Björling et al. 2013). In short, pressure was increased with an increment of 20 mmHg starting from 20 mmHg to 120 mmHg. The vessel was allowed to rest for 5 min at each step. Between the control and intervention periods, K75 was again used to constrict the vessel followed by 15 min of restitution. Subsequently, the vessel was exposed to 30 μm NiCl2 for 10 min before repeating the pressure curve in the presence of NiCl2. Before constructing the passive pressure curve in Ca2+‐free Krebs buffer + EGTA, the vessel was constricted to K75 in the presence of 30 μm NiCl2 to test for unspecific effects of NiCl2.
Vessels were excluded from further analysis in FMVD and MR experiments: (1) if the pressure in the outflow pipette dropped by >2 mmHg as a result of leaks; (2) if non‐uniform constrictions of the vessel were seen; (3) if the diameter measurements were not stable or fluctuated continuously because of technical problems or vasomotion; or (4) if <40% dilatation to ACh was observed in FMVD experiments.
Immunofluorescence microscopy
Immunostaining and fluorescence microscopy was performed as described previously (Björling et al. 2013). In brief, mouse mesenteric arteries were dissected, fixated for 15 min in 2% paraformaldehyde in PBS and subsequently kept in 30% sucrose in PBS for 1 h before they were snap‐frozen in liquid nitrogen when embedded in Tissue‐Tek® (Sakura Finetek Denmark Aps, Denmark). Frozen sections (5 μm) of the vessels were cut using a Cryostat (Leica CM 1950) and subsequently transferred to SuperFrost Plus microscope slides (Thermo Scientific, Fisher Scientific GmbH, Germany) where they were treated as described previously (Björling et al. 2013). A polyclonal antibody raised in rabbit against an epitope of the CaV3.2 T‐type Ca2+ channel (anti‐CaV3.2, Alomone Labs, Jerusalem, Israel) was diluted 1:100 or 1:200 in PBS and applied to the sections. Peptide pre‐incubation controls were included for CaV3.2 Alomone antibody using its short epitopic peptide as described previously for rats (Braunstein et al. 2009). In some WT vessels, we also tested the intensity of CaV3.1 T‐type channel staining, using a CaV3.1 antibody (1:1500) kindly provided by Dr Leanne Cribbs (Loyola University, IL, USA) and characterized previously (Björling et al. 2013; Brueggemann et al. 2005). After treatment with primary and secondary antibodies (dilution 1:800; Donkey anti‐rabbit Alexa Fluor 594) and 4′,6‐diamidino‐2‐phenylindole (dilution 1:10.000; both Life Technologies, Grand Island, NY, USA), a cover slip was mounted using anti‐fade medium (Dako, Glostrup, Denmark ). A BX50 fluorescence microscope (Olympus) equipped with a UPlanApo 40×/1.00 oil iris high‐quality quartz objective was used for inspection and images were acquired using a Retiga 6000 cooled monochrome CCD camera and Q‐Capture Pro7 software (Q‐Imaging Inc., Surrey, BC, Canada). All images were acquired using the exact same camera settings and the same type of final adjustments were applied using Photoshop CS6 (Adobe Systems Inc., San Jose, CA, USA). Background‐corrected total tissue fluorescence of CaV3.1‐specific staining was quantitated using ImageJ, version 1.48 (NIH, Bethesda, MD, USA) and compared in images acquired using simultaneous incubations of young and adult SMAs and the same settings of the fluorescence microscope and CCD camera.
qPCR
Second‐ and third‐order branches of both young and adult mouse mesenteric arteries were stored until use in RNAlater (Thermo Scientific) at 4°C. Endothelial tubes were isolated from 1–3 mm long segments of young mouse SMAs as described previously for mouse superior epigastric arteries (Socha & Segal, 2013). However, the SMA incubation time with collagenase (1.5 mg mL−1) + papain (0.62 mg mL−1) + 1,4‐dithioerythritol (1 mg mL−1) in dispersion buffer was increased to 60–70 min at 37°C to facilitate rapid isolation of endothelial tubes without scattered remaining VSMCs. Ten endothelial tubes from each mouse were transferred directly to the homogenization buffer. Tissue homogenization and extraction of RNA were performed as described previously using TissueLyzer and a RNEasy Micro kit (Qiagen, Hildenberg, Germany) (Björling et al. 2013). Subsequently, cDNA was made using a kit obtained from Promega (Madison, WI, USA) in accordance with the manufacturer's instructions. RT+ and RT– samples were prepared. Primer sets used are shown in Table 1. qPCR was performed using LightCycler 480 (Roche Diagnostics A/S, Hvidovre, Denmark) and SYBR Green PCR Master Mix (Qiagen). The qPCR protocol comprised annealing at 60°C for 10 s and elongation at 72°C for 20 s. Efficiencies were in the range 1.83–1.98 and slopes in the range –3.34 to –3.67. Melting curve analysis showed only single peaks. Samples with H2O or RT– did not show any product or the product appeared at least 10 cycles later than for RT+ samples. Expression of β‐actin (i.e. the reference gene) did not vary significantly between samples.
Table 1.
Primer sets used in qPCR
| Gene name | Forward primer (5′‐ 3′) | Reverse primer (5′‐ 3′) | Product size (bp) | Accession number |
|---|---|---|---|---|
| Kcnma1 | CAGTTTGACCACAACGCTGG | TGTGGGTACTCATGGGCTTG | 233 | NM001253378 |
| Kcnmb1 | CTCAACAGGTCCCTATCCATCT | CCAAAGTCACTGGGAATCGGA | 200 | NM031169 |
| Cacna1c | CGTTCTCATCCTGCTCAACA | GGTGTACCTCGGTGATTGCT | 236 | BC138031 |
| Cacna1g | TCATAGCCGTGCTGATGAAG | AAGGGAGAAGCCTGAAGAGG | 236 | NM009783.3 |
| Cacna1h | CCTTTCTCAGCGTCTCCAAC | GCCACAATGATGTCAACCAG | 169 | BC138026 |
| Cacna1a | TGCTGTGCTCACTGTTTTCC | TTCTCCACACGTTCCCTTTC | 198 | NM007578 |
| Trpc1 | GACATTCCAGGTTTCGTCTTG | GTCAGCACTAAGTTCAAACGC | 107 | NM011643 |
| Trpc3 | GATCGAGGATGACAGTGATGTAG | TCCATCATCGAAGTAGGAGAGC | 73 | NM019510 |
| Trpc6 | TACAATCTGGCCAGGATAAAGTG | CACAGCGATTGCATAAAGACC | 75 | NM013838 |
| Actb | AGCCATGTACGTAGCCATCC | CTCTCAGCTGTGGTGGTGAA | 228 | NM007393 |
Solutions and chemicals
Krebs buffer (in mm): 118 NaCl, 4.7 KCl, 1.2 MgSO4, 2 CaCl2, 25 NaHCO3 and 1.2 KH2PO4. Ca2+‐free solution for passive diameter measurements was prepared as described for Krebs buffer, except that CaCl2 was replaced by 2 mM EGTA. In Krebs buffer with 9.5 or 75 mm KCl, the additional amount of K+ replaced equimolar amounts of Na+. Ca2+‐free Hepes‐buffered PSS (mounting buffer; in mm): 140 NaCl, 5 KCl, 1.2 MgSO4 and 10 Hepes, with the pH being adjusted to 7.4 with NaOH. Note that 5 mm glucose was added to the above‐mentioned buffers before use. Dissection buffer for endothelial tube isolation was Ca2+‐free Hepes‐buffered PSS with 10 mm glucose and 0.1% BSA added. The dissociation buffer for endothelial tube isolation was Hepes‐buffered PSS with 2 mm CaCl2 + 10 mm glucose + 0.1% BSA. The pH of all solutions except the PSS buffers was adjusted continuously by equilibration with 5% CO2/95% O2. Low‐endotoxin BSA, NiCl2, U46619, NA, apamin, 1‐[(2‐chlorophenyl)diphenylmethyl]‐1H‐pyrazole (TRAM‐34), l‐NAME, S‐nitroso‐N‐acetyl‐dl‐penicillamine (SNAP), paxilline, collagenase, papain, 1,4‐dithioerythritol and chemicals of reagent grade were obtained from Sigma‐Aldrich (St Louis, MO, USA), unless stated otherwise.
Statistical analysis
Percentage of dilatation in the FMVD, ACh, SNAP and Krebs buffer with 9.5 mm KCl (K9.5) experiments was calculated as:
where D baseline is the diameter when pre‐constricted with U46619, D is the peak diameter measured at ΔP = 20 mmHg or with ACh, SNAP or K9.5, and D max is the diameter in Ca2+‐free Krebs’ buffer + EGTA.
The MT was calculated as the percentage difference in active lumen diameter in Krebs buffer and in Ca2+‐free Krebs buffer + EGTA at each pressure step using:
Statistical testing of differences between mouse genotypes within age‐groups was performed using two‐way ANOVA with a Bonferroni post hoc test for pairwise comparison of the MR and SNAP data. Student's t test was used for comparison of dilatations to flow, ACh and K9.5 between genotypes. One‐way ANOVA with the Bonferroni post hoc test was used for comparison of the effects of genotype and age for the structural remodelling and vasoconstrictor effects of K75 and NA. A Kruskal–Wallis test with Dunn's post hoc test was used for comparison of mRNA expression across genotype and age. P < 0.05 was considered statistically significant.
Results
MR
Because a previous study reported that the coronary arteries of CaV3.2 deficient mice (CaV3.2KO) were chronically more contracted than their matching WT mice (Chen et al. 2003), we hypothesized that the MR and MT would be enhanced in CaV3.2KO. We first tested, in SMAs from young WT mice (8–15 weeks), the effect on MR of a low concentration of NiCl2 (30 μm) considered to be specific for CaV3.2 channels (Lee et al. 1999). The active curve in Fig. 1 A shows that a MR was elicited at pressures >60 mmHg in contrast to the passive distension of the vessels shown in the passive curve. However, the presence of 30 μm NiCl2 caused the SMAs to be significantly more constricted than the active curve in the pressure range 40–100 mmHg. Next, we employed SMAs from young CaV3.2KO mice (8–15 weeks) to test the validity of this finding. As shown in Fig. 1 B, the active curve is more constricted throughout the pressure range tested and there was no significant effect of adding 30 μm NiCl2. At low pressure (20‐40 mmHg), the NiCl2‐treated CaV3.2KO vessels were somewhat more constricted than the active curve, although this was not significant. Nevertheless, it could indicate a CaV3.2 independent effect of NiCl2. Because ageing in humans is associated with stiffening of major arteries and an increase in systolic blood pressure, we decided to test the above effects in older mice. When we tested the MR in adult, middle‐aged WT mice (32–56 weeks), we observed active constriction at pressures >40 mmHg and generally more constriction of SMAs than in young mice (Fig. 1 C), and there was no significant effect as a result of adding 30 μm NiCl2. When we tested this in age‐matched CaV3.2KO mice, we observed a pattern similar to that in WT mice (i.e. the active curve shows constriction at pressures >40 mmHg), with no additional effect of adding NiCl2 (Fig. 1 D). Thus, the impact of CaV3.2 deletion with respect to enhancing MT in young mice (Fig. 1 E) is not present in mature adult (middle‐aged) mice (Fig. 1 F). Using NiCl2 as a tool, the results of the original study by Chen et al. (2003) and a recent study by Harraz et al. (2014 a) suggested that CaV3.2 channels and BKCa channels in arterial myocytes are functionally coupled to promote vasodilatation. To tests this hypothesis with our knockout model, we repeated the MR experiments in young mice in the presence of 1 μm paxilline (a specific blocker of BKCa channels). We found that the MT curves were significantly different before and after the addition of paxilline in young WT mice (Fig. 1 G), whereas no significant effect was seen in CaV3.2 knockout (CaV3.2KO) mice (Fig. 1 F). These data demonstrate that an interaction between CaV3.2 channels and BKCa channels at the level of individual VSMCs is possible.
Figure 1. Myogenic responsiveness in young vs. mature WT and CaV3.2KO mice .
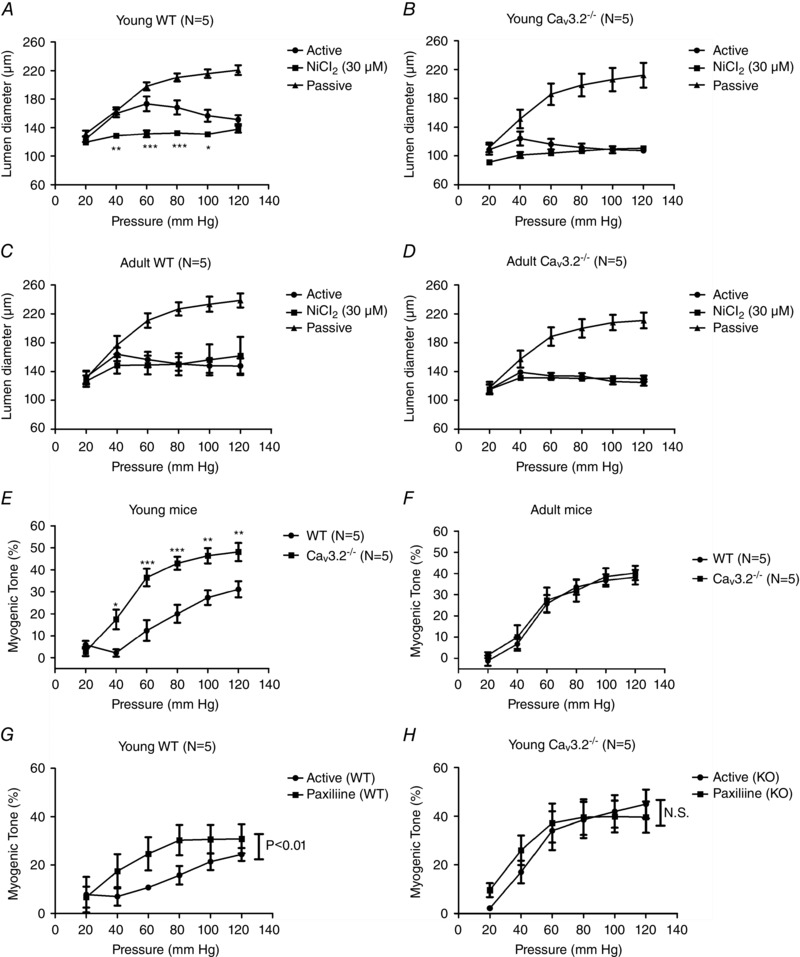
Myogenic response (and effects of 30 μm Ni2+) in young WT (A), young CaV3.2KO (B), mature adult WT (C) and mature adult CaV3.2KO (D) mouse mesenteric artery. Myogenic tone (MT, %) in young WT vs. CaV3.2KO mice (E, same mice as in A and B), and mature adult WT vs. CaV3.2KO mice (F, same mice as in C and D). Effects of paxilline on MT in young WT (G) vs. CaV3.2KO mice (H). * P < 0.05; ** P < 0.01; *** P < 0.001 at individual pressures.
KCl‐ and NA‐induced vasoconstriction
To complete our characterization of the contractile properties of the VSMC in SMA of CaV3.2KO and WT mice, we also performed tests of electromechanical and neurohormonal vasoconstriction using high‐KCl (75 mm) and NA (1 μm). These responses were evaluated as the percentage increase in vascular tone (ΔTone) upon exposure to the stimulus and are shown in Table 2. There were no differences between KCl‐induced vasoconstriction between WT and CaV3.2KO in young mice or adult mice. Furthermore, there were no significant effects on KCl‐induced responses to the addition of 30 μm NiCl2. For NA responses, no differences were detected between WT and CaV3.2KO mice in either of the two age groups; however, we observed a statistically significant increase in percentage tone in adult vs. young WT mice. Accordingly, we found a main effect of ageing with respect to increasing KCl‐ and NA‐induced vasoconstriction (P < 0.01 and P < 0.001, respectively) when including all mice in the analysis.
Table 2.
KCl‐ and NA‐induced vasoconstriction in mouse SMAs
| Young WT (n = 5) | Adult WT (n = 5) | Young KO (n = 5) | Adult KO (n = 5) | |
|---|---|---|---|---|
| Delta tone (%) | (8–17 weeks) | (28–56 weeks) | (8–17 weeks) | (28–56 weeks) |
| KCl (75 mm) | 74.6 ± 3.7 | 87.5 ± 4.4 | 70.0 ± 4.5 | 87.4 ± 4.8 |
| KCl + NiCl2 (30 μm) | 61.9 ± 3.6 | 64.6 ± 13.8 | 59.6 ± 6.1 | 81.2 ± 5.5 |
| NA (1 μm) | 22.8 ± 4.0 | 47.0 ± 6.6* | 30.1 ± 5.7 | 49.5 ± 5.9 |
* P < 0.05 vs. young WT.
FMVD
Because increased basal tone in CaV3.2KO mice could also be a result of endothelial dysfunction, we decided to test the vasodilator response to an increase in shear stress, which is a physiologically important stimulator of endothelium‐dependent vasodilatation. First, we established a protocol for measuring FMVD responses in the pressure myograph. We initially used rat SMAs because CaV3.2 channels were previously shown to be expressed in ECs of rat SMAs and arterioles (Braunstein et al. 2009) and because FMVD has previously been measured in this vascular bed (Thorsgaard et al. 2003;Christensen et al. 2007). Figure 2A shows an original recording in which a reversible FMVD response in a pre‐constricted SMA was elicited at a mean pressure of 60 mmHg to flow/shear stress induced by a longitudinal pressure gradient of 20 mmHg. When rat SMAs were exposed to the NOS inhibitor l‐Name (100 μm), no significant inhibition of FMVD was observed (Fig. 2 B). In a parallel set of experiments, incubation for 30–40 min with the SKCa Ca2+‐activated K+ channel blocker apamin (50 nm) and the IKCa channel blocker Tram‐34 (1 μm) almost abolished the FMVD responses in rat SMAs (Fig. 2 C). In preliminary experiments, we could not detect any effects of incubation with indomethacin and, indeed, previous studies have shown potent FMVD responses in the presence of this drug in rat SMAs (Thorsgaard et al. 2003; Christensen et al. 2007). Next, we tested the effect of 100 μm NiCl2 with respect to blocking the CaV3.2 channels and, unexpectedly, we found that the dilatation induced by intraluminal flow was converted to a constriction (Fig. 2 D). We speculated that this reversal of the response could partially be the result of an unspecific effect of NiCl2 and we therefore aimed to test the hypothesis that endothelial CaV3.2 channels is part of the mechanism in the FMVD response using WT vs. CaV3.2KO mice. In WT mice, the percentage dilatation to flow was less than in rats (compare Figs 2 and 3), whereas the FMVD response was significantly reduced in young CaV3.2KO mice compared to young WT mice (Fig. 3 A). In young WT and CaV3.2KO mice, the effect of 30 μm NiCl2 (IC50 = 12 μm for CaV3.2) (Lee et al, 1999) was not significant, although there was an apparent reversal of FMVD to a constriction in young CaV3.2KO mice. We also investigated FMVD in mature adult mice, and we found no significant difference between the groups (Fig. 3 B). When the main effect of NiCl2 was tested in all mice (both genotypes, both ages), there was a significant reduction in FMVD in the presence of NiCl2 (P < 0.05; n = 29–30). These data appear to suggest that NiCl2 exerted effects that were not mediated solely by CaV3.2 channel inhibition. There was no difference in FMVD between the young and adult WT mice, nor between the young and adult CaV3.2KO mice. There was also no difference when age as a main effect was tested.
Figure 2. Flow‐mediated vasodilatation in rat small arteries .
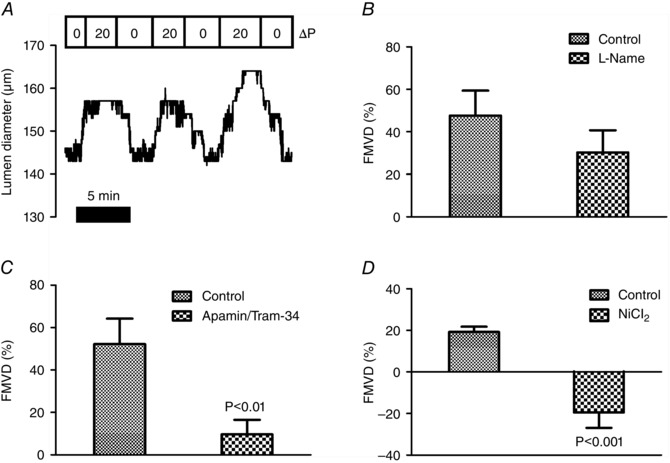
Original recording showing reversibility of FMVD (%) in SMAs induced by a longitudinal pressure gradient (ΔP = 20 mmHg) at a mean pressure of 60 mmHg in a cannulated vessel preconstricted using U46619 (A). Effects of l‐Name (100 μm) (B); apamin (50 nm) + Tram‐34 (1 μm) (C); and NiCl2 (100 μm) (D) on FMVD responses in rat mesenteric artery.
Figure 3. FMVD measurements in WT vs. CaV3.2KO mice .
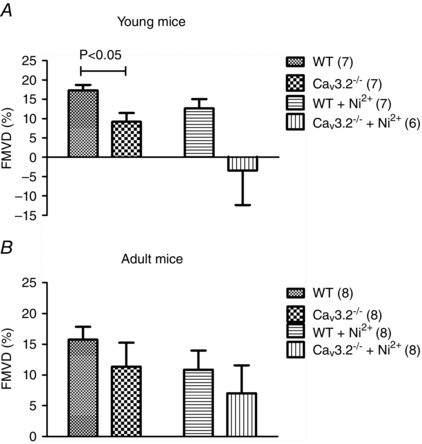
FMVD (and effects of 30 μm Ni2+) in young WT vs. CaV3.2KO mouse mesenteric arteries (A) and in mature adult WT vs. CaV3.2KO arteries (B).
ACh‐induced vasodilatation
In pressurized (60 mmHg), pre‐constricted (U46619) mouse SMAs, we tested the capacity for full endothelium‐dependent vasodilatation by adding a high concentration (10 μm) of ACh to the pressure myograph bath. In young WT mice, ACh induced an ∼70% vasodilatation, which was partially blocked by the addition of 30 μm NiCl2 (Fig. 4 A, left). In young CaV3.2KO, we observed an ∼80% ACh‐dilatation, which was not significantly different from WT. Again, NiCl2 induced block of ACh‐dilatation (Fig. 4 A, right). When we tested the same experimental conditions in adult WT and CaV3.2KO mice, we observed results that were qualitatively and quantitatively similar to those obtained in the young mice (Fig. 4 B). Thus, the effect of NiCl2 on ACh‐dilatation in pressurized mouse SMAs was not caused by CaV3.2 channels, which appears to support the unspecific role of NiCl2, as suggested by the FMVD results reported above.
Figure 4. Endothelium‐dependent (A, B) and ‐ independent (C, D) vasodilatation in WT vs. CaV3.2KO mice .
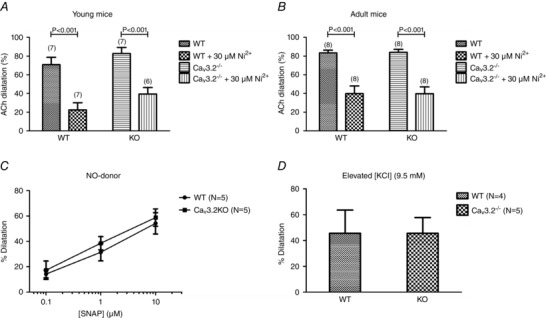
Dilatations (%) to ACh (ACh, 10 μm) in young WT vs. CaV3.2KO mouse mesenteric arteries (A) and in mature adult WT vs. CaV3.2KO arteries (B). C, concentration‐dependent dilatations to NO‐donor SNAP in age‐matched WT vs. CaV3.2KO arteries. D, dilatations to elevated bath [KCl] = 9.5 mM in age‐matched WT vs. CaV3.2KO arteries.
Endothelium independent vasodilatation
Because T‐type channel activity has been shown to be regulated by cGMP/PKG (Harraz et al. 2014 b) downstream of NO release, we decided to circumvent EC activation and to test the effect of activating the cGMP/PKG pathway in VSMC using the specific and potent NO‐donor SNAP. In concentrations from 10−7 to 10−5 m, we observed a highly similar degree of vasodilatation in pressurized, pre‐constricted mouse SMAs from WT vs. age‐matched CaV3.2KO mice (Fig. 4 C). Because CaV3.2 channels are more active at hyperpolarized membrane potentials, we tested the vasodilatation to a moderate elevation of bath [KCl] (to 9.5 mm), which is known to activate KIR channels and Na/K‐ATPase and also cause hyperpolarization of VSMCs and vasodilatation (Edwards et al., 1998). However, exposure of pressurized, pre‐constricted mouse SMAs to 9.5 mm K+ in Krebs buffer resulted in a highly similar degree of vasodilatation in WT and age‐matched CaV3.2KO mice (Fig. 4 D). We did not test the effects of NiCl2 in these experiments. These data clearly demonstrate that CaV3.2 channels do not participate in endothelium independent vasodilatation.
Acute effects of Ni2+ on resting diameter
The possible contribution of CaV3.2 channels to baseline diameter at an in vivo‐like intravascular pressure of 60 mmHg was evaluated as the absolute constrictions seen directly after NiCl2 addition to the myograph bath. As shown in Fig. 5, there was a constriction in young WT mice upon acute exposure to 30 μm NiCl2, and a similar significant effect was seen in age‐matched CaV3.2KO mice. Qualitatively and quantitatively similar results were obtained with the same protocol in adult WT mice and age‐matched CaV3.2KO mice. These data clearly demonstrate that, in pressurized resistance vessels, Ni2+ cannot be used as a specific blocker of CaV3.2 channels even at this low concentration.
Figure 5. Acute vasoconstriction to NiCl2 .
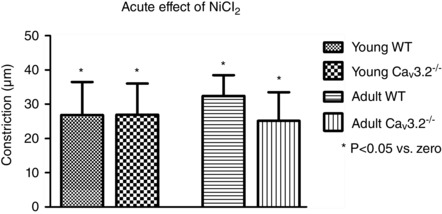
Acute vasoconstriction upon exposure to 30 μM NiCl2 in pressurized (60 mmHg) mesenteric arteries in young WT vs. CaV3.2KO arteries and in mature adult WT vs. CaV3.2KO arteries.
Structural remodelling of the vascular wall
Because both calcium and ageing are known to play a role in remodelling processes in the vascular wall, we tested the passive structural properties of the arterial wall in SMAs from young and adult WT and CaV3.2KO mice. We evaluated passive lumen diameter, passive vessel (outer) diameter, passive wall thickness, passive media/lumen‐ratio and passive media CSA at 60 mmHg. Fig. 6 shows the data obtained for passive lumen diameter, media/lumen‐ratio and media CSA only. Overall, there were no statistical differences between the WT and CaV3.2KO arteries in either young or mature adult mice. However, in arteries from both WT and CaV3.2KO mice, we found a main effect of age on passive media CSA, which was significantly increased in mature adult mice (P < 0.05; n = 24–26) (Fig. 6 C).
Figure 6. Passive structural properties of SMAs .
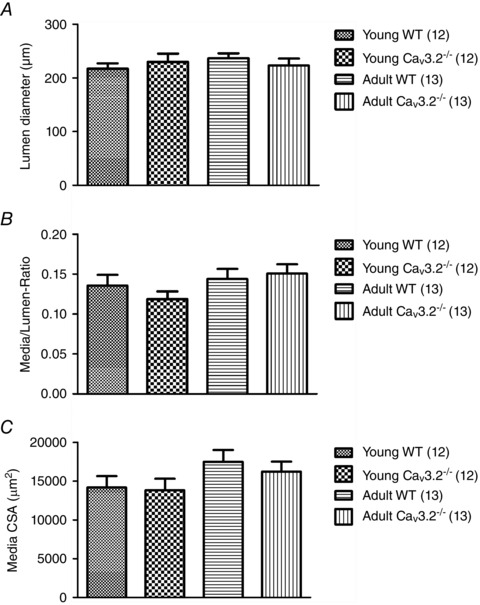
Passive lumen diameter (A), media/lumen‐ratio (B) and media CSA (C) in WT vs. CaV3.2KO arteries for both young and mature adult mice.
CaV3.2 localization
In a previous study using immunofluorescence microscopy, we reported the expression of CaV3.2 protein in both vascular smooth muscle and endothelium in rat SMAs and arterioles (Braunstein et al. 2009). In SMAs from young mice (n = 3), we found a homogeneous distribution of red CaV3.2 fluorescence in the media layer of the vessels corresponding to abundant CaV3.2 expression in VSMCs. A faint red staining corresponding to a weak CaV3.2 expression is seen in the intima layer next to EC nuclei on the lumen side of the green autofluorescent internal elastic lamina (Fig. 7 A). In the peptide pre‐incubation control (Fig. 7 B), we did not observe any red fluorescence, thus clearly demonstrating the specificity of the antibody. We also compared the CaV3.2 staining in young (n = 2) vs. adult (n = 2) mesenteric arteries and found no difference in the staining pattern or intensity (data not shown). Finally, we tested the mRNA expression of Cacna1H by qPCR on ∼10 isolated endothelial tubes (Fig. 7 C) from each of four young WT mice. In these experiments, crossing point (C t) values in the range 35–40 cycles for Cacna1H indicated a low gene expression level in endothelial tubes. ActB gene (β‐actin) was consistently amplified in all endothelial samples, with C t values in the range 25–29 cycles (data not shown). The gel in Fig. 7 D shows bands for the qPCR using Cacna1H primers in endothelial tubes (EC1–EC4) corresponding to the correct Cacna1H product size of 169 bp. To check for contamination with VSMCs in our qPCR using endothelial tubes, we quantitated gene expression of Cacna1C (L‐type channel) in these four mice; however, this was found to be negative, confirming the purity of our endothelial tube isolation (data not shown). Thus, transcription of Cacna1H into mRNA appears to occur at a low level in ECs, which is only barely detectable as translated protein expression in the endothelium of mouse mesenteric arteries in situ.
Figure 7. Expression of CaV3.2 at the mRNA and protein level in young mouse SMAs .
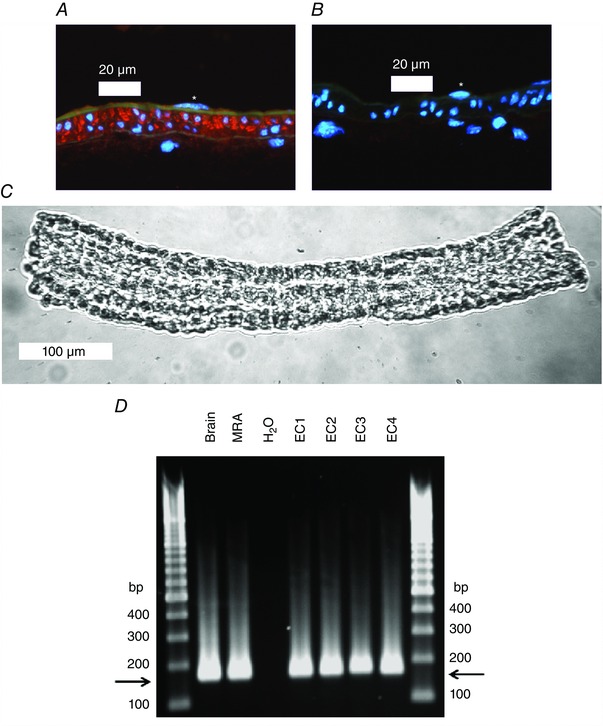
A, mouse SMA (young) stained with primary antibody against CaV3.2 (red colour). The internal and external elastic laminas are visible as faint green bands using the inherent autofluorescence of the tissue. Nuclei are stained blue with 4′,6‐diamidino‐2‐phenylindole. An endothelial cell nucleus is marked with an asterisk in the vessel lumen. Clear CaV3.2‐specific red staining is present in the media layer and faint red staining is visible in the intima layer on the luminal side of the green internal elastic lamina. B, negative peptide pre‐absorption control staining for CaV3.2 primary antibody of mouse mesenteric artery (young). Note the absence of a positive red signal. An endothelial cell nucleus is marked with an asterisk in the vessel lumen. C, image of isolated endothelial tube from a young mouse SMA. The image was acquired with a Axiovert S‐100 microscope (Carl Zeiss, Oberkochen, Germany) through a Neofluar 20× objective using a USB camera and Myoview II software. D, agarose gel showing electrophoresis of Cacna1H qPCR products from mouse brain (n = 1), mouse whole SMA (n = 1) and SMA endothelial tubes isolated from four young WT mice (EC1–EC4). Horizontal arrows depict the product size corresponding to Cacna1H (169 bp). Note the similar product size in brain, whole SMA and EC tubes.
Ca2+ and K+ channel expression profile
Because we have compared four different groups of mice (young WT, young knockout, adult WT, adult knockout), we investigated the level of expression of several Ca2+ channel and K+ channel genes in SMAs to check for transcriptional changes that could potentially interfere with our conclusions. As shown in Table 3, we did not find any significant differences between the four groups with respect to the expression of the BKCa α1‐ and β1‐subunits, CaV1.2, CaV2.1, CaV3.2 (using WT, only), TRPC1, TRPC3 or TRPC6. Surprisingly, for CaV3.1, we found a highly significant down‐regulation of mRNA expression in adult WT SMA compared to young WT, whereas only a tendency for the same change was observed between young vs. adult CaV3.2KO mice. To check the relevance of this dramatic difference (>50%) in CaV3.1 mRNA, we tested the hypothesis that it should be visible in immunostaining experiments. We therefore performed immunofluorescence microscopy using a previously characterized CaV3.1 antibody (Björling et al. 2013; Brueggemann et al. 2005) and found much brighter fluorescence in SMAs from young (Fig. 8 A) compared to adult SMAs (Fig. 8 B). When we measured the arbitrary fluorescence intensity corrected for background fluorescence and normalized it to the area of the tissue examined, we observed a lower intensity in the adult SMAs (Fig. 8 D). We performed similar measurements in SMAs from CaV3.2 KO mice aged 2 months (young; 4 vessels in one mouse), 8 months (adult; 7 vessels in 2 mice) and 21 months (old; 7 vessels in 2 mice) but we did not observe any difference in the staining intensity between these age groups (data not shown). Thus, these findings are consistent with an age‐dependent transcriptional down‐regulation of CaV3.1 channels in WT mice detectable at the mRNA and protein levels.
Table 3.
mRNA expression of selected ion channels in mouse sMAs
| Gene name (protein name) | Young WT (8–17 weeks) | Adult WT (28–56 weeks) | Young KO (8–17 weeks) | Adult KO (28–56 weeks) |
|---|---|---|---|---|
| Kcnma1 (BKCa α1) | 0.036 ± 0.002 | 0.040 ± 0.005 | 0.032 ± 0.004 | 0.035 ± 0.003 |
| Kcnmb1 (BKCa β1) | 19.35 ± 0.63 | 20.84 ± 0.70 | 15.96 ± 1.14 | 18.98 ± 0.42 |
| Cacna1c (CaV1.2) | 0.241 ± 0.012 | 0.286 ± 0.020 | 0.235 ± 0.025 | 0.224 ± 0.016 |
| Cacna1a (CaV2.1) | 0.022 ± 0.002 | 0.022 ± 0.001 | 0.023 ± 0.002 | 0.026 ± 0.003 |
| Cacna1g (CaV3.1) | 0.072 ± 0.007 | 0.030 ± 0.003 *** | 0.053 ± 0.006 | 0.038 ± 0.003 |
| Cacna1h (CaV3.2) | 0.151 ± 0.017 | 0.118 ± 0.013 | − | − |
| Trpc1 (TRPC1) | 0.100 ± 0.008 | 0.143 ± 0.011 | 0.110 ± 0.006 | 0.136 ± 0.009 |
| Trpc3 (TRPC3) | 0.078 ± 0.007 | 0.086 ± 0.011 | 0.061 ± 0.007 | 0.077 ± 0.009 |
| Trpc6 (TRPC6) | 0.131 ± 0.010 | 0.146 ± 0.013 | 0.163 ± 0.015 | 0.164 ± 0.013 |
*** P < 0.001 in young vs. adult WT.
Figure 8. Age‐dependent intensity of CaV3.1‐specific staining in mouse SMAs .
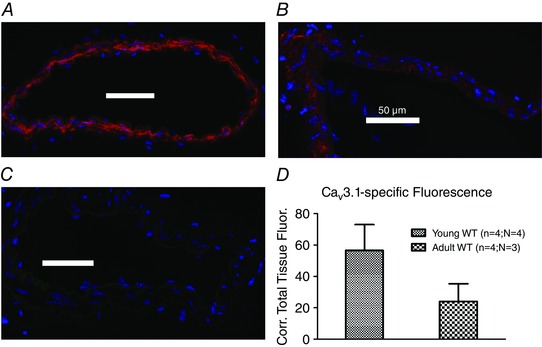
Immunostaining of the CaV3.1 T‐type isoform (red colour) in young (A) vs. mature adult (B) WT mouse mesenteric arteries, and in staining without primary antibody (C). Note the lower fluorescence intensity visible in the mature adult artery compared to the young artery. Images were acquired using same settings of microscope, camera and software. Quantitation of background‐corrected total tissue fluorescence in young vs. mature adult arteries using ImageJ (D).
Discussion
The present study addresses the putative role of CaV3.2 T‐type Ca2+ channels in vasomotor tone responses induced by changes in mechanical forces that are continuously acting on the vascular wall in vivo, such as flow (shear stress) and pressure (wall stress). We eliminated the potential influence of neurohormonal regulation using excised small resistance arteries, and we attempted to approximate physiological conditions using pressure myography with an in vivo‐like temperature, [CO2/HCO3 −] and pH. To avoid basing our conclusions on unspecific pharmacological T‐type antagonists, such as mibefradil, NNC 55‐0396 and Ni2+ (Björling et al. 2013; Moosmang et al. 2006; Stockand et al. 1993;), we carried out a rigorous comparison of age‐matched WT mice and mice with a global knockout of the Cacna1h gene encoding the CaV3.2 T‐type Ca2+ channels (Chen et al. 2003), which were previously found expressed at the mRNA (Braunstein et al. 2009; Gustafsson et al. 2001) and protein levels (Braunstein et al. 2009) in rat SMAs and arterioles. Because cardiovascular defects are often aggravated with advancing age, we compared all functional and structural responses in small arteries from young vs. mature adult mice of both genotypes.
First, we compared myogenic responsiveness in young vs. mature WT and CaV3.2KO mice. The WT arteries from young mice developed a MR at pressures >60 mmHg and this was significantly enhanced by 30 μm Ni2+, which was previously shown to be more specific for inhibition of CaV3.2 over CaV3.1 and CaV3.3 currents (Lee et al. 1999). In young CaV3.2KO arteries, the active pressure‐induced constriction was enhanced compared to WT, and no further effect was achieved by adding Ni2+ (Figs 1 A and B), showing that Ni2+ interfered with the CaV3.2 channels. In mature adult WT arteries, the MR developed at pressures >40 mmHg, and there was no effect as a result of adding Ni2+ in the WT. Furthermore, highly similar effects were obtained in the absence and presence of Ni2+ in the mature adult CaV3.2KO arteries (Figs 1 C and D). Taken together, our data suggest that CaV3.2 channels act as a vasodilator to oppose myogenic constriction in young mice (median age of 12 weeks), although this effect vanishes in adult mice at a median age of 42 weeks, which, in humans, corresponds to between the mature adult and middle age (Flurkey et al. 2007). The lack of an effect of CaV3.2 deletion in mature adult mice could not be explained by a lower expression level of CaV3.2 channels because the transcript level of this channel was not significantly different in young and adult mouse mesenteric arteries (Table 3) and also because we did not observe an age‐dependent difference in CaV3.2 immunofluorescence imaging experiments. This does not rule out the possibility that the channel function could be decreased in adult compared to young WT mice. The effect on MT observed in young mice is in agreement with several independent observations in the literature. First, the original study of CaV3.2KO mice performed by Chen et al. (2003) showed that CaV3.2KO mouse coronary arterioles were significantly more constricted than WT, and also that this was coincident with a higher prevalence of cardiac fibrosis. Second, recent studies have shown that MT in rodent and human cerebral arteries was enhanced by the addition of 50 μm Ni2+ (Harraz et al. 2014 a; Harraz et al. 2015 b). Finally, a very recent study has duplicated our findings showing that MT was enhanced in SMAs from young (8–16 weeks) CaV3.2KO mice compared to WT (Harraz et al. 2015 a). The latter study reported that 50 μm Ni2+ suppressed Ca2+ spark frequency and caused depolarization in VSMCs from the SMA of WT mice but not those from CaV3.2KO mice. However, the Ca2+ spark frequency and resting membrane potential under control conditions were similar in SMAs from WT and CaV3.2KO mice. It would have been interesting to evaluate whether the resting cytosolic Ca2+ concentration and/or the Ca2+ sensitivity is increased in the CaV3.2KO mice because this would explain the increased MT that was observed. Because cardiac fibrosis was more prevalent in CaV3.2KO mice compared to WT, and because this effect was dramatically increased in 1‐year‐old mice (Chen et al. 2003), our data showing an age‐dependent role of CaV3.2 channels in negative feedback on myogenic constriction could indicate a cardioprotective role of CaV3.2 channel expression in small resistance vessels in young individuals. This effect should not be confused with the reappearance of CaV3.2 channel expression in adult ventricular myocytes in mice with pressure overload‐induced cardiac hypertrophy (Chiang et al. 2009). It is very intriguing that a previous study found young (8–10 weeks) CaV3.2KO mice to be normotensive and to demonstrate a normal heart rate compared to age‐matched WT mice (Thuesen et al. 2014). With a higher basal tone at rest in the coronary, cerebral and mesenteric vasculature, the total peripheral resistance must be increased. Furthermore, with no indication of a lower sympathetic drive, the mean arterial pressure should be increased. We suggest that compensatory vasodilatation (e.g. by metabolic coupling) counteracts the increased basal tone in a major vascular bed such as the skeletal muscle. Alternatively, the cardiac stroke volume, which to our knowledge has not been measured in CaV3.2KO mice, is decreased, such that the mean arterial pressure is unchanged.
The proposed effect of 50 μm Ni2+ with respect to enhancing MT via CaV3.2 inhibition, as shown in several recent studies (Harraz et al. 2014 a; Harraz et al. 2015 a; Harraz et al. 2015 b), is consistent with the lack of an effect of 30 μm Ni2+ in arteries from the CaV3.2KO mice in the present study. However, we also demonstrated that 30 μm Ni2+ induced highly similar and significant acute vasoconstrictions in arteries maintained at 60 mmHg in both young and mature WT mice, as well as CaV3.2KO mice (Fig. 5). In addition, Ni2+ inhibited endothelium‐dependent vasodilatation to ACh in the CaV3.2KO mice (Fig. 4 A and B), and there was a significant main effect of Ni2+ with respect to inhibiting FMVD in all mice. Thus, the effects of Ni2+, as used to extrapolate the significance of CaV3.2 channels in the vasculature in many previous studies, must be interpreted with caution because it is evident from the results of the present study that Ni2+ bypasses CaV3.2 channels to promote vasoconstriction/inhibit vasodilatation via other mechanisms. It was previously shown that BKCa channel currents were inhibited by <1 μm Ni2+ in bovine mesenteric artery myocytes (Stockand et al. 1993) and this may partially explain the effects of Ni2+ observed in WT and CaV3.2 KO mice in the present study. Nevertheless, these observations highlight the poor selectivity of T‐type pharmacological tools and the need to employ knockout or knockdown studies to infer the function of T‐type channels in the vasculature. As a result of the frequent use of T‐type channel modulators, it is important to re‐evaluate a proposed link between CaV3.2 channels and BKCa channels and the negative modulation of myogenic constriction. This link was originally proposed as a result of the demonstration of a physical interaction between CaV3.2/BKCa in mouse brain microsomes using co‐immunoprecipitation (Chen et al. 2003). It was developed further as a model to explain negative feedback in the MR in cerebral arteries (Harraz et al. 2014 a; Harraz et al. 2015 a). We therefore tested the effect of BKCa inhibition in VSMCs in situ using 1 μm paxilline (Li & Cheung, 1999) and found that MT was significantly enhanced by paxilline in young WT mice, and also that this effect was absent in CaV3.2KO arteries (Fig. 1 G and H). Although this effect certainly argues for the relevance of a coupling between CaV3.2/BKCa with respect to modifying MT development, it does not show the universal importance of this coupling because it must be absent in mature adult arteries in which we showed no role of CaV3.2 channels.
To further our understanding of the role of CaV3.2 channels in arteriolar tone regulation, we investigated both endothelium‐dependent and independent vasodilator mechanisms, with the primary focus being on FMVD. In rat SMAs, FMVD responses to a driving pressure (ΔP) of 20 mmHg along the vessel were robust and reversible. Because FMVD responses were only insignificantly affected by l‐Name (Fig. 2 B), we tested the effect of SKCa and IKCa channel blockers. Indeed, the addition of a cocktail of 50 nm apamin + 1 μm Tram‐34 strongly inhibited the FMVD (Fig. 2 C), indicating that the EDH mechanism plays a prominent role in FMVD in this vascular bed. This observation is supported by a previous study showing that an EDH‐like mechanism is involved in FMVD in SMAs (Takamura et al. 1999), whereas other studies have shown that it is most probably the KCa2.3 (SKCa) channels that are involved in the FMVD responses by being co‐localized with shear stress‐activated TRPV4 channels in endothelial submembranous Ca2+ compartments, such as caveolae (Ma et al. 2013; Goedicke‐Fritz et al. 2015). Because we found CaV3.2 to be expressed in EC in rat mesenteric arteries and arterioles (Braunstein et al. 2009), we hypothesized that CaV3.2 could be involved in activation of endothelial KCa channels during FMVD; for example, by being co‐localized with TRPV4/SKCa in caveolae. Indeed, our first pharmacological observations using 100 μm Ni2+ appeared to confirm that CaV3.2 channels were involved because the flow‐mediated dilatation was reverted to a constriction by Ni2+ (Fig. 2 D). Thus, we tested the FMVD responses with and without Ni2+ in young vs. mature adult WT and CaV3.2KO mice (Fig. 3 A–B). However, Ni2+ had no significant effects on the FMVD response in either of the mouse strains. It is therefore difficult to determine whether the effects of Ni2+ on FMVD in rats were the result of blockade of CaV3.2 channels; however, the effects might be partially CaV3.2 independent because, in the young CaV3.2KO mice, we observed an apparent reversal of flow‐induced dilatation to a constriction, and we also found a main effect of Ni2+ to inhibit FMVD in all mice. Nevertheless, the most important observation from these experiments is that the FMVD response was significantly reduced (∼50%) in young CaV3.2KO mice compared to age‐matched WT mice, whereas no difference was observed for mature adult mice. This is very interesting because the age‐dependency is similar to that observed for the MR (Fig. 1 A–F), confirming our hypothesis that CaV3.2 channels are involved in either the sensing of shear stress or in the downstream signalling cascade to produce vasodilatation. There are no reports that CaV3.2 channels are mechano‐sensitive per se, although they are required for normal D‐hair receptor excitability and mechano‐sensitivity in dorsal root ganglia in mice (Shin et al. 2003). Another possibility is that they might be stimulated by shear stress‐evoked Ca2+ entry into EC either through TRPV4 channels or P2X4 receptors. Such stimulation of channel activity could arise if Ca2+‐dependent CamKII‐kinase is allowed to phosphorylate CaV3.2 channels (Welsby et al., 2003). Alternatively, the CaV3.2 channels are activated downstream of the shear stress‐dependent production of vasodilators such as PGI2, epoxyeicosatrienoic acids or hydrogen peroxide. Indeed, PGI2 activates PKA, which was shown to augment CaV3.2 channel activity (Kim et al. 2006). Because these vasodilators can diffuse to the underlying VSMCs, the CaV3.2 channels localized in this cell layer could be indirectly involved in the FMVD responses. Our finding that CaV3.2 channels were strongly expressed in the VSMC layer but only weakly expressed in the endothelium (Fig. 7) may not be consistent with a primary role of endothelial CaV3.2 channels. In the present study, we cannot definitively clarify the mechanism responsible for how CaV3.2 is involved in flow responses, and this requires further dissection of the signalling mechanisms. ACh (10 μm) was used at the end of all the FMVD experiments to confirm the presence of an intact endothelium. There was no difference between WT and CaV3.2KO in either young or mature adult mice, although there was a highly similar and significant reduction of ACh dilatation by Ni2+ in WT and CaV3.2KO mice, again showing that Ni2+ has CaV3.2 independent effects in native vascular tissues (Figs 4 A and B). Furthermore, endothelium independent vasodilatation to SNAP or as a result of raising extracellular [KCl] was not different between WT and CaV3.2KO mice, indicating the specific involvement of CaV3.2 in flow‐induced responses (Figs 4 C–D). This also allows exclusion of the effects of CaV3.2 via the cGMP/PKG pathway, as well as via Kir channel activation, because this would have affected our results using SNAP and raised extracellular [KCl], respectively.
By quantitating the mRNA expression profile in WT vs. CaV3.2KO mice (Table 3), we were able to rule out transcriptional changes in the expression of an array of important ion channels that could have compromised our conclusions. However, we did find a highly significant reduction in the transcript of the Cacna1g gene encoding the CaV3.1 T‐type channel in mature adult mice compared to young WT mice. This age‐dependent effect did not reach statistical significance in CaV3.2 KO mice. We found that the specific CaV3.1‐fluorescence immunostaining was much weaker in vessels from mature adult WT mice (Fig. 8 A–C) and, using a semi‐quantitative image‐based approach, we could detect a reduction in the corrected total tissue fluorescence intensity by >50% (Fig. 8 D). In CaV3.2KO mice, we could not detect such age‐dependent reduction of specific CaV3.1‐fluorescence immunostaining. An age‐related down‐regulation of CaV3.1 mRNA and protein expression was previously demonstrated in the brains of humans and mice (Rice et al. 2014). This down‐regulation of CaV3.1 protein might be considered to play a role in the age‐dependent effect of CaV3.2 channels in both MT and FMVD (e.g. by co‐localization and/or functional linkage of CaV3.1/CaV3.2 in vascular cells). The CaV3.1 down‐regulation coincides with the increased noradrenergic tone in mature adult WT arteries observed in the present study (Table 2). This could indicate a further role of CaV3.1 in vasodilatation, in addition to a role in MT at hyperpolarized voltages (Björling et al. 2013), and such a role in vasodilatation has been proposed previously (Svenningsen et al. 2014).
Using mice deficient in the CaV3.2 T‐type Ca2+ channel, we showed an age‐dependent role of CaV3.2 in MT and FMVD, which was present only at a young age. With a dual role in myogenic and flow‐induced tone regulation, CaV3.2 channels may have an important modulatory effect on blood pressure and flow‐regulation in young individuals. We suggest that the function of CaV3.2 in small arteries at a young age offers a protective role against excess arterial tone and cardiovascular disease, which is lost with advancing age. Finally, our discovery of an age‐dependent decline in mRNA and protein expression of the CaV3.1 T‐type isoform indicates a general role of T‐type channels in the process of ageing and perhaps in senescence in small arteries, which must be pursued in future studies.
Additional information
Competing interests
The authors declare that they have no competing interests.
Funding
The present study receieved support from The Danish Council for Independent Research | Medical Sciences, The Novo Nordisk Foundation, The Danish Heart Foundation and The A.P. Møller Foundation for the Advancement of Medical Science.
Author contributions
MFM, KB and LJJ carried out conceptual design of the study and performed experiments. LJJ carried out breeding of knockout mice and secured funding for the project. MFM, KB, and LJJ drafted the final version of the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
Vibeke Grøsfjeld Christensen is gratefully acknowledged for providing expert technical assistance. Professor Pernille B. L. Hansen is gratefully acknowledged for providing the CaV3.2 knockout mice used in the study, as well as for reading the manuscript. We also thank Professor Niels‐Henrik Holstein‐Rathlou for carefully reading the manuscript.
References
- Ando J & Yamamoto K (2013). Flow detection and calcium signalling in vascular endothelial cells. Cardiovasc Res 99, 260–268. [DOI] [PubMed] [Google Scholar]
- Ball CJ, Wilson DP, Turner SP, Saint DA & Beltrame JF (2009). Heterogeneity of L‐ and T‐channels in the vasculature: rationale for the efficacy of combined L‐ and T‐blockade. Hypertension 53, 654–660. [DOI] [PubMed] [Google Scholar]
- Bhagyalakshmi A & Frangos JA (1989). Mechanism of shear‐induced prostacyclin production in endothelial cells. Biochem Biophys Res Commun 158, 31–37. [DOI] [PubMed] [Google Scholar]
- Bijlenga P, Liu JH, Espinos E, Haenggeli CA, Fischer‐Lougheed J, Bader CR & Bernheim L (2000). T‐type alpha 1H Ca2+ channels are involved in Ca2+ signaling during terminal differentiation (fusion) of human myoblasts. Proc Natl Acad Sci USA 97, 7627–7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björling K, Morita H, Olsen MF, Prodan A, Hansen PB, Lory P, Holstein‐Rathlou NH & Jensen LJ (2013). Myogenic tone is impaired at low arterial pressure in mice deficient in the low‐voltage‐activated CaV 3.1 T‐type Ca(2+) channel. Acta Physiol (Oxf) 207, 709–720. [DOI] [PubMed] [Google Scholar]
- Blanks AM, Zhao ZH, Shmygol A, Bru‐Mercier G, Astle S & Thornton S (2007). Characterization of the molecular and electrophysiological properties of the T‐type calcium channel in human myometrium. J Physiol 581, 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de WC, Hoyer J & Kohler R (2009). Genetic deficit of SK3 and IK1 channels disrupts the endothelium‐derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119, 2323–2332. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Schmitz‐Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, Fleming I & Busse R (2000). An endothelium‐derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium‐dependent vasodilator in resistance vessels of wild‐type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA 97, 9747–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein TH, Inoue R, Cribbs L, Oike M, Ito Y, Holstein‐Rathlou NH & Jensen LJ (2009). The role of L‐ and T‐type channels in local and remote calcium responses in rat mesenteric terminal arterioles. J Vasc Res 46, 138–151. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Martin BL, Barakat J, KL Byron, & Cribbs LL (2005). Low voltage‐activated calcium channels in vascular smooth muscle: T‐type channels and AVP‐stimulated calcium spiking. Am J Physiol Heart Circ Physiol 288, H923–H935. [DOI] [PubMed] [Google Scholar]
- Bubolz AH, Mendoza SA, Zheng X, Zinkevich NS, Li R, Gutterman DD & Zhang DX (2012). Activation of endothelial TRPV4 channels mediates flow‐induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol 302, H634–H642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Bourinet E, Nargeot J & Lory P (2001). Alternatively spliced alpha(1 G) (Ca(V)3.1) intracellular loops promote specific T‐type Ca(2+) channel gating properties. Biophys J 80, 1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA, Hill JA & Campbell KP (2003). Abnormal coronary function in mice deficient in alpha1H T‐type Ca2+ channels. Science 302, 1416–1418. [DOI] [PubMed] [Google Scholar]
- Chiang CS, Huang CH, Chieng H, Chang YT, Chang D, Chen JJ, Chen YC, Chen YH, Shin HS, Campbell KP & Chen CC (2009). The Ca(v)3.2 T‐type Ca(2+) channel is required for pressure overload‐induced cardiac hypertrophy in mice. Circ Res 104, 522–530. [DOI] [PubMed] [Google Scholar]
- Christensen FH, Hansen T, Stankevicius E, Buus NH & Simonsen U (2007). Elevated pressure selectively blunts flow‐evoked vasodilatation in rat mesenteric small arteries. Br J Pharmacol 150, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs LL (2006). T‐type Ca2+ channels in vascular smooth muscle: multiple functions. Cell Calcium 40, 221–230. [DOI] [PubMed] [Google Scholar]
- Dora KA, Gallagher NT, McNeish A & Garland CJ (2008). Modulation of endothelial cell KCa3.1 channels during endothelium‐derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res 102, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA, Hinton JM, Walker SD & Garland CJ (2000). An indirect influence of phenylephrine on the release of endothelium‐derived vasodilators in rat small mesenteric artery. Br J Pharmacol 129, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Emerick MC, Stein R, Kunze R, McNulty MM, Regan MR, Hanck DA & Agnew WS (2006). Profiling the array of Ca(v)3.1 variants from the human T‐type calcium channel gene CACNA1G: alternative structures, developmental expression, and biophysical variations. Proteins 64, 320–342. [DOI] [PubMed] [Google Scholar]
- Fleming I & Busse R (1999). Signal transduction of eNOS activation. Cardiovasc Res 43, 532–541. [DOI] [PubMed] [Google Scholar]
- Flurkey K, JM Currer, & Harteneck C (2007). Mouse Models in Aging Research In The Mouse in Biomedical Research: Normative Biology, Husbandry, and Models, eds Fox J. G., Barthold S. W., Davisson M. T., Newcomer C. E., Quimby F. W. & Smith A. L., pp. 637–672. Academic Press, Burlington, MA. [Google Scholar]
- Goedicke‐Fritz S, Kaistha A, Kacik M, Markert S, Hofmeister A, Busch C, Banfer S, Jacob R, Grgic I & Hoyer J (2015). Evidence for functional and dynamic microcompartmentation of Cav‐1/TRPV4/K in caveolae of endothelial cells. Eur J Cell Biol 94, 391–400. [DOI] [PubMed] [Google Scholar]
- Gustafsson F, Andreasen D, Salomonsson M, Jensen BL & Holstein‐Rathlou N (2001). Conducted vasoconstriction in rat mesenteric arterioles: role for dihydropyridine‐insensitive Ca(2+) channels. Am J Physiol Heart Circ Physiol 280, H582–H590. [DOI] [PubMed] [Google Scholar]
- Harraz OF, Abd El‐Rahman RR, Bigdely‐Shamloo K, Wilson SM, Brett SE, Romero M, Gonzales AL, Earley S, Vigmond EJ, Nygren A, Menon BK, Mufti RE, Watson T, Starreveld Y, Furstenhaupt T, Muellerleile PR, Kurjiaka DT, Kyle BD, Braun AP & Welsh DG (2014. a). Ca(V)3.2 channels and the induction of negative feedback in cerebral arteries. Circ Res 115, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz OF, Brett SE & Welsh DG (2014. b). Nitric oxide suppresses vascular voltage‐gated T‐type Ca2+ channels through cGMP/PKG signaling. Am J Physiol Heart Circ Physiol 306, H279–H285. [DOI] [PubMed] [Google Scholar]
- Harraz OF, Brett SE, Zechariah A, Romero M, Puglisi JL, Wilson SM & Welsh DG (2015. a). Genetic ablation of CaV3.2 channels enhances the arterial myogenic response by modulating the RyR‐BKCa axis. Arterioscler Thromb Vasc Biol 35, 1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz OF, Visser F, Brett SE, Goldman D, Zechariah A, Hashad AM, Menon BK, Watson T, Starreveld Y & Welsh DG (2015. b). CaV1.2/CaV3.x channels mediate divergent vasomotor responses in human cerebral arteries. J Gen Physiol 145, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA & Meininger GA (2012). Arteriolar vascular smooth muscle cells: mechanotransducers in a complex environment. Int J Biochem Cell Biol 44, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Meininger GA, Davis MJ & Laher I (2009). Therapeutic potential of pharmacologically targeting arteriolar myogenic tone. Trends Pharmacol Sci 30, 363–374. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Jacobson A, Carroll MA, Falck JR & Kaley G (2005). Epoxyeicosatrienoic acids are released to mediate shear stress‐dependent hyperpolarization of arteriolar smooth muscle. Circ Res 96, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ & Holstein‐Rathlou NH (2009). Is there a role for T‐type Ca2+ channels in regulation of vasomotor tone in mesenteric arterioles? Can J Physiol Pharmacol 87, 8–20. [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Salomonsson M, Jensen BL & Holstein‐Rathlou NH (2004). Depolarization‐induced calcium influx in rat mesenteric small arterioles is mediated exclusively via mibefradil‐sensitive calcium channels. Br J Pharmacol 142, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Park JY, Kang HW, Huh SU, Jeong SW & Lee JH (2006). Augmentation of Cav3.2 T‐type calcium channel activity by cAMP‐dependent protein kinase A. J Pharmacol Exp Ther 318, 230–237. [DOI] [PubMed] [Google Scholar]
- Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T & Hoyer J (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress‐induced vasodilatation. Arterioscler Thromb Vasc Biol 26, 1495–1502. [DOI] [PubMed] [Google Scholar]
- Koller A, Sun D & Kaley G (1993). Role of shear stress and endothelial prostaglandins in flow‐ and viscosity‐induced dilation of arterioles in vitro. Circ Res 72, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Kuo IY, Ellis A, Seymour VA, Sandow SL & Hill CE (2010). Dihydropyridine‐insensitive calcium currents contribute to function of small cerebral arteries. J Cereb Blood Flow Metab 30, 1226–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo IY, Howitt L, Sandow SL, McFarlane A, Hansen PB & Hill CE (2014). Role of T‐type channels in vasomotor function: team player or chameleon? Pflügers Arch 466, 767–779. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL & Perez‐Reyes E (1999). Nickel block of three cloned T‐type calcium channels: low concentrations selectively block alpha1H. Biophys J 77, 3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G & Cheung DW (1999). Effects of paxilline on K+ channels in rat mesenteric arterial cells. Eur J Pharmacol 372, 103–107. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bubolz AH, Mendoza S, Zhang DX & Gutterman DD (2011). H2O2 is the transferrable factor mediating flow‐induced dilation in human coronary arterioles. Circ Res 108, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF, Li RA, Huang Y, Jin J & Yao X (2013). Functional role of TRPV4‐KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin‐induced diabetic rats. Hypertension 62, 134–139. [DOI] [PubMed] [Google Scholar]
- Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M & Zhang DX (2010). TRPV4‐mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298, H466–H476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Wachtel RE, Liu Y, Loberiza FR, Jr. , Saito T, Miura M & Gutterman DD (2001). Flow‐induced dilation of human coronary arterioles: important role of Ca(2+)‐activated K(+) channels. Circulation 103, 1992–1998. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Bruderl B, Welling A & Hofmann F (2006). Antihypertensive effects of the putative T‐type calcium channel antagonist mibefradil are mediated by the L‐type calcium channel Cav1.2. Circ Res 98, 105–110. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F & Klugbauer N (2003). Dominant role of smooth muscle L‐type calcium channel Cav1.2 for blood pressure regulation. EMBO J 22, 6027–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF & Standen NB (1990). Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol 259, C3–C18. [DOI] [PubMed] [Google Scholar]
- Nilius B & Droogmans G (2001). Ion channels and their functional role in vascular endothelium. Physiol Rev 81, 1415–1459. [DOI] [PubMed] [Google Scholar]
- Parkington HC, Chow JA, Evans RG, Coleman HA & Tare M (2002). Role for endothelium‐derived hyperpolarizing factor in vascular tone in rat mesenteric and hindlimb circulations in vivo. J Physiol 542, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E (2003). Molecular physiology of low‐voltage‐activated t‐type calcium channels. Physiol Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Potocnik SJ, McSherry I, Ding H, Murphy TV, Kotecha N, Dora KA, Yuill KH, Triggle CR & Hill MA (2009). Endothelium‐dependent vasodilation in myogenically active mouse skeletal muscle arterioles: role of EDH and K(+) channels. Microcirculation 16, 377–390. [DOI] [PubMed] [Google Scholar]
- Poulsen CB, Al‐Mashhadi RH, Cribbs LL, Skott O & Hansen PB (2011). T‐type voltage‐gated calcium channels regulate the tone of mouse efferent arterioles. Kidney Int 79, 443–451. [DOI] [PubMed] [Google Scholar]
- Qiu W, Kass DA, Hu Q & Ziegelstein RC (2001). Determinants of shear stress‐stimulated endothelial nitric oxide production assessed in real‐time by 4,5‐diaminofluorescein fluorescence. Biochem Biophys Res Commun 286, 328–335. [DOI] [PubMed] [Google Scholar]
- Rice RA, Berchtold NC, Cotman CW & Green KN (2014). Age‐related downregulation of the CaV3.1 T‐type calcium channel as a mediator of amyloid beta production. Neurobiol Aging 35, 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ & Ahluwalia A (2005). Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase‐1 double‐knockout mice: key role for endothelium‐derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation 111, 796–803. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M & Takeshita A (1996). The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium‐dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol 28, 703–711. [DOI] [PubMed] [Google Scholar]
- Shin JB, Martinez‐Salgado C, Heppenstall PA & Lewin GR (2003). A T‐type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci 6, 724–730. [DOI] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de WC, Hoyer J & Kohler R (2006). Impaired endothelium‐derived hyperpolarizing factor‐mediated dilations and increased blood pressure in mice deficient of the intermediate‐conductance Ca2+‐activated K+ channel. Circ Res 99, 537–544. [DOI] [PubMed] [Google Scholar]
- Smirnov SV & Aaronson PI (1992). Ca2+ currents in single myocytes from human mesenteric arteries: evidence for a physiological role of L‐type channels. J Physiol 457, 455–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov SV, Loutzenhiser K & Loutzenhiser R (2013). Voltage‐activated Ca(2+) channels in rat renal afferent and efferent myocytes: no evidence for the T‐type Ca(2+) current. Cardiovasc Res 97, 293–301. [DOI] [PubMed] [Google Scholar]
- Socha MJ & Segal SS (2013). Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp e50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockand J, Sultan A, Molony D, DuBose T, Jr. & Sansom S (1993). Interactions of cadmium and nickel with K channels of vascular smooth muscle. Toxicol Appl Pharmacol 121, 30–35. [DOI] [PubMed] [Google Scholar]
- Svenningsen P, Andersen K, Thuesen AD, Shin HS, Vanhoutte PM, Skott O, Jensen BL, Hill C & Hansen PB (2014). T‐type Ca(2+) channels facilitate NO‐formation, vasodilatation and NO‐mediated modulation of blood pressure. Pflügers Arch 466, 2205–2214. [DOI] [PubMed] [Google Scholar]
- Takamura Y, Shimokawa H, Zhao H, Igarashi H, Egashira K & Takeshita A (1999). Important role of endothelium‐derived hyperpolarizing factor in shear stress–induced endothelium‐dependent relaxations in the rat mesenteric artery. J Cardiovasc Pharmacol 34, 381–387. [DOI] [PubMed] [Google Scholar]
- Thorsgaard M, Lopez V, Buus NH & Simonsen U (2003). Different modulation by Ca2+‐activated K+ channel blockers and herbimycin of acetylcholine‐ and flow‐evoked vasodilatation in rat mesenteric small arteries. Br J Pharmacol 138, 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuesen AD, Andersen H, Cardel M, Toft A, Walter S, Marcussen N, Jensen BL, Bie P & Hansen PB (2014). Differential effect of T‐type voltage‐gated Ca2+ channel disruption on renal plasma flow and glomerular filtration rate in vivo. Am J Physiol Renal Physiol 307, F445–F452. [DOI] [PubMed] [Google Scholar]
- Walsh MP & Cole WC (2013). The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 33, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML & Barrett PQ (2003). A mechanism for the direct regulation of T‐type calcium channels by Ca2+/calmodulin‐dependent kinase II. J Neurosci 23, 10116–10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Korenaga R, Kamiya A & Ando J (2000). Fluid shear stress activates Ca(2+) influx into human endothelial cells via P2×4 purinoceptors. Circ Res 87, 385–391. [DOI] [PubMed] [Google Scholar]