

Abstract
Zona glomerulosa cells (ZG) of the adrenal gland constantly integrate fluctuating ionic, hormonal and paracrine signals to control the synthesis and secretion of aldosterone. These signals modulate Ca2+ levels, which provide the critical second messenger to drive steroid hormone production. Angiotensin II is a hormone known to modulate the activity of voltage‐dependent L‐ and T‐type Ca2+ channels that are expressed on the plasma membrane of ZG cells in many species. Because the ZG cell maintains a resting membrane voltage of approximately −85 mV and has been considered electrically silent, low voltage‐activated T‐type Ca2+ channels are assumed to provide the primary Ca2+ signal that drives aldosterone production. However, this view has recently been challenged by human genetic studies identifying somatic gain‐of‐function mutations in L‐type CaV1.3 channels in aldosterone‐producing adenomas of patients with primary hyperaldosteronism. We provide a review of these assumptions and challenges, and update our understanding of the state of the ZG cell in a layer in which native cellular associations are preserved. This updated view of Ca2+ signalling in ZG cells provides a unifying mechanism that explains how transiently activating CaV3.2 channels can generate a significant and recurring Ca2+ signal, and how CaV1.3 channels may contribute to the Ca2+ signal that drives aldosterone production.
Abbreviations
- APA
aldosterone producing adenoma
- CDI
Ca2+‐dependent inactivation
- IHA
idiopathic bilateral zona glomerulosa hyperplasia
- PA
primary hyperaldosteronism
- RAS
renin–angiotensin system
- ZG
zona glomerulosa
Primary hyperaldosteronism
Circulating aldosterone is synthesized and secreted by zona glomerulosa (ZG) cells of the adrenal cortex. When under the control of the renin–angiotensin system (RAS), aldosterone production is commensurate with the level of circulating renin. By promoting Na+ retention at multiple sites along the nephron, aldosterone contributes to the physiological regulation of electrolytes and water balance. Excessive aldosterone production independent of the RAS can occur, as in primary hyperaldosteronism (PA), and is characterized by a large increase in the aldosterone/renin ratio (Montori & Young, 2002). Because aldosterone overproduction in PA has additional adverse effects on the heart (Catena et al. 2008) and kidneys (Rossi et al. 2006) that are independent of blood pressure elevation, this most common type of endocrine hypertension is associated with increased cardiovascular risk (Stowasser, 2001; Savard et al. 2013). The most common forms of PA result from two major pathologies: unilateral aldosterone producing adenoma (APA) and idiopathic bilateral zona glomerulosa hyperplasia (IHA). With a prevalence of only 4% in the general population, the incidence of PA rises significantly among hypertensive patients (> 10%) (Plouin et al. 2004; Rossi et al. 2006) and markedly among those with resistant hypertension (20%) (Calhoun et al. 2002 a,b; Stowasser, 2014).
Pathogenic mechanisms: insights from human genetic analysis of channels and pumps
Since 2011, great progress has been made in our understanding of the genetic bases for APA. Choi et al. (2011) were the first group to identify recurrent somatic mutations in the coding region of the KCNJ5 G‐protein coupled, inwardly rectifying K+ channel in DNA extracted from aldosterone producing adenomas. These mutations occur in or near the selectivity filter of the channel rendering it permeable to Na+. Originally identified in a small cohort of 22 severely hypertensive patients, these or functionally equivalent mutations in exon 2 of the KCNJ5 gene are now the most frequently identified genetic variants (∼38%) in aldosterone‐producing adenomas (Azizan et al. 2012; Boulkroun et al. 2012; Mulatero et al. 2012; Dekkers et al. 2014; Fernandes‐Rosa et al. 2014; Kuppusamy et al. 2014). Less frequent variants have been identified in the P‐type ATPase gene family, ATP1A1 and ATP2B3, encoding Na+/K+‐ATPase and Ca2+‐ATPase3, respectively (∼7%) (Azizan et al. 2013; Beuschlein et al. 2013; Fernandes‐Rosa et al. 2014). Mutations residing in transmembrane helices M1 and M4 of the Na+/K+‐ATPase cause loss of functional pump activity, the appearance of new ouabain‐insensitive Na+ or H+ inward leak currents and cell depolarization (Azizan et al. 2013; Beuschlein et al. 2013). Cell depolarization is also induced by mutations in the Ca2+‐ATPase3 found in the M4 transmembrane helix that forms the Ca2+ binding pocket of the Ca2+ pump (Beuschlein et al. 2013). By contrast, multiple and more frequent (5–14%) somatic mutations have been identified in the CACNA1D gene encoding the CaV1.3 voltage‐dependent Ca2+ channel (Azizan et al. 2013; Scholl et al. 2013; Fernandes‐Rosa et al. 2014). These variants are scattered broadly throughout the large α1 subunit of the channel protein, and are found in the activation gate (S6 helices), the voltage sensor (S4 helices) and in the S4‐S5 cytoplasmic linkers connecting the voltage sensor to the channel pore. In general, these mutations result in a gain of Ca2+ channel function, increasing the probability for channel opening at hyperpolarized potentials, either alone or in combination with a shift in the voltage dependence of channel inactivation. Some mutations also dramatically slow the time dependence of inactivation. In addition, several germline mutations in the KCNJ5 gene outside the selectivity filter were recently described in patients with apparent sporadic PA (Murthy et al. 2014), and a rare recurrent mutation in the CACNA1H gene encoding the CaV3.2 voltage‐dependent Ca2+ channel was identified in children presenting with severe hyperaldosteronism and hypertension (Scholl et al. 2015). The latter mutation found in the IIIS6 domain markedly slows the time dependence of inactivation of this rapidly inactivating channel without altering the voltage dependencies of channel gating.
Interestingly, the mutational status of tumours among different patient cohorts does not support the hypothesis that APA is associated with a consistent histological phenotype; mutations do not segregate according to cell type (zona glomerulosa, zona fasciculata or mixed). Neither was there a consistent molecular signature (e.g. the overexpression of CYP11B2, KCNK5, CACNA1D, ATP1A1 genes) (Enberg et al. 2004; Lenzini et al. 2007; Fernandes‐Rosa et al. 2014; Boulkroun et al. 2015). One potential exception may include the CACNA1H gene whose level of mRNA expression significantly associates with peripheral blood aldosterone levels, CYP11B2 gene expression levels and KCNJ5 mutational status in a cohort of 74 Japanese APA patients (Felizola et al. 2014). A second exception may involve the KCNK5 gene whose mRNA expression level inversely correlates with aldosterone synthesis and microRNA expression in a subcohort of 32 confirmed APA patients of Italian origin (Rossi et al. 2006 a; Lenzini et al. 2014). While the universality of these findings awaits their replication in multiethnic cohorts, the data in aggregate indicate that APA is a disorder with highly variable cellular and molecular phenotypes with diverse genetic underpinnings.
Despite the lack of consistent genotype–phenotype correlations (Boulkroun et al. 2015; Zennaro et al. 2015), the bulk of evidence supports the hypothesis that independent, mutational‐based mechanisms can support a persistent elevation in Ca2+ that drives the overproduction of aldosterone. Specifically, all of these mutations – directly or indirectly – are expected to raise intracellular Ca2+ in ZG cells, either by an increase in Ca2+ channel open probability (CaV1.3, CaV3.2), by a loss of PMCA3 Ca2+ pump activity, or by a depolarization in cell membrane potential, an action predicted to reduce Na+–Ca2+ exchange activity and/or activate voltage‐dependent Ca2+ channels.
Calcium sites of action and regulation of the Ca2+ signal
In the ZG cell, Ca2+ is the critical second messenger that regulates the production of aldosterone. The steroidogenic, intracellular Ca2+ signal is generated by the opening of Ca2+‐selective, voltage‐dependent channels found at the plasma membrane, or by the release of Ca2+ from intracellular stores (Fakunding & Catt, 1980; Kojima et al. 1984; Barrett et al. 1989; Rasmussen et al. 1989). The subsequent increase in cytosolic Ca2+ facilitates the delivery of cholesterol to the mitochondria (Cherradi et al. 1996) for its conversion to pregnenolone, a precursor in the aldosterone biosynthetic pathway. To persistently increase steroidogenesis, the Ca2+ signal must also be transferred to the mitochondrial matrix (Lalevee et al. 2003) where it stimulates matrix dehydrogenases to generate NADH. NADH, in turn, is converted to NADPH, a cofactor that is required for two critical regulatory steps in the biosynthetic pathway: (1) the conversion of cholesterol to pregnenolone catalysed by CYP11A1, and (2) the conversion of deoxycorticosterone to aldosterone catalysed by CYP11B2 (Rossier et al. 1996; Wiederkehr et al. 2011). Thus, a Ca2+ increase in both the cytosolic and the mitochondrial compartments of the ZG cell is required to increase the production of aldosterone that persists for minutes to hours (Spat & Hunyady, 2004).
Voltage‐gated calcium currents
Early electrophysiological recordings in rat, bovine and human ZG cells consistently identified two components of the Ca2+ current that could be attributed to distinct Ca2+ channel classes based on voltage‐dependent gating properties, kinetics and pharmacology (Matsunaga et al. 1987; Cohen et al. 1988; Durroux et al. 1988; Payet et al. 1994). Typically, the more persistent current that was elicited by strong depolarization (from resting V m of −85 mV to > −50 mV (CaV1.3), or > −30 mV (CaV1.2)) (Xu & Lipscombe, 2001) and inhibited by dihydropyridines characterized the high voltage‐activated component (HVA). By contrast, the rapid, transient current that was evoked by weak depolarization (from resting V m to > −65 mV) and inhibited by Ni2+ characterized the low voltage‐activated component (LVA) (Tsien et al. 1988). Later, the molecular correlates of these currents were identified as HVA, L‐type (i.e. CaV1.3 and CaV1.2) and LVA, T‐type (i.e. CaV3.2 and CaV3.1) channels. The relative mRNA abundance of L‐ and T‐type channel isoforms in adrenal ZG varies among studies and species (Lesouhaitier et al. 2001; Schrier et al. 2001; Rossier et al. 2003) although their biophysical properties remain similar. Additionally, functional N‐type channels (CaV2.0) have been recorded in isolated rat ZG cells (Durroux et al. 1988). Thus, the repertoire of Ca2+ channels found in ZG cells is rich.
More recently, the expression profiles for voltage‐gated Ca2+ channels in normal and pathological human adrenocortical tissue were compared (Scholl et al. 2013; Felizola et al. 2014). mRNA for all three Ca2+ channel classes was detected in human aldosterone‐producing adenomas, with greater relative abundance of CaV1.3 and CaV3.2 mRNA than CaV1.2 and CaV2.2 mRNA (Felizola et al. 2014). Surprisingly, the protein expression of CaV1.3 and CaV3.2 channels is consistent regardless of tissue health, as anti‐CaV1.3 or anti‐CaV3.2 immunoreactivity is equivalent in APA, IHA and normal adrenocortical tissue samples. However, whether all detected protein is functional remains a critical unanswered question.
Calcium homeostasis: lessons learned from isolated cells
From the late 1970s to early 1990s, methods to dissociate and isolate ZG cells from bovine and rat adrenal tissues led to tractable preparations suitable for measuring ZG cell Ca2+/K+ currents, intracellular Ca2+ signals and aldosterone production. Numerous studies document that the major regulators of aldosterone production, angiotensin II and extracellular K+, raise intracellular Ca2+ in ZG cells (Capponi et al. 1984, 1987; Connor et al. 1987; Kramer, 1988; Johnson et al. 1989; Pratt et al. 1989; Rossig et al. 1996) and that changes in cell Ca2+ are commensurate with a striking sustained depolarization of the ZG cell. Angiotensin II elicits ZG cell depolarization by inhibiting hyperpolarizing conductances, predominantly those mediated by leak and voltage‐gated K+ channels (Quinn et al. 1987 b; Brauneis et al. 1991; Lotshaw, 1997 a; Guagliardo et al. 2012), whereas elevated extracellular K+ induces a shift in the K+ equilibrium potential across the highly K+‐conductive ZG plasma membrane (Quinn et al. 1987 a,b; Lotshaw, 1997 b). As depolarization recruits the activation of voltage‐gated Ca2+ channels, these early findings suggested the possibility that voltage‐gated Ca2+ entry is an important contributor to ZG cell activation and aldosterone production.
With the subsequent development of pharmacological agents to reduce the open‐state probability of selective Ca2+ channel classes, as well as hormonal and molecular approaches to alter Ca2+ channel gating mechanisms, the relative contribution of T‐type and L‐type Ca2+ currents to the regulation of aldosterone production was confirmed. Given the extremely hyperpolarized resting membrane potential of isolated ZG cells (∼−85 mV), the relatively modest ∼5–20 mV depolarizing shifts in voltage evoked by physiological concentrations of angiotensin ll or K+, coupled with robust T‐type channel currents recorded in all preparations, it is not surprising that T‐type and not L‐type Ca2+ current amplitude (Barrett et al. 1995; Rossier et al. 1996; Lotshaw, 2001) strongly correlated with aldosterone output in these studies (data from 9 previous studies compiled and reviewed by Rossier (2006)). Thus, voltage‐gated Ca2+ channels have assumed a privileged role in ZG cell Ca2+ homeostasis.
Framed by the aforementioned and other studies, the ZG cell came to be regarded as electrically quiescent, operating over a narrow, relatively hyperpolarized voltage range in which the open probability of T‐type Ca2+ channels is low, but also in which the steady‐state inactivation of the channel is incomplete. The latter, it was argued, promoted a small‐yet‐discernible Ca2+ conductance into the ZG cell via a T channel ‘window current’ that was functionally sufficient to sustain the production of aldosterone (Cohen et al. 1988; Lotshaw, 2001; Wolfe et al. 2002). However, at odds with this description of an electrically silent ZG cell were several findings: (1) angiotensin II elicited oscillatory changes in cytosolic free Ca2+ in a small percentage of isolated cells that persisted for several minutes before transitioning to a tonic elevation (Johnson et al. 1989); (2) oscillatory [Ca2+]i responses preferentially evoked by low concentrations of angiotensin II (Quinn et al. 1988) depended only on voltage‐gated Ca2+ entry and not IP3‐induced Ca2+ release (Rossig et al. 1996); (3) spontaneous Ca2+‐dependent voltage spike potentials were recorded in ZG cells retained within cat adrenal slices (Natke & Kabela, 1979); and (4) spike potentials in isolated ZG cells could be evoked by depolarizing current injection or by pharmacological blockade of K+ currents (Quinn et al. 1987). More recently, our laboratory extended these findings and discovered that ZG cells are indeed electrically excitable when their cellular connectivity is preserved within a tissue slice (Hu et al. 2012). Specifically, ZG cells organized within cortical rosette structures spontaneously generate periodic (∼0.5 Hz), large depolarizing amplitude changes (∆ +75 mV from −85 mV) in membrane potential (V m oscillations) that are modulated in frequency by angiotensin II and extracellular K+. Collectively, these observations demonstrate that Ca2+ levels within ZG cells are highly dynamic and, therefore, motivated a further evaluation of the excitability of the ZG cell.
Pacemakers in the zona glomerulosa
Many excitable cells, ranging from those found in invertebrate (Lewis, 1988; Lamb & Calabrese, 2012; Marder et al. 2015) to mammalian (Steriade et al. 1993; Marcantoni et al. 2010; Vandael et al. 2010) systems, have the capacity to generate electrical oscillations, even when isolated. In many cases, these intrinsic oscillations are driven by a small ensemble of distinct ion channels that operate in concert to generate periodic changes in voltage. These so‐called pacemaker channel conductances often rely on voltage‐gated Ca2+ channels to produce the large, rising phase that characterizes the depolarizing component of the voltage oscillation. For example, a well‐characterized oscillator reliant upon T‐type Ca2+ channels is found in thalamocortical neurons of the thalamus, in which an interplay between T‐type Ca2+ channels and hyperpolarization‐activated cyclic nucleotide‐gated (HCN) channels drives voltage oscillations likely to be important for memory consolidation during sleep (Gais et al. 2002; Eschenko et al. 2006; Fogel et al. 2011). A second, well‐characterized oscillator reliant upon voltage‐gated Ca2+ channels is in the catecholamine secreting adrenal chromaffin cell in which there is an interplay between CaV1.3 channels and Ca2+‐activated K+ channels (BK, SK). In this cell, the relatively low threshold for activation of CaV1.3 currents at interspike potentials (∼−50 mV) drives depolarization and Ca2+ entry which is sufficient to activate BK channels within restricted Ca2+ domains and highly Ca2+‐sensitive SK channels at more distal sites. Once active these K+ channels hyperpolarize the membrane, remove CaV1.3 channel inactivation and enable the initiation of a new oscillatory cycle (Marcantoni et al. 2010; Vandael et al. 2010; Vandael & Carbone, 2015).
The recurrent observation that biological pacemakers often recruit voltage‐gated Ca2+ channels to produce oscillations motivated our laboratory to identify ion channels that endow the ZG cell with the capacity to operate as a pacemaker. While such identification is incomplete, we have learned that targeting NaV1.x channels with TTX or CaV1.x channels with nifedipine fails to alter either the amplitude or the frequency of ZG V m oscillations. In contrast, low concentrations of Ni2+ that selectively target CaV3.x channels halt V m oscillations, indicating an important contribution of these low voltage‐activated Ca2+ channels to ZG pacemaking (Hu et al. 2012). Considering that CaV3.2 is the primary CaV3.x member expressed in the ZG layer, we hypothesize that activation of CaV3.2 channels underlies the rapidly depolarizing phase of the ZG cell oscillation. The identification of the ion channel(s) that contribute to the falling phase remains less well delineated. However, silencing V m oscillations with Ni2+ does not cause a hyperpolarization as would be predicted from block of T‐type Ca2+ current, but rather causes a 20 mV depolarization from baseline (to ∼−60 mV), suggestive of a functional link between CaV3.2 and a Ca2+‐activated K+ current(s). In this model, Ca2+ entry through CaV3.2 channels both depolarizes the ZG cell and recruits Ca2+‐activated K+ channels which serve to hyperpolarize the cell; thus, the Ni2+‐evoked depolarization likely reflects the closure of the latter. It is notable that the peak amplitude of CaV3.2 pacemaker current is during the repolarization phase of the oscillatory cycle; this temporal dependence would allow CaV3.2 current to activate Ca2+‐activated K+ channels to complete the oscillatory cycle. Together these data provide evidence, albeit indirect, that a Ca2+‐activated K+ conductance may participate in the falling phase (hyperpolarization phase) of the ZG cell oscillation (Fig. 1).
Figure 1. A working model of the ZG pacemaker .
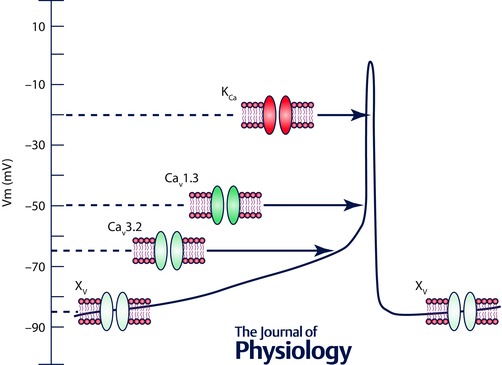
At the start of the oscillatory cycle (V m = −85 mV), depolarizing conductance(s) (Xv) elicit the opening of low voltage‐activated Cav3.2 channels which further depolarize V m and enable the opening of high voltage‐activated Cav1.3 channels. Ca2+ influx and a reduced V m recruits the activity of Ca2+‐dependent K+ channels that return V m to −85 mV to begin another oscillatory cycle.
The aforementioned ZG pacemaker model is developed from observations amassed from normal, wild‐type mice. In adapting these findings to human ZG cells, or to ZG cell behaviour during pathological conditions, it is important to consider the potential contribution of L‐type Ca2+ channels (CaV1.3), the mRNA of which is also found in normal human adrenals, aldosterone‐producing adenomas (Azizan et al. 2013; Scholl et al. 2013; Fernandes‐Rosa et al. 2014) and recently in aldosterone‐producing clusters (Nishimoto et al. 2015) in addition to that for CaV3.2. Two salient features of CaV1.3 channels are worth noting when considering their contribution to ZG function. First, the voltage range over which CaV1.3 channels carry Ca2+ current is ∼25 mV more hyperpolarized than prototypical L‐type Ca2+ channels, potentially enabling them to be active within the voltage range of the ZG cell oscillation (−85 to −10 mV). Although CaV1.3 channels would not be active at rest (−85 mV), they would be recruited in the oscillatory cycle following activation of lower threshold depolarizing conductances, including CaV3.2 channels. However, because CaV1.3 channels undergo rapid Ca2+‐dependent inactivation (CDI) (Xu & Lipscombe, 2001), it remains unclear to what extent these channels contribute to ZG pacemaker function or aldosterone production under non‐pathological conditions. Indeed, it is worth noting that in animal species that express T‐type and L‐type Ca2+ channels (rat, bovine), T‐type current amplitude but not L‐type current correlates with aldosterone production (Barrett et al. 1995; Rossier et al. 1996 b; Lotshaw, 2001; Rossier, 2006).
By contrast, it is likely that the contribution of mutant CaV1.3 channels to exaggerated aldosterone production is substantial. As discussed above, many gain‐of‐function mutations in the CACNA1D gene encoding CaV1.3 channels are observed in aldosterone‐producing adenomas (Azizan et al. 2013; Scholl et al. 2013; Fernandes‐Rosa et al. 2014) and aldosterone producing clusters (Nishimoto et al. 2015). As many of these mutations shift CaV1.3 gating properties to substantially more hyperpolarized membrane voltages, the reliance of Cav1.3 channel activation on lower threshold conductances would be mitigated, thereby allowing channels to open at rest and also to conduct Ca2+ under a greater inward driving force. In principle Ca2+ entry could also be augmented by disrupting CDI (Xu & Lipscombe, 2001). Based on structure–function analysis of CaV1.3 channels, mutations that alter activation gating but spare voltage‐dependent inactivation (VDI) would be most likely to alter CDI (Tadross et al. 2010). However, to date most mutations in APAs have not been experimentally tested for disrupted CDI and in the one study that has, CDI remained unperturbed (Scholl et al. 2013). Nevertheless, the modulation of this channel gating property of CaV1.3 could provide an additional mechanism for regulating Ca2+ entry and cellular excitability by altering the rate of spontaneous and evoked oscillations independent of changes in the voltage gating properties of CaV1.3 channels (see Fig. 1; Scharinger et al. 2015).
Potential relevance of ZG oscillations
If we accept the new place of the ZG cell among pacemakers, what then is the physiological relevance of ZG voltage oscillations? Do ZG cell voltage oscillations simply exist to recruit a range of voltage‐dependent Ca2+ conductances that collectively ensure a significant rise in cytosolic Ca2+ such that aldosterone is produced? In such a context, ZG Ca2+ oscillations would be epiphenomenal, i.e. a large, non‐periodic increase in cytosolic Ca2+ could achieve the same effect. Or do periodic oscillations provide a unique opportunity for Ca2+ channels with divergent biophysical properties to subserve singular functions in regulating ZG cell activity and aldosterone production? Support for such a possibility comes directly from ZG studies and also from many more summarized and formalized by McCobb and Beam (1991).
First, as determined by oscillatory voltage‐clamp experiments conducted in ZG cells, ZG V m oscillations are sufficient in magnitude and correct in frequency to drive repetitive, large‐amplitude CaV3.2‐mediated currents known as low threshold calcium spikes. Thus, V m oscillations in ZG cells provide a mechanism for rapidly inactivating CaV3.2 channels to generate long‐lasting, periodic Ca2+ signals in ZG cells that would be much larger than those associated with T‐channel window currents reliant upon incomplete channel inactivation (Hu et al. 2012). Second, because T‐type Ca2+ channels have the lowest threshold for activation and have a relatively slow rate of closure (deactivation), Ca2+ entry through these channels is driven by more negative driving forces. Thus, Ca2+ entry can be quite large and is favoured in response to a rapidly repolarizing oscillatory wave‐form (McCobb & Beam, 1991), such as recorded in ZG cells. Third, because L‐type Ca2+ channels have a more depolarized inactivation threshold and slower inactivation kinetics, entry through L‐type Ca2+ channels would be favoured in response to oscillatory waveforms that decay/repolarize slowly and have extended durations. Thus, as has been elegantly demonstrated and formalized by McCobb and colleagues, because of the high sensitivity of L‐type Ca2+ channels to the width of the oscillatory waveform, L‐type Ca2+ channels would provide the ZG cell with a mechanism to report activity changes in K+ channels that control the rate of repolarization. By contrast, given the relative insensitivity of T‐type Ca2+ channels to oscillatory waveform width (McCobb & Beam, 1991), Ca2+ entry through T‐type channels would better report ZG oscillation frequency. Thus, although each channel type can convert an electrical signal into a chemical Ca2+ response, when presented with an oscillatory electrical signal they do so advantaged by their biophysical properties. Thus, ZG V m oscillations may allow the cell to functionally compartmentalize the contribution of distinct voltage‐gated Ca2+ channels.
Finally, it is important to consider how ZG cells might process a periodic Ca2+ signal and how such a signal could control the rate of aldosterone production. In this regard the biochemical and computational studies of Ca2+/calmodulin‐dependent protein kinase (CaM kinase II) are noteworthy. In this multimeric enzyme complex, individual subunits of CaMKII are activated by high threshold periodic pulses of Ca2+. After the threshold for kinase activation is attained, some subunits are trans‐phosphorylated at Thr286 to generate a persistent, subthreshold kinase activity that is autonomous of the Ca2+ signal (Hanson et al. 1989). This positive feedback loop augments responses to low frequency stimuli and allows CaMKII to act as a decoder of the frequency of oscillatory Ca2+ signals (De Koninck & Schulman, 1998). In a similar manner, it is formally possible that periodic Ca2+ signals within ZG cells are more potent drivers of aldosterone production than static Ca2+ signals, because in ZG cells CaMKII activation induces a hyperpolarizing shift in the V 1/2 of activation of T‐type channels (Welsby et al. 2003; Yao et al. 2006). Such an increase in T‐current amplitude could thus provide a greater depolarizing current for L‐type channel recruitment further increasing extracellular Ca2+ entry. This possibility awaits future examination.
Final remarks
Here, we provide a brief review of recent mutational analyses performed on human adrenal tissues associated with excessive aldosterone production, and address how the newly defined ZG pacemaker cell, by driving the cell to membrane potentials that support channel activity, may unmask these mutations, many of which are associated with voltage‐dependent ion channels. In attempting to address the latter, we are confronted with many uncertainties. Clearly, to completely understand how a mutated ion channel contributes to excessive steroidogenic signalling, we must first better define the collection of ionic conductances that govern ZG cell excitability in the normal, healthy state. And yet, our current knowledge of the ionic conductances that drive ZG pacemaker activity is primarily limited to how the low threshold, T‐type Ca2+ channel (CaV3.2) drives ZG membrane potential to depolarized levels. To this end, we must also define all currents that mediate the rapid repolarization phase of the ZG pacemaker oscillation. Only when all the relevant channels comprising the ZG pacemaker are identified and the pacemaker regulation of aldosterone production is known, will we be able to fully understand by what means specific mutations support greater aldosterone production. Clearly, the future remains an exciting time for ion channel physiology within the realm of adrenal function.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
This work was supported by NIH grants to P.Q.B. (HL036977 and HL089717).
Biography
Mark P. Beenhakker received his PhD from University of Pennsylvania and is currently Assistant Professor in the Department of Pharmacology. Currently his group focuses on thalamocortical oscillations in epilepsy. Paula Q. Barrett received her PhD from the University of Rochester and is currently Professor in the Department of Pharmacology. She has had a long standing interest in the ionic regulation of aldosterone production. Nick Guagliardo and Peter Kein, neuroendocrine physiologists, Changlong Hu, an electrophysiologist, and David Breault, a developmental biologist, have provided additional insight. Despite their divergent research foci, their recent work has converged towards the goal of better understanding cellular oscillations in aldosterone producing cells of the adrenal cortex.

This review was presented at the symposium “Voltage‐gated calcium channels ‐ from basic mechanisms to disease”, which took place at Physiology 2015 in Cardiff, UK, 6–8 July 2015.
References
- Azizan EA, Lam BY, Newhouse SJ, Zhou J, Kuc RE, Clarke J, Happerfield L, Marker A, Hoffman GJ & Brown MJ (2012). Microarray, qPCR, and KCNJ5 sequencing of aldosterone‐producing adenomas reveal differences in genotype and phenotype between zona glomerulosa‐ and zona fasciculata‐like tumors. J Clin Endocrinol Metab 97, E819–E829. [DOI] [PubMed] [Google Scholar]
- Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, Maniero C, Garg S, Bochukova EG, Zhao W, Shaikh LH, Brighton CA, Teo AE, Davenport AP, Dekkers T, Tops B, Kusters B, Ceral J, Yeo GS, Neogi SG, McFarlane I, Rosenfeld N, Marass F, Hadfield J, Margas W, Chaggar K, Solar M, Deinum J, Dolphin AC, Farooqi IS, Striessnig J, Nissen P & Brown MJ (2013). Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 45, 1055–1060. [DOI] [PubMed] [Google Scholar]
- Barrett PQ, Bollag WB, Isales CM, McCarthy RT & Rasmussen H (1989). Role of calcium in angiotensin II‐mediated aldosterone secretion. Endocr Rev 10, 496–518. [DOI] [PubMed] [Google Scholar]
- Barrett PQ, Ertel EA, Smith MM, Nee JJ & Cohen CJ (1995). Voltage‐gated calcium currents have two opposing effects on the secretion of aldosterone. Am J Physiol Cell Physiol 268, C985–C992. [DOI] [PubMed] [Google Scholar]
- Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson‐Couterie B, Benecke A, Quinkler M, Fallo F, Plouin PF, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro MC, Strom TM & Reincke M (2013). Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone‐producing adenomas and secondary hypertension. Nat Genet 45, 440–444. [DOI] [PubMed] [Google Scholar]
- Boulkroun S, Beuschlein F, Rossi GP, Golib‐Dzib JF, Fischer E, Amar L, Mulatero P, Samson‐Couterie B, Hahner S, Quinkler M, Fallo F, Letizia C, Allolio B, Ceolotto G, Cicala MV, Lang K, Lefebvre H, Lenzini L, Maniero C, Monticone S, Perrocheau M, Pilon C, Plouin PF, Rayes N, Seccia TM, Veglio F, Williams TA, Zinnamosca L, Mantero F, Benecke A, Jeunemaitre X, Reincke M & Zennaro MC (2012). Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension 59, 592–598. [DOI] [PubMed] [Google Scholar]
- Boulkroun S, Fernandes‐Rosa FL & Zennaro MC (2015). Molecular and cellular mechanisms of aldosterone producing adenoma development. Front Endocrinol (Lausanne) 6, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauneis U, Vassilev PM, Quinn SJ, Williams GH & Tillotson DL (1991). ANG II blocks potassium currents in zona glomerulosa cells from rat, bovine, and human adrenals. Am J Physiol Endocrinol Metab 260, E772–E779. [DOI] [PubMed] [Google Scholar]
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB & Weissmann P (2002. a). Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 40, 892–896. [DOI] [PubMed] [Google Scholar]
- Calhoun DA, Zaman MA & Nishizaka MK (2002. b). Resistant hypertension. Curr Hypertens Rep 4, 221–228. [DOI] [PubMed] [Google Scholar]
- Capponi AM, Lew PD, Jornot L & Vallotton MB (1984). Correlation between cytosolic free Ca2+ and aldosterone production in bovine adrenal glomerulosa cells. Evidence for a difference in the mode of action of angiotensin II and potassium. J Biol Chem 259, 8863–8869. [PubMed] [Google Scholar]
- Capponi AM, Lew PD & Vallotton MB (1987). Quantitative analysis of the cytosolic‐free‐Ca2+‐dependency of aldosterone production in bovine adrenal glomerulosa cells. Different requirements for angiotensin II and K+ . Biochem J 247, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catena C, Colussi G, Nadalini E, Chiuch A, Baroselli S, Lapenna R & Sechi LA (2008). Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch Intern Med 168, 80–85. [DOI] [PubMed] [Google Scholar]
- Cherradi N, Rossier MF, Vallotton MB & Capponi AM (1996). Calcium stimulates intramitochondrial cholesterol transfer in bovine adrenal glomerulosa cells. J Biol Chem 271, 25971–25975. [DOI] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson‐Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Akerstrom G, Wang W, Carling T & Lifton RP (2011). K+ channel mutations in adrenal aldosterone‐producing adenomas and hereditary hypertension. Science 331, 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, McCarthy RT, Barrett PQ & Rasmussen H (1988). Ca channels in adrenal glomerulosa cells: K+ and angiotensin II increase T‐type Ca channel current. Proc Natl Acad Sci USA 85, 2412–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Cornwall MC & Williams GH (1987). Spatially resolved cytosolic calcium response to angiotensin II and potassium in rat glomerulosa cells measured by digital imaging techniques. J Biol Chem 262, 2919–2927. [PubMed] [Google Scholar]
- Dekkers T, ter Meer M, Lenders JW, Hermus AR, Schultze Kool L, Langenhuijsen JF, Nishimoto K, Ogishima T, Mukai K, Azizan EA, Tops B, Deinum J & Kusters B (2014). Adrenal nodularity and somatic mutations in primary aldosteronism: one node is the culprit? J Clin Endocrinol Metab 99, E1341–E1351. [DOI] [PubMed] [Google Scholar]
- De Koninck P & Schulman H (1998). Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279, 227–230. [DOI] [PubMed] [Google Scholar]
- Durroux T, Gallo‐Payet N & Payet MD (1988). Three components of the calcium current in cultured glomerulosa cells from rat adrenal gland. J Physiol 404, 713–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enberg U, Volpe C, Hoog A, Wedell A, Farnebo LO, Thoren M & Hamberger B (2004). Postoperative differentiation between unilateral adrenal adenoma and bilateral adrenal hyperplasia in primary aldosteronism by mRNA expression of the gene CYP11B2. Eur J Endocrinol 151, 73–85. [DOI] [PubMed] [Google Scholar]
- Eschenko O, Molle M, Born J & Sara SJ (2006). Elevated sleep spindle density after learning or after retrieval in rats. J Neurosci 26, 12914–12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakunding JL & Catt KJ (1980). Dependence of aldosterone stimulation in adrenal glomerulosa cells on calcium uptake: effects of lanthanum and verapamil. Endocrinology 107, 1345–1353. [DOI] [PubMed] [Google Scholar]
- Felizola SJ, Maekawa T, Nakamura Y, Satoh F, Ono Y, Kikuchi K, Aritomi S, Ikeda K, Yoshimura M, Tojo K & Sasano H (2014). Voltage‐gated calcium channels in the human adrenal and primary aldosteronism. J Steroid Biochem Mol Biol 144, 410–416. [DOI] [PubMed] [Google Scholar]
- Fernandes‐Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, Strom TM, Monticone S, Amar L, Meatchi T, Mantero F, Cicala MV, Quinkler M, Fallo F, Allolio B, Bernini G, Maccario M, Giacchetti G, Jeunemaitre X, Mulatero P, Reincke M & Zennaro MC (2014). Genetic spectrum and clinical correlates of somatic mutations in aldosterone‐producing adenoma. Hypertension 64, 354–361. [DOI] [PubMed] [Google Scholar]
- Fogel SM, Smith CT, Higginson CD & Beninger RJ (2011). Different types of avoidance behavior in rats produce dissociable post‐training changes in sleep. Physiol Behav 102, 170–174. [DOI] [PubMed] [Google Scholar]
- Gais S, Molle M, Helms K & Born J (2002). Learning‐dependent increases in sleep spindle density. J Neurosci 22, 6830–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guagliardo NA, Yao J, Hu C & Barrett PQ (2012). Minireview: aldosterone biosynthesis: electrically gated for our protection. Endocrinology 153, 3579–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Kapiloff MS, Lou LL, Rosenfeld MG & Schulman H (1989). Expression of a multifunctional Ca2+/calmodulin‐dependent protein kinase and mutational analysis of its autoregulation. Neuron 3, 59–70. [DOI] [PubMed] [Google Scholar]
- Hu C, Rusin CG, Tan Z, Guagliardo NA & Barrett PQ (2012). Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J Clin Invest 122, 2046–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EI, Capponi AM & Vallotton MB (1989). Cytosolic free calcium oscillates in single bovine adrenal glomerulosa cells in response to angiotensin II stimulation. J Endocrinol 122, 391–402. [DOI] [PubMed] [Google Scholar]
- Kojima K, Kojima I & Rasmussen H (1984). Dihydropyridine calcium agonist and antagonist effects on aldosterone secretion. Am J Physiol Endocrinol Metab 247, E645–E650. [DOI] [PubMed] [Google Scholar]
- Kramer RE (1988). Angiotensin II‐stimulated changes in calcium metabolism in cultured glomerulosa cells. Mol Cell Endocrinol 60, 199–210. [DOI] [PubMed] [Google Scholar]
- Kuppusamy M, Caroccia B, Stindl J, Bandulik S, Lenzini L, Gioco F, Fishman V, Zanotti G, Gomez‐Sanchez C, Bader M, Warth R & Rossi GP (2014). A novel KCNJ5‐insT149 somatic mutation close to, but outside, the selectivity filter causes resistant hypertension by loss of selectivity for potassium. J Clin Endocrinol Metab 99, E1765–E1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalevee N, Resin V, Arnaudeau S, Demaurex N & Rossier MF (2003). Intracellular transport of calcium from plasma membrane to mitochondria in adrenal H295R cells: implication for steroidogenesis. Endocrinology 144, 4575–4585. [DOI] [PubMed] [Google Scholar]
- Lamb DG & Calabrese RL (2012). Small is beautiful: models of small neuronal networks. Curr Opin Neurobiol 22, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzini L, Caroccia B, Campos AG, Fassina A, Belloni AS, Seccia TM, Kuppusamy M, Ferraro S, Skander G, Bader M, Rainey WE & Rossi GP (2014). Lower expression of the TWIK‐related acid‐sensitive K+ channel 2 (TASK‐2) gene is a hallmark of aldosterone‐producing adenoma causing human primary aldosteronism. J Clin Endocrinol Metab 99, E674–E682. [DOI] [PubMed] [Google Scholar]
- Lenzini L, Seccia TM, Aldighieri E, Belloni AS, Bernante P, Giuliani L, Nussdorfer GG, Pessina AC & Rossi GP (2007). Heterogeneity of aldosterone‐producing adenomas revealed by a whole transcriptome analysis. Hypertension 50, 1106–1113. [DOI] [PubMed] [Google Scholar]
- Lesouhaitier O, Chiappe A & Rossier MF (2001). Aldosterone increases T‐type calcium currents in human adrenocarcinoma (H295R) cells by inducing channel expression. Endocrinology 142, 4320–4330. [DOI] [PubMed] [Google Scholar]
- Lewis DV (1988). Calcium‐activated inward spike after‐currents in bursting neurone R15 of Aplysia . J Physiol 395, 285–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotshaw DP (1997. a). Characterization of angiotensin II‐regulated K+ conductance in rat adrenal glomerulosa cells. J Membr Biol 156, 261–277. [DOI] [PubMed] [Google Scholar]
- Lotshaw DP (1997. b). Effects of K+ channel blockers on K+ channels, membrane potential, and aldosterone secretion in rat adrenal zona glomerulosa cells. Endocrinology 138, 4167–4175. [DOI] [PubMed] [Google Scholar]
- Lotshaw DP (2001). Role of membrane depolarization and T‐type Ca2+ channels in angiotensin II and K+ stimulated aldosterone secretion. Mol Cell Endocrinol 175, 157–171. [DOI] [PubMed] [Google Scholar]
- McCobb DP & Beam KG (1991). Action potential waveform voltage‐clamp commands reveal striking differences in calcium entry via low and high voltage‐activated calcium channels. Neuron 7, 119–127. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger‐Brauns MJ, Striessnig J & Carbone E (2010). Loss of Cav1.3 channels reveals the critical role of L‐type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci 30, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Goeritz ML & Otopalik AG (2015). Robust circuit rhythms in small circuits arise from variable circuit components and mechanisms. Curr Opin Neurobiol 31, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga H, Maruyama Y, Kojima I & Hoshi T (1987). Transient Ca2+‐channel current characterized by a low‐threshold voltage in zona glomerulosa cells of rat adrenal cortex. Pflugers Arch 408, 351–355. [DOI] [PubMed] [Google Scholar]
- Montori VM & Young WF Jr (2002). Use of plasma aldosterone concentration‐to‐plasma renin activity ratio as a screening test for primary aldosteronism. A systematic review of the literature. Endocrinol Metab Clin North Am 31, 619–632, xi. [DOI] [PubMed] [Google Scholar]
- Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, Fischer E, Tizzani D, Pallauf A, Viola A, Amar L, Williams TA, Strom TM, Graf E, Bandulik S, Penton D, Plouin PF, Warth R, Allolio B, Jeunemaitre X, Veglio F & Reincke M (2012). KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 59, 235–240. [DOI] [PubMed] [Google Scholar]
- Murthy M, Xu S, Massimo G, Wolley M, Gordon RD, Stowasser M & O'Shaughnessy KM (2014). Role for germline mutations and a rare coding single nucleotide polymorphism within the KCNJ5 potassium channel in a large cohort of sporadic cases of primary aldosteronism. Hypertension 63, 783–789. [DOI] [PubMed] [Google Scholar]
- Natke E Jr & Kabela E (1979). Electrical responses in cat adrenal cortex: possible relation to aldosterone secretion. Am J Physiol Endocrinol Metab 237, E158–E162. [DOI] [PubMed] [Google Scholar]
- Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez‐Sanchez CE, Nanba K & Rainey WE (2015). Aldosterone‐stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci USA 112, E4591–E4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payet MD, Durroux T, Bilodeau L, Guillon G & Gallo‐Payet N (1994). Characterization of K+ and Ca2+ ionic currents in glomerulosa cells from human adrenal glands. Endocrinology 134, 2589–2598. [DOI] [PubMed] [Google Scholar]
- Plouin PF, Amar L & Chatellier G (2004). Trends in the prevalence of primary aldosteronism, aldosterone‐producing adenomas, and surgically correctable aldosterone‐dependent hypertension. Nephrol Dial Transplant 19, 774–777. [DOI] [PubMed] [Google Scholar]
- Pratt JH, Rothrock JK & Dominguez JH (1989). Evidence that angiotensin‐II and potassium collaborate to increase cytosolic calcium and stimulate the secretion of aldosterone. Endocrinology 125, 2463–2469. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Cornwall MC & Williams GH (1987. a). Electrical properties of isolated rat adrenal glomerulosa and fasciculata cells. Endocrinology 120, 903–914. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Cornwall MC & Williams GH (1987. b). Electrophysiological responses to angiotensin II of isolated rat adrenal glomerulosa cells. Endocrinology 120, 1581–1589. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Williams GH & Tillotson DL (1988). Calcium oscillations in single adrenal glomerulosa cells stimulated by angiotensin II. Proc Natl Acad Sci USA 85, 5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen H, Barrett P, Zawalich W, Isales C, Stein P, Smallwood J, McCarthy R & Bollag W (1989). Cycling of Ca2+ across the plasma membrane as a mechanism for generating a Ca2+ signal for cell activation. Ann N Y Acad Sci 568, 73–80. [DOI] [PubMed] [Google Scholar]
- Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M, Mallamaci F, Mannelli M, Mattarello MJ, Moretti A, Palumbo G, Parenti G, Porteri E, Semplicini A, Rizzoni D, Rossi E, Boscaro M, Pessina AC, Mantero F & Investigators PS (2006. a). A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 48, 2293–2300. [DOI] [PubMed] [Google Scholar]
- Rossi GP, Bernini G, Desideri G, Fabris B, Ferri C, Giacchetti G, Letizia C, Maccario M, Mannelli M, Matterello MJ, Montemurro D, Palumbo G, Rizzoni D, Rossi E, Pessina AC, Mantero F & Participants PS (2006. b). Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension 48, 232–238. [DOI] [PubMed] [Google Scholar]
- Rossier MF (2006). T channels and steroid biosynthesis: in search of a link with mitochondria. Cell Calcium 40, 155–164. [DOI] [PubMed] [Google Scholar]
- Rossier MF, Burnay MM, Brandenburger Y, Cherradi N, Vallotton MB & Capponi AM (1996. a). Sources and sites of action of calcium in the regulation of aldosterone biosynthesis. Endocr Res 22, 579–588. [DOI] [PubMed] [Google Scholar]
- Rossier MF, Burnay MM, Vallotton MB & Capponi AM (1996. b). Distinct functions of T‐ and L‐type calcium channels during activation of bovine adrenal glomerulosa cells. Endocrinology 137, 4817–4826. [DOI] [PubMed] [Google Scholar]
- Rossier MF, Lesouhaitier O, Perrier E, Bockhorn L, Chiappe A & Lalevee N (2003). Aldosterone regulation of T‐type calcium channels. J Steroid Biochem Mol Biol 85, 383–388. [DOI] [PubMed] [Google Scholar]
- Rossig L, Zolyomi A, Catt KJ & Balla T (1996). Regulation of angiotensin II‐stimulated Ca2+ oscillations by Ca2+ influx mechanisms in adrenal glomerulosa cells. J Biol Chem 271, 22063–22069. [DOI] [PubMed] [Google Scholar]
- Savard S, Amar L, Plouin PF & Steichen O (2013). Cardiovascular complications associated with primary aldosteronism: a controlled cross‐sectional study. Hypertension 62, 331–336. [DOI] [PubMed] [Google Scholar]
- Scharinger A, Eckrich S, Vandael DH, Schonig K, Koschak A, Hecker D, Kaur G, Lee A, Sah A, Bartsch D, Benedetti B, Lieb A, Schick B, Singewald N, Sinnegger‐Brauns MJ, Carbone E, Engel J & Striessnig J (2015). Cell‐type‐specific tuning of Cav1.3 Ca2+‐channels by a C‐terminal automodulatory domain. Front Cell Neurosci 9, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson‐Williams C, Libutti SK, Mane S, Hellman P, Westin G, Akerstrom G, Bjorklund P, Carling T, Fahlke C, Hidalgo P & Lifton RP (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone‐producing adenomas and primary aldosteronism. Nat Genet 45, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Stolting G, Nelson‐Williams C, Vichot AA, Choi M, Loring E, Prasad ML, Goh G, Carling T, Juhlin CC, Quack I, Rump LC, Thiel A, Lande M, Frazier BG, Rasoulpour M, Bowlin DL, Sethna CB, Trachtman H, Fahlke C & Lifton RP (2015). Recurrent gain of function mutation in calcium channel CACNA1H causes early‐onset hypertension with primary aldosteronism. Elife 4, e06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier AD, Wang H, Talley EM, Perez‐Reyes E & Barrett PQ (2001). α1H T‐type Ca2+ channel is the predominant subtype expressed in bovine and rat zona glomerulosa. Am J Physiol Cell Physiol 280, C265–C272. [DOI] [PubMed] [Google Scholar]
- Spat A & Hunyady L (2004). Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 84, 489–539. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA & Sejnowski TJ (1993). Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685. [DOI] [PubMed] [Google Scholar]
- Stowasser M (2001). New perspectives on the role of aldosterone excess in cardiovascular disease. Clin Exp Pharmacol Physiol 28, 783–791. [DOI] [PubMed] [Google Scholar]
- Stowasser M (2014). Aldosterone excess and resistant hypertension: investigation and treatment. Curr Hypertens Rep 16, 439. [DOI] [PubMed] [Google Scholar]
- Tadross MR, Ben Johny M & Yue DT (2010). Molecular endpoints of Ca2+/calmodulin‐ and voltage‐dependent inactivation of Cav1.3 channels. J Gen Physiol 135, 197–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR & Fox AP (1988). Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci 11, 431–438. [DOI] [PubMed] [Google Scholar]
- Vandael DH & Carbone E (2015). Cav1.3 channels as key regulators of neuron‐like firings and catecholamine release in chromaffin cells. Curr Mol Pharmacol 8, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A, Mahapatra S, Caro A, Ruth P, Zuccotti A, Knipper M & Carbone E (2010). Cav1.3 and BK channels for timing and regulating cell firing. Mol Neurobiol 42, 185–198. [DOI] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML & Barrett PQ (2003). A mechanism for the direct regulation of T‐type calcium channels by Ca2+/calmodulin‐dependent kinase II. J Neurosci 23, 10116–10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederkehr A, Szanda G, Akhmedov D, Mataki C, Heizmann CW, Schoonjans K, Pozzan T, Spat A & Wollheim CB (2011). Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab 13, 601–611. [DOI] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Perez‐Reyes E & Barrett PQ (2002). Stimulation of recombinant Cav3.2, T‐type, Ca2+ channel currents by CaMKIIγC . J Physiol 538, 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W & Lipscombe D (2001). Neuronal CaV1.3α1 L‐type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21, 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, Howell N, Carey RM, Colbran RJ & Barrett PQ (2006). Molecular basis for the modulation of native T‐type Ca2+ channels in vivo by Ca2+/calmodulin‐dependent protein kinase II. J Clin Invest 116, 2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennaro MC, Boulkroun S & Fernandes‐Rosa F (2015). An update on novel mechanisms of primary aldosteronism. J Endocrinol 224, R63–R77. [DOI] [PubMed] [Google Scholar]