

Abstract
Key points
Women born small are at an increased risk of developing pregnancy complications. Stress may further increase a woman's likelihood for an adverse pregnancy.
Adverse pregnancy adaptations can lead to long‐term diseases even after her pregnancy.
The current study investigated the effects of stress during pregnancy on the long‐term adrenal, metabolic and cardio‐renal health of female rats that were born small.
Stress programmed increased adrenal Mc2r gene expression, a higher insulin secretory response to glucose during intraperitoneal glucose tolerance test (+36%) and elevated renal creatinine clearance after pregnancy.
Females that were born small had increased homeostatic model assessment‐insulin resistance and elevated systolic blood pressure after pregnancy, regardless of stress exposure.
These findings suggest that being born small or being stressed during pregnancy programs long‐term adverse health outcomes after pregnancy. However, stress in pregnancy does not exacerbate the long‐term adverse health outcomes for females that were born small.
Abstract
Females born small are more likely to experience complications during their pregnancy, including pregnancy‐induced hypertension, pre‐eclampsia and gestational diabetes. The risk of developing complications is increased by stress exposure during pregnancy. In addition, pregnancy complications may predispose the mother to diseases after pregnancy. We determined whether stress during pregnancy would exacerbate the adrenal, metabolic and cardio‐renal dysfunction of growth‐restricted females in later life. Late gestation bilateral uterine vessel ligation was performed in Wistar Kyoto rats to induce growth restriction. At 4 months, growth‐restricted and control female offspring were mated with normal males. Those allocated to the stressed group had physiological measurements [metabolic cage, tail cuff blood pressure, intraperitoneal glucose tolerance test (IPGTT)] conducted during pregnancy whilst the unstressed groups were unhandled. After the completion of pregnancy, dams were aged to 12 months and blood pressure, and metabolic and renal function were assessed. At 13 months, adrenal glands, pancreases and plasma were collected at post‐mortem. Females stressed during pregnancy had increased adrenal Mc2r gene expression (+22%), higher insulin secretory response to glucose during IPGTT (+36%) and higher creatinine clearance (+29%, indicating increased estimated glomerular filtration rate). In contrast, females that were born small had increased homeostatic model assessment‐insulin resistance (+54%), increased water intake (+23%), urine output (+44%) and elevated systolic blood pressure (+7%) regardless of exposure to stress. Our findings suggest that low maternal birth weight and maternal stress exposure during pregnancy are both independently detrimental for long‐term adrenal, metabolic and cardio‐renal health of the mother, although their effects were not exacerbated.
Key points
Women born small are at an increased risk of developing pregnancy complications. Stress may further increase a woman's likelihood for an adverse pregnancy.
Adverse pregnancy adaptations can lead to long‐term diseases even after her pregnancy.
The current study investigated the effects of stress during pregnancy on the long‐term adrenal, metabolic and cardio‐renal health of female rats that were born small.
Stress programmed increased adrenal Mc2r gene expression, a higher insulin secretory response to glucose during intraperitoneal glucose tolerance test (+36%) and elevated renal creatinine clearance after pregnancy.
Females that were born small had increased homeostatic model assessment‐insulin resistance and elevated systolic blood pressure after pregnancy, regardless of stress exposure.
These findings suggest that being born small or being stressed during pregnancy programs long‐term adverse health outcomes after pregnancy. However, stress in pregnancy does not exacerbate the long‐term adverse health outcomes for females that were born small.
Abbreviations
- ACTH
adrenocorticotropic hormone
- Agtr1b
angiotensin II receptor, type 1b
- AUC
area under curve
- AUGC
area under glucose curve
- AUIC
area under insulin curve
- Cyp11b1
cytochrome P450, family 11, subfamily b, polypeptide 1
- Cyp11b2
cytochrome P450, family 11, subfamily b, polypeptide 2
- Cyp21a1
cytochrome P450, family 21, subfamily a, polypeptide 1
- E
embryonic day
- eGFR
estimated glomerular filtration rate
- F0
generation zero
- F1
first‐generation
- F2
second‐generation
- HOMA‐IR
Homeostatic model assessment‐insulin resistance
- HPA
hypothalamic–pituitary–adrenal
- Hsd11b2
hydroxysteroid 11‐beta dehydrogenase 2
- IC
insulin challenge
- Igf1r
insulin‐like growth factor 1 receptor
- IPGTT
intraperitoneal glucose tolerance test
- Mc2r
melanocortin 2 receptor
- Pnmt
phenylethanolamine‐N‐methyltransferase
- Star
steroidogenic acute regulatory protein
- Th
tyrosine hydroxylase
Introduction
Pregnancy is one of the greatest physiological challenges that a woman can experience in her life, as it involves significant metabolic, renal and cardiovascular adaptations to support the growth of the developing fetus (Torgersen & Curran, 2006; Weissgerber & Wolfe, 2006). This challenge often unmasks a myriad of underlying health conditions which may impact not only on the health of the developing fetus, but also on the health of the mother during and after pregnancy. Stress often acts as a catalyst in the development of complications during pregnancy (László et al. 2015; Oni et al. 2015), with these health problems potentially persisting well after pregnancy concludes (Damm, 2009; Hermes et al. 2013). Although the impact of maternal stress leading to hypothalamic–pituitary–adrenal (HPA) axis dysregulation (Entringer et al. 2009; Jellyman et al. 2015; O'Sullivan et al. 2015) and altered in utero development (Fowden et al. 2015) of the offspring has been well characterised, its effects on long‐term maternal health outcomes remain largely unknown. Interestingly, both the risk of developing pregnancy complications and the permanent disease outcomes in females can be programmed whilst in utero.
A reduced birth weight is often used as a surrogate marker of impaired fetal development. Low birth weight, which is often caused by intrauterine growth restriction (IUGR), affects ∼10% of all pregnancies (Martin et al. 2011, 2013). In the Western world, IUGR is most commonly caused by uteroplacental insufficiency, which impairs blood flow through the uterine vessels and disrupts oxygen and nutrient supply to the fetus (Henriksen & Clausen, 2002). Low birth weight increases the risk of developing metabolic, renal and cardiovascular diseases (Gluckman et al. 2007). There is evidence to suggest that females who are small at birth are at a greater risk of adverse pregnancy adaptations including gestational diabetes and pre‐eclampsia (Innes et al. 2002; Zetterström et al. 2007; á Rogvi et al. 2012).
We have previously investigated the impact of being born small on maternal physiology in a rat model of uteroplacental insufficiency. Our findings revealed that females that were born small had compensatory glomerular hypertrophy but normal renal physiology during pregnancy (Gallo et al. 2012 b). However, female growth‐restricted rats developed glucose intolerance for the first time during pregnancy, despite compensatory increases in pancreatic β‐cell mass and normal first and second phase insulin responses (Gallo et al. 2012 b). Previous research has demonstrated that women who develop gestational diabetes have faster deterioration in insulin sensitivity and compensatory pancreatic β‐cell function after pregnancy compared to women who did not develop gestational diabetes (Xiang et al. 2013). Other studies have reported that women who develop gestational diabetes have an increased lifetime risk of developing overt diabetes (Kim et al. 2002; Damm, 2009) and their health can be further compromised by second‐hits such as obesity (Lauenborg et al. 2005). As our model of uteroplacental insufficiency programs glucose intolerance in pregnancy, we investigated the long‐term consequences of maternal glucose intolerance on maternal health after pregnancy. However, our studies in the rat have not established any association between maternal low birth weight and long‐term impairment of maternal metabolic health following pregnancy (Tran et al. 2012), suggesting that additional risk factors such as stress exposure during pregnancy may be necessary to unmask long‐term disease after pregnancy.
Intriguingly, by performing physiological measures to characterise physiological adaptations during pregnancy, we observed detrimental effects on fetal growth whereby fetuses whose mothers were born small and had physiological measurements performed throughout her pregnancy were lighter than their counterparts during late gestation (Gallo et al. 2012 b). We concluded that this series of measurements caused a ‘second‐hit’ of stress in the pregnant dams that were small at birth, ultimately compromising growth of the developing fetuses. However, long‐term effects on the mothers’ health have not been investigated. As stress often acts as a catalyst for pregnancy complications, we aimed to investigate if stress during pregnancy could contribute to maternal disease after pregnancy in both normally grown and growth‐restricted females. We hypothesised that stress during pregnancy would lead to sustained and long‐term impairments in maternal health after pregnancy which would be exacerbated in females that were born small.
Methods
Animal procedures
All experiments were approved by The University of Melbourne Animal Ethics Committee (Ethics no. 1011865) and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. The following experiments comply with the policies and regulations of The Journal of Physiology described by Grundy (2015). Female Wistar Kyoto rats were housed in an environmentally controlled room and allocated into a Control (sham surgery) or Restricted (uteroplacental insufficiency) group. Pregnant dams were anaesthetised with 4% isoflurane and 650 ml min−1 oxygen flow (reduced to 3.2% isoflurane and 250 ml min−1 oxygen flow when suturing). Uteroplacental insufficiency surgery was performed to induce growth restriction by bilateral uterine artery and vein ligation on embryonic day 18 (E18) of pregnancy (term E22) as previously described (O'Dowd et al. 2008). Following surgical procedures, animals were monitored until fully awake and alert and then checked hourly for 6–8 h. Visual checks (eyes clear, no secretions, smooth hair, normal behaviour) were made to ensure full recovery. Pregnant rats were allowed to deliver naturally at term and birth weights of F1 female offspring were recorded. Two female offspring per litter from both the Control and the Restricted groups (n = 12 litters per group) were used for subsequent studies. One of each of these two females was allocated to an Unstressed pregnancy or a Stressed pregnancy and mated with a normal male at 17–23 weeks. Those allocated to the Stressed group were subjected to a series of physiological measurements as previously described (Gallo et al. 2012 b) while their Unstressed counterparts were left unhandled except for husbandry purposes throughout pregnancy. Briefly, this comprised a tail cuff blood pressure measurement (E18), non‐fasted intraperitoneal glucose tolerance test (IPGTT) (E18) and 24 h metabolic cage experiment (E19). A subset of animals were anaesthetised at E20 with an intraperitoneal injection of ketamine (100 mg kg body weight−1) and Illium Xylazil‐20 (30 mg kg body weight−1) and a cardiac puncture performed for the rapid collection of plasma for corticosterone analysis (no other parameters were measured at this age) while all remaining females from all groups delivered naturally at term.
Post‐pregnancy physiological measurements
Body weights of F1 Control and Restricted females were measured at postnatal days 1, 7, 14 and 35 and 4 months and after Unstressed and Stressed pregnancies at 7, 9 and 12 months and at post‐mortem (13 months). Tail cuff blood pressure was measured at 6, 9 and 12 months using the protocol previously described (Gallo et al. 2012 b).
At 12 months of age, F1 females were fasted overnight before insulin challenge (IC). A tail vein blood sample (300 μl) was taken before and 20, 40, 60 and 90 min after a subcutaneous bolus injection of insulin (Actrapid, Novo Nordisk Pharmaceuticals, North Rocks, NSW, Australia; 1 U kg body weight−1). A week later, a fasted IPGTT was performed. Blood samples were collected via tail slice 5 min prior to and at 5, 10, 20, 30, 45, 60, 90 and 120 min following an intraperitoneal bolus injection of 50% (w/v) glucose (Pharmalab, Lane Cove, NSW, Australia; 1 g kg body weight−1) as previously described (Siebel et al. 2008; Laker et al. 2011). Plasma was stored at –20°C until further analysis.
Plasma glucose and insulin concentrations were measured in duplicate using enzymatic fluorometric analysis and a rat insulin radioimmunoassay kit (Millipore, Abacus ALS, Brisbane, QLD, Australia), respectively, as described previously (Siebel et al. 2008; Wadley et al. 2008). Fasting plasma glucose and insulin was taken as the average of two time points (10 and 5 min before injection). Glucose and insulin area under the curve (AUC) were calculated as the total area under curve from basal to 120 min for an IPGTT and glucose AUC from basal to 90 min for an IC (Matthews et al. 1990). The insulin‐to‐glucose ratio was calculated by dividing the total insulin AUC by the total glucose AUC. First phase insulin secretion during IPGTT was calculated as the incremental area under the insulin curve from basal to 5 min whilst second phase insulin secretion was calculated as the incremental area under the insulin curve from 5 to 120 min (Siebel et al. 2008; Laker et al. 2011). Homeostasis model assessment for insulin resistance (HOMA‐IR) was calculated using the following formula: fasting plasma insulin (μU ml−1) × fasting plasma glucose (mmol l−1) ÷ 22.5 (Matthews et al. 1985).
One week after IPGTT, female rats were weighed and individually placed in metabolic cages for 24 h measurements of food and water intake and urine production. Measurements of urinary albumin, total protein, sodium, potassium, creatinine and plasma creatinine were performed using a COBAS Integra 400 (Roche Diagnostics, Castle Hill, NSW, Australia). Plasma samples were collected from tail vein blood samples collected immediately upon the conclusion of 24 h metcage sampling and analysed for creatinine to be used in creatinine clearance calculations [urinary creatinine (μmol l−1) × 24 h urine production (ml)]÷[plasma creatinine–1 (μmol l−1) × 1440 (min)].
Post‐mortem tissue collection
Two weeks after IPGTT, rats were anaesthetized with an intraperitoneal injection of ketamine (100 mg kg body weight−1) and Illium Xylazil‐20 (30 mg kg body weight−1) and a cardiac puncture was performed for the rapid collection of plasma for corticosterone analysis. Adrenal glands, pancreas, kidneys, heart, liver and dorsal white adipose tissue were excised and weighed, then snap frozen in liquid nitrogen and stored at –80°C. A piece of pancreatic tissue (∼1 cm) from the head of the pancreas was fixed in 10% neutral buffer formalin for histological analysis.
Plasma corticosterone concentrations were measured both at E20 and at 13 months using 10 μl of plasma using a radioimmunoassay as described (Cuffe et al. 2014).
Pancreatic islet, β‐cell morphology and immunohistochemistry
Pancreatic tissue was exhaustively sectioned at 5 μm. Three sections of equal distance apart were selected and immunostained and random systematic point counting used to determine relative islet and β‐cell volume density to determine the absolute islet and β‐cell mass as previously described (Bonner‐Weir, 2001; Laker et al. 2011).
RT2 profiler PCR array analysis and Western blotting
Total RNA and protein were extracted from whole adrenal glands as described previously (Cuffe et al. 2012). In total, 200 ng of RNA was reverse transcribed using the RT2 First Strand cDNA synthesis kit (Qiagen, Melbourne, Australia). mRNA levels of melanocortin 2 receptor (Mc2r, NM_001100491), steroidogenic acute regulatory protein (Star, NM_031558), cytochrome P450, family 21, subfamily a, polypeptide 1 (Cyp21a1, NM_057101), cytochrome P450, family 11, subfamily b, polypeptide 1 (Cyp11b1, NM_012537), cytochrome P450, family 11, subfamily b, polypeptide 2 (Cyp11b2, NM_012538), hydroxysteroid 11‐beta dehydrogenase 2 (Hsd11b2, NM_017081), angiotensin II receptor, type 1b (Agtr1b, NM_031009), phenylethanolamine‐N‐methyltransferase (Pnmt, NM_031526), insulin‐like growth factor 1 receptor (Igf1r, NM_052807) and tyrosine hydroxylase (Th, NM_012740) were analysed using quantitative PCR (qPCR) on RT2 Profiler Arrays (Qiagen, Cat. No. 330131). mRNA levels were normalised to the average of the Unstressed Control group and determined using the comparative CT (2−ΔΔCT) method.
In total, 20 μg of denatured protein was loaded per sample for analysis of relative protein expression using SDS‐PAGE and Western blotting as described previously (Cuffe et al. 2011). Membranes were incubated overnight with either a rabbit anti‐MC2R (1: 500, Santa Cruz Biotechnology, Cat. No. sc‐13107, Santa Cruz, CA, USA) or rabbit anti‐STAR (1:1000, Abcam, Cat. No. ab96637, Melbourne, VIC., Australia) antibody. A mouse anti‐β‐actin (1:10,000, Sigma‐Aldrich, Cat. No. A5441, Castle Hill, NSW, Australia) antibody was used as loading control. Blots were incubated for 1 h with LI‐COR (LI‐COR Biosciences, Lincoln, NE, USA) IRDye 680 goat anti‐rabbit and IRDye 800CW goat anti‐mouse secondary antibodies. Protein expression was quantified using a LI‐COR Odyssey infrared imaging system.
Statistical analysis
All data were analysed using a two‐way ANOVA to assess the main effects of uteroplacental insufficiency (Group) and maternal stress (Stress) or Student's unpaired t test. If significant interactions were observed, Student's unpaired t test was performed to compare individual group means. All data are presented as means ± SEM and P < 0.05 was considered statistically significant.
Results
Body and organ weights
Uteroplacental insufficiency in F0 females reduced F1 female weights (–16%) at postnatal day 1 (P < 0.01; Table 1). Restricted females remained lighter at postnatal days 7, 14 and 35 (P < 0.01; Table 1) and at mating (4 months; P = 0.023; Table 1). Following pregnancy, F1 females that were born small remained lighter at 7 and 9 months (P Group < 0.01; Table 1) and post‐mortem at 13 months (P Group = 0.03; Table 1), regardless of stress exposure during pregnancy. At 12 months, only Restricted females that were Unstressed during pregnancy were lighter than control counterparts (P = 0.02; Table 1). Relative pancreas, liver, dorsal white adipose tissue, heart, kidney and adrenal weights were not affected by F1 growth restriction or maternal stress (Table 1).
Table 1.
Body and organ weights relative to body weight (% BW)
| Unstressed | Stressed | 2‐way ANOVA | |||||
|---|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Group | Stress | Interaction | |
| Body weight (g) | |||||||
| Postnatal day 1 | 4.32 ± 0.04 | 3.62 ± 0.05γ | |||||
| Postnatal day 7 | 10.8 ± 0.3 | 8.8 ± 0.3γ | |||||
| Postnatal day 14 | 23.3 ± 0.4 | 20.1 ± 0.6γ | |||||
| Postnatal day 35 | 78 ± 1 | 72 ± 2γ | |||||
| 4 months | 219 ± 3 | 210 ± 2γ | |||||
| Post‐pregnancy body weight (g) | |||||||
| 7 months | 268 ± 5 | 250 ± 5 | 267 ± 4 | 260 ± 3 | P = 0.006 | NS | NS |
| 9 months | 262 ± 3 | 247 ± 4 | 259 ± 3 | 254 ± 3 | P = 0.006 | NS | NS |
| 12 months | 271 ± 3 | 252 ± 3* | 264 ± 3 | 261 ± 3 | P = 0.001 | NS | P = 0.02 |
| Post‐mortem (13 months) | 270 ± 4 | 258 ± 4 | 264 ± 4 | 261 ± 3 | P = 0.03 | NS | NS |
| Organ weights (% BW) | |||||||
| Pancreas | 0.299 ± 0.014 | 0.295 ± 0.015 | 0.321 ± 0.016 | 0.323 ± 0.020 | NS | NS | NS |
| Liver | 2.81 ± 0.07 | 2.85 ± 0.06 | 2.81 ± 0.06 | 2.84 ± 0.05 | NS | NS | NS |
| Dorsal white adipose tissue | 1.52 ± 0.07 | 1.57 ± 0.09 | 1.59 ± 0.12 | 1.42 ± 0.05 | NS | NS | NS |
| Heart | 0.368 ± 0.003 | 0.367 ± 0.003 | 0.367 ± 0.005 | 0.374 ± 0.003 | NS | NS | NS |
| Kidneys | 0.614 ± 0.009 | 0.600 ± 0.006 | 0.613 ± 0.006 | 0.609 ± 0.010 | NS | NS | NS |
| Adrenals | 0.0212 ± 0.0005 | 0.026 ± 0.0010 | 0.0209 ± 0.0009 | 0.0214 ± 0.0003 | NS | NS | NS |
Effects of growth restriction and maternal stress on body weights and organ weights relative to body weight (mean ± SEM; n = 10–14 per group). γ P < 0.05 vs. Control; * P < 0.05 vs. Control counterpart (following significant interaction); NS, not significant.
Corticosterone, adrenal steroidogenic gene and protein expression
Corticosterone concentrations at E20 were increased in Stressed pregnant females (P Stress = 0.008; Fig. 1 A). Plasma collected at post‐mortem at 13 months revealed that while corticosterone concentrations were reduced by 45%, this did not reach statistical significance (P Stress = 0.054; Fig. 1 B). Females that were exposed to stress during their pregnancy had increased adrenal mRNA expression of Mc2r (+22%; P Stress = 0.026; Fig. 1 C). Although MC2R protein levels were also increased by 22% in those females exposed to stress during pregnancy (Fig. 1 E), this did not reach statistical significance (P Stress = 0.12). Neither maternal stress during pregnancy nor uteroplacental insufficiency had any effect on adrenal Star (Fig. 1 D), Cyp21a1, Cyp11b1, Cyp11b2, Hsd11b2, Agtr1b, Pnmt, Igf1r and Th gene expression (Table 2).
Figure 1. Plasma corticosterone concentrations, adrenal gene and protein levels .
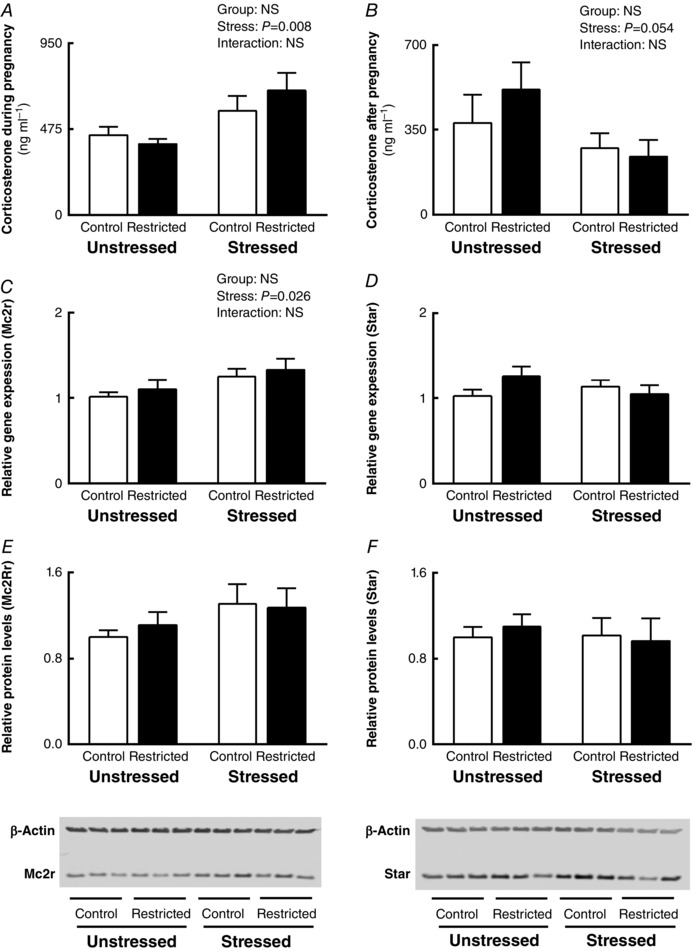
Effects of growth restriction and maternal stress on: A, plasma corticosterone concentrations during pregnancy at E20; B, plasma corticosterone concentrations after pregnancy at 13 months (post‐mortem); C, adrenal Mc2r mRNA expression; D, adrenal Star mRNA expression; E, adrenal Mc2r protein levels; and F, adrenal Star protein levels (mean ± SEM; n = 7–8 per group for corticosterone concentration and qPCR and n = 6 per group for Western blotting).
Table 2.
Adrenal steroidogenic gene expression levels
| Unstressed | Stressed | 2‐way ANOVA | |||||
|---|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Group | Stress | Interaction | |
| Cyp21a1 | 1.08 ± 0.17 | 1.23 ± 0.18 | 1.42 ± 0.18 | 1.03 ± 0.08 | NS | NS | NS |
| Cyp11b1 | 1.00 ± 0.04 | 1.10 ± 0.08 | 1.14 ± 0.07 | 1.18 ± 0.05 | NS | NS | NS |
| Cyp11b2 | 1.01 ± 0.06 | 1.06 ± 0.09 | 1.15 ± 0.1 | 1.13 ± 0.12 | NS | NS | NS |
| Hsd11b2 | 1.03 ± 0.10 | 1.10 ± 0.10 | 1.08 ± 0.10 | 1.22 ± 0.13 | NS | NS | NS |
| Agtr1b | 1.06 ± 0.15 | 1.20 ± 0.15 | 1.52 ± 0.19 | 1.30 ± 0.14 | NS | NS | NS |
| Pnmt | 1.21 ± 0.26 | 1.43 ± 0.27 | 1.44 ± 0.25 | 0.97 ± 0.13 | NS | NS | NS |
| Igf1r | 1.01 ± 0.06 | 1.02 ± 0.05 | 1.07 ± 0.07 | 1.14 ± 0.09 | NS | NS | NS |
| Th | 1.04 ± 0.11 | 1.19 ± 0.14 | 1.27 ± 0.16 | 1.31 ± 0.04 | NS | NS | NS |
Effects of growth restriction and maternal stress on adrenal mRNA expression of steroidogenenic genes (mean ± SEM; n = 7–8 per group). NS, not significant.
Metabolic function
Plasma glucose concentrations were not different between Control and Restricted groups or Unstressed and Stressed groups (Fig. 2 A and B) during IPGTT at 12 months. Similarly, insulin secretion in response to IPGTT was not affected by uteroplacental insufficiency or stress (Fig. 2 C and D). However, the insulin secretory response to glucose, expressed as the ratio of area under the insulin curve (AUIC) to area under the glucose curve (AUGC), was increased in females that experienced stress during pregnancy compared to Unstressed females (+36%; P Stress = 0.037), but was not different between Control and Restricted groups (Fig. 2 E). HOMA‐IR was increased in females that were born small (+54%; P Group = 0.012) independent of stress exposure (Fig. 2 F), indicating impaired basal hepatic insulin sensitivity. First and second phase insulin secretory responses to IPGTT were not different across groups (Table 3).
Figure 2. Plasma glucose and insulin response during IPGTT .
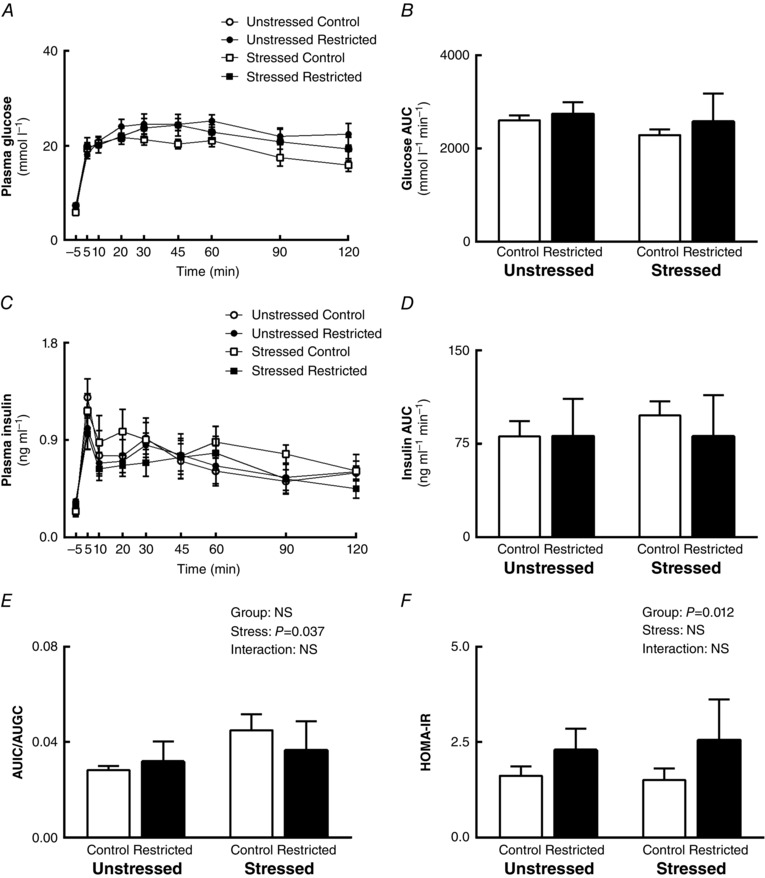
Effects of growth restriction and maternal stress on: A, plasma glucose concentration over 120 min; B, area under glucose curve; C, plasma insulin concentration over 120 min; D, area under insulin curve; E, ratio of area under insulin curve to area under glucose curve; and F, HOMA‐IR during IPGTT (mean ± SEM; n = 6–7 per group).
Table 3.
Metabolic parameters and pancreatic morphology measured in F1 females at 12 months
| Unstressed | Stressed | 2‐way ANOVA | |||||
|---|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Group | Stress | Interaction | |
| Fasting glucose | 6.8 ± 0.5 | 6.4 ± 0.5 | 5.9 ± 0.3 | 7.5 ± 0.7 | NS | NS | NS |
| Fasting insulin | 0.24 ± 0.03 | 0.34 ± 0.03 | 0.24 ± 0.05 | 0.3 ± 0.05 | P = 0.058 | NS | NS |
| Fasting insulin/glucose ratio | 0.04 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 | NS | NS | NS |
| 1st phase insulin AUC | 3.8 ± 0.5 | 3.4 ± 0.5 | 3.5 ± 0.4 | 3.2 ± 0.5 | NS | NS | NS |
| 2nd phase insulin AUC | 77 ± 12 | 78 ± 12 | 94 ± 11 | 77 ± 15 | NS | NS | NS |
| Insulin challenge | |||||||
| Glucose AUC | 241 ± 13 | 263 ± 11 | 242 ± 6 | 242 ± 12 | NS | NS | NS |
| Pancreatic morphology | |||||||
| β‐Cell mass (mg) | 3.4 ± 1.0 | 5.1 ± 1.1 | 5.7 ± 1.3 | 6.9 ± 1.5 | NS | NS | NS |
| Relative β‐cell mass (g g−1) | 12.6 ± 3.6 | 19.6 ± 4.0 | 20.8 ± 4.8 | 26.9 ± 6.0 | NS | NS | NS |
| Islet mass | 5.7 ± 1.4 | 8.0 ± 1.5 | 8.9 ± 1.9 | 10.8 ± 2.4 | NS | NS | NS |
| Relative islet mass | 20.9 ± 5.5 | 30.5 ± 5.6 | 32.4 ± 6.9 | 41.9 ± 9.1 | NS | NS | NS |
| Islet proportion/pancreas (%) | 0.66 ± 0.17 | 0.91 ± 0.14 | 1.06 ± 0.23 | 1.23 ± 0.24 | NS | NS | NS |
Effects of growth restriction and maternal stress on metabolic parameters during IPGTT and insulin challenge and pancreatic morphology (mean ± SEM; n = 5–8 per group). NS, not significant.
Post‐pregnancy fasting plasma glucose, insulin and the ratio of fasting insulin to glucose were not affected by stress during pregnancy or being born small (Table 3). Whole‐body insulin sensitivity determined through the glucose AUC response to an insulin challenge was not influenced by low birth weight or stress during pregnancy (Table 3). Absolute and relative pancreatic β‐cell and islet mass were not different across groups (Table 3). Similarly, islet proportion per pancreas was unchanged between groups (Table 3).
Cardio‐renal function
At 12 months of age, Restricted females consumed more water (+23%; P Group = 0.018; Fig. 3 A) that was associated with higher 24 h urine output compared to Control counterparts (+12%; P Group = 0.013; Fig. 3 B). Subsequent post hoc analyses revealed that urine flow rate was increased in Unstressed Restricted (+44%; P = 0.005) and Stressed Control (+34%; P = 0.005) animals when compared to Unstressed Control (Fig. 3 B). Food intake was similar across all groups over a 24 h period (Table 4). Urinary excretion of total protein, albumin, sodium and potassium were not different regardless of growth restriction or maternal stress (Fig. 3 E, F and Table 4). However, urinary creatinine excretion was increased in Unstressed Restricted (+32%; P = 0.017) and Stressed Control females (+35%; P = 0.026) when compared to Unstressed Control females (Fig. 3 C). Creatinine clearance, which is indicative of estimated glomerular filtration rate (eGFR), was increased in females that were exposed to stress during pregnancy regardless of their birth weight (+29%; P Stress = 0.031; Fig. 3 D).
Figure 3. Renal function .
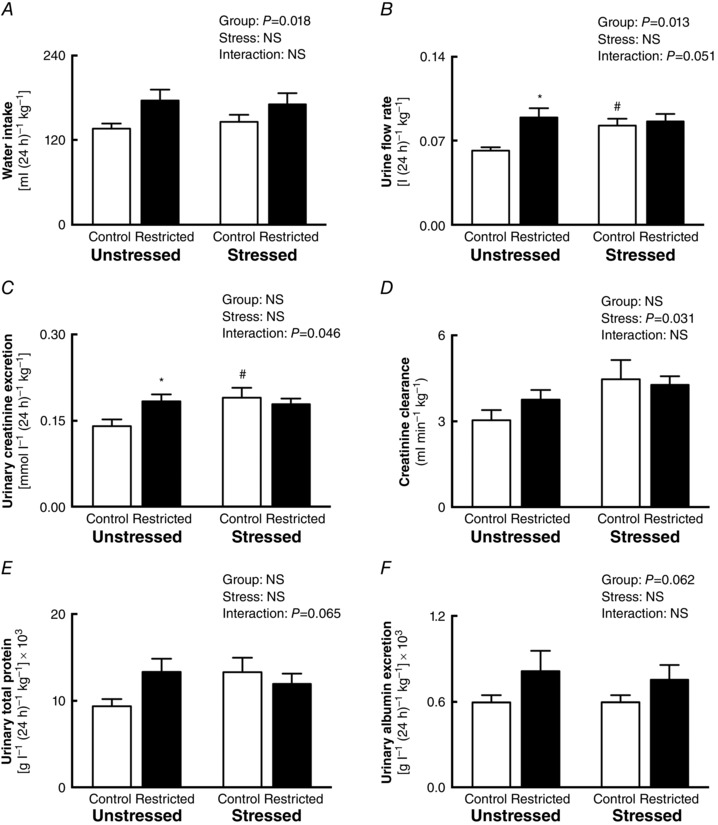
Effects of growth restriction and maternal stress on: A, water intake; B, urinary flow rate; C, urinary creatinine excretion; D, creatinine clearance; E, urinary micro‐total protein excretion; and F, urinary albumin excretion over 24 h (mean ± SEM; n = 8–14 per group). * P < 0.05 vs. Control counterpart (following significant interaction); # P < 0.05 vs. Unstressed counterpart (following significant interaction).
Table 4.
Systolic blood pressure, 24 h metabolic cage and renal measurements
| Unstressed | Stressed | 2‐way ANOVA | |||||
|---|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Group | Stress | Interaction | |
| Systolic blood pressure 6 months (mmHg) | 137 ± 4 | 133 ± 7 | 129 ± 3 | 137 ± 3 | NS | NS | NS |
| Systolic blood pressure 9 months (mmHg) | 130 ± 3 | 133 ± 3 | 130 ± 3 | 129 ± 3 | NS | NS | NS |
| Systolic blood pressure 12 months (mmHg) | 121 ± 3 | 133 ± 3 | 125 ± 3 | 129 ± 3 | P = 0.01 | NS | NS |
| Food intake [g (24 h)−1 kg−1] | 65 ± 3 | 64 ± 3 | 65 ± 4 | 58 ± 3 | NS | NS | NS |
| Urinary sodium excretion [mmol l−1 (24 h)−1 kg−1] | 2.5 ± 0.4 | 3.0 ± 0.6 | 3.0 ± 0.3 | 2.8 ± 0.2 | NS | NS | NS |
| Urinary potassium excretion [mmol l−1 (24 h)−1 kg−1] | 4.8 ± 0.6 | 5.9 ± 0.9 | 6.3 ± 0.5 | 6.1 ± 0.4 | NS | NS | NS |
Effects of growth restriction and maternal stress on systolic blood pressure, food intake and urinary excretion over 24 h (mean ± SEM; n = 8–14 per group). NS, not significant.
Systolic blood pressure was not different between Control and Restricted females in Unstressed and Stressed groups at 6 and 9 months (Table 4). At 12 months, Restricted females had increased systolic blood pressure compared to Control females (+7%; P Group = 0.01; Table 4). Subsequent post hoc analysis revealed that Restricted females that were Unstressed had higher blood pressure compared to Controls (+10%; +12 mmHg; P < 0.012), but this was not observed between Control and Restricted females that were exposed to stress.
Discussion
We have previously demonstrated that growth‐restricted female rats develop glucose intolerance for the first time during pregnancy without any impact on cardiovascular or renal function (Gallo et al. 2012 b). Interestingly, the glucose intolerance observed did not persist after pregnancy (Tran et al. 2012), but cardiovascular and renal function was impaired (Gallo et al. 2012 a). As stress is known to unmask various pregnancy complications, this led us to hypothesise that stress during pregnancy could act as a ‘second‐hit’ in addition to growth restriction, and may unmask long‐term adrenal, metabolic and cardio‐renal outcomes in the mothers. Here, we report that stress in pregnancy resulted in impaired adrenal steroidogenesis and insulin secretory response to glucose during IPGTT in mothers after pregnancy. Furthermore, creatinine clearance was increased suggesting renal hyperfiltration in females that were stressed during pregnancy, regardless of their birth weight. In contrast, females born small at birth had different metabolic and cardio‐renal dysfunction after pregnancy regardless of whether they experienced stress during pregnancy. Hepatic insulin sensitivity was reduced if mothers were born small. In addition, blood pressure, water intake and urine output were elevated at 12 months in Restricted mothers independent of stress. These specific phenotypes observed following pregnancy were not present during pregnancy, but it is likely that the other pregnancy complications we have previously demonstrated in females born small contributed to the long‐term development of these outcomes. Our study demonstrated for the first time that stress during pregnancy induced through routine physiological measurements can independently lead to health complications in rats. It is thus important for investigators studying physiological outcomes using these techniques to consider it as an additional stressor in their studies. Importantly, the effects of maternal stress exposure during pregnancy were not additive to the effects of being born small and appeared to act independently, which was contrary to our initial hypothesis.
Corticosterone and adrenal steroidogenesis
The elevated corticosterone concentrations observed at day 20 of pregnancy confirmed that conducting physiological measurements (tail cuff blood pressure, glucose tolerance test, metabolic cage experiment) resulted in moderate maternal stress as expected. The 48% increase in corticosterone concentrations observed was slightly lower than concentrations demonstrated following maternal corticosterone administration (Cuffe et al. 2012; Vaughan et al. 2012) but similar to exposure to a hypoxic stress (Cuffe et al. 2014) in the mouse. Intriguingly, 8 months after pregnancy, plasma corticosterone concentrations tended to decrease (–45%) in mothers that were exposed to stress during pregnancy. This suggests impaired adrenal glucocorticoid output (Moisiadis & Matthews, 2014). To investigate this further, the expression of several key genes involved in steroidogenesis were examined. While factors other than that examined may have been affected by stress in pregnancy, the only gene found to be affected by stress was Mc2r, which was elevated in mothers that experienced stress during their pregnancy 9 months earlier. This highlights the long‐term impact of stress on HPA axis function, as Mc2r encodes the adrenocorticotropic hormone (ACTH) receptor, which regulates overall adrenal corticosterone synthesis (Moisiadis & Matthews, 2014). This compensatory change in gene expression was mirrored by similar increases in protein levels although it was not statically significant, which could potentially be due to the smaller sample size in our Western blots. While the impact of stress in pregnancy on maternal adrenal function after pregnancy has not been previously studied, exposure to glucocorticoids in utero has been known to program increased corticosterone production in association with elevated Mc2R mRNA expression in the offspring (Waddell et al. 2010). Additionally, it would be beneficial for future studies to subject these females to a stressful challenge to determine if the HPA axis response to acute stress may be impaired.
Metabolic function
Interestingly, both growth restriction and stress during pregnancy independently led to metabolic dysfunction after pregnancy although there were distinct differences in metabolic profiles depending on the parameters measured. We have previously demonstrated that females born small develop glucose intolerance for the first time during pregnancy (Gallo et al. 2012 b), which was not sustained following pregnancy (Tran et al. 2012). Here, we hypothesised that an additional hit of stress during pregnancy may lead to long‐term metabolic disturbances. However, we observed that hepatic insulin resistance, reflected by HOMA‐IR, increased in mothers that were born small but was not affected by stress during pregnancy. Often used in epidemiological studies as an index of insulin resistance, HOMA‐IR in rats has been shown to correlate well with insulin sensitivity as assessed by the gold standard euglycaemic–hyperinsulinaemic clamp for assessing in vivo insulin sensitivity (Cacho et al. 2008). Furthermore, the close to significant increase in basal fasting insulin concentrations supports the fact that these growth‐restricted females have developed some degree of insulin resistance. In contrast, mothers that were exposed to stress during pregnancy had an increased ratio of AUIC to AUGC in response to an IPGTT regardless of birth weight, indicating that they had increased insulin secretory response to glucose. However, this was not attributed to changes in pancreatic β‐cell or islet mass, as they were similar across groups. This may be due to either impaired insulin secretory capacity of individual pancreatic islets or reduced insulin‐stimulated glucose uptake in metabolic tissues. Corticosterone is known to stimulate glucose release and suppress glucose utilisation, which together result in increased plasma glucose (Kuo et al. 2013). As such, the 45% reduction in plasma corticosterone concentrations in dams exposed to stress during pregnancy may have maintained glucose concentrations within the normal physiological range.
We have previously reported a sustained and heightened plasma glucose concentration up to 120 min during IPGTT in females after pregnancy (Tran et al. 2012), and we again observe that plasma glucose concentrations did not return to basal after 120 min in the current study. The delay in glucose returning to basal concentrations could be attributed to advanced age, as ageing is often associated with insulin resistance and glucose intolerance (DeFronzo, 1979; Qiang et al. 2007). It is possible that studying these females at this later age of 12 months may have masked any effects of growth restriction or maternal stress on these parameters as even the females from the control groups may have developed disease outcomes.
Furthermore, the fact that our growth‐restricted females did not become overweight or obese may have protected them against developing overt metabolic phenotypes. Uteroplacental insufficiency resulted in female offspring that were born small and remained slightly small throughout life although by 12 months of age this deficit was less than 5%. This suggests catch‐up growth during the postnatal period, but it is important to note that these rats did not become obese. Inappropriate catch‐up growth after birth is known to increase risk of developing metabolic and cardiovascular outcomes later in life in both the human population and in rodents (Forsén et al. 2000; Hales & Ozanne, 2002; Barker et al. 2005; Gallo et al. 2013). If these females became obese, it may well unmask more severe disease phenotypes, and those previously programmed either through growth restriction or the experience of a stressful pregnancy may respond to this additional stressor in a different manner.
Cardio‐renal function
We have previously demonstrated that Control and Restricted females had increased creatinine clearance during pregnancy compared to their non‐pregnant counterparts, highlighting renal hyperfiltration to compensate for the increased renal demands during pregnancy regardless of their birth weight (Gallo et al. 2012 b). Following the conclusion of pregnancy, eGFR returned to normal (Gallo et al. 2012 a). In the current study, we again observed no effect of growth restriction on eGFR following pregnancy. However, we demonstrated that exposure to stress during pregnancy resulted in higher creatinine clearance and thus eGFR 8 months after the conclusion of pregnancy. Although a low GFR is usually the hallmark of chronic kidney disease (Levey et al. 2010; Johnson et al. 2012), an increased GFR suggests hyperfiltration and is an early indication of renal insufficiency (Palatini et al. 2012). If these females continue to progress towards more severe renal phenotypes, chronic kidney disease may eventually emerge. Mothers born small also had increased urinary albumin excretion, although this did not reach significance, supporting our suggestions of early renal damage. In humans, albuminuria is associated with cardiovascular risk and increased mortality (Hillege, 2002). Importantly, even a slight increase in albuminuria is sufficient to increase a person's risk of cardiovascular complications (Klausen, 2004). Our findings suggest that stress during pregnancy reveals renal phenotypes that persist after pregnancy, with the potential to lead to long‐term and significant renal damage.
We have previously demonstrated that growth‐restricted females do not develop elevated systolic blood pressure even at 18 months of age (Moritz et al. 2009). Follow‐up studies demonstrated these females did not develop increased blood pressure either when pregnant (Gallo et al. 2012 b) or after pregnancy (Gallo et al. 2012 a) when measured by tail cuff plethysmography, and nor was it different between virgin and pregnant females (Gallo et al. 2012 a). However, when mean arterial pressure was assessed through indwelling tail‐artery catheter, both control and growth‐restricted females that previously experience pregnancy had elevated mean arterial pressure (Gallo et al. 2012 a). Our current study supports this, and at 12 months of age, systolic blood pressure by tail cuff was elevated in mothers that were born small. Although statistical analysis suggests that this is an effect of growth restriction, we believe that the elevated blood pressure observed in the Restricted group was mainly driven by the 12 mmHg increase in blood pressure only in the Restricted group that remained unstressed during pregnancy. While it was hypothesised that stress during pregnancy would further exacerbate and increase post‐pregnancy blood pressure, this was not observed when measured by tail cuff. It is important to note that blood pressure can be regulated by multiple physiological systems, and studies have demonstrated that offspring of female rodents exposed to glucocorticoids develop alterations in adrenal outcomes (Quinn et al. 2014), renal function (Singh et al. 2007) and blood pressure regulation (O'Sullivan et al. 2013). It is therefore likely that the impairments in adrenal and renal function may contribute to the elevated blood pressure observed in growth‐restricted females after pregnancy. Future studies should include gold standard blood pressure and GFR measurements through radiotelemetry and insulin clearance, respectively, which may reveal changes we were unable to identify. Given the invasive nature of these techniques and hence the stress it may cause, separate additional cohorts would be required to achieve this.
Conclusion
The current study investigated if stress during pregnancy could lead to long‐term health dysfunction in female rats that were born small. While previous studies have focused on offspring outcomes following stress in pregnancy, this is the first time that maternal health has been investigated. We report that being born small or being stressed while pregnant independently programmed impaired the adrenal, metabolic and cardio‐renal health of mothers up to 9 months after pregnancy. Mothers who experienced stress during pregnancy had higher insulin secretory response to glucose during IPGTT, increased eGFR and also increased mRNA expression of the ACTH receptor Mc2r, while those that were born small had increased HOMA‐IR and elevated blood pressure. Although multiple physiological systems were impaired, overt disease was not present in any one system. However, collectively this may have major implications for the future health of the mother. This study highlights that mothers who were small at birth or have experienced stress during pregnancy may be at a greater risk of developing a broad range of long‐term pathologies, although being affected by both low birth weight and stress during pregnancy do not appear to place her at greater risk than those with only one risk factor. The different mechanisms in which the two insults act to program future diseases may explain the different phenotypes we observed, as growth restriction can result in disease risk being programmed even prior to these females being born, in contrast to the acute impact of stress during pregnancy, although this remains to be elucidated. Given our findings, it is important that women who experience adverse pregnancy events be monitored after pregnancy and appropriate intervention considered, ensuring the best outcomes for her health during pregnancy and in later life. Follow‐up studies in offspring of these mothers is also warranted to fully understand the long‐term impact of an adverse pregnancy.
Additional information
Competing interests
None.
Author contributions
J.N.C., J.S.M.C., A.J.J., K.M.M. and M.E.W. were responsible for the conception and design of the experiments. J.N.C., J.S.M.C. and A.J.J. were responsible for the collection of data. All authors were involved in the analysis and interpretation of the data. All authors were involved in drafting the manuscript and revising it critically for intellectual content. All authors approve of the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by the National Health and Medical Research Council of Australia (Grant no. 1025426) awarded to M.E.W. and K.M.M.
This is an Editor's Choice article from the 15 October 2016 issue.
References
- á Rogvi R, Forman JL, Damm P & Greisen G (2012). Women born preterm or with inappropriate weight for gestational age are at risk of subsequent gestational diabetes and pre‐eclampsia. PLoS ONE 7, e34001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Forsén TJ, Kajantie E & Eriksson JG (2005). Trajectories of growth among children who have coronary events as adults. N Engl J Med 353, 1802–1809. [DOI] [PubMed] [Google Scholar]
- Bonner‐Weir S (2001). β‐Cell turnover: its assessment and implications. Diabetes 50, S20–S24. [DOI] [PubMed] [Google Scholar]
- Cacho J, Sevillano J, de Castro J, Herrera E & Ramos MP (2008). Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague‐Dawley rats. Am J Physiol Endocrinol Metab 295, E1269–E1276. [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, Dickinson H, Simmons DG & Moritz KM (2011). Sex specific changes in placental growth and MAPK following short term maternal dexamethasone exposure in the mouse. Placenta 32, 981–989. [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, O'Sullivan L, Simmons DG, Anderson ST & Moritz KM (2012). Maternal corticosterone exposure in the mouse has sex‐specific effects on placental growth and mRNA expression. Endocrinology 153, 5500–5511. [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, Walton SL, Singh RR, Spiers JG, Bielefeldt‐Ohmann H, Wilkinson L, Little MH & Moritz KM (2014). Mid‐ to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex‐specific manner. J Physiol 592, 3127–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm P (2009). Future risk of diabetes in mother and child after gestational diabetes mellitus. Int J Gynaecol Obstet 104, S25–S26. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA (1979). Glucose intolerance and aging: evidence for tissue insensitivity to insulin. Diabetes 28, 1095–1101. [DOI] [PubMed] [Google Scholar]
- Entringer S, Kumsta R, Hellhammer DH, Wadhwa PD & Wüst S (2009). Prenatal exposure to maternal psychosocial stress and HPA axis regulation in young adults. Horm Behav 55, 292–298. [DOI] [PubMed] [Google Scholar]
- Forsén T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C & Barker D (2000). The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med 133, 176–182. [DOI] [PubMed] [Google Scholar]
- Fowden AL & Forhead AJ (2015). Glucocorticoids as regulatory signals during intrauterine development. Exp Physiol 100, 1477–1487. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Denton KM, Moritz KM, Tare M, Parkington HC, Davies M, Tran M, Jefferies AJ & Wlodek ME (2012. a). Long‐term alteration in maternal blood pressure and renal function after pregnancy in normal and growth‐restricted rats. Hypertension 60, 206–213. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Tran M, Moritz KM & Wlodek ME (2013). Developmental programming: variations in early growth and adult disease. Clin Exp Pharmacol Physiol 40, 795–802. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Tran M, Moritz KM, Mazzuca MQ, Parry LJ, Westcott KT, Jefferies AJ, Cullen McEwen LA & Wlodek ME (2012. b). Cardio‐renal and metabolic adaptations during pregnancy in female rats born small: implications for maternal health and second generation fetal growth. J Physiol 590, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA & Beedle AS (2007). Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol 19, 1–19. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . Exp Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CN & Ozanne SE (2002). The dangerous road of catch‐up growth. J Physiol 547, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen T & Clausen T (2002). The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well‐nourished populations. Acta Obstet Gynecol Scand 81, 112–114. [DOI] [PubMed] [Google Scholar]
- Hermes W, Franx A, van Pampus MG, Bloemenkamp KWM, Bots ML, van der Post JA, Porath M, Ponjee GAE, Tamsma JT, Mol BWJ & de Groot CJM (2013). Cardiovascular risk factors in women who had hypertensive disorders late in pregnancy: a cohort study. Am J Obstet Gynecol 208, 474.e1–8. [DOI] [PubMed] [Google Scholar]
- Hillege HL (2002). Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 106, 1777–1782. [DOI] [PubMed] [Google Scholar]
- Innes KE, Byers TE, Marshall JA, Barón A, Orleans M & Hamman RF (2002). Association of a woman's own birth weight with subsequent risk for gestational diabetes. JAMA 287, 2534–2541. [DOI] [PubMed] [Google Scholar]
- Jellyman JK, Valenzuela OA, Allen VL, Forhead AJ, Holdstock NB & Fowden AL (2015). Neonatal glucocorticoid overexposure programs pituitary–adrenal function in ponies. Domest Anim Endocrinol 50, 45–49. [DOI] [PubMed] [Google Scholar]
- Johnson DW, Jones GRD, Mathew TH, Ludlow MJ, Chadban SJ, Usherwood T, Polkinghorne K, Colagiuri S, Jerums G, MacIsaac R & Martin H (2012). Chronic kidney disease and measurement of albuminuria or proteinuria: a position statement. Med J Aust 197, 224–225. [DOI] [PubMed] [Google Scholar]
- Kim C, Newton KM & Knopp RH (2002). Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care 25, 1862–1868. [DOI] [PubMed] [Google Scholar]
- Klausen K (2004). Very low levels of microalbuminuria are associated with increased risk of coronary heart disease and death independently of renal function, hypertension, and diabetes. Circulation 110, 32–35. [DOI] [PubMed] [Google Scholar]
- Kuo T, Harris CA & Wang J‐C (2013). Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol 380, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker RC, Gallo LA, Wlodek ME, Siebel AL, Wadley GD & McConell GK (2011). Short‐term exercise training early in life restores deficits in pancreatic β‐cell mass associated with growth restriction in adult male rats. Am J Physiol Endocrinol Metab 301, E931–E940. [DOI] [PubMed] [Google Scholar]
- Lauenborg J, Mathiesen E, Hansen T, Glümer C, Jørgensen T, Borch‐Johnsen K, Hornnes P, Pedersen O & Damm P (2005). The prevalence of the metabolic syndrome in a Danish population of women with previous gestational diabetes mellitus is three‐fold higher than in the general population. J Clin Endocrinol Metab 90, 4004–4010. [DOI] [PubMed] [Google Scholar]
- László KD, Olsen J, Li J, Persson M, Vestergaard M, Svensson T, Obel C & Cnattingius S (2015). The risk of gestational diabetes mellitus following bereavement: a cohort study from Denmark and Sweden. Paediatr Perinat Epidemiol 29, 271–280. [DOI] [PubMed] [Google Scholar]
- Levey AS, de Jong PE, Coresh J, Nahas El M, Astor BC, Matsushita K, Gansevoort RT, Kasiske BL & Eckardt K‐U (2010). The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 80, 17–28. [DOI] [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Ventura SJ, Osterman MJ & Matthews TJ (2013). Births: final data for 2011. Natl Vital Stat Rep 62, 1–90. [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Ventura SJ, Osterman MJ, Kirmeyer S, Mathews TJ & Wilson EC (2011). Births: final data for 2009. Natl Vital Stat Rep 60, 1–70. [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF & Turner RC (1985). Homeostasis model assessment: insulin resistance and β‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419. [DOI] [PubMed] [Google Scholar]
- Matthews JN, Altman DG, Campbell MJ & Royston P (1990). Analysis of serial measurements in medical research. BMJ 300, 230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisiadis VG & Matthews SG (2014). Glucocorticoids and fetal programming part 1: outcomes. Nature 10, 391–402. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, Owens JA & Wlodek ME (2009). Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol 587, 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dowd R, Kent JC, Moseley JM & Wlodek ME (2008). Effects of uteroplacental insufficiency and reducing litter size on maternal mammary function and postnatal offspring growth. Am J Physiol Regul Integr Comp Physiol 294, R539–R548. [DOI] [PubMed] [Google Scholar]
- Oni O, Harville E, Xiong X & Buekens P (2015). Relationships among stress coping styles and pregnancy complications among women exposed to Hurricane Katrina. J Obstet Gynecol Neonatal Nurs 44, 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan L, Cuffe JSM, Koning A, Singh RR, Paravicini TM & Moritz KM (2015). Excess prenatal corticosterone exposure results in albuminuria, sex‐specific hypotension, and altered heart rate responses to restraint stress in aged adult mice. Am J Physiol Renal Physiol 308, F1065–F1073. [DOI] [PubMed] [Google Scholar]
- O'Sullivan L, Cuffe JSM, Paravicini TM, Campbell S, Dickinson H, Singh RR, Gezmish O, Black MJ & Moritz KM (2013). Prenatal exposure to dexamethasone in the mouse alters cardiac growth patterns and increases pulse pressure in aged male offspring. PLoS ONE 8, e69149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatini P (2012). Glomerular hyperfiltration: a marker of early renal damage in pre‐diabetes and pre‐hypertension. Nephrol Dial Transplant 27, 1708–1714. [DOI] [PubMed] [Google Scholar]
- Qiang W, Weiqiang K, Qing Z & Pengju Z (2007). Aging impairs insulin‐stimulated glucose uptake in rat skeletal muscle via suppressing AMPKα. Exp Mol Med 39, 535–543. [DOI] [PubMed] [Google Scholar]
- Quinn TA, Ratnayake U, Castillo‐Melendez M, Moritz KM, Dickinson H & Walker DW (2014). Adrenal steroidogenesis following prenatal dexamethasone exposure in the spiny mouse. J Endocrinol 221, 347–362. [DOI] [PubMed] [Google Scholar]
- Siebel AL, Mibus A, De Blasio MJ, Westcott KT, Morris MJ, Prior L, Owens JA & Wlodek ME (2008). Improved lactational nutrition and postnatal growth ameliorates impairment of glucose tolerance by uteroplacental insufficiency in male rat offspring. Endocrinology 149, 3067–3076. [DOI] [PubMed] [Google Scholar]
- Singh RR, Cullen McEwen LA, Kett MM, Boon W‐M, Dowling J, Bertram JF & Moritz KM (2007). Prenatal corticosterone exposure results in altered AT 1/AT 2, nephron deficit and hypertension in the rat offspring. J Physiol 579, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgersen CKL & Curran CA (2006). A systematic approach to the physiologic adaptations of pregnancy. Crit Care Nurs Q 29, 2–19. [DOI] [PubMed] [Google Scholar]
- Tran M, Gallo LA, Wadley GD, Jefferies AJ, Moritz KM & Wlodek ME (2012). Effect of pregnancy for females born small on later life metabolic disease risk. PLoS ONE 7, e45188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan OR, Sferruzzi‐Perri AN & Fowden AL (2012). Maternal corticosterone regulates nutrient allocation to fetal growth in mice. J Physiol 590, 5529–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell BJ, Bollen M, Wyrwoll CS, Mori TA & Mark PJ (2010). Developmental programming of adult adrenal structure and steroidogenesis: effects of fetal glucocorticoid excess and postnatal dietary omega‐3 fatty acids. J Endocrinol 205, 171–178. [DOI] [PubMed] [Google Scholar]
- Wadley GD, Siebel AL, Cooney GJ, McConell GK, Wlodek ME & Owens JA (2008). Uteroplacental insufficiency and reducing litter size alters skeletal muscle mitochondrial biogenesis in a sex‐specific manner in the adult rat. Am J Physiol Endocrinol Metab 294, E861–E869. [DOI] [PubMed] [Google Scholar]
- Weissgerber TL & Wolfe LA (2006). Physiological adaptation in early human pregnancy: adaptation to balance maternal–fetal demands. Appl Physiol Nutr Metab 31, 1–11. [DOI] [PubMed] [Google Scholar]
- Xiang AH, Takayanagi M, Black MH, Trigo E, Lawrence JM, Watanabe RM & Buchanan TA (2013). Longitudinal changes in insulin sensitivity and beta cell function between women with and without a history of gestational diabetes mellitus. Diabetologia 56, 2753–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterström K, Lindeberg S, Haglund B, Magnuson A & Hanson U (2007). Being born small for gestational age increases the risk of severe pre‐eclampsia. BJOG 114, 319–324. [DOI] [PubMed] [Google Scholar]