

Abstract
Key points
Hyperfibrinogenaemia (HFg) results in vascular remodelling, and fibrinogen (Fg) and amyloid β (Aβ) complex formation is a hallmark of Alzheimer's disease. However, the interconnection of these effects, their mechanisms and implications in cerebrovascular diseases are not known.
Using a mouse model of HFg, we showed that at an elevated blood level, Fg increases cerebrovascular permeability via mainly caveolar protein transcytosis.
This enhances deposition of Fg in subendothelial matrix and interstitium making the immobilized Fg a readily accessible substrate for binding Aβ and cellular prion protein (PrPC), the protein that is thought to have a greater effect on memory than Aβ.
We showed that enhanced formation of Fg–Aβ and Fg–PrPC complexes are associated with reduction in short‐term memory.
The present study delineates a new mechanistic pathway for vasculo‐neuronal dysfunctions found in inflammatory cardiovascular and cerebrovascular diseases associated with an elevated blood level of Fg.
Abstract
Many cardiovascular diseases are associated with inflammation and as such are accompanied by an increased blood level of fibrinogen (Fg). Besides its well‐known prothrombotic effects Fg seems to have other destructive roles in developing microvascular dysfunction that include changes in vascular reactivity and permeability. Increased permeability of brain microvessels has the most profound effects as it may lead to cerebrovascular remodelling and result in memory reduction. The goal of the present study was to define mechanisms of cerebrovascular permeability and associated reduction in memory induced by elevated blood content of Fg. Genetically modified, transgenic hyperfibrinogenic (HFg) mice were used to study cerebrovascular transcellular and paracellular permeability in vivo. The extent of caveolar formation and the role of caveolin‐1 signalling were evaluated by immunohistochemistry (IHC) and Western blot (WB) analysis in brain samples from experimental animals. Formation of Fg complexes with amyloid β (Aβ) and with cellular prion protein (PrPC) were also assessed with IHC and WB analysis. Short‐term memory of mice was assessed by novel object recognition and Y‐maze tests. Caveolar protein transcytosis was found to have a prevailing role in overall increased cerebrovascular permeability in HFg mice. These results were associated with enhanced formation of caveolae. Increased formation of Fg–PrPC and Fg–Aβ complexes were correlated with reduction in short‐term memory in HFg mice. Using the model of hyperfibrinogenaemia, the present study shows a novel mechanistic pathway of inflammation‐induced and Fg‐mediated reduction in short‐term memory.
Key points
Hyperfibrinogenaemia (HFg) results in vascular remodelling, and fibrinogen (Fg) and amyloid β (Aβ) complex formation is a hallmark of Alzheimer's disease. However, the interconnection of these effects, their mechanisms and implications in cerebrovascular diseases are not known.
Using a mouse model of HFg, we showed that at an elevated blood level, Fg increases cerebrovascular permeability via mainly caveolar protein transcytosis.
This enhances deposition of Fg in subendothelial matrix and interstitium making the immobilized Fg a readily accessible substrate for binding Aβ and cellular prion protein (PrPC), the protein that is thought to have a greater effect on memory than Aβ.
We showed that enhanced formation of Fg–Aβ and Fg–PrPC complexes are associated with reduction in short‐term memory.
The present study delineates a new mechanistic pathway for vasculo‐neuronal dysfunctions found in inflammatory cardiovascular and cerebrovascular diseases associated with an elevated blood level of Fg.
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer's disease
- AOI
area of interest
- APP
amyloid β precursor protein
- BSA
bovine serum albumin
- BSA‐647
Alexa Fluor 647‐conjugated bovine serum albumin
- Cav‐1
caveolin‐1
- Co‐IP
co‐immunoprecipitation
- DAPI
4,6‐diamidino‐2‐phenyl‐indole HCl
- DR
discrimination ratio
- EC
endothelial cell
- Fg
fibrinogen
- FITC
fluorescein isothiocyanate
- FIU
fluorescence intensity units
- Fluor
fluorescein reference standard
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HFg
hyperfibrinogenaemia, hyperfibrinogenic
- HMW
high molecular weight
- IHC
immunohistochemistry
- IOD
integrated optical density
- JP
junction protein
- LEA
Lycopersicon esculentum agglutinin tomato lectin
- LMW
low molecular weight
- NDS
normal donkey serum
- NORT
novel object recognition test
- pCav‐1
phosphorylated Cav‐1
- PrPC
prion protein (cellular)
- PV‐1
plasmalemma vesicle associated protein‐1
- RIPA
radioimmunoprecipitation assay buffer
- SEM
subendothelial matrix
- siRNA
small interfering ribonucleic acid
- TBS
Tris‐buffered saline
- TBS‐T
Tris‐buffered saline with Triton X‐100
- TXR
Texas Red
- WB
Western blot
- WT
wild‐type, C57BL/6J
Introduction
Many diseases such as stroke, hypertension, Alzheimer's disease (AD), diabetes, atherosclerosis and traumatic brain injury are associated with inflammation. Inflammation is accompanied by elevation of inflammatory mediators including plasma adhesion glycoprotein fibrinogen (Fg), which is considered a high risk factor for many cardiovascular and cerebrovascular diseases (Danesh et al. 2005). If the blood level of Fg is higher than normal (∼2 mg ml−1) this is called hyperfibrinogenaemia (HFg ≥ 4 mg ml−1); it is considered not only a marker of inflammation (Danesh et al. 2005) but also a cause of inflammatory responses (Tyagi et al. 2008; Patibandla et al. 2009; Lominadze et al. 2010; Davalos & Akassoglou, 2012; Muradashvili et al. 2012 a).
One of the indications of inflammation is an increase in vascular permeability that results in the movement of blood plasma components out of the bloodstream to the interstitium and may cause oedema. The brain oedema may be characterized by intra‐ and/or extracellular fluid accumulation and is mostly featured by the accumulation of proteins in the tissue (Mehta & Malik, 2006). The blood–brain barrier impairment is considered as a main cause of oedema. It occurs in brain tumours, infections, cerebral ischaemia, traumas, metabolic disorders, intoxications and hypertensive crisis (Mehta & Malik, 2006).
Plasma components may pass through the endothelial barrier via two major transcellular and paracellular transport pathways (Mehta & Malik, 2006; Simionescu et al. 2009). The paracellular transport occurs through the gaps between the endothelial cells (ECs) and involves alterations in junction proteins (JPs) (Mehta & Malik, 2006). It is considered that this pathway is taken mainly by low molecular weight (LMW) molecules contrary to the transcellular transport which is used by high molecular weight (HMW) molecules, such as proteins. The latter process occurs through ECs involving the formation of functional caveolae in ECs and their enhanced motility (Stan et al. 1999). It is defined as a caveolar protein transcytosis.
It has been found that at high blood level Fg enhances EC layer permeability (Patibandla et al. 2009) to proteins, and can itself leak through the EC layer (Tyagi et al. 2008). After extravasation at sites of inflammation Fg is immobilized and then converted to fibrin (Rybarczyk et al. 2003). Since Fg is synthesized in hepatocytes and circulates in blood, it may appear in the extravascular space only after crossing the vascular wall.
HFg is associated with an increased risk of dementia and AD (van Oijen et al. 2005). An interaction between Aβ and Fg in brain tissue (Ahn et al. 2010) and the possible formation of Fg–Aβ plaque may be linked to cognitive dysfunction (Cortes‐Canteli & Strickland, 2009; Ahn et al. 2010; Cortes‐Canteli et al. 2010). It is known that there is a strong connection between the Aβ pathology and loss of memory (Johnson et al. 2010). Defects in Aβ along with the Aβ precursor protein (APP) are considered a cause of AD and dementia. However, some studies indicate that Aβ has a limited effect on memory and point to a greater role of cellular prion protein (PrPC) (Chung et al. 2010; Gimbel et al. 2010). It has been shown that Fg interacts with non‐digested scrapie prion protein (PrPSc) (Fischer et al. 2000). Our previous study also points to the role of PrPC in memory impairment (Muradashvili et al. 2015). Therefore, formation of Fg–Aβ and/or Fg–PrPC complexes may indicate a mechanism for memory reduction seen in diseases associated with cerebrovascular impairment. As a result, these findings highlight a new role of Fg during inflammation‐induced dysfunction of a microvascular bed. In the present study we define possible mechanisms of HFg‐induced vasculo‐neuronal dysfunction using a transgenic mouse model of HFg.
Methods
Animals
All animal procedures for the study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Louisville in accordance with National Institute of Health Guidelines for animal research.
Wild‐type (WT) C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). HFg transgenic mice on C57BL/6J background (strain name: C57BL/6‐Tg(Fga,Fgb,Fgg)1Unc/Mmnc; Stock Number 004104) were purchased from Mutant Mouse Regional Resource Centre at the University of North Carolina at Chapel Hill (Chapel Hill, NC, USA). Homozygote HFg mice were crossbred and the offspring genotyped (Muradashvili et al. 2012 b). Age‐matched, 14‐week‐old (29–32 g) male WT and HFg mice were used in all experiments.
Reagents and antibodies
Fluorescein (Fluor) reference standard, Alexa Fluor 647‐conjugated bovine serum albumin (BSA‐647) and secondary antibodies conjugated with Alexa Fluor 488, Alexa Fluor 594, Alexa Fluor 647, and Dynabeads Protein A for immunoprecipitation were purchased from Life Technologies/Invitrogen (Grand Island, NY, USA). Predesigned in vivo silencer small interfering RNA (siRNA) against caveolin‐1 (Cav‐1) (cat. no.: IVF3001) was from Life Technologies/Ambion (Grand Island, NY, USA). Goat polyclonal anti‐collagen antibody COL4A1/5 (C‐19, epitope near the C‐terminus of collagen α type IV, detects collagen α1 and α5 type IV in mouse) and 4,6‐diamidino‐2‐phenyl‐indole HCl (DAPI) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rat anti‐mouse plasmalemma vesicle associated protein‐1 (PV‐1) monoclonal antibody (clone: MECA‐32; isotype: IgG2a) was from AbD Serotec (Raleigh, NC, USA). Rabbit polyclonal antibody against Cav‐1 was obtained from Novus Biological (Littleton, CO, USA). Purified mouse anti‐phospho‐caveolin‐1 (pY14) was purchased from BD Biosciences (San Diego, CA, USA). Polyclonal rabbit anti‐human Fg (detects native Fg as well as Fg fragments D and E) was from Dako (Carpinteria, CA, USA). For immunohistochemistry, rabbit polyclonal anti‐Aβ antibody, which identifies amino acid residues 1–14 of Aβ and stains extracellular aggregates of Aβ 40–42 peptides as well as APP, was obtained from Abcam (ab2539, Cambridge, MA, USA). For Western blot (WB) analysis and co‐immunoprecipitation (Co‐IP) tests Aβ monoclonal antibody (2C8) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Rabbit IgG was also from Thermo Fisher Scientific. Monoclonal anti‐prion protein antibody, phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail and β‐actin were from Sigma‐Aldrich Co. (St Louis, MO, USA). Radioimmunoprecipitation assay (RIPA) buffer was obtained from Boston Biomedical (Cambridge, MA, USA). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was purchased from EMD Millipore/Life Science (Billerica, MA, USA). Normal donkey serum (NDS) was obtained from Jackson ImmunoResearch (West Grove, PA, USA). Texas Red (TXR)‐ or fluorescein isothiocyanate (FITC)‐conjugated Lycopersicon esculentum agglutinin (LEA) tomato lectin were from Vector Laboratories (Burlingame, CA, USA). Artificial cerebrospinal fluid was purchased from Harvard Apparatus (Holliston, MA, USA).
Cranial window preparation
Animals were anaesthetized with sodium pentobarbital (70 mg kg−1, i.p.). Supplemental anaesthesia was given as required during the experiment. The left carotid artery was cannulated for blood pressure monitoring and necessary infusions. The trachea was intubated to maintain a patent airway. Body temperature of the mouse was kept at 37 ± 1°C with a heating pad. Mean arterial blood pressure and heart rate were continuously monitored through a carotid artery cannula connected to a transducer and a blood pressure analyser (CyQ 103/302; Cybersense, Lexington, KY, USA).
Brain pial microcirculation was prepared for observations according to the method described previously (Muradashvili et al. 2012 a,b, 2014 c, 2015). Briefly, a mouse was placed on a stereotaxic apparatus (World Precision Instruments, Sarasota, FL, USA). The scalp and connective tissues were removed over the parietal cranial bone above the left hemisphere. A craniotomy (∼4 mm in diameter) was done with a high‐speed micro‐drill (Fine Science Tools, Foster City, CA, USA). The dura mater was lifted with the bone disc. The surface of the exposed pial circulation was continuously superfused with cerebrospinal fluid at 37°C.
Microvascular leakage observation
After a 1 h equilibration period following the surgical preparation, autofluorescence of the observed area was recorded over a standard range of camera gains before each experiment. A mixture of 100 μl of Fluor (300 μg ml−1) and 20 μl of BSA‐647 (3.3 mg ml−1) in PBS was infused through the cannulated carotid artery with a syringe pump at a speed of 30 μl min−1 and allowed to circulate for up to 10 min (Muradashvili et al. 2012 b). After ensuring that there was no spontaneous leakage of BSA, venules were identified by the topology of the pial circulation and direction of blood flow. Images of the selected third‐order venular segments were recorded and used as a baseline. After obtaining the baseline reading, images of the chosen venular segments were recorded at 10, 20, 40, 60 and 90 min of observation.
An epi‐illumination system was used to observe intravascular and extravascular Fluor and BSA‐647. The area of interest (AOI) was exposed to blue light (488 nm) and then red light (647 nm) for 10–15 s with a power density of 3.5 μW cm−2. The microscope images were acquired by an electron‐multiplying charge‐coupled device camera (Quantem 512SC; Photometrics, Tucson, AZ, USA) and image acquisition system (Slidebook 5.0, Intelligent Imaging Innovations, Inc.; Philadelphia, PA, USA). The camera output was standardized with 50 ng ml−1 Fluor for each experiment. The light intensity and camera gain settings were held constant during experiments, and the camera response was verified to be linear over the range used for these acquisitions. The magnification of the system with Olympus ×20/0.40 (UPlanSApo) objective was determined with a stage micrometer.
Images of the pial venular circulation were analysed by image analysis software (Image‐Pro Plus 7.0; Media Cybernetics, Bethesda, MD, USA). Two or three pial vessels in each animal were observed and analysed. In each analysed image, a 30 μm in length line profile probe was positioned in the middle of a selected vessel and outside of the venular wall, parallel to the vessel. Mean fluorescence intensities along the line profile probes were measured for each dye and leakage of Fluor and BSA‐647 to the interstitium was assessed by changes in the ratio of fluorescence intensity of each dye in the interstitium to that inside the venule for the respective dye. The results were averaged for each animal and then for each experimental group, and presented as a percentage of baseline.
Collection and preparation of plasma and brain samples
At the end of each experiment, blood was withdrawn by venipuncture of the vena cava using a 23‐gauge needle and polypropylene syringe containing sodium citrate anticoagulant (10.9 mmol l−1) to provide a ratio of 1 part citrate to 9 parts of blood (Lominadze et al. 1998). Blood was transferred to Eppendorf tubes and centrifuged at 1000 g for 5 min to obtain plasma samples. Plasma was used to assess content of Fg by WB analysis (Muradashvili et al. 2012 a).
Animals were infused with PBS and exsanguinated to collect brain samples to assess Aβ content with WB analysis. Brain samples were digested in RIPA buffer (20 g of tissue/1 ml of RIPA) in the presence of protease inhibitor cocktail. The samples were homogenized using a Tenbroek Tissue Grinder (Corning Inc., Corning, NY, USA) at 4°C. After homogenization, samples were centrifuged at 16,000 g for 10 min. The supernatant was centrifuged again at 16,000 g for 10 min to separate leftover tissue debris. The supernatant was collected and total protein content was determined by the Bradford method.
In a separate series of experiments, mice were infused with FITC‐ or TXR‐LEA conjugates via the external jugular vein to fluorescently label vascular endothelium for further analysis with immunohistochemistry (IHC) (Muradashvili et al. 2012 a). After the animals were euthanized with an anaesthetic overdose, they were immediately infused with PBS and then 4% paraformaldehyde solution through the left ventricle for exsanguination. After the cranium was opened, the brain was gently dissected and removed for fresh tissue processing. Brain samples mounted in protective matrix (Polyscience, Inc., Warrington, PA, USA) were cryosectioned with a Leica CM‐1850 Cryocut (Bannockburn, IL, USA) into 25 μm thick slices (Muradashvili et al. 2012 a) and stored at −80°C until further analysis.
Treatment with siRNA against Cav‐1
To define the role of caveolar transcytosis in overall cerebrovascular permeability, HFg mice were treated with in vivo silencer siRNA against Cav‐1 (1.0 mg kg−1). The siRNA dissolved in PBS (total volume ∼200 μl) was administered through the jugular vein. It was shown that downregulation of Cav‐1 expression was effective during 72–144 h after treatment with siRNA against Cav‐1 (Miyawaki‐Shimizu et al. 2006). Therefore, cerebrovascular permeability to two tracers was assessed 96 h after administration of siRNA against Cav‐1 (details are described above in ‘Microvascular leakage observation’). The choice of the specific type of siRNA against Cav‐1 was based on its robust downregulatory effect on Cav‐1 expression in cultured mouse brain ECs detected in a preliminary study.
Western blot analysis
An equal volume (30 μl) of protein from brain sample of each animal was loaded onto a 4–20% Mini‐Protean TGX (Tris‐glycine extended) gradient gel (Bio‐Rad Laboratories, Inc., Hercules, CA, USA) and electrophoresed under reducing conditions and then transferred onto nitrocellulose membranes. After blocking membranes with 5% non‐fat dried milk in Tris‐buffered saline (TBS) with Triton X‐100 (TBS‐T), membranes with plasma or brain samples were incubated with anti‐Fg or anti‐Aβ antibodies overnight at 4°C. Then, after probing with appropriate secondary antibodies for 2 h at room temperature, the blots were developed using a ChemiDoc XRS+ imager system (Bio‐Rad). The blot images were analysed with Image‐Pro Plus. The levels of protein expressions were assessed by measuring an integrated optical density (IOD) of bands of interest and GAPDH or β‐actin bands in the respective lane profile. The latter two were used as loading controls. Results were presented as a ratio of the IOD of Fg bands to the IOD of the respective GAPDH band and a ratio of the IOD of Aβ bands to the IOD of the respective β‐actin band.
Immunohistochemistry
Mouse brain tissue IHC analysis was done according to the method described previously (Muradashvili et al. 2012 a). Briefly, after warming the slides at 37°C for 20 min and removing the mounting matrix, the sections were fixed in ice‐cold 100% methanol for 10 min, washed three times in TBS, and blocked against non‐specific epitope binding in a mixture of 0.1% TBS‐T, 0.5% BSA, and 10% NDS for 1 h at room temperature.
When used, primary antibodies such as anti‐Cav‐1 (dilution 1:100), anti‐PV‐1 (dilution 1:100), anti‐Aβ (dilution 1:150), anti‐collagen (dilution 1:150), anti‐Fg (dilution 1:200), anti‐PrPC (dilution 1:100), or pY14 (dilution 1:150) were applied to the brain slices overnight at 4°C. After washing, respective fluorescent dye‐conjugated secondary antibodies (dilution 1:500) were applied to the brain slices for 1 h at room temperature. Cell nuclei were labelled with DAPI (1:1000). The laser‐scanning confocal microscope (Olympus FluoView1000, with objective ×60) was used to capture images (Muradashvili et al. 2012 a). TXR‐LEA were visualized using a HeNe‐Green laser (543 nm) to excite the dye, while emission was observed above 620 nm. FITC‐LEA or Aβ were visualized using a multiline argon‐ion laser (458 nm/488 nm/515 nm) to excite the dye, while emission was observed above 519 nm. Fg and collagen were visualized using a HeNe‐Red laser (633 nm), while emission was observed above 667 nm. Fluorescence intensity (for each colour) was adjusted to its saturation point in an experimental group with the maximum fluorescence intensity for the colour of interest and the laser and multipliers’ settings were kept unaltered during measurements in each experimental series. Prior to assessing expressions or co‐localizations of proteins of interest by confocal microscopy, levels of autofluorescence for each specific primary antibody used in the study were measured in control samples. Results showed that autofluorescence of samples were neglectable. Therefore, although included in image analyses, autofluorescence values of samples are not reported. In addition, levels of fluorescence generated by secondary antibodies alone were measured and found to be almost non‐existent.
Off‐line image analysis software (Image‐Pro Plus) was used to assess the expression of Cav‐1, PV‐1, phosphorylation of Cav‐1 (pCav‐1), deposition of Fg, Aβ and PrPC, and formation of collagen. Cav‐1, the most abundant protein of the caveolae wall (Yu et al. 2006) and PV‐1, another marker of caveolae (Hnasko et al. 2002), were used in combination to define the formation of caveoli in the vessels of brain samples as described previously (Muradashvili et al. 2014 a,c; 2015). Fluorescence intensity was measured in AOI along the vessel wall and data were normalized by the length of the respective vascular segment. Fluorescence intensity in six randomly placed constant size AOIs was measured. The results were averaged for each experimental group and values were presented as fluorescence intensity units (FIU). Co‐localizations of Cav‐1 and PV‐1, Fg and Aβ, Aβ and collagen, and Fg and PrPC in brain vessels were assessed by measuring the number of spots generated after co‐localization of respective colours in the images formed after deconvolution of the original images as described earlier (Muradashvili et al. 2014 a). The resolution of presented images is 2.84 pixel mm−1 (width, 279 pixels, height, 259 pixels).
Co‐immunoprecipitation
The following protocol was similar to that used by others for testing the presence of Aβ in mouse brain protein precipitates (Manczak & Reddy, 2013). The brain tissue samples were homogenized in ice‐cold digestion buffer (RIPA) supplemented with PMSF (0.005 μl ml−1) and a protease inhibitor cocktail (0.002 μl ml−1) using a Dounce homogenizer. The tissue lysate was collected in a centrifuge tube and kept on a rotator for at least 16 h at 4°C. After centrifugation at 12,000 g for 10 min the supernatant was collected. Protein content in samples was estimated by the Bradford method. Dynabeads were conjugated with IgG or anti‐Fg antibody according to the manufacturer's recommendation. A total of 25 mg ml−1 of sample protein in 200 μl of TBS‐T was added to the conjugated Dynabeads and incubated on a rotator for 1 h at room temperature. Then tubes were placed on a magnet and the solution was aspirated. The remaining Dynabeads were washed 3 times with 200 μl of TBS‐T each time. For elution of a target protein the washed Dynabeads were suspended in 20 μl 2xSDS sample dilution buffer and incubated for 1 h at room temperature. Then the samples were boiled for 2 min and the Dynabeads were separated by a magnet. The remaining sample was used to define the content of Aβ by WB analysis.
Memory assessment
To assess the memory‐related behaviour of mice a novel object recognition test (NORT) and two Y‐maze (spontaneous alternation and two‐trial recognition) tests were performed.
NORT was used to assess visual short‐term memory (Jadavji et al. 2015) and was performed as described previously (Muradashvili et al. 2014 c, 2015). Briefly, after acclimatization and prior to the test, mice were trained for 2 days in the test box for 10 min twice a day. On the day of the test, each mouse was placed in the box at the mid‐point of the wall opposite to two similar objects and allowed to investigate the objects for 5 min. After 1 h one of the objects was replaced with a new, different‐shaped object and the animal was returned to the box for 3 min. TopScan behavioural analysing system (Version 3.00; Clever Sys Inc., Reston, VA, USA) was used to record and analyse the behaviour of the mouse and calculate a discrimination ratio (DR, time spent at the novel object/time spent at both objects). Lower DR indicates an impairment of memory.
The spontaneous alternation test was used to assess spatial working short‐term memory (Yamada et al. 1996) and was performed according to the method described elsewhere (Yamada et al. 1996; Kutiyanawalla et al. 2012; Jadavji et al. 2015). Age‐matched animals from a separate set were placed in the middle of Y‐maze with all three arms opened. All arm entries were sequentially recorded for 8 min. The percentage of alternation was calculated as the ratio of actual alternations (subsequent entries into three arms on overlapping triplet sets) to possible alternations (the total number of arm entries minus two), multiplied by 100 (Maurice et al. 1994; Jadavji et al. 2015).
The two‐trial recognition memory test was conducted according to the method described elsewhere (Kutiyanawalla et al. 2012). Each mouse from yet another set of age‐matched animals was placed in one of the three arms (start arm) while one of the remaining two arms was blocked and allowed to explore the maze for 10 min. After 1 h the mouse was returned to the maze with all arms opened and its behaviour was recorded for 5 min. The choice of first entry and the number of all entries to each arm were recorded. To define possible changes in short‐term memory, a discrimination index was calculated as the number of entries to the novel arm and expressed as a percentage of the total number of entries to all arms. This variable defines place recognition memory (Dellu et al. 1997) and can also be called spatial discrimination memory (Conrad et al. 1997). The number of mice in each group that entered the novel arm first is presented as a percentage of all tested mice in the group. This variable would indicate discrimination memory (Conrad et al. 1997).
Data analysis
All data are expressed as mean ± standard error of the mean. The experimental groups were compared by one‐way ANOVA. Differences in cerebrovascular permeability to two tracers were analysed with repeated measures ANOVA. If ANOVA indicated a significant difference (P < 0.05), Tukey's multiple comparison test was used to compare group means. Differences were considered significant if P < 0.05.
Results
HFg increases macromolecular leakage of pial venules in mice
In HFg mice the plasma content of Fg was greater than that in WT mice (Fig. 1, inset). Mean arterial blood pressure did not change after Fluor and BSA‐647 infusion in WT (−1 ± 0.5 mmHg) or in HFg (−3 ± 0.5 mmHg) mice. Baseline diameters of chosen vessels averaged 38 ± 5 μm in both groups of animals. There were no significant changes in average vascular diameters during the observation period in WT (+1 ± 0.2 μm) animals compared to that in HFg (+2 ± 0.6 μm) mice.
Figure 1. Hyperfibrinogenaemia‐induced leakage of mouse pial venules .
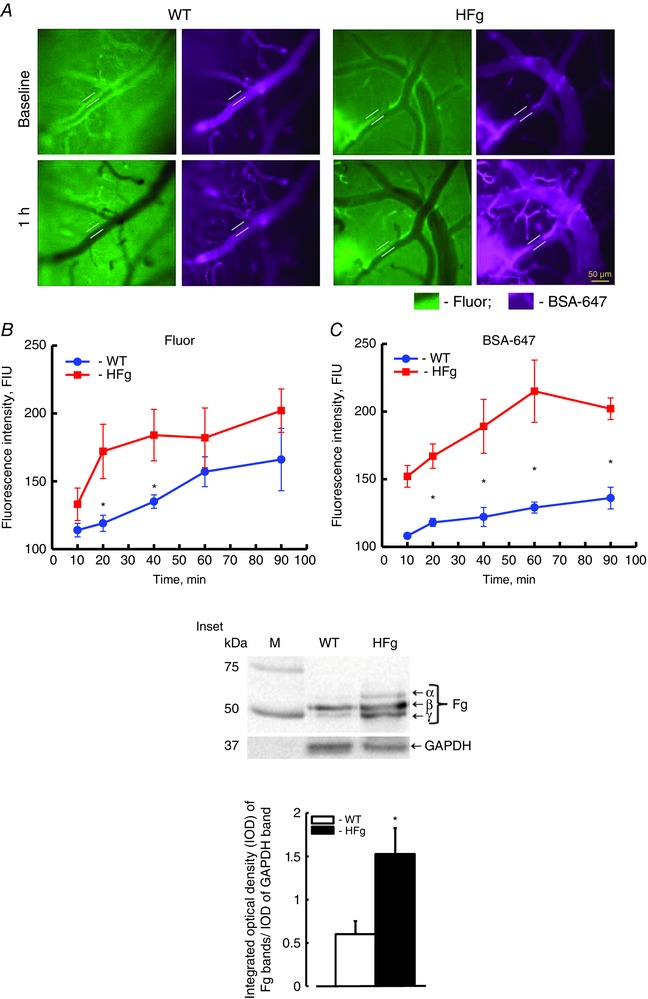
A dual‐tracer probing method was used to define the prevailing role of transcellular vs. paracellular transport in mouse brain pial venules. A, examples of images of cerebral vessels recorded immediately (baseline, upper row) and 1 h after (lower row) infusion of Fluor (green) and BSA‐647 (violet) dyes in wild‐type (WT, left) and hyperfibrinogenic (HFg, right) mice. Cerebrovascular permeability was assessed by comparison of ratios of fluorescence intensities of dyes measured along the line profile probe (shown as a yellow line segment) outside to that inside of the vessel. Summaries of changes in fluorescence intensity ratios of Fluor (B) and BSA‐647 tracers (C). * P < 0.05 vs. WT; n = 8 for all groups. Inset: top, Western blot analysis of plasma samples from WT and HFg mice shows content of Fg (α, β and γ chains); bottom, summary of ratios of integrated optical density (IOD) of Fg bands to IOD of respective GAPDH (used as a loading control) bands. * P < 0.05 vs. WT; n = 6 for all groups. [Colour figure can be viewed at wileyonlinelibrary.com]
To define the prevailing role of transcellular or paracellular transport in overall cerebrovascular permeability at elevated levels of Fg, pial venular permeability to two, LMW and HMW, tracers (Fluor and BSA‐647, respectively) were compared as previously described (Muradashvili et al. 2012 b). Pial venular permeability to both tracers was increased in HFg mice compared to that in WT mice (Fig. 1). However, pial venular leakage of Fluor increased steadily in WT mice from the beginning of observation while in HFg mice it was greater than that in WT mice only at the 20th and 40th minutes of observation (Fig. 1 B). After 40 min of observation there was no difference in leakage of Fluor between the animals (Fig. 1 B). This effect was accompanied by greater accumulation of BSA‐647 in the interstitium of HFg mice starting from the 20th minute of observation and it remained higher than that in WT mice for the duration of observation (Fig. 1 C). Thus, while Fluor leakage was only transiently enhanced in HFg mice, the crossing of BSA‐647 of vascular wall was greater in HFg mice even at the time when there was no difference in Fluor leakage between the animal groups (Fig. 1).
HFg increases caveolae formation in mouse brain vessels
To define the extent of caveolae formation in mice brain microvessels the expressions of its markers Cav‐1 and PV‐1, and their co‐localization, were assessed by IHC. Expressions of Cav‐1 (red) and PV‐1 (green) in HFg mice (30.5 ± 7.3 and 38.6 ± 8.4 FIU, respectively) were greater compared to those (16.9 ± 1.1 and 21.1 ± 2.4 FIU, respectively) in WT animals (Fig. 2 A, B and C). The formation of caveolae (yellow) was defined by spots with co‐localized Cav‐1 and PV‐1 and was greater in HFg mice (64 ± 3) compared to that (19 ± 5) in WT mice (Fig. 2 A and D).
Figure 2. Hyperfibrinogenaemia‐induced increased expression of Cav‐1 and PV‐1, their co‐localization, and expression of pCav‐1 in mouse brain vessels .
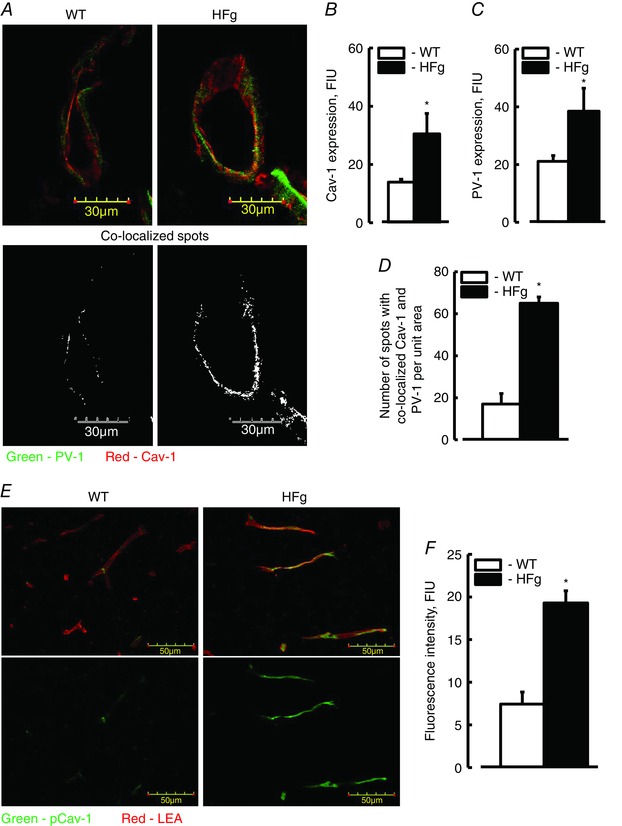
A, examples of images showing expression of PV‐1 (green) and Cav‐1 (red), and their co‐localization (shown in yellow) as a marker of caveolae formation in brain cortical vessels of WT and HFg mice. Summaries of Cav‐1 (B) and PV‐1 (C) expression, and Cav‐1 and PV‐1 co‐localization (D). E, examples of images showing the level of Cav‐1 phosphorylation (pCav‐1, green) as a signalling mechanism for caveolae formation; LEA, a marker of endothelium is shown in red. F, summary of the data analysis shows levels of fluorescence intensity (presented as fluorescence intensity units, FIU) of pCav‐1 in brain samples of WT and HFg mice. * P < 0.05 vs. WT, n = 6 for all groups. [Colour figure can be viewed at wileyonlinelibrary.com]
To confirm the effect of a high blood level of Fg on caveolae formation, phosphorylation of Cav‐1 protein, the main component of caveolae walls, was evaluated.
The expression of pCav‐1 (green) in brain vessels of HFg mice was greater (19.3 ± 1.4 FIU) compared to that (7.4 ± 1.2 FIU) in WT animals (Fig. 2 E and F). The presence of endothelial marker LEA (red) clearly indicated a well‐perfused brain vasculature (Fig. 2 E).
Reduction of Cav‐1 protein content with siRNA against Cav‐1 ameliorates cerebrovascular protein leakage
To define the role of caveolar protein transcytosis in overall cerebrovascular permeability, HFg mice were treated with in vivo silencer siRNA against Cav‐1. Treatment with the silencer siRNA against Cav‐1 decreased transcellular transport of BSA‐647 (Fig. 3 A and B) but had no effect on Fluor leakage. Since the siRNA was injected into the mice systemic circulation, reduction of Cav‐1 was expected to occur primarily in vascular endothelium. The effectiveness of the in vivo silencer siRNA against Cav‐1 treatment was confirmed by observing a decrease in Cav‐1 protein level in cerebral vessels of treated mice compared to that in vehicle‐treated mice (Fig. 3 C and D).
Figure 3. Effect of siRNA against Cav‐1 on cerebrovascular permeability in HFg mice .
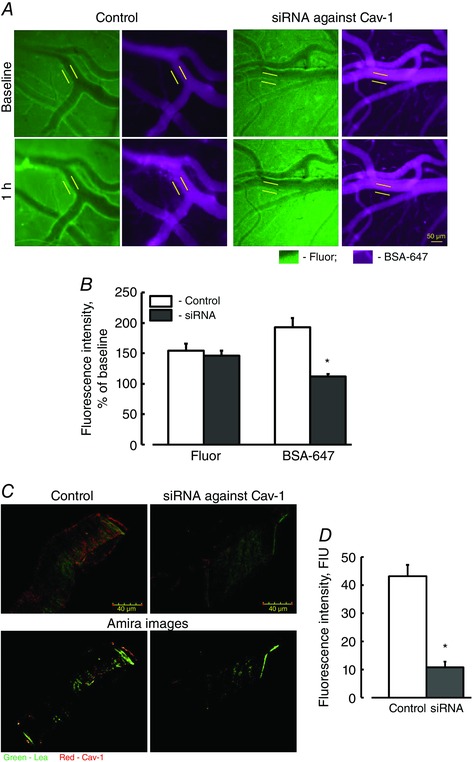
A, examples of images of pial venules recorded immediately (baseline, upper row) and 1 h (lower row) after infusion of Fluor (green) and BSA‐647 (violet) in hyperfibrinogenic mice with (right two columns) or without (left two columns) treatment with in vivo select siRNA against Cav‐1. Cerebrovascular permeability was assessed by comparison of ratios of fluorescence intensities of each dye measured along the line profile probe (shown as a yellow line segment) outside to that of inside the vascular segment. B, summary of changes in ratios of fluorescence intensity values of Fluor and BSA‐647 tracers. C, examples of images showing expression of Cav‐1 (red) in brain cortical vessels in HFg mice treated with siRNA against Cav‐1 (lower row shows images analysed by 3‐D visualization Amira software, which was used in addition to Image‐Pro Plus). LEA, a marker of endothelium is shown in green. D, summary of Cav‐1 expression analysis. The levels of Cav‐1 expression are presented in fluorescence intensity units (FIU). * P < 0.05 vs. WT, n = 4 for all groups. [Colour figure can be viewed at wileyonlinelibrary.com]
HFg increases deposition of Fg and Aβ and promotes Fg–Aβ complex formation in mouse brain vessels
Depositions of Fg and Aβ and the resultant formation of the Fg–Aβ complex were studied in brain samples from WT and HFg mice by IHC. The deposition of Fg (green) in brain vessels of HFg mice was greater than that in WT mice (Fig. 4 A and B).
Figure 4. Hyperfibrinogenaemia‐induced increased deposition of fibrinogen and amyloid β in mouse brain vessels .
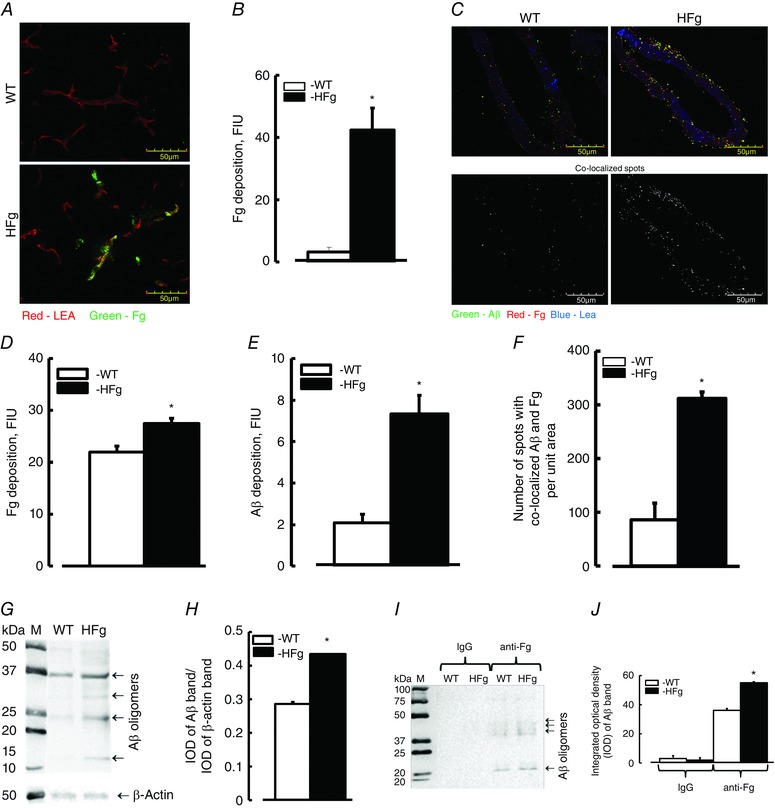
A, examples of images showing fibrinogen (Fg, green) deposition in brain vessels (labelled with LEA and shown in red) in samples obtained from WT and HFg mice. B, summary of Fg fluorescence intensity levels presented as fluorescence intensity units (FIU). C, examples of images showing expression of Fg (shown as red dots) and amyloid β (Aβ, shown as green dots), and their co‐localization (shown in yellow) in brain vessels of WT and HFg mice (upper row). Images in the lower row show results of co‐localized spots identified after deconvolution of respective upper images by Image‐Pro Plus. Summaries of fluorescence intensities of Fg (D) and Aβ (E) are presented as FIU. F, number of co‐localized Fg and Aβ spots are presented. G, example of WB analysis for content of Aβ in brain samples from WT and HFg mice. H, summary of ratios of integrated optical density (IOD) of Aβ bands to IOD of the respective β‐actin bands. I, example of IgG‐ and anti‐Fg antibody‐precipitated brain proteins from WT and HFg mice tested for presence of Aβ. Note: IgG was used as a negative control. J, summary of IOD of Aβ bands in lanes. * P < 0.05 vs. WT, n = 8 (IHC) and n = 5 (WB and Co‐IP). [Colour figure can be viewed at wileyonlinelibrary.com]
In a separate series of experiments we found that depositions of Fg (red) and Aβ (green) were greater (27.4 ± 0.9 and 7.3 ± 0.9 FIU, respectively) in brain vessels of HFg mice compared to those (21.9 ± 1.1 and 2.2 ± 0.4 FIU) in WT animals (Fig. 4 C, D and E). Co‐localization of Fg and Aβ (shown in yellow) in brain vasculature indicated the formation of Fg–Aβ complexes (Fig. 4 C). The number of co‐localized spots of Fg and Aβ were more in brain samples from HFg mice (311 ± 12) compared to those in the samples from WT (89 ± 21) mice (Fig. 4 C and F).
WB analysis confirmed an increased (0.44 ± 0.01 IOD) expression of Aβ in mouse brain of HFg mice compared to that (0.29 ± 0.03 IOD) in brain samples from WT mice (Fig. 4 G and H).
A method of Co‐IP was used to further define the association of Fg and Aβ in brain samples of mice. Data showed that while Aβ was not found in precipitates with IgG (served as a control for anti‐Fg antibody), it was greater in precipitates with anti‐Fg antibody in samples from HFg mice compared to that in samples from WT mice (Fig. 4 I and J).
HFg increases association of Aβ with collagen in mouse brain vessels
To confirm the deposition of Aβ in the subendothelial matrix (SEM) of a vessel, we tested if Aβ was deposited on brain vascular collagen matrix. Data showed that the deposition of Aβ (green) and formation of collagen (red) were more in HFg mice (102 ± 5.6 and 77.4 ± 6.2 FIU, respectively) compared to those (10.1 ± 0.5 and 22.3 ± 1.5 FIU, respectively) in WT animals (Fig. 5 A, B and C). Aβ–collagen complex formation was defined by spots of co‐localized Aβ and collagen (shown in yellow) and was greater in HFg mice (624 ± 52) compared to that (97 ± 100) in WT animals (Fig. 5 A and D).
Figure 5. Hyperfibrinogenaemia‐induced increased deposition of amyloid β on vascular collagen in mouse brain vessels .

A, examples of brain vessel images in samples obtained from WT and HFg mice. Deposition of amyloid β (Aβ, green) and collagen (red) and their co‐localization (yellow) was assessed by measuring fluorescence intensities of Aβ and collagen and number of spots with co‐localized green and red colours after deconvolution of images. Brain vessels identified by LEA are shown in blue. Summaries of levels of Aβ deposition (B), levels of collagen formation (C) and number of co‐localized Aβ and collagen spots (D). * P < 0.05 vs. WT, n = 8 for all groups. [Colour figure can be viewed at wileyonlinelibrary.com]
HFg promotes Fg and PrPC complex formation in mouse brain vessels
Since it is considered that PrPC has a greater effect on loss of memory, the deposition of PrPC and its association with Fg in brain samples from WT and HFg mice were evaluated by IHC. Depositions of Fg (red) and PrPC (green), and Fg–PrPC complex formation defined by spots with co‐localized Fg and PrPC (shown in yellow) were greater in HFg mice compared to those in the WT group (Fig. 6 A, B, C and D).
Figure 6. Hyperfibrinogenaemia‐induced deposition of fibrinogen and cellular prion protein in mouse brain vessels .
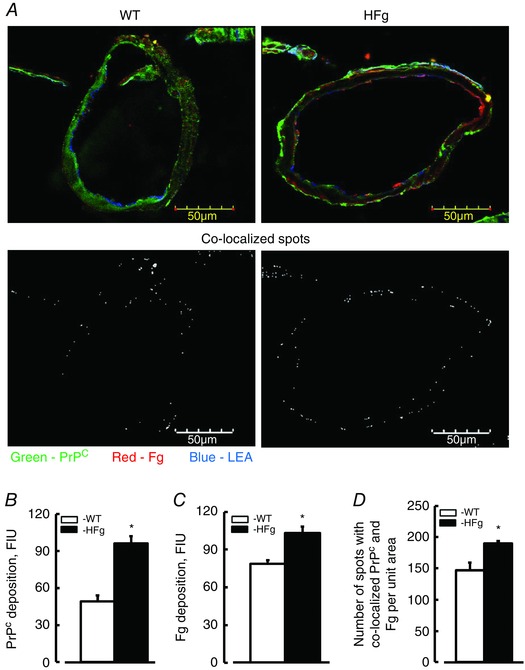
A, examples of brain vessel images in samples obtained from WT and HFg mice. Deposition of fibrinogen (Fg, red) and cellular prion protein (PrPC, green) and their co‐localization (Fg–PrPC complex, yellow) was assessed by measuring fluorescence intensities of Fg and PrPC and number of spots with co‐localized green and red colours after deconvolution of respective upper images. Brain vessels identified by LEA are shown in blue. Summaries of levels of deposition of PrPC (B), Fg (C) and number of co‐localized spots of Fg and PrPC (Fg–PrPC complex) (D) are presented. * P < 0.05 vs. WT, n = 8 for all groups. [Colour figure can be viewed at wileyonlinelibrary.com]
HFg leads to a reduction in short‐term memory in mice
To define the effect of elevated blood levels of Fg on cognition, short‐term memory was assessed in mice with NORT and Y‐maze tests. Reduced short‐term memory was found in HFg mice compared to that in WT mice (Fig. 7). This was confirmed by the lower DR (NORT) and percentage of alternation (Y‐maze) in HFg mice compared to those in WT animals (Fig. 7 B and C). HFg mice also showed a poor performance compared to control animals in the two‐trial recognition test (Fig. 7 D). In addition, while 75% of WT mice demonstrated a high level of discrimination memory (the first choice of entry to a novel arm), only 15% of HFg mice went to the novel arm first.
Figure 7. Hyperfibrinogenaemia‐induced short‐term memory reduction .
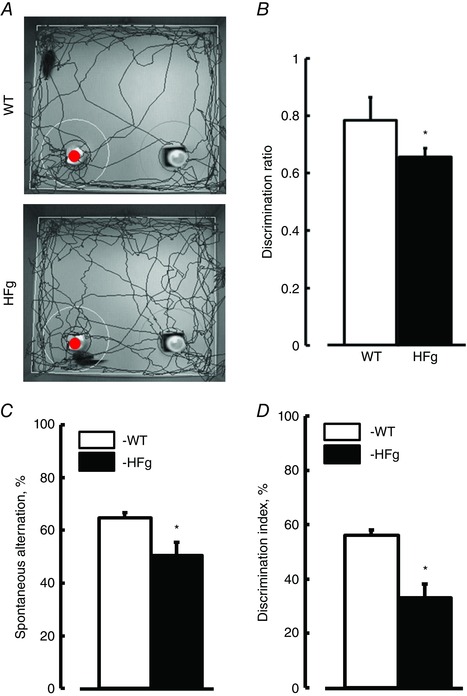
Short‐term memory of mice was assessed by a novel object recognition test (NORT) and Y‐maze (spontaneous alternation and two‐trial recognition) tests. A, examples of movement traces of WT and HFg mice assessed by NORT. The red circle indicates the new object. B, values of discrimination ratios for WT and HFg mice are shown. Summaries of Y‐maze tests: spontaneous alternation (C) and two‐trial recognition (D). * P < 0.05 vs. WT; n = 26 (NORT) and n = 19 (Y‐maze tests). [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
It is known that LMW molecules (e.g. Fluor) can readily pass through the cell junctions (Little et al. 1995), while HMW molecules, such as BSA, are too large to cross the EC layer easily through their junctions. Comparison of leakages of our tracers showed that when leakage of Fluor in HFg mice was no longer significantly different (after 40 min) from that in WT mice, the crossing of the vascular wall by BSA‐647 in HFg mice was increasing compared to that in WT mice. Thus, when paracellular transport became similar in HFg and WT mice (after 40 min), transcellular protein transport remained greater in HFg mice suggesting the prevailing role of the transcellular transport. In other words, our data indicated that HFg affected JPs and led to a transient opening of gaps between the ECs temporarily enhancing the leakage of Fluor, while the movement of BSA‐647 through EC cells, via most likely caveolar protein transcytosis, was constantly greater in pial venules of HFg mice. Thus, during HFg, BSA‐647 may move through the pial venular wall first via both transport pathways and later, when EC gap openings return to the level in WT mice, primarily by transcytosis. Our study indicated that this transport mechanism was activated during HFg, suggesting that targeting the caveolar transcytosis in addition to preventing the paracellular transport should be considered during inflammatory cerebrovascular diseases. This finding can be useful during treatment of increased cerebrovascular permeability. For example, since sphingolipid signalling is involved in caveolae formation (Muradashvili et al. 2014 b) to effectively reduce the formation of caveolae, as an additional step, an inhibition of sphingolipid synthesis can be considered.
Greater expression of caveolae markers Cav‐1 and PV‐1 was found in brain vessels of HFg mice than in vessels of WT animals. However, the co‐localization of these two proteins, which is an indication of caveolae formation, has a greater physiological relevance (Muradashvili et al. 2014 a). Co‐localization of Cav‐1 and PV‐1 in brain vessels of HFg mice was exceeding that in vessels of WT mice. These results suggest that formation of caveolae was increased during HFg. Formation of functional caveoli, their motility, and therefore caveolar protein transcytosis was confirmed by enhanced phosphorylation of Cav‐1. Phosphorylation of Cav‐1 plays an essential role in the process of caveolae formation (Parton et al. 1994; Minshall et al. 2000; Hu et al. 2008; Sun et al. 2009; Wei et al. 2009). The motility of caveolae is mediated by Cav‐1 phosphorylation‐dependent signalling events (Minshall et al. 2000). We already showed that increased content of Fg enhanced the formation of functional caveoli that had taken up albumin (Muradashvili et al. 2014 a) and increased their motility (Muradashvili et al. 2014 b). According to our findings an elevated blood level of undegraded Fg has a considerable role in the formation of functional caveoli in mouse brain vessels and in cultured mouse brain ECs (Muradashvili et al. 2014 a). Our data suggest that an increase in blood level of Fg may play a significant role in microvascular remodelling during inflammatory cerebrovascular diseases.
Treatment of animals with siRNA against Cav‐1 confirmed that caveolar transcytosis is the main route for BSA‐647 leakage in pial vessels of mice with chronic elevation of Fg. Our data showed that a decrease in Cav‐1 expression, which significantly diminished caveolae formation, resulted in reduction of transcellular transport without affecting the paracellular pathway. These results confirmed that elevated blood level of Fg affects mainly the caveolar protein transcytosis, and the HFg‐induced increase in overall vascular permeability occurs primarily via the transcellular transport pathway.
We showed that an increase in Fg content increased EC layer permeability to albumin and Fg itself crossed the EC layer (Tyagi et al. 2008). In normal conditions, deposited in SEM and immobilized, Fg is more prone to degradation. However, during many inflammatory diseases associated with increased blood content of Fg (e.g. during hypertension) fibrinolysis is decreased (Landin et al. 1990). In AD tissue plasminogen activator is decreased resulting in a lowered level of plasminogen, the Fg digestive enzyme, in brains of patients or mouse models of the disease (Ledesma et al. 2000). Therefore, during elevated levels of Fg, its deposition in SEM and in extravascular tissue is increased exacerbating vascular remodelling (Lominadze et al. 2010) and resulting in favourable conditions for the formation of complexes such as Fg–Aβ and Fg–PrPC seen in the present study.
It is known that all the forms of Aβ (including APP) are well pronounced during AD and/or amyloidosis. During other inflammatory disorders the level of Aβ may be less or non‐pronounced. In our case, during HFg (which indicates the inflammatory condition) there is an enhanced expression of Aβ oligomers compared to that in control animals. The major constituent of Aβ plaques is the neurotoxic Aβ peptide (oligomers of Aβ), derived from the APP. When immobilized in SEM, Fg becomes readily available for binding to Aβ oligomers and even APP. Thus, the appearance of the Fg–Aβ (or Fg–APP) complex in SEM can be a result of vascular hyper‐permeability leading to transcytosis of Fg to SEM (Muradashvili et al. 2014 c). It has been shown that the binding of Aβ to Fg leads to its oligomerization (Ahn et al. 2010), and the Fg–Aβ complex is highly resistant to degradation (Cortes‐Canteli et al. 2010). Our data indicate that there is an increased deposition of Fg during HFg. This enhanced Fg deposition promotes the binding of Fg to Aβ (and possibly to APP) and the formation of the Fg–Aβ (and possibly Fg–APP) complex presumably leading to a Fg–Aβ plaque formation later. Increased levels of Aβ and its oligomers and enhanced association of Fg and Aβ during HFg was also confirmed by WB and Co‐IP analyses, respectively. These results provide strong evidence of the effects of the blood protein Fg, which is found immobilized in extravascular space and can be associated with other proteins, such as collagen, Aβ and PrPC. Alteration of the collagen content in SEM is one of the indications of vascular remodelling. An increased collagen level in cerebral microvessels during AD has been shown (Kalaria & Pax, 1995). Earlier we found that collagen can serve as a substrate for Fg–Aβ complex deposition in SEM (Muradashvili et al. 2014 c). The present results indicate an increased formation of collagen along with increased expression of Aβ and enhanced deposition of Fg and Aβ. These data suggest that HFg‐induced increased cerebrovascular permeability leads to an enhanced deposition of Fg on SEM collagen through formation of the Fg–Aβ–collagen complex, which was found to be involved in memory deficits (Muradashvili et al. 2014 c).
Enhanced protein transcytosis, which leads to an increased deposition of Fg in SEM of brain vasculature during HFg can cause vascular remodelling. Changes in vasculature were reported by others showing that HFg results in augmented intimal hyperplasia and vascular lumen narrowing (Kerlin et al. 2004). Our data show that HFg is associated with increased collagen formation in the extracellular matrix of brain vessels. These changes, along with an increased deposition of Fg, can lead to vascular stiffening and overall vascular dysfunction. Combined, our results suggest a mechanism of HFg‐induced vascular remodelling that can exacerbate vascular dysfunction during various cardiovascular and cerebrovascular diseases associated with an increased blood content of Fg.
Another well‐known protein associated with memory changes is PrPC (Chung et al. 2010; Gimbel et al. 2010). Previously we showed that Fg may bind PrPC and result in Fg–PrPC complex formation, which can be associated with memory impairment (Muradashvili et al. 2015). Our present data confirm the formation of the Fg–PrPC complex during elevated blood content of Fg and the associated memory impairment. Thus, formation of Fg–Aβ and/or Fg–PrPC complexes can be a result of the process triggered by impaired vascular wall permeability leading to neuronal dysfunction indicated by the reduction in short‐term memory found in the present study.
The effect of increased deposition of Fg and formation of complexes with Aβ and/or PrPC in extravascular space on short‐term memory was demonstrated by changes in cognition of HFg mice compared to that of WT animals. The cognition tests used in the present work were based on the natural tendency of mice to explore/investigate a novel object (NORT) and/or place (Y‐maze, two‐trial recognition test) and served as effective tools to analyse responses to novelty and recognition (Dellu et al. 1997). The data showed that HFg mice had a lesser short‐term memory than age‐matched control mice. The other, spontaneous alternation test relies on the latent learning processes involved during exploration of the Y‐maze and allows the assessment of the spatial working short‐term memory (Yamada et al. 1996). This test also showed that HFg mice have a lower spontaneous alternation compared to that in control animals, which indicates their poor spatial working memory. Thus, an increase in blood content of Fg and the resultant formation of Fg–Aβ and/or Fg–PrPC complexes are associated with a reduction in short‐term memory in mice indicating a possible cause and effect in cognition deficiency during inflammation.
The presented data clarify the pathway for vasculo‐neuronal dysfunction initiated by an elevated blood level of Fg, which increases cerebrovascular permeability via mainly the transcellular transport as was found previously (Muradashvili et al. 2012 b). Here we show that this transcellular transport occurs primarily by the caveolar protein transcytosis leading to accumulation of Fg in SEM. Enhanced deposition of Fg in the vascular wall and SEM can lead to the formation of Fg–Aβ and/or Fg–PrPC complexes, which have a higher resistance to degradation and can contribute to the short‐term memory impairment (schematic representation of this hypothesis is shown in Fig. 8).
Figure 8. Possible mechanism of hyperfibrinogenaemia‐induced vasculo‐neuronal dysfunction .
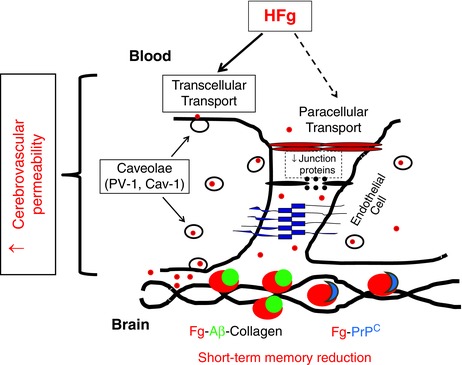
HFg enhances cerebrovascular permeability via mainly the transcellular transport pathway that involves caveolar protein transcytosis, which is mediated through Cav‐1 signalling (phosphorylation of Cav‐1). This leads to an increased accumulation of Fg in subendothelial matrix and promotes its binding to Aβ and PrPC resulting in formation of Fg–Aβ–collagen and Fg–PrPC complexes. Formed Fg–Aβ and Fg–PrPC complexes can be involved in short‐term memory reduction found during HFg indicating a path for vasculo‐neuronal dysfunction. [Colour figure can be viewed at wileyonlinelibrary.com]
In summary, our data provide direct evidence for a mechanism showing that an elevation in blood content of Fg enhances caveolar protein transcytosis leading to increased deposition of Fg mainly in SEM of cerebral vessels. These changes can lead to vascular remodelling and subsequent vascular dysfunction. Greater availability of immobilized Fg creates favourable conditions for its binding to Aβ and/or PrPC and the formation of Fg–Aβ and/or Fg–PrPC complexes highly resistant to degradation. The formation of these complexes can be associated with a reduction in short‐term memory. Thus, these data indicate that HFg is not only a marker of inflammation and cardiovascular or cerebrovascular diseases, but can contribute to the severity and/or progression of the disease and particularly to its accompanying vasculo‐neuronal dysfunction.
Additional information
Competing interests
None declared.
Author contributions
The experiments were performed in the laboratories of D. Lominadze and S. C. Tyagi, Department of Physiology, University of Louisville, Louisville, KY, USA. N.M.: performed studies related to vascular permeability, immunohistochemistry and memory assessments; collected and analysed data, and provided interpretation of results; drafted the article. R.T.: performed co‐immunoprecipitation and Western blot analyses; collected and analysed data; drafted the section related to the Western blot methodology. N.T.: performed data analyses and interpretation of results related to co‐immunoprecipitation, and drafted the co‐immunoprecipitation methodology section. S.C.T.: provided interpretation of results related to the study on the role of collagen. D.L.: overall conception and design, interpretation of results, writing and revising the manuscript critically for important intellectual content. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
N. Tyagi was supported by NIH Heart, Lung, and Blood Institute (HL‐107640). S. C Tyagi was supported by NIH Institute of Neurological Disorders and Stroke (NS‐084823) and Heart, Lung, and Blood Institute (HL‐108621 and HL‐074185). D. Lominadze was supported by NIH Institute of General Medical Sciences (P30 GM‐103507), Institute of Neurological Disorders and Stroke (NS‐084823), Heart, Lung, and Blood Institute (HL‐107640), and Institute of Diabetes and Digestive and Kidney Diseases (DK‐104653).
References
- Ahn HJ, Zamolodchikov D, Cortes‐Canteli M, Norris EH, Glickman JF & Strickland S (2010). Alzheimer's disease peptide β‐amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci USA 107, 21812–21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak R, Kascsak R, Mehta P, Strittmatter S & Wisniewski T (2010). Anti‐PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, Lupien SJ, Thanasoulis LC & McEwen BS (1997). The effects of Type I and Type II corticosteroid receptor agonists on exploratory behavior and spatial memory in the Y‐maze. Brain Res 759, 76–83. [DOI] [PubMed] [Google Scholar]
- Cortes‐Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, Bhuvanendran S, Fenz KM & Strickland S (2010). Fibrinogen and β‐amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer's disease. Neuron 66, 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes‐Canteli M & Strickland S (2009). Fibrinogen, a possible key player in Alzheimer's disease. J Thromb Haemost 7, 146–150. [DOI] [PubMed] [Google Scholar]
- Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB, Wilson AC, Folsom AR, Wu K, Benderly M, Goldbourt U, Willeit J, Kiechl S, Yarnell JW, Sweetnam PM, Elwood PC, Cushman M, Psaty BM, Tracy RP, Tybjaerg‐Hansen A, Haverkate F, de Maat MP, Fowkes FG, Lee AJ, Smith FB, Salomaa V, Harald K, Rasi R, Vahtera E, Jousilahti P, Pekkanen J, D'Agostino R, Kannel WB, Wilson PW, Tofler G, Arocha‐Pinango CL, Rodriguez‐Larralde A, Nagy E, Mijares M, Espinosa R, Rodriquez‐Roa E, Ryder E, Diez‐Ewald MP, Campos G, Fernandez V, Torres E, Coll E, Marchioli R, Valagussa F, Rosengren A, Wilhelmsen L, Lappas G, Eriksson H, Cremer P, Nagel D, Curb JD, Rodriguez B, Yano K, Salonen JT, Nyyssonen K, Tuomainen TP, Hedblad B, Lind P, Loewel H, Koenig W, Meade TW, Cooper JA, De Stavola B, Knottenbelt C, Miller GJ, Bauer KA, Rosenberg RD, Sato S, Kitamura A, Naito Y, Iso H, Rasi V, Palosuo T, Ducimetiere P, Amouyel P, Arveiler D, Evans AE, Ferrieres J, Juhan‐Vague I, Bingham A, Schulte H, Assmann G, Cantin B, Lamarche B, Despres JP, Dagenais GR, Tunstall‐Pedoe H, Woodward M, Ben Shlomo Y, Davey SG, Palmieri V, Yeh JL, Rudnicka A, Ridker P, Rodeghiero F, Tosetto A, Shepherd J, Ford I, Robertson M, Brunner E, Shipley M, Feskens EJ, Kromhout D; Fibrinogen Studies Collaboration (2005). Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta‐analysis. JAMA 294, 1799–1809. [DOI] [PubMed] [Google Scholar]
- Davalos D & Akassoglou K (2012). Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol 34, 43–62. [DOI] [PubMed] [Google Scholar]
- Dellu F, Fauchey V, Moal ML & Simon H (1997). Extension of a new two‐trial memory task in the rat: influence of environmental context on recognition processes. Neurobiol Learn Mem 67, 112–120. [DOI] [PubMed] [Google Scholar]
- Fischer MB, Roeckl C, Parizek P, Schwarz HP & Aguzzi A (2000). Binding of disease‐associated prion protein to plasminogen. Nature 408, 479–483. [DOI] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA & Strittmatter SM (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 30, 6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko R, McFarland M & Ben‐Jonathan N (2002). Distribution and characterization of plasmalemma vesicle protein‐1 in rat endocrine glands. J Endocrinol 175, 649–661. [DOI] [PubMed] [Google Scholar]
- Hu G, Vogel SM, Schwartz DE, Malik AB & Minshall RD (2008). Intercellular adhesion molecule‐1–dependent neutrophil adhesion to endothelial cells induces caveolae‐mediated pulmonary vascular hyperpermeability. Circ Res 102, e120–e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadavji NM, Deng L, Malysheva O, Caudill MA & Rozen R (2015). MTHFR deficiency or reduced intake of folate or choline in pregnant mice results in impaired short‐term memory and increased apoptosis in the hippocampus of wild‐type offspring. Neuroscience 300, 1–9. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W & Smith DH (2010). Traumatic brain injury and amyloid‐β pathology: a link to Alzheimer's disease? Nat Rev Neurosci 11, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN & Pax AB (1995). Increased collagen content of cerebral microvessels in Alzheimer's disease. Brain Res 705, 349–352. [DOI] [PubMed] [Google Scholar]
- Kerlin B, Cooley BC, Isermann BH, Hernandez I, Sood R, Zogg M, Hendrickson SB, Mosesson MW, Lord S & Weiler H (2004). Cause‐effect relation between hyperfibrinogenemia and vascular disease. Blood 103, 1728–1734. [DOI] [PubMed] [Google Scholar]
- Kutiyanawalla A, Promsote W, Terry A & Pillai A (2012). Cysteamine treatment ameliorates alterations in GAD67 expression and spatial memory in heterozygous reeler mice. Int J Neuropsychopharmacol 15, 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landin K, Tengborn L & Smith U (1990). Elevated fibrinogen and plasminogen activator inhibitor (PAI‐1) in hypertension are related to metabolic risk factors for cardiovascular disease. J Intern Med 227, 273–278. [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B & Dotti CG (2000). Brain plasmin enhances APP α‐cleavage and Aβ degradation and is reduced in Alzheimer's disease brains. EMBO Rep 1, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little TL, Xia J & Duling BR (1995). Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circ Res 76, 498–504. [DOI] [PubMed] [Google Scholar]
- Lominadze D, Dean WL, Tyagi SC & Roberts AM (2010). Mechanisms of fibrinogen‐induced microvascular dysfunction during cardiovascular disease. Acta Physiol Scand 198, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominadze D, Joshua IG & Schuschke DA (1998). Increased erythrocyte aggregation in spontaneously hypertensive rats. Am J Hypertens 11, 784–789. [DOI] [PubMed] [Google Scholar]
- Manczak M & Reddy PH (2013). Abnormal interaction of oligomeric amyloid‐β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. J Alzheimers Dis 36, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T, Hiramatsu M, Itoh J, Kameyama T, Hasegawa T & Nabeshima T (1994). Behavioral evidence for a modulating role of σ ligands in memory processes. I. Attenuation of dizocilpine (MK‐801)‐induced amnesia. Brain Res 647, 44–56. [DOI] [PubMed] [Google Scholar]
- Mehta D & Malik AB (2006). Signaling mechanisms regulating endothelial permeability. Physiol Rev 86, 279–367. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE & Malik AB (2000). Endothelial cell‐surface gp60 activates vesicle formation and trafficking via Gi‐coupled Src kinase signaling pathway. J Cell Biol 150, 1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki‐Shimizu K, Predescu D, Shimizu J, Broman M, Predescu S & Malik AB (2006). siRNA‐induced caveolin‐1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am J Physiol Lung Cell Mol Physiol 290, L405–L413. [DOI] [PubMed] [Google Scholar]
- Muradashvili N, Benton RL, Saatman KE, Tyagi SC & Lominadze D (2015). Ablation of matrix metalloproteinase‐9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab Brain Dis 30, 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Benton RL, Tyagi R, Tyagi SC & Lominadz D (2014. a). Elevated level of fibrinogen increases caveolae formation; Role of matrix metalloproteinase‐9. Cell Biochem Biophys 69, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Khundmiri SJ, Tyagi R, Gartung A, Dean WL, Lee M‐J & Lominadze D (2014. b). Sphingolipids affect fibrinogen‐induced caveolar transcytosis and cerebrovascular permeability. Am J Physiol Cell Physiol 307, C169–C179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Qipshidze N, Munjal C, Givvimani S, Benton RL, Roberts AM, Tyagi SC & Lominadze D (2012. a). Fibrinogen‐induced increased pial venular permeability in mice. J Cereb Blood Flow Metab 32, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi R & Lominadze D (2012. b). A dual‐tracer method for differentiating transendothelial transport from paracellular leakage in vivo and in vitro. Front Physiol 3, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi R, Metreveli N, Tyagi SC & Lominadze D (2014. c). Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen–amyloid beta complex formation during hyperhomocysteinemia. J Cereb Blood Flow Metab 34, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Joggerst B & Simons K (1994). Regulated internalization of caveolae. J Cell Biol 127, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patibandla PK, Tyagi N, Dean WL, Tyagi SC, Roberts AM & Lominadze D (2009). Fibrinogen induces alterations of endothelial cell tight junction proteins. J Cell Physiol 221, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybarczyk BJ, Lawrence SO & Simpson‐Haidaris PJ (2003). Matrix‐fibrinogen enhances wound closure by increasing both cell proliferation and migration. Blood 102, 4035–4043. [DOI] [PubMed] [Google Scholar]
- Simionescu M, Popov D & Sima A (2009). Endothelial transcytosis in health and disease. Cell Tissue Res 335, 27–40. [DOI] [PubMed] [Google Scholar]
- Stan R‐V, Marion K & Palade GE (1999). PV‐1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proc Natl Acad Sci USA 96, 13203–13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Hu G, Zhang X & Minshall RD (2009). Phosphorylation of caveolin‐1 regulates oxidant‐induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res 105, 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Roberts AM, Dean WL, Tyagi SC & Lominadze D (2008). Fibrinogen induces endothelial cell permeability. Mol Cell Biochem 307, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oijen M, Witteman JC, Hofman A, Koudstaal PJ & Breteler MMB (2005). Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke 36, 2637–2641. [DOI] [PubMed] [Google Scholar]
- Wei E, Hamm R, Baranova A & Povlishock J (2009). The long‐term microvascular and behavioral consequences of experimental traumatic brain injury after hypothermic intervention. J Neurotrauma 26, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Hiramatsu M, Noda Y, Mamiya T, Murai M, Kameyama T, Komori Y, Nikai T, Sugihara H & Nabeshima T (1996). Role of nitric oxide and cyclic GMP in the dizocilpine‐induced impairment of spontaneous alternation behavior in mice. Neuroscience 74, 365–374. [DOI] [PubMed] [Google Scholar]
- Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV & Sessa WC (2006). Direct evidence for the role of caveolin‐1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]