

Abstract
Key points
The cortical collecting duct (CCD) plays an essential role in sodium homeostasis by fine‐tuning the amount of sodium that is excreted in the urine.
Ex vivo, the microperfused CCD reabsorbs sodium in the absence of lumen‐to‐bath concentration gradients.
In the present study, we show that, in the presence of physiological lumen‐to‐bath concentration gradients, and in the absence of endocrine, paracrine and neural regulation, the mouse CCD secretes sodium, which represents a paradigm shift.
This secretion occurs via the paracellular route, as well as a transcellular pathway that is energized by apical H+/K+‐ATPase type 2 pumps operating as Na+/K+ exchangers.
The newly identified transcellular secretory pathway represents a physiological target for the regulation of sodium handling and for anti‐hypertensive therapeutic agents.
Abstract
In vitro microperfusion experiments have demonstrated that cortical collecting ducts (CCDs) reabsorb sodium via principal and type B intercalated cells under sodium‐depleted conditions and thereby contribute to sodium and blood pressure homeostasis. However, these experiments were performed in the absence of the transepithelial ion concentration gradients that prevail in vivo and determine paracellular transport. The present study aimed to characterize Na+, K+ and Cl− fluxes in the mouse CCD in the presence of physiological transepithelial concentration gradients. For this purpose, we combined in vitro measurements of ion fluxes across microperfused CCDs of sodium‐depleted mice with the predictions of a mathematical model. When NaCl transport was inhibited in all cells, CCDs secreted Na+ and reabsorbed K+; Cl− transport was negligible. Removing inhibitors of type A and B intercalated cells increased Na+ secretion in wild‐type (WT) mice but not in H+/K+‐ATPase type 2 (HKA2) knockout mice. Further inhibition of basolateral NaCl entry via the Na+‐K+‐2Cl− cotransporter in type A intercalated cells reduced Na+ secretion in WT mice to the levels observed in HKA2−/− mice. With no inhibitors, WT mouse CCDs still secreted Na+ and reabsorbed K+. In vivo, HKA2−/− mice excreted less Na+ than WT mice after switching to a high‐salt diet. Taken together, our results indicate that type A intercalated cells secrete Na+ via basolateral Na+‐K+‐2Cl− cotransporters in tandem with apical HKA2 pumps. They also suggest that the CCD can mediate overall Na+ secretion, and that its ability to reabsorb NaCl in vivo depends on the presence of acute regulatory factors.
Keywords: H‐K‐ATPase, ion transport, mathematical model, microperfusion, sodium
Key points
The cortical collecting duct (CCD) plays an essential role in sodium homeostasis by fine‐tuning the amount of sodium that is excreted in the urine.
Ex vivo, the microperfused CCD reabsorbs sodium in the absence of lumen‐to‐bath concentration gradients.
In the present study, we show that, in the presence of physiological lumen‐to‐bath concentration gradients, and in the absence of endocrine, paracrine and neural regulation, the mouse CCD secretes sodium, which represents a paradigm shift.
This secretion occurs via the paracellular route, as well as a transcellular pathway that is energized by apical H+/K+‐ATPase type 2 pumps operating as Na+/K+ exchangers.
The newly identified transcellular secretory pathway represents a physiological target for the regulation of sodium handling and for anti‐hypertensive therapeutic agents.
Abbreviations
- ANF
atrial natriuretic factor
- CCD
cortical collecting duct
- ENaC
epithelial Na+ channel
- CFTR
cystic fibrosis transmembrane conductance regulator
- HCTZ
hydrochlorothiazide
- HKA2
H+/K+‐ATPase type 2
- IMCD
inner medullary collecting duct
- NDCBE
Na+‐driven Cl−/HCO3 − exchanger
- NET
non‐equilibrium thermodynamics
- ROMK
renal outer medullary K+ channel
- ΔVte
transepithelial electric potential difference
- WT
wild‐type
Introduction
The urinary excretion of sodium, potassium and chloride is fine‐tuned in the aldosterone‐sensitive distal nephron, which consists of the distal convoluted tubule, the connecting tubule and the collecting duct. Results based on the microperfusion technique have shown that the cortical collecting duct (CCD) mediates NaCl reabsorption in response to aldosterone or sodium depletion. The CCD epithelium consists of principal cells and intercalated cells; the latter are subdivided into type A, type B and non‐A non‐B. Until recently, sodium reabsorption in the CCD was assumed to be mediated exclusively by principal cells, via the amiloride‐sensitive epithelial Na+ channel (ENaC) on the apical side and Na+/K+‐ATPase on the basolateral side. In 1990, an additional, thiazide‐sensitive NaCl transport pathway was identified in the rat CCD (Terada & Knepper, 1990). Several groups subsequently characterized this pathway in the mouse CCD. Wall et al. (2004) first showed that Cl− reabsorption is abolished in mice lacking the apical Cl−/HCO3 − exchanger pendrin (slc26a4). Subsequently, it was demonstrated that type B intercalated cells can mediate NaCl reabsorption under the concerted action of several transporters, including pendrin, the apical Na+‐driven Cl−/HCO3 − exchanger (NDCBE) (slc4a8) and the basolateral Na+/HCO3 − cotransporter AE4 (Slc4a9) (Leviel et al. 2010). It was also reported that this NaCl reabsorption is energized by the basolateral H+‐ATPase (Chambrey et al. 2013).
In addition, a recent study showed that, following aldosterone administration, type A intercalated cells in the CCD of sodium‐replete mice express the bumetanide‐sensitive Na+‐K+‐2Cl− cotransporter 1 NKCC1 (slc12a2) in the basolateral region. Moreover, ENaC inhibition stimulates Cl− secretion through a NKCC1‐dependent mechanism (Pech et al. 2012). Because NKCC1 carries one Na+ ion and one K+ ion together with 2 Cl− ions into the cell, type A cells could also mediate Na+ secretion across the CCD, via NKCC1 on the basolateral side and the yet‐to‐be‐determined Na+ transporters on the apical side. Indeed, bumetanide‐sensitive Na+ secretion was observed in the outer medullary collecting ducts of deoxycorticosterone pivalate‐treated rats that were perfused in vitro (Wall & Fischer, 2002), although the underlying pathways were not determined.
Because CCDs are not accessible to in vivo micropuncture and microperfusion studies, our understanding of their transport function derives from in vitro microperfusion experiments. However, ion fluxes across the CCD are usually measured under symmetric conditions, that is, with identical solutions in the perfusion solution and the bath, whereas, in vivo, the composition of the tubular fluid entering the CCD differs significantly from that of the surrounding interstitium. The luminal concentration of Na+ and Cl− in the late superficial distal tubule is 0.2 to 0.5 times that in plasma; conversely, the concentration of K+ is 2–4 times greater in the lumen than in the interstitium (Elalouf et al. 1984; Beck et al. 1988). The luminal concentration of HCO3 − was reported to be 5 mm (Buerkert et al. 1983). In the presence of these transepithelial concentration gradients, the paracellular pathway should mediate Na+ secretion, thereby counterbalancing, at least partly, Na+ reabsorption across principal and type B cells.
The present study aimed to determine the net direction of NaCl transport across the CCD under asymmetric conditions as encountered in vivo, and also to characterize the contribution of each pathway to overall fluxes. Accordingly, we combined measurements of ion fluxes across microperfused cortical collecting ducts with the predictions of a mathematical model of transepithelial transport across the CCD.
Methods
Ethical approval
Experimental protocols were approved by the Darwin's Ethic Committee and the French Ministère de la Recherche (Agreement # 2289.01).
Experimental procedures
All experiments were performed on male 8–10‐week‐old mice. A colony of H+/K+‐ATPase type 2 (HKA2) knockout mice (C57BL/6J background) was obtained from P. Meneton (Meneton et al. 1998). Control (wild‐type; WT) mice were either commercial C57BL/6J mice (Charles River, L'arbresle, France) or littermate homozygous HKA2+/+ mice; there was no difference between these mice and their results were pooled. All animals were fed a Na+‐depleted diet (containing 0.11 g of Na+ per kg) for 2 weeks before the microperfusion experiments.
Microperfusion studies
Mice were killed by cervical dislocation and their right kidney was removed. CCDs were microdissected from cortico‐medullary rays and microperfused under asymmetrical conditions. The bath contained (in mm): 118 NaCl, 23 NaHCO3, 1.2 MgSO4, 2 K2HPO4, 2 calcium lactate, 1 sodium citrate, 5.5 glucose and 12 creatinine (pH 7.4). The perfused solution contained (in mm): 74 NaCl, 1 KCl, 2 K2HPO4, 5 K‐gluconate, 5 KHCO3, 1.2 MgSO4, 2 calcium lactate, 1 sodium citrate, 5.5 glucose and 12 creatinine (pH 7.04). The bath and perfusate were continuously gassed with 95% O2/5% CO2 at 37°C. The transepithelial voltage difference (ΔV te) was measured via microelectrodes connected through an Ag/AgCl half‐cell to an electrometer. Measurements were performed during the first 90 min of perfusion. During four periods of 15 min each, 30–35 nl of fluid was collected. The collection volume was determined under water‐saturated mineral oil with calibrated volumetric pipettes. Concentrations of Na+, K+ and creatinine were determined by HPLC using 26 nl of collected fluid. Concentrations of Cl– were measured by microcoulometry on 2–3 nl of collected fluid. For each collection period, the flux of ion X per unit length of tubule (J X) was calculated as J X = ([X]p × V p – [X]c × V c)/(L × t), where [X]p and [X]c are the concentrations of ion X in the perfusate and collectate, respectively; V p and V c are the perfusion and collection rates, respectively; L is the tubule length; and t is the collection time. V p was calculated as V p = V c × [creat]c/[creat]p, where [creat]c and [creat]p are the concentrations of creatinine in the collectate and perfusate, respectively. For each tubule, ion fluxes were averaged over the four collection periods. Microperfusion results were analysed using a one‐way ANOVA analysis followed by Bonferroni's multiple comparison test in Prism (GraphPad Software Inc., San Diego, CA, USA).
Metabolic studies
Mice were individually housed in metabolic cages (Techniplast, Seclin, France) with free access to a standard laboratory diet (2.8 g of Na+ /kg; Safe, Augy, France). After 3 days of acclimatization, 24 h urine was collected. The mice were then switched to a high sodium diet (3% NaCl solution as drinking water) and urine was collected during the next 24 h. Urinary creatinine concentrations were determined using an automatic analyser (Konelab 20i; Thermo, Illkirch‐Graffenstaden, France) and the urinary Na+ concentration was determined by flame photometry (IL943; Servilab, Le Mans, France). The results were analysed using a two‐way ANOVA analysis followed by Bonferroni's multiple comparison test in Prism.
Mathematical model description
The main Na+, K+, and Cl− transport pathways in the mouse CCD are shown in Fig. 1. In principal cells, the basolateral Na+/K+‐ATPase generates the gradients needed to sustain Na+ reabsorption via ENaC, as well as K+ secretion via the renal outer medullary K+ channel (ROMK) or K+ recycling via basolateral K+ channels. In type B intercalated cells, as proposed by Chambrey et al. (2013), the net reabsorption of NaCl is energized by basolateral vacuolar‐ATPase pumps. Na+ enters the cell on the apical side via NDCBE and exits via AE4 on the basolateral side. The apical uptake of Cl− is mediated by the Cl−/HCO3 − exchanger pendrin; a fraction of Cl− is recycled via NDCBE, whereas the remainder exits via the basolateral Cl− channel ClC‐K2. Note that this scheme assumes that AE4, the transport properties of which remain controversial, operates as a Na+‐HCO3 − cotransporter, with a Na+:HCO3 − stoichiometry of 1:3 as required for a sodium extruder (Boron, 2004). Type A intercalated cells express NKCC1 (Pech et al. 2012), ClC‐K2 (Kobayashi et al. 2001; Nissant et al. 2006) and the K+‐Cl− cotransporter KCC4 (Boettger et al. 2002) on the basolateral side. At the apical membrane, we assume the presence of K+ and Cl− channels, as suggested by Pech et al. (2012) and discussed below. We also assume that Na+ can be extruded apically by H+/K+‐ATPase type 2 (HKA2) pumps, as described below, because those pumps may extrude Na+ instead of protons in exchange for K+ (Grishin et al. 1996; Cougnon et al. 1998; Crambert et al. 2002).
Figure 1. Representation of principal and intercalated cells in mouse CCD .
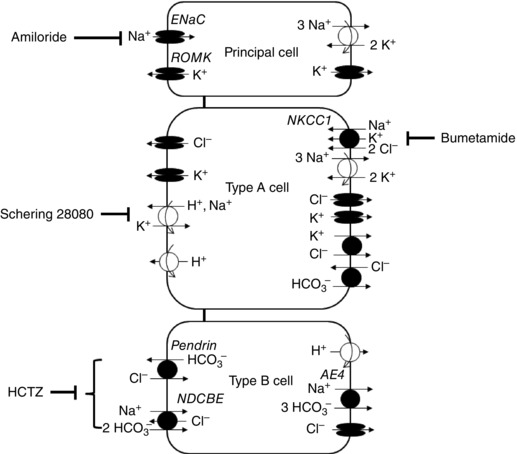
Shown are the main transcellular Na+, K+ and Cl− transporters. The mathematical model accounts for the transport of water and 12 solutes along the full length of the tubule.
The mathematical model describes the transepithelial reabsorption or secretion of water and solutes along the full length of the CCD, via paracellular and transcellular pathways. It is conceptually similar to the rat CCD model developed by Weinstein (2001), with significant differences in morphology and in the presence and/or activity of some transporters, as detailed below. We distinguish six compartments: the lumen (denoted by the superscript L), principal cells (P), type A (A) and B (B) intercalated cells, the extracellular (i.e. lateral) space between cells (E) and the peritubular solution (S). We account for the transport of the following solutes in addition to water: Na+, K+, Cl−, HCO3 −, H2CO3, CO2, NH3, NH4 +, HPO4 2−, H2PO4 − and H+. The composition of the luminal fluid is specified at the CCD entrance, and that of the peritubular space is fixed. At a given position along the tubule (taken to be 1 mm long), the volume, electric potential and solute concentrations in the lumen and cells are obtained by solving steady‐state conservation equations for volume, charge and mass, respectively. A summary of the steady‐state conservation equations for volume, charge and mass is available elsewhere (Weinstein, 2001; Bonny & Edwards, 2013). The specific characteristics of this mouse CCD model are described below.
Morphological parameters
The morphological parameters of the mouse CCD are determined under ex vivo conditions. Based upon our measurements (Morla L, unpublished data), the outer diameter (D o) is estimated as 51 μm and the inner diameter (D i) as 27.5 μm; note that the latter value is used to normalize by the epithelial surface area. The epithelial volume is thus computed as 16.8 μm3 μm–2 epithelium (or 16.8 × 10−4 cm3 cm–2 epithelium). Excluding non‐A non‐B and bipolar cells, the fraction of P, A, and B cells in mouse CCD is taken as 71.5 %, 21.5% and 7.0%, respectively (Kim et al. 1999). In the absence of mouse data, we assume that the lateral space represents 10% of epithelial volume, as in rabbits (Welling et al. 1981), and that individual cells occupy the same volume under basal conditions. With these hypotheses, the aggregate volume of P, A, and B cells is, respectively, computed as 10.8 × 10−4, 3.2 × 10−4 and 1.1 × 10−4 cm3 cm–2 epithelium.
Membrane surface areas are determined assuming that the cuboidal cells are between two co‐axial cylinders of diameters D i and D o, there are approximately four per cross‐section, all cells have the same (axial) height, and the area occupied by tight junctions represents 1% of the epithelial surface area vs. 5% for the basement membrane. Examination of cell shape and surface areas in mouse CCDs (Doucet A, unpublished observations) suggests that membrane infolding increases basal membrane areas approximately by 5‐fold, and lateral membrane areas by 2‐fold, in all cell types. Infolding appears to enhance the apical membrane area more in intercalated cells (by a factor of ∼4) than in principal cells (by a factor of ∼2). Morphological parameter values are summarized in Table 1.
Table 1.
Morphometric parameters of the mouse CCD
| Principal cells | Type A cells | Type B cells | Lateral interspace | |
|---|---|---|---|---|
| Volume (x 10−4 cm3 cm–2 epith) | 10.79 | 3.25 | 1.06 | 1.68 |
| Apical membrane area (cm2 cm–2 epith) | 1.42 | 0.85 | 0.277 | 0.01 |
| Lateral membrane area (cm2 cm–2 epith) | 10.04 | 1.36 | 0.443 | |
| Basal membrane area (cm2 cm–2 epith) | 6.56 | 1.97 | 0.643 | 0.05 |
The reference epithelial surface area is based upon the inner diameter of the lumen.
Permeability values
To match our experimental results, the water permeability per unit surface area (denoted P f) of principal cell apical membranes is set to 10 μm s−1 in the absence of ADH. The basolateral‐to‐apical membrane P f ratio as is taken as 3.5 (Harris et al. 1991) and the P f of intercalated cell membranes is assumed to be 1000 times lower than that of corresponding principal cell membranes. The water permeability of the tight junction is taken as 0.0040 cm3 s−1 per cm2 epithelial area, and that of the basement membrane as 1.80 cm3 s−1 per cm2 epithelial area, as in the rat model (Weinstein, 2001).
Solute permeability values are identical to those of the rat CCD model, except for certain modifications (Table 2). The background H+ permeability of all cell membranes is set to zero. In principal cells, the apical membrane is taken to be impermeable to Cl− and HCO3 −, and the basolateral permeability to Cl− is chosen as 0.002 × 10−5 cm s−1. The permeability of ENaC and ROMK is set to 0.67 × 10−5 and 2.50 × 10−5 cm s−1, respectively. The apical membrane of A cells includes conductive pathways for K+ (0.30 × 10−5 cm s−1) and Cl− (6.0 × 10−5 cm s−1) but not to HCO3 −; the A cell basolateral permeability to Cl− is set to 0.50 × 10−5 cm s−1, and that to K+ is set to 0.125 × 10−5 cm s−1. Note that the contribution of K+ to the basolateral conductance in intercalated cells, assumed to be small in rabbit CCD (Muto et al. 1990), is more significant in rat CCD (Schlatter & Schafer, 1988). In B cells, the basolateral permeability to HCO3 − is zero, and HCO3 − exits the cell via AE4 instead. Note that, unless otherwise noted, the background permeability to HCO3 − is taken as 20% of that to Cl−, and the background permeability to NH4 + as 20% of that to K+ (Weinstein, 2001). The permeabilities of the basement membrane, taken to be the same in mice and rats (per cm2 epithelial area), yield a conductance of ∼1000 mS cm−2.
Table 2.
Solute permeabilities of the mouse CCD
| Principal cells (x 10−5 cm s−1) | Type A cells (x 10−5 cm s −1) | Type B cells (x 10−5 cm s−1) | |||||
|---|---|---|---|---|---|---|---|
| Apical | Basolat | Apical | Basolat | Apical | Basolat | Paracellular pathway (x 10−5 cm s−1) | |
| Na+ | 0.67 | 0 | 0 | 0 | 0 | 0 | 0.35 |
| K+ | 2.50 | 0.60 | 0.3 | 0.125 | 0 | 0.015 | 1.05 |
| Cl− | 0 | 0.002 | 6 | 0.50 | 0 | 0.40 | 0.42 |
| HCO3 − | 0 | 0.0004 | 0 | 0.10 | 0 | 0 | 0.084 |
‘Apical’ and ‘basolat’ denote the apical and basolateral membranes of each cell type. Basolateral chloride permeability values correspond to pHi 7.40; see Eqn (12).
Membrane transporters
By contrast to the rat CCD model (Weinstein, 2001), the present model incorporates the recently identified transporters mediating Na+ reabsorption across B cells, namely NDCBE and AE4. It also includes NKCC1 and KCC4, as well as apical K+ and Cl− channels, in A cells. Conversely, it assumes there are no apical NaCl co‐transporters in principal cells and no basolateral Cl−/HCO3 − exchangers in B cells. Solute fluxes across ClC‐K2, NHE1, pendrin and Na+/K+‐ATPase pumps are also formulated differently compared to the rat model. Corresponding flux expressions are described in the Appendix. Main solute permeability values and transporter densities are given in Tables 2 and 3.
Table 3.
Mouse CCD transporter density or maximum rate
| Principal cell | Type A cell | Type B cell | |
|---|---|---|---|
| Na+/K+‐ATPase | 510 × 10−9 mmol s−1 cm−2 (basolateral) | 75 × 10−9 mmol s−1 cm−2 (basolateral) | 75 × 10−9 mmol s−1 cm−2 (basolateral) |
| H+‐ATPase | 100 × 10−9 mmol cm−2 (apical) | 5,000 × 10−9 mmol cm−2 (basolateral) | |
| H+/K+‐ATPase | 3 × 10−9 mmol cm−2 (gastric, apical) 70 × 10−9 mmol cm−2 (non‐gastric, apical) | 6 × 10−9 mmol cm−2 (gastric, apical) | |
| Basolateral Na+/H+ exchangers | 0.2 × 10−9 mmol cm−2 (NHE1) | 6 × 10−9 mmol2 J−1 s−1 cm−2 | 0.6 × 10−9 mmol2 J−1 s−1 cm−2 |
| Basolateral Na2HPO4 cotransporters | 0.20 × 10−9 mmol2 J−1 s−1 cm−2 | 0.08 × 10−9 mmol2 J−1 s−1 cm−2 | 0.08 × 10−9 mmol2 J−1 s−1 cm−2 |
| Cl−/HCO3 − exchangers | 0.2 × 10−9 mmol2 J−1 s−1 cm−2 (basolateral) | 20 × 10−9 mmol cm−2 (AE1, basolateral) | 0.100 × 10−9 mmol cm−2 (pendrin, apical) |
| KCl cotransporters | 0.06 × 10−9 mmol cm−2 (KCC4, basolateral) | ||
| Na(HCO3 −)3 cotransporters | 2500 × 10−9 mmol2 J−1 s−1 cm−2 (AE4, basolateral) | ||
| Other NaCl transport pathways | 60 × 10−9 mmol2 J−1 s−1 cm−2 (NKCC1, basolateral) | 400 × 10−9 mmol2 J−1 s−1 cm−2 (NDCBE, apical) |
Cortical collecting duct Na+, K+ and Cl− transporter expression was chosen so that model predictions approximately match ionic fluxes measured under asymmetric conditions.
Results
We measured solute transport in isolated, microperfused CCDs of Na+‐depleted mice; a low sodium diet has been shown to stimulate NaCl reabsorption, whereas the net transepithelial fluxes of Na+ and Cl− are insignificant under a normal diet (Leviel et al. 2010). To mimic in vivo conditions, the CCDs were perfused with a hypo‐osmotic solution (184 mOsm kg H2O–1) that contained less Na+, Cl− and HCO3 −, and more K+, than the bath (Buerkert et al. 1983; Elalouf et al. 1984; Beck et al. 1988), as specified above. The contribution of the different transcellular and paracellular pathways to net transepithelial fluxes was assessed by adding specific inhibitors to the perfusion solution or the bath.
Characterization of the paracellular pathway
To assess the contribution of the paracellular route to Na+ and Cl− transport in mouse CCDs, we first inhibited NaCl transport across transcellular routes: ENaC and the NDCBE‐pendrin pathway were respectively blocked by infusing amiloride and hydrochlorothiazide (HCTZ) in the lumen, and transport pathways in type A intercalated cells were blocked by adding Schering 28080 (an inhibitor of H+/K+‐ATPase types 1 and 2) to the perfusate and bumetanide in the bath. We assumed here that HCTZ fully inhibits NaCl reabsorption across the NDCBE‐pendrin pathway, as observed in several experimental studies on sodium‐depleted mice (Leviel et al. 2010; Tokonami et al. 2013). It should be noted that some investigators found no effect of HCTZ on Cl− absorption in aldosterone‐treated mice (Pech et al. 2012) and also that the precise molecular target of HCTZ in type B intercalated cells remains to be determined (Leviel et al. 2010).
Under these conditions (referred to as the ‘paracellular’ case), as shown in Fig. 2 A–D, the CCD secreted Na+ (J Na = −16.8 ± 0.8 pmol min−1 mm−1; n = 5), whereas the Cl− flux was negligible (J Cl = −3.4 ± 2.4 pmol min−1 mm−1); the transepithelial voltage (denoted ΔV te) was –12.8 ± 1.8 mV. By fitting the parameters of our mathematical model to these flux and voltage values, we estimated the permeabilities of the paracellular pathway to Na+ and Cl− as 0.35 × 10−5 and 0.42 × 10−5 cm s−1, respectively. These values are consistent with the Cl−:Na+ permeability ratio of 1.2 measured in rat CCD (Schafer & Troutman, 1990). We should nevertheless recognize that there is some uncertainty in our Cl− permeability estimate because the measured paracellular Cl− flux is small as a result of the counterbalancing effects of the transepithelial electric potential difference and the Cl− concentration gradient, and experimental variations are thus large in relative terms. As shown in Fig. 3, the model predicts that, because of these counteracting effects, the Cl− flux across the tight junction changes direction along the CCD.
Figure 2. Ion transport across WT mouse CCDs in the presence of inhibitors .
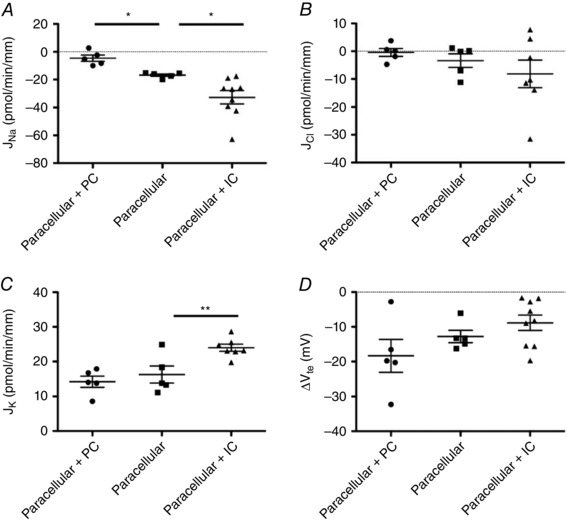
Measured transepithelial fluxes (pmol min–1 mm–1) of Na+ (A), Cl− (B), K+ (C) and transepithelial voltage difference (D, in mV) in the CCDs of wild‐type mice perfused under asymmetric conditions, in three cases: in the presence of amiloride, hydrochlorothiazide, bumetanide and Schering 28080 (‘paracellular’); in the presence of hydrochlorothiazide, bumetanide and Schering 28080 (‘paracellular + PC’); or in the presence of amiloride only (‘paracellular + IC’). Results are shown as individual data and the mean ± SEM.
Figure 3. Predicted fluxes across paracellular and transcellular pathways under asymmetric conditions, in the presence of amiloride, hydrochlorothiazide, bumetanide and Schering 28080 .
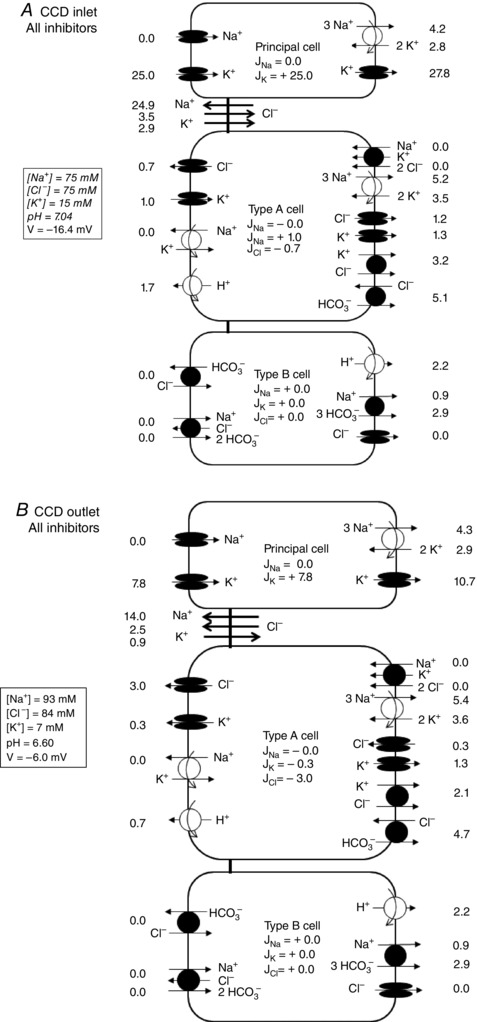
Solute fluxes (pmol min–1 mm–1) are shown at the CCD inlet (A) and outlet (B) for a 1 mm long tubule. Prescribed values (i.e. boundary conditions) are shown in italics. Not shown are basolateral Na+/H+ exchangers, basolateral 2Na+‐HPO4 2− cotransporters and apical H+/K+‐ATPase type 1 pumps.
Under the same conditions, the CCD reabsorbed K+ (J K = 16.3 ± 2.5 pmol min−1 mm−1) (Fig. 2 C). The cocktail of inhibitors that we used does not necessarily abolish all transcellular K+ transport because most cells display apical and basolateral K+ channels. Assuming an intracellular concentration of K+ of 140 mm, our model predicts that, in the presence of 15 mm and 4 mm of potassium at the lumen entrance and in the bath, respectively, the electrochemical gradient of K+ across the apical membrane is favorable to its entry into the cell, whereas that across the basolateral membrane is favorable to its exit (Fig. 3). Our model predictions matched K+ flux measurements well, assuming that the K+ permeability of the paracellular pathway is three times that of Na+ (i.e. 1.05 × 10−5 cm s−1), as previously reported in rats treated with desoxycorticosterone (29).
The tight junctional conductance (G TJ) of the mouse CCD was then computed as 3.3 mS cm−2, which is similar to the reported values for the rabbit CCD (Stoner et al. 1974; Koeppen & Giebisch, 1985). Two other studies of mouse CCD reported larger transepithelial conductance values (G T ∼10 mS cm−2), although they did not distinguish between paracellular and transcellular pathways (Lehrmann et al. 2002; Pech et al. 2007). In rats treated with DOCA, G TJ was determined to be 11–13 mS cm−2; note, however, that a model of the rat CCD had to assume a much lower conductance (i.e. 5 mS cm−2) to avoid unrealistic backflux values (Weinstein, 2001).
Contribution of principal cells
To estimate transport rates across principal cells, we then measured ion fluxes in the presence of HCTZ, Schering 28080, and bumetanide but in the absence of amiloride (conditions referred to as ‘paracellular + PC’). Under the same asymmetric conditions as described above, removal of amiloride abolished sodium secretion (J Na = −4.6 ± 2.2 pmol min−1 mm−1, n = 5; P < 0.05) (Fig. 2 A) but did not significantly alter chloride and potassium fluxes (J Cl = −0.4 ± 1.4 pmol min−1 mm−1; J K = 14.2± 1.6 pmol min−1 mm−1) (Fig. 2 B and C), nor the electric potential difference (ΔV te = −18.3 ± 4.7 mV) (Fig. 2 D). In other words, Na+ reabsorption across principal cells approximately counterbalanced Na+ secretion via the paracellular route under asymmetric conditions.
Sodium secretion in the presence of amiloride
Conversely, we assessed the contribution of intercalated cells by measuring fluxes in the presence of amiloride only, under the same asymmetric conditions (the ‘paracellular + IC’ case). Compared with the ‘paracellular’ case, removing the inhibitors of transport pathways in type A and type B intercalated cells increased the secretion of Na+ (J Na = −32.7 ± 4.7 pmol min−1 mm−1; P < 0.01; n = 9) (Fig. 2 A), as well as the reabsorption of K+ (J K = 22.1 ± 1.5 pmol min−1 mm−1; P < 0.01) (Fig. 2 C), without significantly altering Cl− fluxes (J Cl = −7.8 ± 3.8 pmol min−1 mm−1), nor the transepithelial voltage (ΔV te = −8.8 ± 2.2 mV). These results (Table 4) indicate that type A and/or type B intercalated cells mediate Na+ secretion and K+ reabsorption.
Table 4.
Measured and calculated fluxes in the mouse CCD under asymmetric conditions
| J Na | J Cl | J K | ΔV te | ||
|---|---|---|---|---|---|
| WT, All inhibitors | Microperfusion (n = 5) | −16.8 ± 0.8 [−19.2; −14.4] | −3.4 ± 2.4 [−10.1; 3.4] | 16.3 ± 2.5 [9.5; 23.1] | −12.8 ± 1.8 [−17.8; −7.9] |
| Model | −19.0 [0 + 0 + 0 – 19.0] | −2.0 [0 – 2.0 + 0 – 0.1] | 17.4 [15.3 + 0.3 − 0 + 1.8] | −16.4 | |
| WT, All inhibitors except amiloride | Microperfusion (n = 5) | −4.6 ± 2.3 [−10.9; 1.6] | −0.4 ± 1.4 [−4.4; 3.5] | 14.2 ± 1.6 [9.7; 18.7] | −18.3 ± 4.7 [−31.4; −5.2] |
| Model | −2.2 [24.4 + 0 + 0 – 26.6] | 6.7 [0 + 0.4 + 0 + 6.3] | 9.7 [7.6 + 0.5 + 0 + 1.6] | −21.3 | |
| WT, Amiloride | Microperfusion (n = 5) | −32.7 ± 4.7 [−43.5; −21.9] | −7.8 ± 3.8 [−16.6; 0.9] | 22.1 ± 1.5 [18.7; 25.5] | −8.8 ± 2.2 [−14.0; −3.7] |
| Model | −30.5 [0 – 14.9 +3.5 – 19.1] | −8.7 [0 – 16.6 + 7.0 + 0.9] | 19.4 [11.8 + 6.1 + 0.1 + 1.4] | −16.9 | |
| HKA2−/−, Amiloride | Microperfusion (n = 6) | −17.0 ± 1.5 [−20.9; −13.1] | −1.6 ± 2.1 [−7.1; 3.9] | 20.6 ± 0.8 [18.6; 22.6] | −9.0 ± 2.4 [−15.1; −2.8] |
| Model | −16.3 [0 + 0 + 3.4 – 19.7] | 2.4 [0 – 4.9 + 7.0 +0.3] | 16.7 [15.4 – 0.7 + 0.2 + 1.8] | −16.0 | |
| WT, Amiloride + bumetanide | Microperfusion (n = 5) | −18.4 ± 1.8 [−23.3 ; −13.4] | 1.4 ± 1.8 [−3.5 ; 6.3] | 18.4 ± 1.8 [13.4; 23.3] | −13.0 ± 0.8 [−15.3; −10.6] |
| Model | −23.1 [0 – 9.4 +3.5 – 17.2] | 0.7 [0 – 4.4 + 7.0 – 1.9] | 21.7 [11.7 + 8.4 + 0.1 + 1.4] | −16.3 | |
| WT, No inhibitors | Microperfusion (n = 14) | −8.2 ± 1.6 [−11.6; −4.7] | −1.3 ± 1.3 [−4.2; 1.6] | 12.0 ± 0.9 [10.0; 14.0] | −20.2± 1.6 [−23.7; −16.8] |
| Model | −11.4 [+25.8 – 14.9 + 3.4 – 25.7] | 3.1 [0 – 9.9 + 7.0 + 6.0] | 12.3 [5.3 + 5.5 + 0.1 + 1.4] | −20.9 |
J Na, J Cl, J K: transepithelial fluxes of Na+, Cl− and K+ (pmol min–1 mm–1) (positive flux values denote reabsorption, negative flux values denote secretion). ΔV te, transepithelial electrical potential difference at the tubule inlet (mV). Experimental values are given as the mean ± SEM, followed by the 95% confidence interval. Model values denote predictions from the mathematical model. The overall flux is the sum of four components (shown within brackets); namely, the net fluxes across principal cells, type A and type B intercalated cells, and the paracellular pathway. ‘All inhibitors’: amiloride (10−5 m), HCTZ (10−4 m), and Schering 28080 (10−3 m) in the lumen, and bumetanide (10−4 m) in the bath.
Sodium secretion in the presence of bumetanide and amiloride
According to our hypothesis (see Introduction), type A intercalated cells could secrete Na+ by mediating the basolateral entry of Na+ and Cl− along with K+ via the NKCC1 cotransporter. To confirm the involvement of type A intercalated cells in Na+ secretion, we evaluated the effect of adding bumetanide to the bath. In the presence of both amiloride and bumetanide, the secretion of Na+ was significantly attenuated (J Na = −18.4 ± 1.8 pmol min−1 mm−1; P < 0.05; n = 5) (Fig. 4 A) compared to that in the presence of amiloride only, and the secretion of Cl− was abolished (J Cl = 1.4 ± 1.8 pmol min−1 mm−1) but was not significantly different from that measured in the presence of amiloride only (Fig. 4 B). The addition of bumetamide in the presence of amiloride did not affect K+ reabsorption, nor the transepithelial voltage (Fig. 4 C–D). These results strongly suggest that the secretion of Na+ by intercalated cells originates from type A intercalated cells and is mediated by NKCC1 at the basolateral membrane.
Figure 4. Ion transport across WT and HKA2‐null mouse CCDs .

Measured transepithelial fluxes (pmol min–1 mm–1) of Na+ (A), Cl− (B), K+ (C) and transepithelial voltage difference (D, in mV) in the CCDs of wild‐type mice perfused under asymmetric conditions, in the presence of amiloride only (WT) or in the presence of amiloride and bumetanide (WT+bumetanide), and in the CCDs of HKA2‐null mice perfused under asymmetric conditions with amiloride (HKA2‐KO). Results are shown as individual data and the mean ± SEM.
As predicted by the model, the addition of bumetanide (in the presence of amiloride) has only a small impact on K+ reabsorption across type A cells because of compensatory mechanisms. NKCC1 inhibition leads to a large reduction in intracellular Na+ and Cl−, which downregulates the activity of apical HKA2 pumps, but this is partially offset by a large decrease in apical K+ secretion via K+ channels (Fig. 5).
Figure 5. Predicted fluxes across paracellular and transcellular pathways in the presence of amiloride .
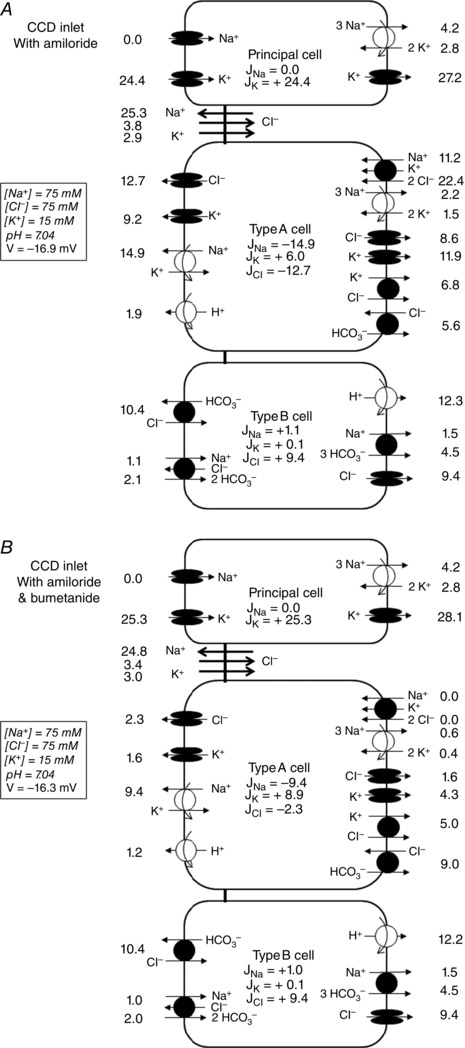
Predicted fluxes (pmol min–1 mm–1) at the CCD inlet, under asymmetric conditions, with amiloride only (A) and with amiloride and bumetanide (B). Not shown are basolateral Na+/H+ exchangers, basolateral 2Na+‐HPO4 2− cotransporters and apical H+/K+‐ATPase type 1 pumps.
Role of H+/K+‐ATPase type 2 in Na+ secretion
We next investigated the apical pathways that might support Na+ secretion in type A intercalated cells. The existence of Cl− channels at the apical membrane was proposed by Pech et al. (2012), although the mechanisms by which Na+ may be extruded apically remain unknown. Our calculations indicate that secondary active Na+ exit is theoretically possible via apical Na+‐HCO3 − cotransporters and/or NCBDE, both of which would couple the extrusion of Na+ to that of HCO3 −. Because the main role of type A cells is to acidify the lumen, a secretion scheme involving these transporters is not probable. Another candidate is H+/K+‐ATPase type 2 (HKA2), which can pump out Na+ instead of protons in exchange for K+ (Grishin et al. 1996; Cougnon et al. 1998; Crambert et al. 2002), and which is expressed apically in type A cells in the rat CCD (Wingo et al. 1990; Bastani, 1995).
To determine whether HKA2 mediates transcellular Na+ secretion, we measured ion fluxes across the CCD of HKA2‐null mice, under the same asymmetric conditions as above, in the presence of amiloride. As shown in Fig. 4 A, Na+ secretion fluxes in HKA2‐null mice were significantly attenuated compared to those in WT mice under identical conditions (J Na = −17.0 ± 1.5 pmol min−1 mm−1; n = 6; P < 0.01) and were similar to those measured in the presence of bumetanide in WT mouse CCDs. Cl− and K+ fluxes, as well as the transepithelial voltage, were not modified in HKA2‐null mice (Fig. 4 B–D). These results demonstrate the involvement of H+(Na+)/K+‐ATPase type 2 in apical Na+ exit in type A intercalated cells.
Sodium transport in the absence of inhibitors
We used the previous results to calibrate the mathematical model, specifically to estimate the density of the transcellular transporters of Na+, K+ and Cl− (Table 3). We then predicted net transepithelial fluxes under asymmetric conditions in the absence of inhibitors (Fig. 6). Assuming a perfusion rate of 2.0 nl min−1 and a fixed tubular length of 1 mm, the predicted fluxes of Na+, Cl− and K+ were equal to −11.4, 3.1 and 12.3 pmol min−1 mm−1, respectively. Subsequent measurements of these fluxes in vitro yielded good agreement between theoretical and experimental values (Fig. 7): the measured fluxes of Na+, Cl− and K+ were respectively – 8.2 ± 1.6, −1.3 ± 1.3 and 12.0 ± 0.9 pmol min−1 mm−1.
Figure 6. Predicted fluxes across paracellular and transcellular pathways in the absence of inhibitors .
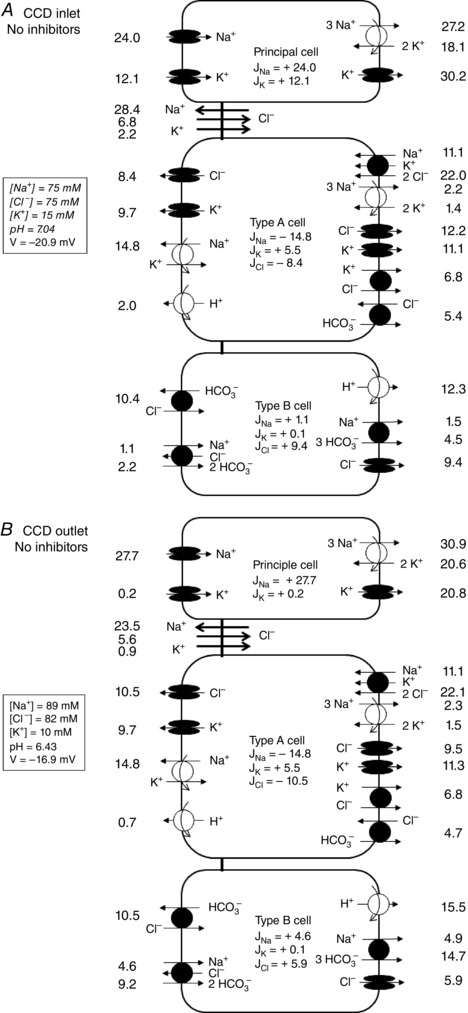
Predicted fluxes (pmol min–1 mm–1) under asymmetric conditions, at the CCD inlet (A) and outlet (B). Not shown are basolateral Na+/H+ exchangers, basolateral 2Na+‐HPO4 2− cotransporters and apical H+/K+‐ATPase type 1 pumps.
Figure 7. Measured and predicted ion transport across WT mouse CCDs in the absence of inhibitors .

Measured (grey circles) and predicted (white circles) values of Na+, Cl−, K+ fluxes (pmol min–1 mm–1) and of the transepithelial voltage difference (in mV) in the CCDs of wild‐type mice perfused under asymmetric conditions, in the absence of inhibitors. Experimental results are shown as individual data and the mean ± 95% confidence interval. [Colour figure can be viewed at wileyonlinelibrary.com]
Note that the model predicts that inhibiting apical proton secretion via H+‐ATPase has a negligible impact on J Na, J K and J Cl (results not shown).
Physiological relevance of the Na+ secretion pathway
To assess the physiological relevance of HKA2‐mediated Na+ secretion in the collecting duct, we measured the renal response to Na+ loading of WT and HKA2‐null mice. There was no difference in the daily Na+ intake of WT and HKA2‐null mice under either a normal or a high‐salt diet (Fig. 8 A). However, whereas urinary Na+ excretion was similar between both strains under normal conditions, 1 day after the switch from a normal to a high‐salt diet, HKA2‐null mice excreted ∼40% less Na+ compared to WT mice (Fig. 8 B).
Figure 8. In vivo impact of HKA2‐mediated Na+ secretion in the CCD .
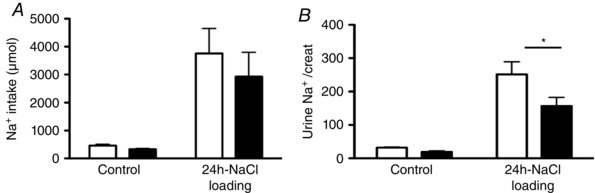
Daily Na+ intake (A) and urinary Na+ excretion (B) in WT (white bars) and HKA2‐null mice (black bars) under normal conditions (control) and 1 day after switching to a high salt diet (3% NaCl in drinking water). Results are shown as the mean ± SEM (n = 5). * P < 0.05.
Discussion
In the present study, first, we demonstrated that type A intercalated cells of the mouse CCD have the ability to avidly secrete Na+; second, we found that ex vivo (i.e. in the absence of acute regulatory factors) and in the presence of physiological transepithelial concentration gradients, the CCD of Na+‐depleted mice paradoxically secretes Na+ and reabsorbs K+; and, third, we developed a mathematical model of ion transport in the mouse CCD that integrates the NaCl transport capacity of type A intercalated cells; model predictions fit fairly well the measured values of ion fluxes under all of the experimental conditions tested. These results raise several questions regarding (i) the mechanism of NaCl secretion in type A intercalated cells and its physiological regulation; (ii) the differences in ion fluxes observed ex vivo between symmetrical and asymmetrical conditions; and (iii) the discrepancy between the ion fluxes measured ex vivo and those expected to prevail in vivo in CCDs from Na+‐depleted mice (i.e. NaCl reabsorption in absence of net K+ transport). These inter‐related questions are addressed below by combining our experimental findings with data from the literature and the predictions of our mathematical model.
Mechanisms and regulation of NaCl secretion by type A intercalated cells
NaCl secretion across the CCD has been already observed under symmetric conditions, such as in the presence of amiloride and thiazide (Leviel et al. 2010) or in slc4a8‐ or slc4a9‐knockout mice (Chambrey et al. 2013); because ΔV te is lumen‐positive in the presence of amiloride and thiazide, the secretion of Na+ cannot occur across the paracellular route. Bumetanide‐sensitive Na+ secretion has also been reported in the rat outer medullary collecting duct (Wall & Fischer, 2002) and inner medullary collecting duct (Rocha & Kudo, 1990 a). In the rabbit CCD, however, bumetanide does not affect net Na+ reabsorption under symmetric conditions (Liu et al. 2011).
Our flux measurements in WT and HKA2−/− mice strongly suggest that HKA2 mediates apical Na+ exit from type A intercalated cells. HKA2 has been localized to the apical membrane of type A intercalated cells in the CCD and also shown to be a versatile transporter regarding its cation specificity (Crambert, 2014). Several experimental studies have demonstrated that HKA2 can extrude Na+ instead of H+ (Grishin et al. 1996; Cougnon et al. 1998; Crambert et al. 2002), and a recent mathematical model of the pump suggests that it mediates mainly Na+/K+ exchange under physiological conditions (Nadal‐Quiros et al. 2015).
The physiological pertinence of Na+ secretion across H+/K+‐ATPase type 2 pumps was demonstrated in the present study by our finding that HKA2−/− mice displayed an impaired ability to excrete a sodium load. In other words, HKA2 pumps play a significant role in excreting excess Na+ following sodium loading.
The secretion of chloride across type A intercalated cells in mouse CCD was demonstrated previously (Pech et al. 2012). In the present study, there was a similar tendency for Cl− secretion across type A cells, although it did not reach statistical significance. The apical pathways mediating the secretion of Cl− in type A cells remain to be characterized. The contribution of Cl− to the apical membrane resistance in intercalated cells appears to be small, at least in rat CCD (Schlatter & Schafer, 1988). However, the study of Pech et al. (2012) indicates that the apical Cl− flux across type A cells is modulated by the transepithelial voltage, suggesting that it is mediated by an electrogenic transporter. One candidate is Slc26a11, an anion transporter that co‐localizes with the vacuolar H+‐ATPase in the mouse CCD (in particular at the apical membrane of type A cells) and regulates its activity (Xu et al. 2011). Slc26a11 was shown to operate both as a Cl− channel and as a Cl−/HCO3 − exchanger in heterologous expression systems (Xu et al. 2011). Its functional role in vivo has not been examined. Another, less probable, candidate is the cystic fibrosis transmembrane conductance regulator (CFTR), which forms apical Cl− channels in cultures of principal cells from the mouse collecting duct (Lu et al. 2010). Whether it is also expressed by intercalated cells remains unclear. Moreover, Pech et al. (2012) found that CFTR inhibition did not affect benzamil‐sensitive Cl− fluxes in microperfused CCDs. As shown in Fig. 6, our model confirms that a two‐step mechanism of NaCl secretion in type A cells involving basolateral NaCl entry via NKCC1 and Na+ and Cl− apical exit via HKA2 and chloride channels, respectively, is thermodynamically feasible. According to this model, type A cells also reabsorb K+ under asymmetrical conditions. Alternatively, apical Cl− secretion in type A cells could be mediated by a cotransporter such as KCC; a chloride‐dependent potassium flux has been described in rabbit CCD (Wingo, 1989). Model predictions were very similar when we replaced the apical K+ and Cl− channels with an apical KCC cotransporter (results not shown).
The existence of a NaCl secreting pathway in the CCD suggests that this nephron segment might be a target of natriuretic hormones such as atrial natriuretic factor (ANF) and guanylin peptides (Sindic & Schlatter, 2007), as well as of their common second messenger cyclic GMP. Classically, ANF is considered to exert natriuretic effects through inhibition of ENaC and nucleotide‐gated channels in the inner medullary collecting duct (IMCD) (Theilig & Wu, 2015). However, the ANF receptor guanylyl cyclase A has been found in the intercalated cells of rat CCDs (Hirsch et al. 2001) and, in microperfused rat IMCDs, ANF not only reduces sodium reabsorption, but also increases the furosemide‐sensitive secretion of NaCl by the Na‐K‐Cl cotransport system (Rocha & Kudo, 1990 b). Thus, ANF might also be natriuretic by stimulating NaCl secretion through type A intercalated cells in the CCD. This conclusion is consistent with the only available studies in CCDs showing that ANF reduces net sodium transport (Nonoguchi et al. 1989) but has no effect on the lumen‐to‐bath Na+ flux (Rouch et al. 1991; Schlatter et al. 1996).
Differences in ion fluxes between symmetrical and asymmetrical conditions
When CCDs from Na+‐depleted mice are microperfused under symmetric conditions, J Na, J Cl and J K are on the order of 35, 15 and −5 pmol min−1 mm−1, respectively, in the absence of inhibitors (Leviel et al. 2010). All else being equal, our mathematical model predicts that, under symmetric conditions, these fluxes would be approximately 24, 3 and −6 pmol min−1 mm−1, respectively (Table 5). This indicates that abolishing the ion concentration gradients and/or hypo‐osmolality of the apical fluid augments substantially the net reabsorption of chloride and sodium.
Table 5.
Model predictions under symmetric conditions
| J Na | J Cl | J K | ΔV te | |
|---|---|---|---|---|
| Measured values | +36.4 ± 3.3 | +13.4 ± 2.0 | −5.3 ± 1.0 | −13.9 ± 2.6 |
| Predictions | ||||
| No adjustment | + 24.3 [37.4 – 14.3 + 12.4 – 11.1] | 3.0 [0 – 6.5 − 0.7 + 10.2] | −6.0 [− 12.1 + 6.0 + 0.1 – 0] | −9.1 |
| NaCl TJ × 0.5 | + 26.6 [35.9 – 14.3 + 12.4 – 7.3] | + 3.6 [0 – 2.6 −0.6 + 6.8] | −7.7 [−13.3 + 5.6 + 0.1 – 0.1] | −11.8 |
| NaCl TJ × 0.5 and ENaC × 1.25 | + 36.8 [48.0 – 14.2 +12.3 – 9.3] | + 9.5 [0 + 1.6 −0.6 + 8.5] | −11.8 [−17.2 + 5.4 +0.1 – 0.1] | −15.5 |
| NaCl TJ × 0.5 and type A cell downregulation | + 34.3 [34.4 – 4.0 +12.3 – 8.4] | + 7.8 [0 + 0.8 −0.6 + 7.7] | −10.8 [−12.3 + 1.4 +0.1 – 0.1] | −12.3 |
| NaCl TJ × 0.5 and pendrin × 5 | + 30.0 [36.9 – 14.3 +15.0 – 7.6] | + 10.8 [0 −2.5 + 6.5 + 6.8] | −8.2 [−13.7 + 5.6 +0.1 – 0.2] | −12.0 |
J Na, J Cl, J K: transepithelial fluxes of Na+, Cl− and K+ (pmol min–1 mm–1). ΔV te: transepithelial electrical potential difference at the tubule inlet (mV). Measured values (in italics, mean ± SEM) are taken from Leviel et al. (2010). Indicated within brackets are the four flux components (i.e. the net fluxes across principal cells, type A and type B intercalated cells, and the paracellular pathway). Model predictions assume varying scenarios: NaCl TJ × 0.5: the tight junction permeability to Na+ and Cl− is divided by 2; ENaC × 1.25: ENaC expression is multiplied by 1.25; pendrin × 5: pendrin expression is multiplied by 5; type A cell downregulation: the expression of HKA2, NKCC1 and apical Cl− channels in type A cells is reduced by 75%.
Data from the literature show that the paracellular permeability of the tight junction to Na+ and Cl− is decreased 2‐fold in symmetric conditions compared to apical hypotonic conditions (Tokuda et al. 2008), probably in response to inhibition of with‐no‐lysine kinase 4 (WNK) (Kahle et al. 2004). Our mathematical model predicts that this increases only slightly the net fluxes of Na+ and Cl− (Table 5).
Intuitively, one would expect that increasing Cl− reabsorption can be achieved either by stimulating ENaC (via its effect on the transepithelial voltage), by stimulating pendrin or by inhibiting NaCl secretion by type A intercalated cells. The model predicts that a slight increase in ENaC activity increases Cl− reabsorption through the paracellular pathway via its effect on ΔV te but simultaneously increases Na+ reabsorption and K+ secretion. Inhibition of transporter activity in type A cells (NKCC1, HKA2 and apical Cl− channels) hardly increases Cl− reabsorption up to the observed level and markedly increases Na+ reabsorption. It is only when we simulate an increase in pendrin activity that all predicted values (Na+, K+ and Cl− fluxes and ΔV te) are close to measured values under symmetric conditions (Table 5). This suggests that pendrin activity might be higher under symmetric relative to asymmetric conditions.
The mechanism by which pendrin activity might be inhibited under asymmetric conditions remains unknown. Under asymmetric conditions, the concentration of Cl− in the cytosol of type B intercalated cells is predicted to be significantly higher than under symmetric conditions (27.7 vs. 15.7 mm at the tubule mid‐point). This might inactivate with‐no‐lysine kinase 3 (previously described as a putative chloride‐sensing kinase; Pacheco‐Alvarez & Gamba, 2011) and, in turn, modulate the activity of pendrin.
Discrepancy between the ion fluxes measured ex vivo and those expected to prevail in vivo in CCDs from Na+‐depleted mice
The most puzzling finding of the present study is that the CCD of Na+‐depleted mice, when perfused with a hypo‐osmotic solution of a composition mimicking in vivo conditions, secretes Na+ and reabsorbs K+. In that sodium‐deprived state, we expected the CCD to mediate net NaCl reabsorption and for there also to be no K+ transport, so as to maintain sodium homeostasis and blood pressure without affecting the potassium balance. The discrepancy between ex vivo and in vivo conditions probably stems from the absence in the former condition of regulatory factors modulating one or several transport pathways.
The most probable candidate is angiotensin 2, the concentration of which is increased in Na+‐depleted states. Angiotensin 2 is known to stimulate pendrin (Pech et al. 2007) and ENaC (Zaika et al. 2013) without altering ROMK activity (Wei et al. 2014). Our model predicts that doubling the activity of ENaC and pendrin induces significant NaCl reabsorption without K+ secretion under asymmetric conditions (J Na = 15.5, J Cl = 20.1, J K = −0.2 pmol min−1 mm−1).
In conclusion, we found that type A intercalated cells in the mouse cortical collecting duct have the capacity to significantly secrete Na+: this secretion is mediated, at least in part, by H+/K+‐ATPase type 2 pumps on the apical side and by a bumetanide‐sensitive pathway on the basolateral side. These results demonstrate for the first time that, in contrast to the classical view, the CCD mediates Na+ secretion in the presence of physiological concentration gradients, and that its ability to reabsorb NaCl in vivo is regulated in an acute manner by hormonal and neural factors. The capacity of the CCD to simultaneously activate reabsorption routes and inhibit secretion pathways, or vice versa, should allow this segment to adapt to a wide range of conditions at a small energetic cost.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
LM, AD, GC and AE conceived and designed the experiments. LM, CL, AD, GC and AE collected, assembled, analysed and interpretated data. AD, GC and AE drafted and/or critically revised the article. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
No funding was received for the present study.
Transepithelial fluxes
General expressions for water and solute fluxes are given in the literature (Weinstein, 2001; Bonny & Edwards, 2013). Described below are the fluxes that differ from the rat CCD model (Weinstein, 2001). Note that is the electric potential in compartment M, is the concentration of a given solute i in M, and is flux of i from compartment M to compartment N.
Coupled transport
Becausee the kinetic behavior of NDCBE, AE4 and NKCC1 has not been characterized experimentally, we use the non‐equilibrium thermodynamics (NET) approach to determine solute fluxes across these transporters. The driving force is given by the net difference in electrochemical potentials across the membrane, and the flux of solute i is computed as:
| (1) |
where is a matrix of coefficients that depend on the stoichiometry and expression of the transporter, and is the electrochemical potential of solute i in compartment M:
| (2) |
The NET formulation was previously used to describe transport across NDCBE in the basolateral membrane of the proximal tubule (Weinstein, 1992). In the CCD, NDCBE fluxes from the lumen to type B cells are determined as:
| (3) |
| (4) |
Similarly, the Na+ and HCO3 − fluxes across AE4, from type B cells to the peritubular solution, are calculated as:
| (5) |
| (6) |
AE4 fluxes across from type B cells to lateral spaces are obtained by replacing ‘S’ with ‘E’ in Eqns (5) and (6).
Using the same approach, NKCC1 fluxes from type A cells to the peritubular solution are given by:
| (7) |
| (8) |
KCC4
Fluxes across the K+‐Cl− cotransporter KCC4 are determined using the kinetic model of Weinstein (2010). The reaction scheme assumes that K+ binds to, and unbinds from, the carrier before Cl−, and it accounts for the competitive replacement of K+ by NH4 +.
Pendrin
Corresponding fluxes are computed based upon our kinetic model of Cl−/HCO3 − and Cl−/OH− exchange across pendrin (Azroyan et al. 2011; Bonny & Edwards, 2013).
Basolateral Na+/K+‐ATPase
The Na+ and K+ fluxes across Na+/K+‐ATPase pumps between cell M (M = P, A, B) and the peritubular solution S are calculated as:
| (9) |
| (10) |
The affinity of the pump for sodium () is fixed at 16.3 mm (Summa et al. 2004), whereas that for potassium increases with the peritubular concentration of Na+ (Strieter et al. 1992):
| (11) |
Luminal H+(Na+)/K+(NH4 +)‐ATPase
The non‐gastric (or colonic, type 2) H+/K+‐ATPase has been shown to mediate the exchange of Na+ and K+. As observed in vitro and in vivo, there is competitive binding between Na+ and H+ on the cytosolic side, and between K+ and NH4 + on the luminal side. The fluxes of K+, H+, Na+ and NH4 + across H+/K+‐ATPase type 2 are determined using the recent kinetic model of the pump developed by Nadal‐Quiros et al. (2015), assuming a 2H+:2K+‐per ATP stoichiometry.
CLC‐K2
The permeability of ClC‐K2 in intercalated cells is taken to decrease with intracellular acidification (Lourdel et al. 2003) and determined as:
| (12) |
where is the ClC‐K2 permeability to Cl− at pHi 7.40, respectively, taken as 0.50 × 10−5 and 0.40 × 10−5 cm s−1 in type A and B cells.
NHE1
The basolateral membrane of principal cells expresses the Na+/H+ exchanger isoform NHE1 (Biemesderfer et al. 1992). NHE1 fluxes are calculated using the parallel coupling model of Fuster et al. (2008).
This is an Editor's Choice article from the 15 October 2016 issue.
Linked articles This article is highlighted by a Perspective by Weinstein. To read this Perspective, visit http://dx.doi.org/10.1113/JP273092.
References
- Azroyan A, Laghmani K, Mordasini D, Doucet A & Edwards A (2011). Regulation of pendrin by pH: dependence on glycosylation. Biochem J 434, 61–72. [DOI] [PubMed] [Google Scholar]
- Bastani B (1995). Colocalization of H‐ATPase and H,K‐ATPase immunoreactivity in the rat kidney. J Am Soc Nephrol 5, 1476–1482. [DOI] [PubMed] [Google Scholar]
- Beck FX, Dorge A, Rick R, Schramm M & Thurau K (1988). The distribution of potassium, sodium and chloride across the apical membrane of renal tubular cells: effect of acute metabolic alkalosis. Pflügers Archiv 411, 259–267. [DOI] [PubMed] [Google Scholar]
- Biemesderfer D, Reilly RF, Exner M, Igarashi P & Aronson PS (1992). Immunocytochemical characterization of Na+‐H+ exchanger isoform NHE‐1 in rabbit kidney. Am J Physiol Renal Physiol 263, F833–F840. [DOI] [PubMed] [Google Scholar]
- Boettger T, Hubner CA, Maier H, Rust MB, Beck FX & Jentsch TJ (2002). Deafness and renal tubular acidosis in mice lacking the K‐Cl co‐transporter Kcc4. Nature 416, 874–878. [DOI] [PubMed] [Google Scholar]
- Bonny O & Edwards A (2013). Calcium reabsorption in the distal tubule: regulation by sodium, pH, and flow. Am J Physiol Renal Physiol 304, F585–F600. [DOI] [PubMed] [Google Scholar]
- Boron WF (2004). Regulation of intracellular pH. Adv Physiol Educ 28, 160–179. [DOI] [PubMed] [Google Scholar]
- Buerkert J, Martin D & Trigg D (1983). Segmental analysis of the renal tubule in buffer production and net acid formation. Am J Physiol Renal Physiol 244, F442–F454. [DOI] [PubMed] [Google Scholar]
- Chambrey R, Kurth I, Peti‐Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hubner CA & Eladari D (2013). Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 110, 7928–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougnon M, Bouyer P, Planelles G & Jaisser F (1998). Does the colonic H,K‐ATPase also act as an Na,K‐ATPase? Proc Natl Acad Sci USA 95, 6516–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crambert G (2014). H‐K‐ATPase type 2: relevance for renal physiology and beyond. Am J Physiol Renal Physiol 306, F693–F700. [DOI] [PubMed] [Google Scholar]
- Crambert G, Horisberger J‐D, Modyanov NN & Geering K (2002). Human nongastric H+‐K+‐ATPase: transport properties of ATP1al1 assembled with different beta‐subunits. Am J Physiol Cell Physiol 283, C305–C314. [DOI] [PubMed] [Google Scholar]
- Elalouf JM, Roinel N & Rouffignac C (1984). Effects of antidiuretic hormone on electrolyte reabsorption and secretion in distal tubules of rat kidney. Pflügers Archiv Eur J Physiol 401, 167–173. [DOI] [PubMed] [Google Scholar]
- Fuster D, Moe OW & Higelmann DW (2008). Steady‐state function of the ubiquitous mammalian Na/H exchanger (NHE1) in relation to dimer coupling models with 2Na/2H stoichiometry. J Gen Physiol 132, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin AV, Bevensee MO, Modyanov NN, Rajendran V, Boron WF & Caplan MJ (1996). Functional expression of the cDNA encoded by the human ATP1AL1 gene. Am J Physiol Renal Physiol 271, F539–F551. [DOI] [PubMed] [Google Scholar]
- Harris HW, Strange K & Zeidel ML (1991). Current understanding of the cellular biology and molecular structure of the anti‐diuretic hormone‐stimulated water transport pathway. J Clin Invest 88, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JR, Kruhoffer M, Adermann K, Heitland A, Maronde E, Meyer M, Forssmann W‐G, Herter P, Plenz G & Schlatter E (2001). Cellular localization, membrane distribution, and possible function of guanylyl cyclases A and 1 in collecting ducts of rat. Cardiovasc Res 51, 553–561. [DOI] [PubMed] [Google Scholar]
- Kahle KT, MacGregor GG, Wilson FH, Van Hoek AN, Brown D, Ardito T, Kashgarian M, Giebisch G, Hebert SC, Boulpaep EL & Lifton RP (2004). Paracellular Cl– permeability is regulated by WNK4 kinase: Insight into normal physiology and hypertension. Proc Natl Acad Sci USA 101, 14877–14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JIN, Kim Y‐H, Cha J‐H, Tisher CC & Madsen KM (1999). Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J Am Soc Nephrol 10, 1–12. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Uchida S, Mizutani S, Sasaki SEI & Marumo F (2001). Intrarenal and cellular localization of CLC‐K2 protein in the mouse kidney. J Am Soc Nephrol 12, 1327–1334. [DOI] [PubMed] [Google Scholar]
- Koeppen B & Giebisch G (1985). Cellular electrophysiology of potassium transport in the mammalian cortical collecting tubule. Pflügers Archiv 405, S143–S146. [DOI] [PubMed] [Google Scholar]
- Lehrmann H, Thomas J, Kim SJ, Jacobi C & Leipziger J (2002). Luminal P2Y2 receptor‐mediated inhibition of Na+ absorption in isolated perfused mouse CCD. J Am Soc Nephrol 13, 10–18. [DOI] [PubMed] [Google Scholar]
- Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, R Chambrey & Eladari D (2010). The Na+‐dependent chloride‐bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Schreck C, Coleman RA, Wade JB, Hernandez Y, Zavilowitz B, Warth R, Kleyman TR & Satlin LM (2011). Role of NKCC in BK channel‐mediated net K+ secretion in the CCD. Am J Physiol Renal Physiol 301, F1088–F1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourdel S, Paulais M, Marvao P, Nissant A & Teulon J (2003). A chloride channel at the basolateral membrane of the distal convoluted tubule: a candidate ClC‐K channel. J Gen Physiol 121, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Dong K, Egan ME, Giebisch GH, Boulpaep EL & Hebert SC (2010). Mouse cystic fibrosis transmembrane conductance regulator forms cAMP‐PKA‐regulated apical chloride channels in cortical collecting duct. Proc Natl Acad Sci USA 107, 6082–6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneton P, Schultheis PJ, Greeb J, Nieman ML, Liu LH, Clarke LL, Duffy JJ, Doetschman T, Lorenz JN & Shull GE (1998). Increased sensitivity to K+ deprivation in colonic H,K‐ATPase‐deficient mice. J Clin Invest 101, 536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto S, Yasoshima K, Yoshitomi K, Imai M & Asano Y (1990). Electrophysiological identification of alpha‐ and beta‐intercalated cells and their distribution along the rabbit distal nephron segments. J Clin Invest 86, 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal‐Quiros M, Moore LC & Marcano M (2015). Parameter estimation for mathematical models of a nongastric H+(Na+)‐K+(NH4+)‐ATPase. Am J Physiol Renal Physiol 309, F434–F446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissant A, Paulais M, Lachheb S, Lourdel S & Teulon J (2006). Similar chloride channels in the connecting tubule and cortical collecting duct of the mouse kidney. Am J Physiol Renal Physiol 290, F1421–F1429. [DOI] [PubMed] [Google Scholar]
- Nonoguchi H, Sands JM & Knepper MA (1989). ANF inhibits NaCl and fluid absorption in cortical collecting duct of rat kidney. Am J Physiol Renal Physiol 256, F179–F186. [DOI] [PubMed] [Google Scholar]
- Pacheco‐Alvarez D & Gamba G (2011). WNK3 is a putative chloride‐sensing kinase. Cell Physiol Biochem 28, 1123–1134. [DOI] [PubMed] [Google Scholar]
- Pech V, Thumova M, Kim YH, Agazatian D, Hummler E, Rossier BC, Weinstein AM, Nanami M & Wall SM (2012). ENaC inhibition stimulates Cl– secretion in the mouse cortical collecting duct through an NKCC1‐dependent mechanism. Am J Physiol Renal Physiol 303, F45–F55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pech VR, Kim YH, Weinstein AM, Everett LA, Pham TD & Wall SM (2007). Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin‐dependent mechanism. Am J Physiol Renal Physiol 292, F914–F920. [DOI] [PubMed] [Google Scholar]
- Rocha AS & LcH Kudo (1990. a). Factors governing sodium and chloride transport across the inner medullary collecting duct. Kidney Int 38, 654–667. [DOI] [PubMed] [Google Scholar]
- Rocha AS & Kudo LH (1990. b). Atrial peptide and cGMP effects on NaCl transport in inner medullary collecting duct. Am J Physiol Renal Physiol 259, F258–268. [DOI] [PubMed] [Google Scholar]
- Rouch AJ, Chen L, Troutman SL & Schafer JA (1991). Na+ transport in isolated rat CCD: effects of bradykinin, ANP, clonidine, and hydrochlorothiazide. Am J Physiol Renal Physiol 260, F86–F95. [DOI] [PubMed] [Google Scholar]
- Schafer JA & Troutman SL (1990). cAMP mediates the increase in apical membrane Na+ conductance produced in rat CCD by vasopressin. Am J Physiol Renal Physiol 259, F823–F831. [DOI] [PubMed] [Google Scholar]
- Schlatter E, Cermak R, Forssmann WG, Hirsch JR, Kleta R, Kuhn M, Sun D & Schafer JA (1996). cGMP‐activating peptides do not regulate electrogenic electrolyte transport in principal cells of rat CCD. Am J Physiol Renal Physiol 271, F1158–F1165. [DOI] [PubMed] [Google Scholar]
- Schlatter E & Schafer JA (1988). Electrophysiological studies in intercalated cells of rat cortical collecting tubules. Pflügers Archiv 411 (Suppl 1), R101. [DOI] [PubMed] [Google Scholar]
- Sindic A & Schlatter E (2007). Renal electrolyte effects of guanylin and uroguanylin. Curr Opin Nephrol Hypertens 16, 10–15. [DOI] [PubMed] [Google Scholar]
- Stoner LC, Burg MB & Orloff J (1974). Ion transport in cortical collecting tubule; effect of amiloride. Am J Physiol 227, 453–459. [DOI] [PubMed] [Google Scholar]
- Strieter J, Stephenson JL, Giebisch G & Weinstein AM (1992). A mathematical model of the rabbit cortical collecting duct. Am J Physiol Renal Physiol 263, F1063–F1075. [DOI] [PubMed] [Google Scholar]
- Summa V, Camargo SMR, Bauch C, Zecevic M & Verrey F (2004). Isoform specificity of human Na+,K+‐ATPase localization and aldosterone regulation in mouse kidney cells. J Physiol 555, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada Y & Knepper MA (1990). Thiazide‐sensitive NaCl absorption in rat cortical collecting duct. Am J Physiol Renal Physiol 259, F519–F528. [DOI] [PubMed] [Google Scholar]
- Theilig F & Wu Q (2015). ANP‐induced signaling cascade and its implications in renal pathophysiology. Am J Physiol Renal Physiol 308, F1047–F1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan SK, Nikolaeva S, Wagner CA, Bonny O, Houillier P, Doucet A & Firsov D (2013). alpha‐Ketoglutarate regulates acid‐base balance through an intrarenal paracrine mechanism. J Clin Invest 123, 3166–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda S, Niisato N, Nakajima K & Marunaka Y (2008). Regulation of the paracellular Na+ and Cl– conductances by the NaCl‐generated osmotic gradient in a manner dependent on the direction of osmotic gradients. Biochem Biophys Res Commun 366, 464–470. [DOI] [PubMed] [Google Scholar]
- Wall SM & Fischer MP (2002). Contribution of the Na+‐K+‐2Cl– cotransporter (NKCC1) to transepithelial transport of H+, NH4+, K+, and Na+ in rat outer medullary collecting duct. J Am Soc Nephrol 13, 827–835. [DOI] [PubMed] [Google Scholar]
- Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED & Verlander JW (2004). NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl– conservation. Hypertension 44, 982–987. [DOI] [PubMed] [Google Scholar]
- Wei Y, Liao Y, Zavilowitz B, Ren J, Liu W, Chan P, Rohatgi R, Estilo G, Jackson EK, Wang WH & Satlin LM (2014). Angiotensin II type 2 receptor regulates ROMK‐like K+ channel activity in the renal cortical collecting duct during high dietary K+ adaptation. Am J Physiol Renal Physiol 307, F833–F843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein AM (1992). Chloride transport in a mathematical model of the rat proximal tubule. Am J Physiol Renal Physiol 263, F784–F798. [DOI] [PubMed] [Google Scholar]
- Weinstein AM (2001). A mathematical model of rat cortical collecting duct: determinants of the transtubular potassium gradient. Am J Physiol Renal Physiol 280, F1072–F1092. [DOI] [PubMed] [Google Scholar]
- Weinstein AM (2010). A mathematical model of rat ascending Henle limb. I. Cotransporter function. Am J Physiol Renal Physiol 298, F512–F524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling LW, Evan AP & Welling DJ (1981). Shape of cells and extracellular channels in rabbit cortical collecting ducts. Kidney Int 20, 211–222. [DOI] [PubMed] [Google Scholar]
- Wingo CS (1989). Reversible chloride‐dependent potassium flux across the rabbit cortical collecting tubule. Am J Physiol Renal Physiol 256, F697–F704. [DOI] [PubMed] [Google Scholar]
- Wingo CS, Madsen KM, Smolka A & Tisher CC (1990). H‐K‐ATPase immunoreactivity in cortical and outer medullary collecting duct. Kidney Int 38, 985–990. [DOI] [PubMed] [Google Scholar]
- Xu J, Barone S, Li H, Holiday S, Zahedi K & Soleimani M (2011). Slc26a11, a chloride transporter, localizes with the vacuolar H+‐ATPase of A‐intercalated cells of the kidney. Kidney Int 80, 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O, Mamenko M, Staruschenko A & Pochynyuk O (2013). Direct activation of ENaC by angiotensin II: recent advances and new insights. Curr Hypertens Rep 15, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]