

Abstract
Brain Cav1.2 and Cav1.3 L‐type Ca2+ channels play key physiological roles in various neuronal processes that contribute to brain function. Genetic studies have recently identified CACNA1C as a candidate risk gene for bipolar disorder (BD), schizophrenia (SCZ), major depressive disorder (MDD) and autism spectrum disorder (ASD), and CACNA1D for BD and ASD, suggesting a contribution of Cav1.2 and Cav1.3 Ca2+ signalling to the pathophysiology of neuropsychiatric disorders. Once considered sole clinical entities, it is now clear that BD, SCZ, MDD and ASD share common phenotypic features, most likely due to overlapping neurocircuitry and common molecular mechanisms. A major future challenge lies in translating the human genetic findings to pathological mechanisms that are translatable back to the patient. One approach for tackling such a daunting scientific endeavour for complex behaviour‐based neuropsychiatric disorders is to examine intermediate biological phenotypes in the context of endophenotypes within distinct behavioural domains. This will better allow us to integrate findings from genes to behaviour across species, and improve the chances of translating preclinical findings to clinical practice.
Abbreviations
- ASD
autism spectrum disorder
- BD
bipolar disorder
- BDNF
brain derived neurotrophic factor
- CPP
conditioned place preference
- DHP
dihydropyridine
- FST
forced swim test
- GWAS
genome‐wide association studies
- LTCCs
L‐type Ca2+ channels
- MDD
major depressive disorder
- NAc
nucleus accumbens
- PFC
prefrontal cortex
- PTSD
post‐traumatic stress disorder
- SCZ
schizophrenia
- SNP
single nucleotide polymorphism
- SVF
semantic verbal fluency
- VTA
ventral tegmental area
Introduction
Recent human genetic studies have raised tremendous interest and excitement for a role of brain voltage‐gated Cav1.2 and Cav1.3 L‐type Ca2+ channels (LTCCs) in neuropsychiatric and neurodevelopmental disorders. Genome‐wide association studies (GWAS) have linked multiple single nucleotide polymorphisms (SNPs) in the CACNA1C and CACNA1D genes to neuropsychiatric disorders (Bhat et al. 2012; Cross‐Disorder Group et al. 2013; Heyes et al. 2015). Several CACNA1C SNPs, and in particular SNP rs1006737 (risk A allele), have been widely reproduced and strongly associated with bipolar disorder (BD) (Ferreira et al. 2008), major depressive disorder (MDD) (Green et al. 2010; Casamassima et al. 2010 b), schizophrenia (SCZ) (Green et al. 2010; Nyegaard et al. 2010) and recently also autism spectrum disorder (ASD) (Li et al. 2015). CACNA1D SNPs have been linked to BD (Ament et al. 2015). Additionally, variants in coding regions of CACNA1C cause Timothy syndrome, a syndromic ASD (Splawski et al. 2004), and coding variants in CACNA1D have been associated with ASD (O'Roak et al. 2012; Pinggera et al. 2015). The contribution of CACNA1C and CACNA1D SNPs to the pathology of neuropsychiatric disorders is further underscored by their presence in biological pathways implicated in these disorders (Psychiatric GWAS Consortium et al. 2011; Network and Pathway Analysis Subgroup et al. 2015).
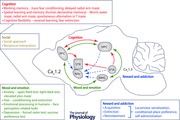
Cav1.2 and Cav1.3 channels are important mediators of Ca2+ entry into excitable cells such as neurons. They are both expressed in neurons, with Cav1.2 expressed at higher levels compared to Cav1.3 in the forebrain (Hell et al. 1993) and Cav1.3 serving as the primary LTCC in the midbrain (Rajadhyaksha et al. 2004; Day et al. 2006). They shape neuronal firing and are present in signalling complexes (Calin‐Jageman & Lee, 2008; Zamponi et al. 2015) primarily at the postsynaptic membrane (Di Biase et al. 2008; Jenkins et al. 2010), where they are poised for excitation–transcription coupling (Ma et al. 2012) by activation of Ca2+ second messenger pathways (Simms & Zamponi, 2014) (Fig. 1). Because of these properties, LTCCs play an important role in neuronal plasticity related to neuronal development, learning and memory, drug addiction, and neuropsychiatric illness (Casamassima et al. 2010 a; Bhat et al. 2012; Berger & Bartsch, 2014). Additionally, L‐type activated signal transduction pathways crosstalk with dopamine and glutamate signalling pathways (Rajadhyaksha & Kosofsky, 2005), important mediators of neuronal processes underlying the neuropathology of multiple brain disorders. (For more in‐depth information, readers are directed to Casamassima et al. 2010 a; Bhat et al. 2012; Berger & Bartsch, 2014; Simms & Zamponi, 2014; Striessnig et al. 2014; Zamponi et al. 2015).
Figure 1.
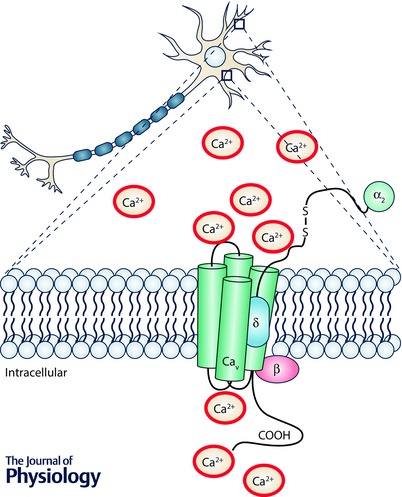
Schematic diagram of the L‐type Ca2+ channels Cav1.2 and Cav1.3, and β and α2δ auxiliary subunits
Given the importance of Cav1.2 and Cav1.3 channels in brain function and the recent human genetic findings establishing CACNA1C and CACNA1D as risk genes for neuropsychiatric disorders, the time is prime to structure scientific studies to begin to identify the biological effects of human genetic variants that are reliable and eventually translatable back to the patient. This requires detailed analyses across multiple levels of complexity from gene to molecule to cell to circuit and behaviour. This is particularly important when studying neuropsychiatric disorders that clinically are behaviour‐based disorders. Additionally, neuropsychiatric disorders (such as BD, SCZ and ASD), once considered unitary entities, share common behavioural phenotypic features most likely due to overlapping neurocircuitry (the primary focus of this review; Millan et al. 2012; Luthi & Luscher, 2014). Thus, the emphasis on utilizing behavioural domains (endophenotypes) as opposed to clinical diagnosis, as proposed in the Research Domain Criteria Framework (RDoC) by the National Institute of Mental Health (NIMH) (Insel, 2014), to explore the impact of CACNA1C and CACNA1D human polymorphisms on neuronal function and on neuronal circuits can be greatly helpful. We believe that such an approach will allow the integration of information from molecule to behaviour and increase the chance of translation of biological findings across species (human to mouse and back to human), towards better treatment outcomes in patients. Such an approach is already underway in multiple laboratories. The availability of new molecular and genetic technologies to study cacna1c and cacna1d in rodent models and the availability of samples derived from patients (such as patient brain tissue and patient‐derived induced pluripotent cells) have already begun to shed light on the contribution of human cacna1c genetic variants on the pathophysiology of disease.
There have been several excellent recent reviews on Cav1.2 and Cav1.3 LTCCs in brain and brain disorders (Casamassima et al. 2010 a; Bhat et al. 2012; Berger & Bartsch, 2014; Striessnig et al. 2014; Zamponi et al. 2015). In this review we aim to integrate recent human behavioural and neuroimaging data with information available from preclinical cacna1c and cacna1d rodent models to begin to gain insight on how dysfunction of Cav1.2 and Cav1.3 channels at the level of brain anatomy and circuitry can lead to behavioural phenotypes associated with neuropsychiatric disease. We hope that this will encourage electrophysiological, cellular and molecular studies in the context of neuropsychiatric‐related behavioural deficits.
Integrating human and mouse findings to understand pathological circuitry underlying intermediate phenotypes of psychiatric disorders
To date, the CACNA1C SNPs linked to neuropsychiatric disorders are present within non‐coding regions (intronic, 5′ and 3′ untranslated regions) (Bhat et al. 2012) suggesting that they would affect CACNA1C gene expression. Consistent with this, both an increase (Bigos et al. 2010; Yoshimizu et al. 2015) and decrease (Gershon et al. 2014; Roussos et al. 2014; Yoshimizu et al. 2015) in CACNA1C mRNA have been reported in human samples (brain tissue and induced pluripotent stem cell (iPSC)‐derived neurons), suggesting that gain or loss of Cav1.2 function can be detrimental. Cav1.2 gain of function mutation causes Timothy syndrome and CACNA1D single nucleotide variants identified in ASD (O'Roak et al. 2012) have recently been shown to be gain of function variants (Pinggera et al. 2015).
Brain imaging and clinical studies in the most replicated CACNA1C neuropsychiatric risk SNP rs1006737 has revealed altered brain structure and function in healthy carriers and patients (Bhat et al. 2012; Berger & Bartsch, 2014), underscoring the functional contribution of CACNA1C to the neuropathology underlying neuropsychiatric disorders.
Rodents (in particular mice) have proved to be useful in exploring human behavioural deficits, specifically for mapping circuitry that underlies behavioural phenotypes. Additionally humans and rodents share emotional behaviours and emotional circuitry that are conserved from mouse to human (Janak & Tye, 2015), thus allowing translation of human polymorphisms to mouse and back to the human. This has been elegantly demonstrated for a polymorphism in the BDNF gene (Glatt & Lee, 2015), a downstream molecular target of LTCCs (Tao et al. 1998; Tabuchi et al. 2000). At this time, studying CACNA1C non‐coding variants in mouse models poses a challenge. However, studying loss of function (such as the use of knockout mice; Moosmang et al. 2005; Dao et al. 2010; Lee et al. 2012) or gain of function (such as the Timothy syndrome mouse model; Barrett & Tsien, 2008; Bader et al. 2011) can be informative.
To begin to gain a greater understanding of the Cav1.2 and Cav1.3 mediated neural mechanisms underlying the pathophysiology of psychiatric disorders, we attempt to integrate anatomical and circuit information that has been obtained from human neuroimaging and clinical studies with findings reported in cacna1c and cacna1d mouse models in the context of the major behavioural endophenotypes of neuropsychiatric disorders (mood and emotion, cognition, and social, in addition to addiction, a co‐morbid endophenotype) associated with the CACNA1C and CACNA1D risk genetic variants.
Mood and emotion
CACNA1C
Changes in mood and emotion are core features that underlie psychiatric disorders. These can be manifested as anxiety and depression that are prominent components of the forms of neuropsychiatric disease in which CACNA1C has been implicated. Interestingly, healthy CACNA1C rs1006737 risk allele carriers had significantly higher scores for depression and anxiety on a self‐report questionnaire (Erk et al. 2010, 2014 a). These findings were replicated in a separate cohort of CACNA1C rs1006737 risk‐allele healthy individuals who displayed increased trait anxiety and proneness to depression (Roussos et al. 2011).
Additionally, dysregulation of negative emotions, such as fear, also forms a core component underlying anxiety disorders (Dymond et al. 2014; Singewald et al. 2015). Human and rodent studies have revealed overlapping circuitry underlying fear, anxiety and depression. Fear‐associative learning protocols have proven to be extremely useful to examine the neurocircuitry underlying anxiety disorders (Hijzen et al. 1995; Ohl et al. 2003; Singewald et al. 2015). In humans and rodents, fear conditioning is a form of emotional learning in which a naturally aversive stimulus (unconditioned) is paired either with a neutral context (contextual‐associated) or tone (cue‐associated) to elicit a fear response measured as freezing behaviour in rodents, or a physiological response in humans. Emotional responses to facial expressions (e.g. fearful faces, happy faces) are additionally used in humans to evaluate emotional processing (Ekman, 1992). In humans, a startle response is often used to measure negative emotional processing during visualization of unpleasant pictures (Lissek et al. 2007; Anokhin & Golosheykin, 2009; Fendt & Koch, 2013). In both humans and rodents, the inability to extinguish the conditioned context or cue fear response is additionally utilized to evaluate dysregulated fear (rodents) and emotional (humans) processing (VanElzakker et al. 2014) as observed in patients with SCZ and post‐traumatic stress disorder (PTSD), an anxiety disorder.
Using fear‐learning protocols, the amygdala, prefrontal cortex (PFC) and hippocampus have been established as the primary fear circuit in both humans and rodents (Phelps & LeDoux, 2005; Sehlmeyer et al. 2009). The amygdala is critical for the acquisition and expression of a conditioned fear response in rodents (LeDoux, 2003; Keifer et al. 2015) and humans (LaBar et al. 1995; Hamm & Weike, 2005). The PFC exerts a top‐down regulation of the amygdala while the hippocampus, with its regional connectivity, can provide a modulatory effect on the amygdala either directly or via the PFC. The hippocampus functions to influence the context‐dependent expression of fear (Knight et al. 2004; Sierra‐Mercado et al. 2011; Sotres‐Bayon et al. 2012), the extinction of cue‐associated fear memories in rodents (Sierra‐Mercado et al. 2011; Maren et al. 2013; Rosas‐Vidal et al. 2014), and the extinction recall of contextual‐associated fear memories in humans (Kalisch et al. 2006; Milad et al. 2007; Ahs et al. 2015). The use of optogenetic studies in rodents has also established the PFC, amygdala and hippocampus in regulating anxiety and depression (Covington et al. 2010; Tye et al. 2011; Warden et al. 2012; Ota et al. 2014; Bagot et al. 2015). In depressed patients, structural and functional alterations in these regions have been reported (Busatto, 2013).
Consistent with a role of the PFC–amygdala–hippocampus circuit in anxiety, depression and fear, neuroimaging studies in both healthy CACNA1C risk allele carriers (rs1006737 having been studied the most to date) and in patients, have identified structural and functional alterations within this circuit (Fig. 2). Structural neuroimaging studies have revealed that CACNA1C rs1006737 risk allele carriers have increased grey matter density in the PFC (Wang et al. 2011) and right amygdala (Perrier et al. 2011), and altered white matter microstructure in the right hippocampal formation (Dietsche et al. 2014). Functional imaging studies have found increased activation in the amygdala and ventral PFC of CACNA1C rs1006737 risk allele carriers during facial affect processing (Dima et al. 2013). Similar findings were obtained in two other independent studies that observed increased activity in the left amygdala during a negative face matching task in both healthy individuals (Jogia et al. 2011; Tesli et al. 2013) and BD patients that carry the CACNA1C rs1006737 risk allele (Tesli et al. 2013). Additionally at the circuit level, Wang et al. (2011) reported decreased functional connectivity between the PFC and amygdala during a facial processing task (fearful and happy faces) in CACNA1C rs1006737 risk allele carriers (Wang et al. 2011). In BD patients that were carriers of the CACNA1C rs1006737 risk allele, Radua et al. (2012) reported decreased outflow of the medial frontal gyrus to the left putamen (part of the striatum) during perception of fearful faces, demonstrating hypo‐connectivity between a prefrontal cortical region and a limbic structure during emotional processing (Radua et al. 2012).
Figure 2. Overlapping neurocircuitry of L‐type Ca2+ channels contributing to endophenotypes of psychiatric disorders .
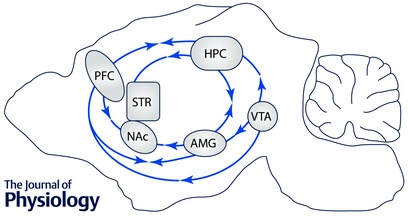
Single arrow denotes unidirectional connectivity in the direction of the arrow and double arrow denotes bidirectional connectivity. PFC, prefrontal cortex; HPC, hippocampus; STR, striatum; NAc, nucleus accumbens; AMG, amygdala; VTA, ventral tegmental area.
Similarly in the hippocampus of CACNA1C risk allele carriers, Bigos et al. (2010) reported increased hippocampal activity in healthy individuals who were homozygote for the CACNA1C rs1006737 risk allele (AA), when measured during a facial processing task (Bigos et al. 2010). In a separate study Erk et al. (2010) showed a significant negative correlation between regional activation in the hippocampus and depression and anxiety scores (Erk et al. 2010) that was confirmed in a later study (Erk et al. 2014 a). Data from these human neuroimaging studies provide clear evidence of structural and functional alterations in the PFC–amygdala–hippocampus circuit as a consequence of the CACNA1C risk allele. The dysregulation of this circuit has been suggested to be the underlying factor modulating the anxiety and depression intermediate phenotypes commonly observed in BD, SCZ and MDD (Erk et al. 2014 b). With regard to the behavioural effects of the CACNA1C rs1006737 risk allele on emotional processing a study by Pasparakis et al. (2015) found reduced affective startle responses to pleasant pictures and exaggerated responses to negative pictures in healthy CACNA1C rs1006737 risk allele carriers (Pasparakis et al. 2015), further supporting the impact of the CACNA1C risk allele on the PFC–amygdala–hippocampus circuit.
In contrast to the human literature there have been few, but significantly important, animal studies that have begun to dissect the role of cacna1c (Cav1.2) in anxiety, depression and fear and their underlying neurocircuitry. To study anxiety in rodents, behavioural protocols that use approach–avoidance conflict to inhibit an ongoing behaviour that is characteristic for the animal have been utilized (Bailey & Crawley, 2009) and include the open‐field test, light–dark test and the elevated plus maze test (Ohl et al. 2003). Using these tests, constitutive heterozygous cacna1c knockout female (Dao et al. 2010) and male mice (Bader et al. 2011; Lee et al. 2012) have been shown to exhibit increased anxiety‐like behaviour. The anxiety phenotype was recapitulated in conditional cacna1c‐deficient male mice that had cacna1c knocked out specifically in excitatory neurons of the forebrain (Lee et al. 2012). Additionally consistent with altered PFC function in CACNA1C risk allele carriers, focal deletion of cacna1c in the PFC of adult mice resulted in an anxiogenic phenotype (Lee et al. 2012). Interestingly, the knock‐in Cav1.2 gain of function TS mouse displayed no alteration in anxiety (Bader et al. 2011). As human studies have revealed altered function of the amygdala and hippocampus in CACNA1C risk allele carriers, additional rodent studies using strategies such as focal deletion of Cav1.2 in these regions would be greatly helpful in elucidating the cacna1c neural circuitry underlying anxiety.
Studying depression in rodents is accomplished using behavioural protocols like the Porsolt forced swim test (FST) (Porsolt et al. 1977; Can et al. 2012), tail suspension test (Bergner et al. 2010) and the sucrose preference test (Katz, 1982; Strekalova et al. 2004) that target the core components of depressive‐like behaviour such as despair (FST and tail suspension test) and anhedonia (sucrose preference test). So far, only one study has been published reporting an anti‐depressive phenotype in constitutive cacna1c heterozygous mice using FST and the tail suspension test (Dao et al. 2010). We have replicated this finding using FST and the sucrose preference test (authors’ unpublished observations) and ongoing studies in our laboratory are currently examining the anatomical and molecular mechanisms mediating the anti‐depressive phenotype in constitutive cacna1c heterozygous mice.
As the behavioural paradigm and underlying neurocircuitry in fear‐learning has been well established, several researchers have focused their attention on the role of Cav1.2 channels in fear processing. Interestingly, conditional knockout of cacna1c in the excitatory neurons of the forebrain had no effect on the conditioning or extinction of a contextual‐associated fear memory (McKinney et al. 2008). Langwieser et al. (2010) similarly found no deficit in the consolidation and recall of a cue‐associated fear memory in brain specific cacna1c knockout mice and identified that the lack of this particular phenotype was a result of compensatory upregulation of Ca2+ permeable glutamate AMPA receptors in the amygdala (Langwieser et al. 2010). Thus, compensatory adaptations in developmental knockout mouse models may override the influence of Cav1.2 LTCCs in fear conditioning protocols and may not be the most appropriate option to study the role of these channels in fear processing. This is highlighted from pharmacological studies where acute inhibition of LTCCs with an LTCC blocker (isradipine or verapamil) delivered either into the intracerebroventricular compartment (Langwieser et al. 2010) or directly into the lateral division of the amygdala (Bauer et al. 2002) of the adult animal was sufficient to induce a deficit in recall of a cue‐associated fear memory. Similarly repeated inhibition of LTCCs with verapamil or nifedipine in the basolateral amygdala of the adult rat impaired cue fear extinction, demonstrating that LTCCs can mediate fear processing (Davis & Bauer, 2012). As pharmacological blockers to date do not differentiate between the two LTCC isoforms, Cav1.2 and Cav1.3, we encourage alternative strategies such as the use of focal brain knockout of Cav1.2 in the adult brain using viral vectors (Lee et al. 2012) to directly test the role of the Cav1.2 LTCCs in fear memory recall. This suggestion is underscored by the finding that knockout of Cav1.2 channels using viral vectors specifically in the anterior cingulate cortex, a brain region that has increased activity during the observation of others’ fear (Olsson et al. 2007), impaired observational fear learning in mice (Jeon et al. 2010). This further suggests that the Cav1.2 isoform can be recruited during fear learning and highlights the importance of examining site‐targeted deletion of these channels. Further support of the role of Cav1.2 in fear processing is provided from the knock‐in Timothy syndrome mouse that displayed significantly increased contextual‐ and cue‐associated fear memories that persisted up to 2 weeks postconditioning (Bader et al. 2011).
Contextual‐associated fear memories are dependent not only on the amygdala but also on the hippocampus that is recruited to form a representation of the fear‐associated context (Phillips & LeDoux, 1992). Since conditional cacna1c knockout mice display a deficit in recall of a hippocampal‐dependent spatial memory 30 days after training (White et al. 2008), it begs the question on whether these mice have a similar deficit in long‐term contextual‐associated fear memories, an area of study that has not been as extensively explored but is highly relevant with regard to PTSD.
CACNA1D
Mouse models of cacna1d have been utilized to study the role of the Cav1.3 LTCCs in anxiety (Lee et al. 2012), depressive‐like behaviour (Sinnegger‐Brauns et al. 2004) and fear processing (McKinney & Murphy, 2006). We have previously shown that focal knock‐down of Cav1.3 in the PFC does not impact anxiety (Lee et al. 2012), ruling out the role of these channels in the PFC in mediating anxiety‐like behaviour. When tested in FST, Cav1.3 knockout mice exhibit an antidepressive‐like phenotype. Using Cav1.2 dihydropyridine (DHP)‐insensitive mutant mice that have a point mutation in the DHP‐binding pocket of the Cav1.2 subunit eliminating the sensitivity of the Cav1.2 channels to DHP blockers, when treated with the DHP LTCC activator BayK8644 resulted in a depressive‐like behavioural phenotype (Sinnegger‐Brauns et al. 2004), suggesting a role for Cav1.3 LTCCs in the modulation of depressive‐like behaviour. In contextual fear conditioning, homozygous cacna1d knockout mice display impaired consolidation of fear memory but normal extinction (McKinney & Murphy, 2006). Given the recent finding of compensatory adaptation in cacna1d knockout mice (Poetschke et al. 2015), the Cav1.2 DHP insensitive mutant mouse line is an excellent mouse model to further explore the role of Cav1.3 channels in fear processing, in addition to the use of viral vectors expressing Cav1.3 short hairpin (sh)RNA (Schierberl et al. 2011 a; Lee et al. 2012).
Cognition
Although alterations in mood and emotion form the main components in neuropsychiatric disorders, deficits in cognition are commonly observed and can be equally debilitating to the lifestyle of an individual (Millan et al. 2012). Several human studies have shown an association between the CACNA1C rs1006737 risk variant and cognitive function. In healthy individuals there are conflicting results over the influence of this gene on cognitive function. Some studies report lower verbal fluency performance (Krug et al. 2010), deficits in attention (Thimm et al. 2011), impaired working memory (Zhang et al. 2012) and poorer learning performance (Dietsche et al. 2014). In contrast, others have reported no significant association between the rs1006737 risk allele in the CACNA1C gene and verbal learning and memory, verbal intelligence (Erk et al. 2010, 2014 a; Roussos et al. 2011), recognition memory (Dietsche et al. 2014), working memory (Paulus et al. 2014) and overall cognitive function (Hori et al. 2012; Soeiro‐de‐Souza et al. 2013). However, in BD and SCZ patients, all studies to date have reported a negative effect of the CACNA1C rs1006737 risk allele on executive function (Arts et al. 2013; Soeiro‐de‐Souza et al. 2013) and working memory (Zhang et al. 2012).
Abnormal neural activity in cortical anatomical regions and concurrent dysregulation in their connectivity to other brain regions have been suggested to be responsible for deficits in cognitive function (Millan et al. 2012) (Fig. 2). From animal studies, it has been shown that the PFC is the central anatomical structure mediating high‐order cognitive function by exerting a top‐down executive control on subcortical structures including the hippocampus, striatum, thalamus and amygdala (Miller, 2000; Riga et al. 2014). Depending on the subcortical structure innervated, the impact of the PFC on distinct cognitive domains varies.
Several functional imaging studies in CACNA1C rs1006737 risk allele carriers have identified alterations in brain activation during multiple cognitive behavioural tasks. During an n‐back working memory task, healthy CACNA1C risk AA homozygote individuals displayed significantly increased activity in the PFC, which they suggested indicated an inefficiency of the PFC (Bigos et al. 2010). In contrast to these findings two separate studies showed that healthy carriers of the CACNA1C rs1006737 risk allele had significantly reduced activation in the dorsolateral PFC during an associative episodic memory task (Erk et al. 2014 a) and the n‐back working memory task (Paulus et al. 2014) with altered connectivity between the dorsolateral PFC and hippocampus (Paulus et al. 2014). In the hippocampus, three independent studies have reported significantly lower activation of the hippocampus in healthy carriers of the CACNA1C rs1006737 risk allele during an episodic memory recall test (Erk et al. 2010, 2014 a,b; Krug et al. 2014) with diminished functional coupling between the left and right hippocampal regions (Erk et al. 2010). Concurrent with this the authors reported decreased activation in the subgenual part of the perigenual anterior cingulate, a brain region that is implicated in mood regulation as well as memory processing (Erk et al. 2014 a,b). Consistent with lower verbal fluency in CACNA1C rs1006737 risk allele carriers (Krug et al. 2010), a test used to measure executive control (Shao et al. 2014), the same study reported increased activation in the left inferior frontal gyrus and left precuneus during a semantic verbal fluency (SVF) task in risk carriers (Krug et al. 2010), while in depressed patients with the CACNA1C rs1006737 risk allele there was increased SVF task‐related activation in the left middle/inferior frontal gyrus and enhanced functional coupling between the left middle/inferior and right superior/middle frontal gyri (Backes et al. 2014).
In the last several decades, distinct behavioural tasks have been developed in animal models to study the underlying neurocircuit and molecular pathways mediating different domains within cognition. To relate these animal models to the deficits in executive function observed in human neuropsychiatric patients, researchers have designed behavioural protocols to mimic the behaviours often used in human studies. Using behavioural tasks in which stimuli are separated in time such as trace fear conditioning (Gilmartin et al. 2013) and the delayed radial arm maze (Floresco et al. 1999), working memory has been shown to be mediated by the prefrontal modulation of the striatum and thalamus (Floresco et al. 1999; Gilmartin et al. 2013). With regard to spatial learning and memory, which has been considered to be equivalent to human declarative abilities (Morellini, 2013), a form of memory that is often disrupted in SCZ (Cirillo & Seidman, 2003), BD (van Gorp et al. 1999) and MDD (Austin et al. 2001), animal behavioural tasks like the Morris water maze, the radial arm maze and spontaneous alternation in the T maze have been utilized to demonstrate the involvement of the PFC, hippocampus and dorsal striatum (Morellini, 2013; Pooters et al. 2015). Another domain within cognition that has often been reported in neuropsychiatric disorders is cognitive flexibility, defined as the ability to flexibly alternate one's behaviour in response to changing environmental contingencies. In humans, this has been measured using the Wisconsin card‐sorting task (Miyake et al. 2000) and the Stroop test (Jensen & Rohwer, 1966) while in animal models this has been tested through reversal learning in a Y maze (Trinh et al. 2012) or operant task (Brady & Floresco, 2015), as well as through extinction of a conditioned fear (Trinh et al. 2012).
While there is significant, though limited, evidence of the negative relationship between the CACNA1C risk allele and cognition in humans, not much has been done in identifying the neuroanatomical basis of these deficits. To date, only one study has identified a deficit in long‐term (30 day) recall of a spatial memory in mice that had a conditional knockout of cacna1c in the excitatory neurons of the forebrain (White et al. 2008), suggesting a role of these channels in cognition. Similar evidence of a role of these channels in cognition has been obtained through studies done in the knock‐in Timothy syndrome mouse (Bader et al. 2011) that displayed normal learning and memory but significant deficits in reversal learning (Bader et al. 2011). Additional studies need to further explore cacna1c animal models to understand the role of Cav1.2 channels in cognitive deficits.
We believe that it is of high priority to explore Cav1.2 as a potential target for treating cognitive impairments in neuropsychiatric patients particularly since the current drugs used to treat the mood‐related symptoms in psychiatric disorders fail to alleviate the cognitive deficits (Millan, 2006; Hill et al. 2010). Unpublished data from our lab suggest a role for Cav1.2 channels in mediating different aspects of cognition that is interactive with distinct neurotransmitter systems.
Social behaviour
Deficits in social behaviour and communication are observed in a range of neuropsychiatric disorders, including SCZ and depression, and represents a core feature observed in ASD (Kennedy & Adolphs, 2012; APA, 2013). In humans, social interaction is complex and heavily dependent on appropriate cognitive and emotional function. Interestingly, in healthy individuals the CACNA1C rs1006737 risk allele was significantly associated with low extraversion, a personality trait that is characterized by reduced social activities and interactions (Roussos et al. 2011).
As with all the other endophenotypes discussed above social behaviour is mediated by a network of brain regions that function in an interdependent manner (Fig. 2). These include the fusiform gyrus that mediates face perception, the amygdala and anterior cingulate cortex that are involved in processing facial expressions of fear, and the PFC that is required for interpreting others’ intentions and motives (Grady & Keightley, 2002). In CACNA1C rs1006737 risk allele carriers, there is increased activation of the fusiform gyrus during a facial affect‐processing task (Dima et al. 2013), suggesting dysregulation within the circuitry that is recruited for facial processing in social interaction.
Rodents, being innately social creatures, have been used to study social behaviour. Through lesion and optogenetic studies, certain key anatomical regions mediating social behaviour in rodents have been delineated and includes the hippocampus (Felix‐Ortiz & Tye, 2014; Hitti & Siegelbaum, 2014; Stevenson & Caldwell, 2014), amygdala (Martel et al. 2008; Katayama et al. 2009), PFC (Jodo et al. 2010; Yizhar et al. 2011; Felix‐Ortiz et al. 2015), nucleus accumbens (NAc) and ventral tegmental area (VTA) (Gunaydin et al. 2014). In rodents, social behaviour is most commonly studied using the social approach test in a 3‐chamber apparatus in which the time that the test mouse spends interacting with a stranger mouse versus an inanimate object is measured. Reciprocal interactions between pairs of mice are also studied in which a variety of parameters, including nose‐to‐nose interactions, anogenital sniffing, allogrooming and play, are measured (Silverman et al. 2010).
Although the CACNA1C gene has been implicated in ASD and SCZ, both of which show significant impairments in social behaviour, no study to date has tested social behaviour in cacna1c deficient mice. However, the knock‐in Timothy syndrome mouse displays a severe deficit in social behaviour (Bader et al. 2011), demonstrating that Cav1.2 channels can regulate social behaviour. The lack of studies points to a need for additional work to further explore the role of cacna1c in social behaviour. Similarly, because of the recent finding of the association between the CACNA1D gene and ASD (O'Roak et al. 2012; Pinggera et al. 2015), preclinical studies need to be pursued using cacna1d mouse models to identify the impact of this gene on social behaviour.
Reward and addictive behaviour
Altered reward processing and reward brain circuitry is often associated with multiple psychiatric disorders (Pechtel et al. 2013; Whitton et al. 2015). Importantly, substance abuse disorders are often co‐morbid with neuropsychiatric disorders (Post & Kalivas, 2013; Luthi & Luscher, 2014; Lai et al. 2015). Cocaine use in particular has been associated with higher incidence of mood and anxiety disorders (Falck et al. 2004; Post & Kalivas, 2013; Luthi & Luscher, 2014). In a study conducted by Herrero et al. (2008), over 40% of young cocaine users recruited in non‐clinical settings presented psychiatric co‐morbidity, with 27% diagnosed with mood disorders and 13% diagnosed with anxiety disorders (Herrero et al. 2008). Furthermore, the severity of anxiety disorders such as PTSD, MDD and BD may be increased when co‐morbid with substance use disorder (Kessler et al. 2005). Conversely, patients with psychiatric disorders may have an increased vulnerability to abuse drugs. Patients with anxiety disorders, BD and SCZ have an increased likelihood of cocaine and stimulant use (Volkow, 2009; Post & Kalivas, 2013; Tang et al. 2014).
Overlapping neurocircuitry and common genetic risk factors have been suggested to underlie the high co‐morbidity in mood and drug abuse disorders (Nestler & Carlezon, 2006; Luthi & Luscher, 2014). Many studies reveal convergence of neuronal circuits (Fig. 2) and molecular mechanisms involved in mood disorders and reward systems (Peters & Buchel, 2009; Russo & Nestler, 2013; Luthi & Luscher, 2014). The NAc is the key nucleus that modulates reward and motivation for drug seeking (Kelley, 2004; Kalivas & Volkow, 2005), by integrating information from other brain regions within the mesolimbic reward circuitry that include, but are not limited to, dopaminergic inputs from the VTA and glutamatergic inputs from the basolateral amygdala, medial PFC and hippocampus (Koob & Volkow, 2010), identical brain regions also recruited for the intermediate phenotypes discussed above. The VTA additionally projects to the medial PFC, hippocampus and basolateral amygdala (Fig. 2).
In rodents, three commonly used behavioural protocols to assess the addictive potential of drugs of abuse are behavioural sensitization, conditioned place preference (CPP) and self‐administration. Behavioural sensitization is a model of drug‐induced long‐term synaptic and behavioural plasticity (Robinson & Berridge, 1993; Pierce & Kalivas, 1997; Vanderschuren & Pierce, 2010). Interestingly, increased behavioural drug responsivity also occurs as a result of repeated stress and repeated occurrence of episodes of BD (Post & Kalivas, 2013). CPP is a simple, non‐invasive procedure wherein animals are trained to associate a specific environment with the rewarding effects of a drug (Bardo & Bevins, 2000). Subsequently when animals are allowed to freely explore the drug‐paired and non‐drug‐paired environment, they prefer the drug‐paired environment, indicating drug–reward associations. The conditioned response is thought to be relevant to human drug‐seeking behaviour and to drug‐ and cue‐induced relapse (Stewart, 2008). The operant reinstatement model of cocaine seeking using the self‐administration procedure wherein animals volitionally respond for delivery of a drug such as cocaine, possesses greatest face validity as a model of addiction, as it most closely resembles human self‐administered drug taking behaviour (Panlilio & Goldberg, 2007).
Using the above behavioural protocols, pharmacological studies have established an important role of LTCCs in addictive behaviour (reviewed in Bhat et al. 2012; Berger & Bartsch, 2014). Below we specifically highlight what has been found for CACNA1C and CACNA1D in human and mouse.
CACNA1C
To the best of our knowledge, to date human CACNA1C genetic studies in a drug‐dependent population have not been reported. However, as outlined above, several of the brain regions that comprise the brain's reward pathway have altered function in CACNA1C rs1006737 risk allele carriers as revealed by neuroimaging. Two studies have examined reward response in CACNA1C rs1006737 risk allele carriers. Lancaster et al. (2014) found that CACNA1C risk allele carriers had a blunted behavioural response in a reward‐based task compared to non‐carriers (Lancaster et al. 2014). Wessa et al. (2010) tested reward reversal learning and reported increased amygdala activity in CACNA1C rs1006737 risk allele carriers in response to delivery of reward (Wessa et al. 2010). Using conditional cacna1c knockout mice in the behavioural sensitization protocol, we have found that Cav1.2 channels in the NAc mediate the long‐term expression of the sensitized response (Schierberl et al. 2011 a). Using the CPP protocol we find that constitutive heterozygous cacna1c mice have a potentiated cocaine reward response (Schierberl et al. 2011 b). Ongoing studies are currently examining the neural circuitry and molecular mechanisms that underlie the enhanced reward response in constitutive heterozygous cacna1c mice.
CACNA1D
A new study from our group has identified CACNA1D SNPs in cocaine dependent individuals. Examination of 947 CACNA1D SNPs has identified three significant SNPs associated with cocaine dependence (Martinez‐Rivera et al. 2015). To date, this is the only human genetic study that has been reported in a cocaine‐dependent population.
Using cacna1d knockout mice and Cav1.2 DHP‐insensitive mutant mice (Sinnegger‐Brauns et al. 2004), we have demonstrated a role for Cav1.3 channels in the VTA in the development of behavioural sensitization (Schierberl et al. 2011 a), in mediating dopamine D2 receptor‐induced molecular changes in the striatum (Schierberl et al. 2012) and in the acquisition of cocaine CPP (Martinez‐Rivera et al. 2015). A new study supporting our data finds that the LTCC blocker isradipine, injected directly into the rat VTA, blocks acquisition of cocaine CPP (potentially via Cav1.3 channels as Cav1.3 is the predominant subunit expressed in VTA neurons; Rajadhyaksha et al. 2004). They further extend these findings to demonstrate that isradipine can enhance extinction of cocaine CPP and block cocaine‐induced reinstatement (Degoulet et al. 2016), a model of relapse to drug taking behaviour. Similarly, the LTCC blockers nifedipine and isradipine, infused into the VTA, attenuate cue‐induced cocaine seeking in the rat cocaine self‐administration model (Nunes et al. 2015). Based on our human genetic findings and a role for cacna1d in addictive behaviour in rodents, additional studies to explore CACNA1D in mood disorders is warranted.
Conclusion
Several recent human genetic studies have identified L‐type Ca2+ channel genes CACNA1C and CACNA1D as candidate risk genes in neuropsychiatric and neurodevelopmental disorders. Human neuroimaging studies, along with cellular and mouse studies, have established an important role for Cav1.2 and Cav1.3 channels, encoded by cacna1c and cacna1d, respectively, in intermediate phenotypes relevant to neuropsychiatric disorders. In this review we have integrated human behavioural and neuroimaging data with neuropsychiatric‐related behavioural domains and endophenotypes, to allow a platform to pose experimental questions towards gaining a better understanding of neural mechanisms that underlie neuropsychiatric disorders in which CACNA1C and CACNA1D have been implicated. Within this framework, the time is ideal to exploit the newly developed genetic, cellular and molecular tools for neuroscience research to move forward in our understanding of the impact of the newly discovered CACNA1C and CACNA1D human polymorphisms on the brain and behaviour.
Additional information
Competing interests
None declared.
Funding
The authors’ work is supported by the National Institute of Drug Abuse (NIDA)/National Institute of Health (NIH) 5R01DA029122(A.M.R.), NIDA/NIH F31 DA032169‐01A1 (A.S.L.), The Hartwell Foundation (A.M.R.) and the Weill Cornell Autism Research Program (A.M.R.).
Biographies
Zeeba Kabir is a postdoctoral fellow in the Rajadhyaksha Lab. Her interest is in the role of Cav1.2 and Cav1.3 L‐type Ca2+ channels in cognition and understanding the underlying anatomical and molecular processes.

Anni Lee recently completed her PhD in the Rajadhyaksha Lab. Her work has contributed to describing the role of the neuronal Cav1.2 L‐type Ca2+ channel and its downstream molecules in anxiety, depression and addictive behaviours.
Anjali Rajadhyaksha is a molecular neuroscientist with scientific interest in understanding the contribution of Cav1.2 and Cav1.3 L‐type Ca2+ channel mechanisms to behaviours related to neuropsychiatric disorders including addiction and mood disorders. The group's research methods include the use of genetically modified mouse models, stereotaxic techniques to generate site‐ and cell‐type‐specific knockouts coupled with biochemical studies and rodent behavioural tests that encompass a range of human neuropsychiatric‐related phenotypes to better understand the molecular mechanisms underlying brain disorders in the context of behavioural deficits.
This review was presented at the symposium “Voltage‐gated calcium channels ‐ from basic mechanisms to disease”, which took place at Physiology 2015 in Cardiff, UK, 6–8 July 2015.
References
- Ahs F, Kragel PA, Zielinski DJ, Brady R & LaBar KS (2015). Medial prefrontal pathways for the contextual regulation of extinguished fear in humans. Neuroimage 122, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ament SA, Szelinger S, Glusman G, Ashworth J, Hou L, Akula N, Shekhtman T, Badner JA, Brunkow ME, Mauldin DE, Stittrich AB, Rouleau K, Detera‐Wadleigh SD, Nurnberger JI Jr, Edenberg HJ, Gershon ES, Schork N, Price ND, Gelinas R, Hood L, Craig D, McMahon FJ, Kelsoe JR & Roach JC (2015). Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc Natl Acad Sci USA 112, 3576–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anokhin AP & Golosheykin S (2009). Startle modulation by affective faces. Biol Psychol 83, 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APA (2013). Diagnostic and Statistical Manual of Mental Disorders, 2nd edn. American Psychiatric Association, Washington, DC, USA. [Google Scholar]
- Arts B, Simons CJ & Os J (2013). Evidence for the impact of the CACNA1C risk allele rs1006737 on 2‐year cognitive functioning in bipolar disorder. Psychiatr Genet 23, 41–42. [DOI] [PubMed] [Google Scholar]
- Austin MP, Mitchell P & Goodwin GM (2001). Cognitive deficits in depression: possible implications for functional neuropathology. Br J Psychiatry 178, 200–206. [DOI] [PubMed] [Google Scholar]
- Backes H, Dietsche B, Nagels A, Konrad C, Witt SH, Rietschel M, Kircher T & Krug A (2014). Genetic variation in CACNA1C affects neural processing in major depression. J Psychiatr Res 53, 38–46.24612926 [Google Scholar]
- Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, Bett GC, Tsien RW, Rasmusson RL & Shamloo M (2011). Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci USA 108, 15432–15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot RC, Parise EM, Pena CJ, Zhang HX, Maze I, Chaudhury D, Persaud B, Cachope R, Bolanos‐Guzman CA, Cheer J, Deisseroth K, Han MH & Nestler EJ (2015). Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun 6, 7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey KR & Crawley JN (2009). Anxiety‐related behaviors in mice In Methods of Behavior Analysis in Neuroscience, 2nd edn, ed. Buccafusco JJ, chap. 5. CRC Press/Taylor & Francis, BocaRaton, FL, USA. [Google Scholar]
- Bardo MT & Bevins RA (2000). Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 153, 31–43. [DOI] [PubMed] [Google Scholar]
- Barrett CF & Tsien RW (2008). The Timothy syndrome mutation differentially affects voltage‐ and calcium‐dependent inactivation of CaV1.2 L‐type calcium channels. Proc Natl Acad Sci USA 105, 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE & LeDoux JE (2002). NMDA receptors and L‐type voltage‐gated calcium channels contribute to long‐term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22, 5239–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SM & Bartsch D (2014). The role of L‐type voltage‐gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 357, 463–476. [DOI] [PubMed] [Google Scholar]
- Bergner CL, Smolinsky AN, Hart PC, Dufour BD, Egan RJ, Laporte JL & Kalueff AV (2010). Mouse models for studying depression‐like states and antidepressant drugs. Methods Mol Biol 602, 267–282. [DOI] [PubMed] [Google Scholar]
- Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM & Gould TD (2012). CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol 99, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B, Hyde TM, Lipska BK, Kleinman JE & Weinberger DR (2010). Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry 67, 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady AM & Floresco SB (2015). Operant procedures for assessing behavioral flexibility in rats. J Vis Exp, e52387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busatto GF (2013). Structural and functional neuroimaging studies in major depressive disorder with psychotic features: a critical review. Schizophr Bull 39, 776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin‐Jageman I & Lee A (2008). Cav1 L‐type Ca2+ channel signaling complexes in neurons. J Neurochem 105, 573–583. [DOI] [PubMed] [Google Scholar]
- Can A, Dao DT, Arad M, Terrillion CE, Piantadosi SC & Gould TD (2012). The mouse forced swim test. J Vis Exp, e3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamassima F, Hay AC, Benedetti A, Lattanzi L, Cassano GB & Perlis RH (2010. a). L‐type calcium channels and psychiatric disorders: A brief review. Am J Med Genet B Neuropsychiatr Genet 153B, 1373–1390. [DOI] [PubMed] [Google Scholar]
- Casamassima F, Huang J, Fava M, Sachs GS, Smoller JW, Cassano GB, Lattanzi L, Fagerness J, Stange JP & Perlis RH (2010. b). Phenotypic effects of a bipolar liability gene among individuals with major depressive disorder. Am J Med Genet B Neuropsychiatr Genet 153B, 303–309. [DOI] [PubMed] [Google Scholar]
- Cirillo MA & Seidman LJ (2003). Verbal declarative memory dysfunction in schizophrenia: from clinical assessment to genetics and brain mechanisms. Neuropsychol Rev 13, 43–77. [DOI] [PubMed] [Google Scholar]
- Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, Neve RL, Deisseroth K & Nestler EJ (2010). Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci 30, 16082–16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics Consortium et al (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome‐wide analysis. Lancet 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao DT, Mahon PB, Cai X, Kovacsics CE, Blackwell RA, Arad M, Shi J, Zandi PP, O'Donnell P, Knowles JA, Weissman MM, Coryell W, Scheftner WA, Lawson WB, Levinson DF, Thompson SM, Potash JB & Gould TD (2010). Mood disorder susceptibility gene CACNA1C modifies mood‐related behaviors in mice and interacts with sex to influence behavior in mice and diagnosis in humans. Biol Psychiatry 68, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SE & Bauer EP (2012). L‐type voltage‐gated calcium channels in the basolateral amygdala are necessary for fear extinction. J Neurosci 32, 13582–13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW & Surmeier DJ (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 9, 251–259. [DOI] [PubMed] [Google Scholar]
- Degoulet M, Stelly CE, Ahn KC & Morikawa H (2016). L‐type Ca channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug‐associated contextual memory. Mol Psychiatry 21, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase V, Obermair GJ, Szabo Z, Altier C, Sanguesa J, Bourinet E & Flucher BE (2008). Stable membrane expression of postsynaptic CaV1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. J Neurosci 28, 13845–13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietsche B, Backes H, Laneri D, Weikert T, Witt SH, Rietschel M, Sommer J, Kircher T & Krug A (2014). The impact of a CACNA1C gene polymorphism on learning and hippocampal formation in healthy individuals: a diffusion tensor imaging study. Neuroimage 89, 256–261. [DOI] [PubMed] [Google Scholar]
- Dima D, Jogia J, Collier D, Vassos E, Burdick KE & Frangou S (2013). Independent modulation of engagement and connectivity of the facial network during affect processing by CACNA1C and ANK3 risk genes for bipolar disorder. JAMA Psychiatry 70, 1303–1311. [DOI] [PubMed] [Google Scholar]
- Dymond S, Dunsmoor J, Vervliet B, Roche B & Hermans D (2014). Fear generalization in humans: Systematic review and implications for anxiety disorder research. Behav Ther 46, 561–582. [DOI] [PubMed] [Google Scholar]
- Ekman P (1992). Facial expressions of emotion: an old controversy and new findings. Philos Trans R Soc Lond B Biol Sci 335, 63–69. [DOI] [PubMed] [Google Scholar]
- Erk S, Meyer‐Lindenberg A, Linden DE, Lancaster T, Mohnke S, Grimm O, Degenhardt F, Holmans P, Pocklington A, Schmierer P, Haddad L, Muhleisen TW, Mattheisen M, Witt SH, Romanczuk‐Seiferth N, Tost H, Schott BH, Cichon S, Nothen MM, Rietschel M, Heinz A & Walter H (2014. a). Replication of brain function effects of a genome‐wide supported psychiatric risk variant in the CACNA1C gene and new multi‐locus effects. Neuroimage 94, 147–154. [DOI] [PubMed] [Google Scholar]
- Erk S, Meyer‐Lindenberg A, Schmierer P, Mohnke S, Grimm O, Garbusow M, Haddad L, Poehland L, Muhleisen TW, Witt SH, Tost H, Kirsch P, Romanczuk‐Seiferth N, Schott BH, Cichon S, Nothen MM, Rietschel M, Heinz A & Walter H (2014. b). Hippocampal and frontolimbic function as intermediate phenotype for psychosis: evidence from healthy relatives and a common risk variant in CACNA1C . Biol Psychiatry 76, 466–475. [DOI] [PubMed] [Google Scholar]
- Erk S, Meyer‐Lindenberg A, Schnell K, Opitz von Boberfeld C, Esslinger C, Kirsch P, Grimm O, Arnold C, Haddad L, Witt SH, Cichon S, Nothen MM, Rietschel M & Walter H (2010). Brain function in carriers of a genome‐wide supported bipolar disorder variant. Arch Gen Psychiatry 67, 803–811. [DOI] [PubMed] [Google Scholar]
- Falck RS, Wang J, Siegal HA & Carlson RG (2004). The prevalence of psychiatric disorder among a community sample of crack cocaine users: an exploratory study with practical implications. J Nerv Ment Dis 192, 503–507. [DOI] [PubMed] [Google Scholar]
- Felix‐Ortiz AC, Burgos‐Robles A, Bhagat ND, Leppla CA & Tye KM (2015). Bidirectional modulation of anxiety‐related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience 321, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix‐Ortiz AC & Tye KM (2014). Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci 34, 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt M & Koch M (2013). Translational value of startle modulations. Cell Tissue Res 354, 287–295. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, O'Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, Smoller JW, Grozeva D, Stone J, Nikolov I, Chambert K, Hamshere ML, Nimgaonkar VL, Moskvina V, Thase ME, Caesar S, Sachs GS, Franklin J, Gordon‐Smith K, Ardlie KG, Gabriel SB, Fraser C, Blumenstiel B, Defelice M, Breen G, Gill M, Morris DW, Elkin A, Muir WJ, McGhee KA, Williamson R, MacIntyre DJ, MacLean AW, St Clair D, Robinson M, Van Beck M, Pereira AC, Kandaswamy R, McQuillin A, Collier DA, Bass NJ, Young AH, Lawrence J, Ferrier IN, Anjorin A, Farmer A, Curtis D, Scolnick EM, McGuffin P, Daly MJ, Corvin AP, Holmans PA, Blackwood DH, Wellcome Trust Case Control Consortium , Gurling HM, Owen MJ, Purcell SM, Sklar P & Craddock N (2008). Collaborative genome‐wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Braaksma DN & Phillips AG (1999). Thalamic‐cortical‐striatal circuitry subserves working memory during delayed responding on a radial arm maze. J Neurosci 19, 11061–11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon ES, Grennan K, Busnello J, Badner JA, Ovsiew F, Memon S, Alliey‐Rodriguez N, Cooper J, Romanos B & Liu C (2014). A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar‐associated common SNP of CACNA1C in brain. Mol Psychiatry 19, 890–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin MR, Miyawaki H, Helmstetter FJ & Diba K (2013). Prefrontal activity links nonoverlapping events in memory. J Neurosci 33, 10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt CE & Lee FS (2015). Common polymorphisms in the age of research domain criteria (RDoC): integration and translation. Biol Psychiatry 79, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL & Keightley ML (2002). Studies of altered social cognition in neuropsychiatric disorders using functional neuroimaging. Can J Psychiatry 47, 327–336. [DOI] [PubMed] [Google Scholar]
- Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S, Gordon‐Smith K, Fraser C, Forty L, Russell E, Hamshere ML, Moskvina V, Nikolov I, Farmer A, McGuffin P, Wellcome Trust Case Control Consortium , Holmans PA, Owen MJ, O'Donovan MC & Craddock N (2010). The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 15, 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, Tye KM, Anikeeva P, Malenka RC & Deisseroth K (2014). Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm AO & Weike AI (2005). The neuropsychology of fear learning and fear regulation. Int J Psychophysiol 57, 5–14. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP & Catterall WA (1993). Identification and differential subcellular localization of the neuronal class C and class D L‐type calcium channel α1 subunits. J Cell Biol 123, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero MJ, Domingo‐Salvany A, Torrens M & Brugal MT; ITINERE Investigators (2008). Psychiatric comorbidity in young cocaine users: induced versus independent disorders. Addiction 103, 284–293. [DOI] [PubMed] [Google Scholar]
- Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ & Dolphin AC (2015). Genetic disruption of voltage‐gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol 134, 36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijzen TH, Houtzager SW, Joordens RJ, Olivier B & Slangen JL (1995). Predictive validity of the potentiated startle response as a behavioral model for anxiolytic drugs. Psychopharmacology (Berl) 118, 150–154. [DOI] [PubMed] [Google Scholar]
- Hill SK, Bishop JR, Palumbo D & Sweeney JA (2010). Effect of second‐generation antipsychotics on cognition: current issues and future challenges. Expert Rev Neurother 10, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti FL & Siegelbaum SA (2014). The hippocampal CA2 region is essential for social memory. Nature 508, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H, Yamamoto N, Fujii T, Teraishi T, Sasayama D, Matsuo J, Kawamoto Y, Kinoshita Y, Ota M, Hattori K, Tatsumi M, Arima K & Kunugi H (2012). Effects of the CACNA1C risk allele on neurocognition in patients with schizophrenia and healthy individuals. Sci Rep 2, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR (2014). The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry 171, 395–397. [DOI] [PubMed] [Google Scholar]
- Janak PH & Tye KM (2015). From circuits to behaviour in the amygdala. Nature 517, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ & Lee A (2010). Ca2+‐dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin‐dependent protein kinase II. J Neurosci 30, 5125–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AR & Rohwer WD Jr (1966). The Stroop color‐word test: a review. Acta Psychol (Amst) 25, 36–93. [DOI] [PubMed] [Google Scholar]
- Jeon D, Kim S, Chetana M, Jo D, Ruley HE, Lin SY, Rabah D, Kinet JP & Shin HS (2010). Observational fear learning involves affective pain system and Cav1.2 Ca2+ channels in ACC. Nat Neurosci 13, 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodo E, Katayama T, Okamoto M, Suzuki Y, Hoshino K & Kayama Y (2010). Differences in responsiveness of mediodorsal thalamic and medial prefrontal cortical neurons to social interaction and systemically administered phencyclidine in rats. Neuroscience 170, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Jogia J, Ruberto G, Lelli‐Chiesa G, Vassos E, Maieru M, Tatarelli R, Girardi P, Collier D & Frangou S (2011). The impact of the CACNA1C gene polymorphism on frontolimbic function in bipolar disorder. Mol Psychiatry 16, 1070–1071. [DOI] [PubMed] [Google Scholar]
- Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B & Dolan RJ (2006). Context‐dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci 26, 9503–9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW & Volkow ND (2005). The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 162, 1403–1413. [DOI] [PubMed] [Google Scholar]
- Katayama T, Jodo E, Suzuki Y, Hoshino KY, Takeuchi S & Kayama Y (2009). Phencyclidine affects firing activity of basolateral amygdala neurons related to social behavior in rats. Neuroscience 159, 335–343. [DOI] [PubMed] [Google Scholar]
- Katz RJ (1982). Animal model of depression: pharmacological sensitivity of a hedonic deficit. Pharmacol Biochem Behav 16, 965–968. [DOI] [PubMed] [Google Scholar]
- Keifer OP Jr, Hurt RC, Ressler KJ & Marvar PJ (2015). The physiology of fear: reconceptualizing the role of the central amygdala in fear learning. Physiology (Bethesda) 30, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AE (2004). Ventral striatal control of appetitive motivation: role in ingestive behavior and reward‐related learning. Neurosci Biobehav Rev 27, 765–776. [DOI] [PubMed] [Google Scholar]
- Kennedy DP & Adolphs R (2012). The social brain in psychiatric and neurological disorders. Trends Cogn Sci 16, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR & Walters EE (2005). Prevalence, severity, and comorbidity of 12‐month DSM‐IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DC, Smith CN, Cheng DT, Stein EA & Helmstetter FJ (2004). Amygdala and hippocampal activity during acquisition and extinction of human fear conditioning. Cogn Affect Behav Neurosci 4, 317–325. [DOI] [PubMed] [Google Scholar]
- Koob GF & Volkow ND (2010). Neurocircuitry of addiction. Neuropsychopharmacology 35, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, Nieratschker V, Markov V, Krach S, Jansen A, Zerres K, Eggermann T, Stocker T, Shah NJ, Treutlein J, Muhleisen TW & Kircher T (2010). Effect of CACNA1C rs1006737 on neural correlates of verbal fluency in healthy individuals. Neuroimage 49, 1831–1836. [DOI] [PubMed] [Google Scholar]
- Krug A, Witt SH, Backes H, Dietsche B, Nieratschker V, Shah NJ, Nothen MM, Rietschel M & Kircher T (2014). A genome‐wide supported variant in CACNA1C influences hippocampal activation during episodic memory encoding and retrieval. Eur Arch Psychiatry Clin Neurosci 264, 103–110. [DOI] [PubMed] [Google Scholar]
- LaBar KS, LeDoux JE, Spencer DD & Phelps EA (1995). Impaired fear conditioning following unilateral temporal lobectomy in humans. J Neurosci 15, 6846–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HM, Cleary M, Sitharthan T & Hunt GE (2015). Prevalence of comorbid substance use, anxiety and mood disorders in epidemiological surveys, 1990–2014: A systematic review and meta‐analysis. Drug Alcohol Depend 154, 1–13. [DOI] [PubMed] [Google Scholar]
- Lancaster TM, Heerey EA, Mantripragada K & Linden DE (2014). CACNA1C risk variant affects reward responsiveness in healthy individuals. Transl Psychiatry 4, e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langwieser N, Christel CJ, Kleppisch T, Hofmann F, Wotjak CT & Moosmang S (2010). Homeostatic switch in Hebbian plasticity and fear learning after sustained loss of Cav1.2 calcium channels. J Neurosci 30, 8367–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux J (2003). The emotional brain, fear, and the amygdala. Cell Mol Neurobiol 23, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Ra S, Rajadhyaksha AM, Britt JK, De Jesus‐Cortes H, Gonzales KL, Lee A, Moosmang S, Hofmann F & Pieper AA (2012). Forebrain elimination of cacna1c mediates anxiety‐like behavior in mice. Mol Psychiatry 17, 1054–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao L, You Y, Lu T, Jia M, Yu H, Ruan Y, Yue W, Liu J, Lu L, Zhang D & Wang L (2015). Schizophrenia related variants in CACNA1C also confer risk of autism. PLoS One 10, e0133247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissek S, Orme K, McDowell DJ, Johnson LL, Luckenbaugh DA, Baas JM, Cornwell BR & Grillon C (2007). Emotion regulation and potentiated startle across affective picture and threat‐of‐shock paradigms. Biol Psychol 76, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi A & Luscher C (2014). Pathological circuit function underlying addiction and anxiety disorders. Nat Neurosci 17, 1635–1643. [DOI] [PubMed] [Google Scholar]
- Ma H, Cohen S, Li B & Tsien RW (2012). Exploring the dominant role of CaV1 channels in signalling to the nucleus. Biosci Rep 33, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney BC & Murphy GG (2006). The L‐type voltage‐gated calcium channel Cav1.3 mediates consolidation, but not extinction, of contextually conditioned fear in mice. Learn Mem 13, 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney BC, Sze W, White JA & Murphy GG (2008). L‐type voltage‐gated calcium channels in conditioned fear: a genetic and pharmacological analysis. Learn Mem 15, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Phan KL & Liberzon I (2013). The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci 14, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel G, Nishi A & Shumyatsky GP (2008). Stathmin reveals dissociable roles of the basolateral amygdala in parental and social behaviors. Proc Natl Acad Sci USA 105, 14620–14625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Hao J, Rice R, Striessnig J, Hans S & Rajadhyaksha AM (2015). Cav1.3 L‐type Ca2+ channels: Role in VTA dopamine neurons on cocaine's behavioral effects and genetic variants in cocaine dependent humans. Program No. 51.18/G10. Neuroscience 2015 Meeting Planner. Society for Neuroscience, Chicago, IL, USA. Online. [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ & Rauch SL (2007). Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry 62, 446–454. [DOI] [PubMed] [Google Scholar]
- Millan MJ (2006). Multi‐target strategies for the improved treatment of depressive states: Conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther 110, 135–370. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Agid Y, Brune M, Bullmore ET, Carter CS, Clayton NS, Connor R, Davis S, Deakin B, DeRubeis RJ, Dubois B, Geyer MA, Goodwin GM, Gorwood P, Jay TM, Joels M, Mansuy IM, Meyer‐Lindenberg A, Murphy D, Rolls E, Saletu B, Spedding M, Sweeney J, Whittington M & Young LJ (2012). Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 11, 141–168. [DOI] [PubMed] [Google Scholar]
- Miller EK (2000). The prefrontal cortex and cognitive control. Nat Rev Neurosci 1, 59–65. [DOI] [PubMed] [Google Scholar]
- Miyake A, Friedman NP, Emerson MJ, Witzki AH, Howerter A & Wager TD (2000). The unity and diversity of executive functions and their contributions to complex ‘Frontal Lobe’ tasks: a latent variable analysis. Cogn Psychol 41, 49–100. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F & Kleppisch T (2005). Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor‐independent synaptic plasticity and spatial memory. J Neurosci 25, 9883–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morellini F (2013). Spatial memory tasks in rodents: what do they model? Cell Tissue Res 354, 273–286. [DOI] [PubMed] [Google Scholar]
- Nestler EJ & Carlezon WA Jr (2006). The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium et al (2015). Psychiatric genome‐wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 18, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes EJ, Hughley SM, Solecki W, Rajadhyaksha AM & Addy NA (2015). L‐type calcium channels in the ventral tegmental area mediate cue‐induced cocaine seeking. Program No. 51.19/G11. Neuroscience 2015 Meeting Planner. Society for Neuroscience, Chicago, IL, USA. Online. [Google Scholar]
- Nyegaard M, Demontis D, Foldager L, Hedemand A, Flint TJ, Sorensen KM, Andersen PS, Nordentoft M, Werge T, Pedersen CB, Hougaard DM, Mortensen PB, Mors O & Borglum AD (2010). CACNA1C (rs1006737) is associated with schizophrenia. Mol Psychiatry 15, 119–121. [DOI] [PubMed] [Google Scholar]
- Ohl F, Roedel A, Binder E & Holsboer F (2003). Impact of high and low anxiety on cognitive performance in a modified hole board test in C57BL/6 and DBA/2 mice. Eur J Neurosci 17, 128–136. [DOI] [PubMed] [Google Scholar]
- Olsson A, Nearing KI & Phelps EA (2007). Learning fears by observing others: the neural systems of social fear transmission. Soc Cogn Affect Neurosci 2, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J & Eichler EE (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KT, Liu RJ, Voleti B, Maldonado‐Aviles JG, Duric V, Iwata M, Dutheil S, Duman C, Boikess S, Lewis DA, Stockmeier CA, DiLeone RJ, Rex C, Aghajanian GK & Duman RS (2014). REDD1 is essential for stress‐induced synaptic loss and depressive behavior. Nat Med 20, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panlilio LV & Goldberg SR (2007). Self‐administration of drugs in animals and humans as a model and an investigative tool. Addiction 102, 1863–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis E, Koiliari E, Zouraraki C, Tsapakis EM, Roussos P, Giakoumaki SG & Bitsios P (2015). The effects of the CACNA1C rs1006737 A/G on affective startle modulation in healthy males. Eur Psychiatry 30, 492–498. [DOI] [PubMed] [Google Scholar]
- Paulus FM, Bedenbender J, Krach S, Pyka M, Krug A, Sommer J, Mette M, Nothen MM, Witt SH, Rietschel M, Kircher T & Jansen A (2014). Association of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto‐hippocampal connectivity. Hum Brain Mapp 35, 1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechtel P, Dutra SJ, Goetz EL & Pizzagalli DA (2013). Blunted reward responsiveness in remitted depression. J Psychiatr Res 47, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier E, Pompei F, Ruberto G, Vassos E, Collier D & Frangou S (2011). Initial evidence for the role of CACNA1C on subcortical brain morphology in patients with bipolar disorder. Eur Psychiatry 26, 135–137. [DOI] [PubMed] [Google Scholar]
- Peters J & Buchel C (2009). Overlapping and distinct neural systems code for subjective value during intertemporal and risky decision making. J Neurosci 29, 15727–15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA & LeDoux JE (2005). Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron 48, 175–187. [DOI] [PubMed] [Google Scholar]
- Phillips RG & LeDoux JE (1992). Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106, 274–285. [DOI] [PubMed] [Google Scholar]
- Pierce RC & Kalivas PW (1997). A circuitry model of the expression of behavioral sensitization to amphetamine‐like psychostimulants. Brain Res Brain Res Rev 25, 192–216. [DOI] [PubMed] [Google Scholar]
- Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, Tuluc P & Striessnig J (2015). CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L‐type calcium channels. Biol Psychiatry 77, 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetschke C, Dragicevic E, Duda J, Benkert J, Dougalis A, DeZio R, Snutch TP, Striessnig J & Liss B (2015). Compensatory T‐type Ca2+ channel activity alters D2‐autoreceptor responses of substantia nigra dopamine neurons from Cav1.3 L‐type Ca2+ channel KO mice. Sci Rep 5, 13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooters T, Van der Jeugd A, Callaerts‐Vegh Z & D'Hooge R (2015). Telencephalic neurocircuitry and synaptic plasticity in rodent spatial learning and memory. Brain Res 1621, 294–308. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M & Jalfre M (1977). Depression: a new animal model sensitive to antidepressant treatments. Nature 266, 730–732. [DOI] [PubMed] [Google Scholar]
- Post RM & Kalivas P (2013). Bipolar disorder and substance misuse: pathological and therapeutic implications of their comorbidity and cross‐sensitisation. Br J Psychiatry 202, 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric GWAS Consortium Bipolar Disorder Working Group et al (2011). Large‐scale genome‐wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radua J, Surguladze SA, Marshall N, Walshe M, Bramon E, Collier DA, Prata DP, Murray RM & McDonald C (2012). The impact of CACNA1C allelic variation on effective connectivity during emotional processing in bipolar disorder. Mol Psychiatry 18, 526–527. [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha A, Husson I, Satpute SS, Kuppenbender KD, Ren JQ, Guerriero RM, Standaert DG & Kosofsky BE (2004). L‐type Ca2+ channels mediate adaptation of extracellular signal‐regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. J Neurosci 24, 7464–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajadhyaksha AM & Kosofsky BE (2005). Psychostimulants, L‐type calcium channels, kinases, and phosphatases. Neuroscientist 11, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riga D, Matos MR, Glas A, Smit AB, Spijker S & Van den Oever MC (2014). Optogenetic dissection of medial prefrontal cortex circuitry. Front Syst Neurosci 8, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE & Berridge KC (1993). The neural basis of drug craving: an incentive‐sensitization theory of addiction. Brain Res Brain Res Rev 18, 247–291. [DOI] [PubMed] [Google Scholar]
- Rosas‐Vidal LE, Do‐Monte FH, Sotres‐Bayon F & Quirk GJ (2014). Hippocampal–prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology 39, 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Giakoumaki SG, Georgakopoulos A, Robakis NK & Bitsios P (2011). The CACNA1C and ANK3 risk alleles impact on affective personality traits and startle reactivity but not on cognition or gating in healthy males. Bipolar Disord 13, 250–259. [DOI] [PubMed] [Google Scholar]
- Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J, Stahl EA, Georgakopoulos A, Ruderfer DM, Charney A, Okada Y, Siminovitch KA, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Plenge RM, Raychaudhuri S, Fromer M, Purcell SM, Brennand KJ, Robakis NK, Schadt EE, Akbarian S & Sklar P (2014). A role for noncoding variation in schizophrenia. Cell Rep 9, 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ & Nestler EJ (2013). The brain reward circuitry in mood disorders. Nat Rev Neurosci 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierberl K, Giordano T, Satpute S, Hao J, Kaur G, Hofmann F, Moosmang S, Striessnig J & Rajadhyaksha A (2012). Cav1.3 L‐type Ca2+ channels mediate long‐term adaptation in dopamine D2L‐mediated GluA1 trafficking in the dorsal striatum following cocaine exposure. Channels (Austin) 6, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q, Garraway SM, Hofmann F, Moosmang S, Striessnig J, Inturrisi CE & Rajadhyaksha AM (2011. a). Cav1.2 L‐type Ca2+ channels mediate cocaine‐induced GluA1 trafficking in the nucleus accumbens, a long‐term adaptation dependent on ventral tegmental area Cav1.3 channels. J Neurosci 31, 13562–13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierberl KC, Lee AS, Moosmang S, Hofmann F, Striessnig J & Rajadhyaksha AM (2011. b). Cav1.2 L‐type Ca2+ channels regulate cocaine reward via epigenetic regulation of BDNF. Program No. 909.09. Neuroscience 2011 Meeting Planner. Society for Neuroscience, Washington, DC, USA. Online. [Google Scholar]
- Sehlmeyer C, Schoning S, Zwitserlood P, Pfleiderer B, Kircher T, Arolt V & Konrad C (2009). Human fear conditioning and extinction in neuroimaging: a systematic review. PLoS One 4, e5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Janse E, Visser K & Meyer AS (2014). What do verbal fluency tasks measure? Predictors of verbal fluency performance in older adults. Front Psychol 5, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra‐Mercado D, Padilla‐Coreano N & Quirk GJ (2011). Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C & Crawley JN (2010). Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms BA & Zamponi GW (2014). Neuronal voltage‐gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45. [DOI] [PubMed] [Google Scholar]
- Singewald N, Schmuckermair C, Whittle N, Holmes A & Ressler KJ (2015). Pharmacology of cognitive enhancers for exposure‐based therapy of fear, anxiety and trauma‐related disorders. Pharmacol Ther 149, 150–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger‐Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N & Striessnig JJ (2004). Isoform‐specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L‐type Ca2+ channels. J Clin Invest 113, 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro‐de‐Souza MG, Bio DS, Dias VV, Vieta E, Machado‐Vieira R & Moreno RA (2013). The CACNA1C risk allele selectively impacts on executive function in bipolar type I disorder. Acta Psychiatr Scand 128, 362–369. [DOI] [PubMed] [Google Scholar]
- Sotres‐Bayon F, Sierra‐Mercado D, Pardilla‐Delgado E & Quirk GJ (2012). Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron 76, 804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager‐Flusberg H, Priori SG, Sanguinetti MC & Keating MT (2004). CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Stevenson EL & Caldwell HK (2014). Lesions to the CA2 region of the hippocampus impair social memory in mice. Eur J Neurosci 40, 3294–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J (2008). Review. Psychological and neural mechanisms of relapse. Philos Trans R Soc Lond B Biol Sci 363, 3147–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strekalova T, Spanagel R, Bartsch D, Henn FA & Gass P (2004). Stress‐induced anhedonia in mice is associated with deficits in forced swimming and exploration. Neuropsychopharmacology 29, 2007–2017. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Pinggera A, Kaur G, Bock G & Tuluc P (2014). L‐type Ca channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3, 15–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi A, Nakaoka R, Amano K, Yukimine M, Andoh T, Kuraishi Y & Tsuda M (2000). Differential activation of brain‐derived neurotrophic factor gene promoters I and III by Ca2+ signals evoked via L‐type voltage‐dependent and N‐methyl‐d‐aspartate receptor Ca2+ channels. J Biol Chem 275, 17269–17275. [DOI] [PubMed] [Google Scholar]
- Tang Y, Martin NL & Cotes RO (2014). Cocaine‐induced psychotic disorders: presentation, mechanism, and management. J Dual Diagn 10, 98–105. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ & Greenberg ME (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor‐dependent mechanism. Neuron 20, 709–726. [DOI] [PubMed] [Google Scholar]
- Tesli M, Skatun KC, Ousdal OT, Brown AA, Thoresen C, Agartz I, Melle I, Djurovic S, Jensen J & Andreassen OA (2013). CACNA1C risk variant and amygdala activity in bipolar disorder, schizophrenia and healthy controls. PLoS One 8, e56970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm M, Kircher T, Kellermann T, Markov V, Krach S, Jansen A, Zerres K, Eggermann T, Stocker T, Shah NJ, Nothen MM, Rietschel M, Witt SH, Mathiak K & Krug A (2011). Effects of a CACNA1C genotype on attention networks in healthy individuals. Psychol Med 41, 1551–1561. [DOI] [PubMed] [Google Scholar]
- Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR & Klann E (2012). Brain‐specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep 1, 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C & Deisseroth K (2011). Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ & Pierce RC (2010). Sensitization processes in drug addiction. Curr Top Behav Neurosci 3, 179–195. [DOI] [PubMed] [Google Scholar]
- VanElzakker MB, Dahlgren MK, Davis FC, Dubois S & Shin LM (2014). From Pavlov to PTSD: the extinction of conditioned fear in rodents, humans, and anxiety disorders. Neurobiol Learn Mem 113, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gorp WG, Altshuler L, Theberge DC & Mintz J (1999). Declarative and procedural memory in bipolar disorder. Biol Psychiatry 46, 525–531. [DOI] [PubMed] [Google Scholar]
- Volkow ND (2009). Substance use disorders in schizophrenia – clinical implications of comorbidity. Schizophr Bull 35, 469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, McIntosh AM, He Y, Gelernter J & Blumberg HP (2011). The association of genetic variation in CACNA1C with structure and function of a frontotemporal system. Bipolar Disord 13, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, Adhikari A, Tye KM, Frank LM & Deisseroth K (2012). A prefrontal cortex‐brainstem neuronal projection that controls response to behavioural challenge. Nature 492, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessa M, Linke J, Witt SH, Nieratschker V, Esslinger C, Kirsch P, Grimm O, Hennerici MG, Gass A, King AV & Rietschel M (2010). The CACNA1C risk variant for bipolar disorder influences limbic activity. Mol Psychiatry 15, 1126–1127. [DOI] [PubMed] [Google Scholar]
- White JA, McKinney BC, John MC, Powers PA, Kamp TJ & Murphy GG (2008). Conditional forebrain deletion of the L‐type calcium channel CaV1.2 disrupts remote spatial memories in mice. Learn Mem 15, 1–5. [DOI] [PubMed] [Google Scholar]
- Whitton AE, Treadway MT & Pizzagalli DA (2015). Reward processing dysfunction in major depression, bipolar disorder and schizophrenia. Curr Opin Psychiatry 28, 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P & Deisseroth K (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, Ongur D, McPhie D, Cohen B, Perlis R & Tsai LH (2015). Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry 20, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shen Q, Xu Z, Chen M, Cheng L, Zhai J, Gu H, Bao X, Chen X, Wang K, Deng X, Ji F, Liu C, Li J, Dong Q & Chen C (2012). The effects of CACNA1C gene polymorphism on spatial working memory in both healthy controls and patients with schizophrenia or bipolar disorder. Neuropsychopharmacology 37, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]