

Abstract
Key point
PGC‐1α pathway activation has been shown to decrease disease severity and can be driven by quercetin.
Oral quercetin supplementation protected respiratory function for 4–6 months during a 12 month dosing regimen.
This transient protection was probably due to a failure to sustain elevated SIRT1 activity and downstream PGC‐1α signalling.
Quercetin supplementation may be a beneficial treatment as part of a cocktail provided continued SIRT1 activity elevation is achieved.
Abstract
Duchenne muscular dystrophy (DMD) impacts 1 : 3500 boys and leads to muscle dysfunction culminating in death due to respiratory or cardiac failure. There is an urgent need for effective therapies with the potential for immediate application for this patient population. Quercetin, a flavonoid with an outstanding safety profile, may provide therapeutic relief to DMD patients as the wait for additional therapies continues. This study evaluated the capacity of orally administered quercetin (0.2%) in 2 month old mdx mice to improve respiratory function and end‐point functional and histological outcomes in the diaphragm following 12 months of treatment. Respiratory function was protected for the first 4–6 months of treatment but appeared to become insensitive to quercetin thereafter. Consistent with this, end‐point functional measures were decreased and histopathological measures were more severe in dystrophic muscle compared to C57 and similar between control‐fed and quercetin‐fed mdx mice. To better understand the transient nature of improved respiratory function, we measured PGC‐1α pathway activity, which is suggested to be up‐regulated by quercetin supplementation. This pathway was largely suppressed in dystrophic muscle compared to healthy muscle, and at the 14 month time point dietary quercetin enrichment did not increase expression of downstream effectors. These data support the efficacy of quercetin as an intervention for DMD in skeletal muscle, and also indicate the development of age‐dependent quercetin insensitivity when continued supplementation fails to drive the PGC‐1α pathway. Continued study is needed to determine if this is related to disease severity, age or other factors.
Key points
PGC‐1α pathway activation has been shown to decrease disease severity and can be driven by quercetin.
Oral quercetin supplementation protected respiratory function for 4–6 months during a 12 month dosing regimen.
This transient protection was probably due to a failure to sustain elevated SIRT1 activity and downstream PGC‐1α signalling.
Quercetin supplementation may be a beneficial treatment as part of a cocktail provided continued SIRT1 activity elevation is achieved.
Abbreviations
- COXIV
cytochrome c oxidase subunit IV
- DMD
Duchenne muscular dystrophy
- ERRα
oestrogen‐related receptor alpha
- GABPA
GA binding protein transcription factor, alpha
- H&E
haematoxylin and eosin
- IRES
internal ribosome entry site
- MYH7
myosin, heavy chain 7
- NK‐κB
nuclear factor of kappa light polypeptide gene enhancer in B cells
- NRF1
nuclear respiratory factor‐1
- p‐ACCS79
phospho‐acetyl‐CoA carboxylase (Ser79)
- p‐p38 MAPK
phospho‐p38 mitogen‐activated protein kinase
- p38 MAPK
p38 mitogen‐activated protein kinase
- PGC‐1α
peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha
- PKA
protein kinase A
- SDHA
succinate dehydrogenase complex, subunit A
- SIRT1
sirtuin 1
- TFAM
transcription factor A, mitochondrial
- TNF
tumour necrosis factor
Introduction
Duchenne muscular dystrophy (DMD) is caused by a dystrophin protein deficiency. Dystrophin serves as a major conduit of force transmission by linking the actin cytoskeleton to large extracellular matrix proteins through the dystrophin glycoprotein complex (Petrof et al. 1993 a; Ervasti et al. 1994). With dystrophin deficiency or insufficiency there is a well‐defined collapse of dystrophin glycoprotein complex localization to the membrane and complex assembly (Ervasti et al. 1990; Hollinger et al. 2014). Skeletal muscle cells become particularly sensitive to contraction‐induced injury, resulting in membrane disruption and ultimately cellular necrosis (Duance et al. 1980; Morris et al. 2010; Selsby et al. 2010; Selsby, 2011). These cellular events define an evolving pathology that leads to whole muscle dysfunction, loss of ambulation and death, generally by the third decade (Duance et al. 1980; Lynch et al. 2001). DMD is modelled by the mdx mouse and the diaphragm of the mdx mouse recapitulates many features of human disease, including steady disease progression as well as fibrosis, necrosis and loss of function (Stedman et al. 1991).
For some time it has been appreciated in animal models that up‐regulation of the dystrophin‐related protein, utrophin, provides therapeutic relief for DMD, although translation to human patients has been slow in coming (Wakefield et al. 2000; Cerletti et al. 2003). Using prevention and rescue paradigms we and others found that up‐regulation of peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) protects dystrophic muscle and leads to increased utrophin abundance as well as increased resistance to fatigue, contraction‐induced injury, and a shift in muscle toward a more oxidative and slower phenotype (Handschin et al. 2007; Godin et al. 2012; Selsby et al. 2012; Hollinger et al. 2013; Hollinger & Selsby, 2015). In many regards this approach mimics the effects of endurance exercise, which is controversial in DMD patients due to the potential for increased injury (Carter et al. 1995; Brussee et al. 1997; Selsby et al. 2013). PGC‐1α may be activated by a variety of exogenous sources via the deacetylase, sirtuin 1 (SIRT1) (Nemoto et al. 2005; Guedes‐Dias & Oliveira, 2013), or 5′ AMP‐activated protein kinase (AMPK) (Jager et al. 2007), among others. Early attempts to drive PGC‐1α pathway activity, however, have been underwhelming and/or equivocal, and in one instance generated a high rate of mortality (Ljubicic et al. 2011, 2014; Selsby et al. 2012; Gordon et al. 2013).
Given the promise of PGC‐1α pathway activation we recently used the SIRT1 activator quercetin to drive PGC‐1α pathway activity in mdx mice. Quercetin is an orally available isoflavone with an outstanding safety profile and was granted Substance Generally Regarded as Safe (GRAS) status (GRAS Notice No. GRN 000341). We found that 6 months of quercetin treatment, ending at the middle of the lifespan, decreased histopathological injury in the diaphragm and hearts in treated mdx mice compared to mdx mice maintained on a control diet (Ballmann et al. 2015; Hollinger & Selsby, 2015). Aside from PGC‐1α pathway activation, quercetin may also protect dystrophic muscle as it functions as an anti‐inflammatory and antioxidant. Indeed, quercetin was successfully used to treat sarcoidosis, a condition, like DMD, with components of inflammation and oxidative stress (Boots et al. 2011). As DMD is a chronic disease therapeutic applications may need to be applied for years to maintain efficacy. Hence, the purpose of this experiment was to determine the extent to which long‐term quercetin treatment decreased histological injury and maintained function in dystrophin‐deficient skeletal muscle. We hypothesized that during a 12 month treatment period mdx mice given a quercetin‐enriched diet would have better respiratory function compared to control‐fed mice. We further hypothesized that following a 12 month treatment period mdx mice given a quercetin‐enriched diet would have better diaphragm function, decreased histological injury and increased PGC‐1α pathway signalling compared to diaphragms from mice maintained on a control diet.
Methods
Ethical approval and animal treatments
All procedures were approved by the Institutional Animal Care and Use Committee at Auburn University. Eight male C57 mice and 16 male mdx mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and allowed to acclimate for 1 week prior to the initiation of the experiment and animal treatments. Baseline measurements were recorded for respiratory function (Quindry et al. 2016) and then the dietary intervention was initiated at 2 months of age. C57 mice were maintained on a standard AIN93 diet (Bioserv, Flemington, NJ, USA) and mdx mice were assigned to either a standard AIN93 diet (mdx; n = 8) or a diet containing 0.2% quercetin (mdxQ; n = 8). Regardless of group, animals were given access to food and water ad libitum. Respiratory function was measured on alternating months throughout the 12 month testing period. Upon completion of the final in vivo measures mice were sent to the Physiological Assessment Core at the University of Pennsylvania so that in vitro muscle function could be performed. Mice were given new numbers upon arrival and only linked to the original numbering code at the conclusion of data collection. Resultant tissues were marked only by the new code and then distributed to the investigators to establish blinded conditions for subsequent histological measures. To ensure appropriate execution, samples were unmasked prior to performance of biochemical measures. Given the long‐term nature of this investigation not all animals reached the intended goal of 14 months of age. Furthermore, several tissues were damaged upon removal or freezing causing omission from the study. Hence, numbers per group used for each measurement are clearly indicated in the figure and table legends.
Respiratory function
Beginning at 2 months of age we measured respiratory function using whole body unrestrained plethysmography using a four‐chamber system according to the manufacturer's instructions, with minor modifications (Buxco, now Data Sciences International, St. Paul, MN, USA). This technique has been previously established to distinguish between healthy and dystrophin‐deficient mice (Huang et al. 2011). Prior to the initiation of data collection we performed validation and repeatability experiments, which resulted in our narrowing of the data collection window to the same 2 h period during the morning based upon published laboratory protocols (Quindry et al. 2016). Briefly, after daily calibration a single mouse was placed into each of the four chambers for a 20 min acclimatization period. We found previously that only 15 min was required for mice to acclimatize to the chambers (Quindry et al. 2016). Data were collected for a period of 25 min, which is at least 5 min longer than the duration required to achieve a steady‐state signal in mdx and C57 mice (Quindry et al. 2016). Mice were assigned to respiratory chambers at random throughout the 12 month testing period. All groups were represented during each heat of data collection. Upon completion of testing mice were returned to their cages.
Tissue collection and in vitro muscle function
Upon conclusion of the 12 month testing period mice were sent to the Physiological Assessment Core of the Wellstone Muscular Dystrophy Cooperative Centre at the University of Pennsylvania. Mice were sedated to a surgical level of anaesthesia, and the costal diaphragm was removed and divided into two hemi‐diaphragms used for histology or for functional and biochemical analyses. The hemi‐diaphragm used for histology was coated in freezing media, rolled so as to create a tube and frozen in melting isopentane. A strip was cut from the second hemi‐diaphragm and the remainder frozen in liquid nitrogen and stored at −80°C for biochemical analyses. The diaphragm strip was used to measure specific tension and resistance to contraction‐induced injury according to standard techniques performed previously (Barton et al. 2005; Selsby, 2011; Selsby et al. 2012; Hollinger et al. 2013). The diaphragm strip was placed in oxygenated Ringer solution and attached with one end to a force transducer and the other end with an anchor. Measurements of function were collected with an Aurora dual mode lever system (Ontario, Canada) using DMC software (version 3.2). Optimum length (L o) was measured using supramaximal stimulation (100 Hz) for 500 ms resulting in tetanic contractions. Tetanic contractions were performed in triplicate with 10 min between contractions. Resistance to contraction‐induced injury was assessed through five contractions at 80 Hz for 500 ms followed by 200 ms at 10% beyond L o. Peak force produced in the final four contractions was made relative to peak force produced in the first contraction. Standard equations were used to calculate cross‐sectional area and specific tension (Brooks & Faulkner, 1988).
Histological analyses
Diaphragms were embedded in OCT and frozen in melting isopentane, cut into 10 μm sections and then stained with haematoxylin and eosin (H&E) according to standard techniques. Prior to use, slides were put in a room temperature PBS bath and rocked for 10 min. Slides were incubated in Mayer's Hematoxylin (MHS32‐1 l, Sigma, St Louis, MO, USA) for 6 min, rinsed for 1 min in a water bath with cold running water, and incubated in 1% eosin for 1 min. Slides were then passed through two successive baths of 95% ethanol and two baths of 100% ethanol. In each bath, slides were submerged in the ethanol and quickly removed from the bath ten times. This repetitive wash technique was also used to rinse slides in three successive baths of CitriSolv. Finally, slides were air dried and mounted with cover slips using Permount Mounting Medium and sealed with nail polish. Once sealed, slides were imaged using an inverted DMI3000 B microscope and QICAM MicroPublisher 5.0 (MP5.0‐RTV‐CLR‐10, Q Imaging, Surrey, BC, Canada) camera using QCapture software. Images were taken at 10× magnification and 3–5 images were captured per diaphragm section. From these images blinded and trained technicians obtained (1) the total number of muscle cells per area by counting all of the muscle cells in a given area, (2) the per cent of cells with a centralized nucleus as an indicator of previous muscle injury and subsequent regeneration, (3) the total number of non‐muscle cell nuclei (extracellular nuclei) as an indicator of immune cell infiltration and (4) the area of necrosis.
An additional set of histological sections were stained with Masson's Trichrome (KTMTR, American MasterTech, Lodi, CA, USA) to quantify fibrosis. Slides were first rinsed with room temperature PBS for 10 min and then placed in Bouin's solution at room temperature overnight. Slides were washed the next day in running tap water for 5 min, incubated in a Weigert's A & B Hematoxylin bath for 10 min and again washed in running tap water for 5 min. Slides were then submerged and removed from distilled water ten times and then placed in a Biebrich scarlet solution for 5 min, and rinsed in distilled water. Slides were moved to a phosphotungstic/phosphomolydbic acid for 10 min and immediately transferred to Aniline blue solution for 5 min and rinsed with distilled water. Slides were placed in 1% acetic acid for 1 min and rinsed with distilled water. Finally, slides were finished in ethanol and CitriSolv as described above, dried, covered with coverslips using Permount, and sealed with nail polish. Slides were imaged at 10× magnification as done with H&E imaging. Trained technicians under blinded conditions measured area of fibrosis by quantifying 3–5 non‐overlapping images per animal.
Lastly, fibre area distribution was determined using an immunohistochemical approach. Diaphragm sections were exposed to an anti‐laminin antibody (1:100, Thermo Scientific, Waltham, MA, USA) overnight at 4°C and then donkey anti‐rabbit rhodamine (1:200, Millipore, Billerica, MA, USA) for 1 h at room temperature in the dark. Images were captured from a Leica DMI3000 B microscope with a QICAM 12‐bit Mono Fast 1394 Cooled (QIC‐F‐M‐12‐C, Q Imaging) camera at 20× magnification. Trained and blinded technicians quantified fibre area distribution of 3–5 non‐overlapping images of each animal.
Gene expression
mRNA was isolated from the remainder of the hemi‐diaphragm used for muscle function with TriZol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer instructions. Concentration and purity were assured using a Nanodrop (Thermo Scientific). cDNA was then made using the QuantiTect Reverse Transcriptase Kit (Qiagen) according to the manufacturer instructions, except random hexamers (IDT PreMade Primers) were used instead of the RT Primer Mix (Qiagen, Valencia, CA, USA). Measurements of gene abundance were collected using a Fluidigm approach as we have done previously (Hollinger et al. 2013). Primer pairs were prepared by Fluidigm as shown in Table 1. Samples were then applied to a 96 × 96 chip allowing the simultaneous detection of gene expression from 96 genes.
Table 1.
Primers for Fluidigm
| Gene | Forward | Reverse |
|---|---|---|
| 18 s | CTCTAGATAACCTCGGGCCG | GTCGGGAGTGGGTAATTTGC |
| Ak1 | CAATGCCACAGAACCTGTCA | TGCAGACCTCAGAGAAGACA |
| Akt1 | AGAACTCTAGGCATCCCTTCC | CGTTGGCATACTCCATGACA |
| Apaf1 | CACAGACCTTTCCATCCTTCA | CGTTTCCAAGTCCCAGAGAA |
| Atg12 | CCCAGACCAAGAAGTTGGAAC | CATGCCTGGGATTTGCAGTA |
| Atg5 | CAGCTCTTCCTTGGAACATCAC | GCATCCTTGGATGGACAGTGTA |
| Atp1a2 | AGCTGGGCCGAAAATACCAA | TGGGTCCATCTCTAGCCAGAA |
| Bax | GCGTGGTTGCCCTCTTCTA | CTGATCAGCTCGGGCACTTTA |
| Bcl2 | ATGTGTGTGGAGAGCGTCAA | GATGCCGGTTCAGGTACTCA |
| Becn1 | TGTCTTCAATGCCACCTTCC | CAAGCGACCCAGTCTGAAA |
| Bnip2 | CTCAGGAGATGCTACCAGCAA | AACCAAGAGGGGTGCACAA |
| Casp3 | AGTCTGACTGGAAAGCCGAAA | TCTGTCTCAATGCCACAGTCC |
| Cat | GGGATCTTGTGGGAAACAACAC | CTGTGGGTTTCTCTTCTGGCTA |
| Ckm | TCCATGGAGAAGGGAGGCAATA | GGGTGACCAGCCTTCTTGAA |
| Cs | ACCGAGCTCATGCGTTTGTA | ATGGCTTGTGTGGGCACTTA |
| Cybb | CCCAACTGGGATAACGAGTTCA | TTCAGGGCCACACAGGAAAA |
| Cycs | TGGAGAAAGACCTCATCGTGAC | CACAAAGTGCTGCCTCTGAC |
| Dag1 | GCCTCCAGTGGGGAGATCA | ACTGTGTGGGTCCCAGTGTA |
| Dtna | ACAGAACAGTCAGCCAGATCC | TCTGACATGAGCGTGTCCAA |
| Dysf | TGGTGGATCCCTTTGTGGAA | TGAGGGTTAGCTGTCTTCTCC |
| Esrra | GCCTGCAAAGCCTTCTTCAA | GTCTCCGCTTGGTGATCTCA |
| Fbl | GTGTCTTTATCTGTCGCGGAAA | CTCTCCATACACAGACTCTCCA |
| Fnip1 | GTGTGGGCATGTTGGCAAA | AGGAGATCCAATAGCGCCATAC |
| Gabarap | CGGATAGGAGACCTGGACAAA | GATGAATTCGCTTCCGGATCA |
| Gata2 | CACCCCTAAGCAGAGAAGCAA | TGTGGCACCACAGTTGACA |
| Gpx1 | TCGGTTTCCCGTGCAATCA | GTCGGACGTACTTGAGGGAA |
| Gpx4 | TTACGAATCCTGGCCTTCCC | TAGCCGGCTGCAAACTCC |
| Gsr | TATGTGAGCCGCCTGAACA | TCTGCGAATGTTGCATAGCC |
| Hspa1a | CACCATCGAGGAGGTGGATTA | ACAGTCCTCAAGGCCACATA |
| Hspa5 | TGCTGAGGCGTATTTGGGAA | TCGCTGGGCATCATTGAAGTA |
| Hspb1 | CGGAGATCACCATTCCGGTTA | TGGCTCCAGACTGTTCAGAC |
| Il1b | TGGCAACTGTTCCTGAACTCA | GGGTCCGTCAACTTCAAAGAAC |
| Il5 | GATGAGGCTTCCTGTCCCTA | TTCAGTATGTCTAGCCCCTGAA |
| Il6 | CGATGATGCACTTGCAGAAA | ACTCCAGAAGACCAGAGGAA |
| Map1lc3a | TCCGACCGGCCTTTCAA | ATCTGCTGCACCTCCTTACA |
| Mb | AAGCACAAGATCCCGGTCAA | CCCCGGAATGTCTCTTCTTCA |
| Mdh1 | TGTCCCTGATGACCTGCTCTA | AATGGGGAGGCCTTCAACAA |
| Mef2c | CTTATCCCACCTGGCAGCAA | ACTGAGCCGACTGGGAGTTA |
| Mstn | TCACGCTACCACGGAAACA | AACATTTGGGCTTGCCATCC |
| Mt‐atp6 | CCACACACCAAAAGGACGAA | GGCCTAGGAGATTTGTTGATCC |
| Mt‐cox1 | GACCGCAACCTAAACACAAC | GGGTGCCCAAAGAATCAGAA |
| Mt‐cox2 | ACCGAGTCGTTCTGCCAATA | ACTGCTCATGAGTGGAGGAC |
| Mt‐cyb | TTCATGTCGGACGAGGCTTA | TGCGAACAGTAGAAGTACTCCA |
| Mt‐nd1 | AGCCTGACCCATAGCCATAA | TTCTCCTTCTGTCAGGTCGAA |
| Mt‐nd4 | GGAACCAAACTGAACGCCTA | GAGGGCAATTAGCAGTGGAA |
| Mtor | AACCCGGGCGTGATCAATAA | ACCCACTTCCGCATTTCCA |
| Myf5 | CAGCAGCTTTGACAGCATCTAC | AGCTGGACACGGAGCTTTTA |
| Myh1 | GCAATCAAAGGTCAAGGCCTAC | CGGAATTTGGCCAGGTTGAC |
| Myh2 | CAAGACCGTGAGGAATGACAAC | AGTTTCCCCGTAGTGCCAAA |
| Myh7 | AGGCAAAGAAAGGCTCATCC | TGGAGCGCAAGTTTGTCATA |
| Myocd | CCGATGGATTCTTCCGTGAAA | CATCCTCAAAGGCGAATGCA |
| Myod1 | GCTACGACACCGCCTACTAC | ACACAGCCGCACTCTTCC |
| Myof | CAGCATGTTTGTCCTGGGAA | TCTCCACGTCATCACTGTCA |
| Myog | TGCCCAGTGAATGCAACTCC | GCAGATTGTGGGCGTCTGTA |
| Nfe2l2 | AGTGGATCCGCCAGCTAC | CTCTGCCAAAAGCTGCATACA |
| Nfkb1 | ACCGTATGAGCCTGTGTTCA | GTAGCCTCGTGTCTTCTGTCA |
| Nos2 | GAGGAGCAGGTGGAAGACTA | GGAAAAGACTGCACCGAAGATA |
| Nrf1 | GTCCGCACAGAAGAGCAAAA | TCCCGCCCATGTTGCTTATA |
| Nrip1 | ATTCGGGAGGAACACATCCA | TGAGAAGGCTGTTGAAAAGCAA |
| Pfkm | CACCAGAGGACGTTTGTGTTA | ACAGGACAGAGAGGTGACAA |
| Poldip2 | AAGCCCAGAATTCCCACGTCTA | AGCTGTACCACATCACTGTCCAA |
| Pparg | ACCCAATGGTTGCTGATTACA | AGGTGGAGATGCAGGTTCTA |
| Ppargc1a | AAACCACACCCACAGGATCA | GCTCTTCGCTTTATTGCTCCA |
| Prdx2 | TGTCCGGCTCTTGCTCAC | AAGTCAGGAGCCGACTTTCC |
| Prkaa1 | ATGTCTCTGGAGGAGAGCTA | GGAAAGGATCTGCTGGAACA |
| Sgca | TCCAGAAGATCGTGGGTACCA | AGCCTCTGTCTAGTGGTGTCA |
| Sirt1 | CTGAAAGTGAGACCAGTAGCA | GATGAGGCAAAGGTTCCCTA |
| Sod1 | CTCACTCTCAGGAGAGCATTCC | TTCCACCTTTGCCCAAGTCA |
| Sod2 | AAGGAGCAAGGTCGCTTACA | AATCCCCAGCAGCGGAATAA |
| Tfam | TTTCCACAGAACAGCTACCC | GGGCTGCAATTTTCCTAACC |
| Tfb1m | GAGCGGTGAAACAGCTTTCC | CTAGACTGCCAGCTTTCCTTACA |
| Tfb2m | GGCCTAGTGTGGTGGTGAAA | TAAGGTAGCCTCTGCGAAGAAC |
| Tgfb1 | GCTGCGCTTGCAGAGATTAA | GTAACGCCAGGAATTGTTGCTA |
| Tlr4 | GTTCTTCTCCTGCCTGACAC | GCTGAGTTTCTGATCCATGCA |
| Tnf | GGGTGATCGGTCCCCAAA | TGAGGGTCTGGGCCATAGAA |
| Traf1 | CCCAGGAAGCCCTCTGAC | GAACAGCCAACACCTGCAAA |
| Traf2 | TCTGTCCCAATGATGGATGCA | GCAGGAATGGGCAAAGTCC |
| Trp53 | CACAGCGTGGTGGTACCTTA | CCCATGCAGGAGCTATTACACA |
| Ucp1 | AGCTGTGCGATGTCCATGTA | ACCCGAGTCGCAGAAAAGAA |
| Ucp3 | TGTGCTGAGATGGTGACCTA | GCTCCAAAGGCAGAGACAAA |
| Uqcrc1 | AGGTGACTCGGGGCAAAA | GCGACCAATGTCTTCACACA |
| Utrn 3’ | AAGGGGAGAGCCATGATCAC | GCGGCATATAAAGCTATGTCCAA |
| Utrn 5’ | CAGCAGACAAACAGCGAGAA | GGTGAAGTTGAGGACGTTGAC |
| Xiap | CAAGTGAAGACCCTTGGGAAC | TTCTTGCCCCTTCTCATCCA |
Western blot
Protein was extracted from the portion of the hemi‐diaphragm designated for biochemical analyses and Western blotting was conducted as previously described (Hollinger et al. 2014). Samples were homogenized in 200 ml of whole muscle extraction buffer (10 mm sodium phosphate buffer, pH 7.0, 2% SDS), then centrifuging at 20,000 relative centrifugal force for 15 min. Supernatant containing the protein was removed and protein concentration was determined using a Nanodrop (Thermo Scientific). Samples were then diluted in 2× Laemmli buffer and warmed to 95°C for 5 min. Then, 26 μg of protein was loaded into 4–20% gradient gels (Lonza, Basel, Switzerland), separated by mass (60 V, 20 min then 120 V, 60 min) and transferred to a nitrocellulose membrane (100 V, 60 min). Ponceau stain was used to confirm equal loading. Membranes were probed overnight with the following primary (P) antibodies diluted in 5% milk, unless otherwise indicated, and incubated with anti‐rabbit secondary (S); all antibodies were purchased from Cell Signaling (Danvers, MA, USA) unless otherwise stated: phosphorylated (p‐) AMPKα (thr172) (P 1:500, S 1:2000), p‐ACCS79 (P 1:1000, S 1:2000, Millipore), p38 mitogen‐activated protein kinase (MAPK) (P 1:1000, S 1:2000), p‐p38 MAPK (Thr180/Tyr182) (P 1:1000, S 1:2000), SIRT1 (P 1:500, S 1:2000, Millipore), Histone 3 Lysine 9 Acetylation (H3K9ac) (P 1:1000, S 1:5000 in 0% milk), PGC‐1α (P 1:1000, S 1:2000, Abcam, Cambridge, MA, USA), oestrogen‐related receptor alpha (ERRα) (P 1:1000, S 1:2000), nuclear respiratory factor‐1 (NRF1) (P 1:250, S 1:5000), transcription factor A, mitochondrial (TFAM) (P 1:375 in 1% milk, S 1:1000 in 0% milk), GA binding protein transcription factor, alpha (GABPA) (P 1:1000, S 1:2000, Abcam), Utrophin (P 1:500, S 1:2000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), myosin, heavy chain 7 (MYH7) (type I) (P 1:1000, S 1:2000, Proteintech, Manchester, UK), Cytochrome C (P 1:1000, S 1:2000), cytochrome c oxidase subunit IV (COXIV) (P 1:500, S 1:2000), succinate dehydrogenase complex, subunit A (SDHA) (P 1:500, S 1:2000).
Statistics
Respiratory function and diaphragm fatigue were compared over time using an ANOVA with repeated measures in SAS with time and treatment as main effects. The remaining in vitro muscle function measures, histological analyses and biochemical analyses were compared using an ANOVA with a Newman–Keuls post‐hoc test. Significance was set P < 0.05 and data are shown as means ± SEM unless otherwise noted.
Results
To determine the extent to which dietary quercetin enrichment protected dystrophic skeletal muscle, mdx mice were treated with quercetin for a period of 12 months (2–14 months). We measured food consumption over the course of a week and body weight several times throughout the study period (Fig. 1). Animals fed a diet containing 0.2% quercetin ate slightly, but significantly, less food than control fed mdx mice in several instances. However, when considered in aggregate, daily consumption throughout the study was similar between groups. As predicted in growing animals, body weight increased as the study progressed but was similar between groups with the exception of 12 months of age during which the mdxQ group was slightly smaller than the mdx group. For the duration of the study animals in the mdxQ group ate 3.9 g day−1 food containing 0.2% quercetin, yielding 204 mg kg−1 day−1 quercetin, which is less than the approximately 285 mg kg−1 day−1 consumed in our previous work. This degree of exposure was fairly constant throughout the study period ranging from a nadir of 179 mg kg−1 day−1 at 5 months to a peak of 232 mg kg−1 day−1 at 3 months. Serum creatine kinase activity examined from samples collected when the animals were killed was similar between groups (data not shown).
Figure 1. Food consumption and body weight .
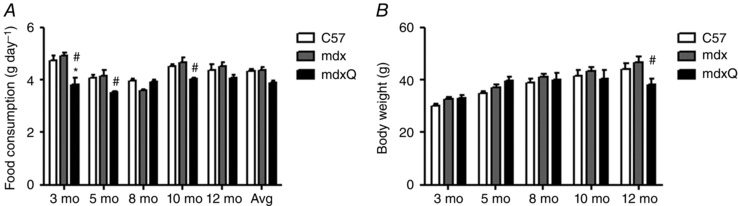
A, animals in the mdxQ group occasionally ate less food than animals in the mdx group and ate less than the C57 group at 3 months of age. In aggregate, animals ate similar amounts of food throughout the testing period. B, animals grew at approximately the same rate throughout the testing period and the main effect of time was predictably significant such that each time point was significantly larger than the one preceding it. Only at the 12 month time point were body weights different within a time point such that mdxQ was significantly less than mdx. *Significantly different from C57; #significantly different from mdx. C57 (n = 7), mdx (n = 7), mdxQ (n = 7).
Respiratory and in vitro function
Despite random assignment of mdx mice to control or quercetin‐fed groups there were significant differences between mdx and mdxQ at baseline for several respiratory function measures. Hence, respiratory function variables were normalized to each group's baseline value and set at 100%. In this fashion a value of more than 100% for a given variable indicates greater than baseline while a value below 100% indicates a value less than baseline. Throughout the first 6–8 months of age respiratory function for many of the selected dependent variables was frequently higher in the mdxQ group compared to the mdx group. For example, tidal volume, minute ventilation, peak inspiratory flow and peak expiratory flow were significantly greater in mdxQ compared to mdx during this time period (Fig. 2). However, beyond the 8 month data collection time point, the mdxQ group appeared to develop an insensitivity to the quercetin treatment such that the mdx and mdxQ groups had similar function at 14 months of age and often significantly decreased function compared to corresponding C57 mice.
Figure 2. Quercetin dietary enrichment transiently improved respiratory function in mdx mice .
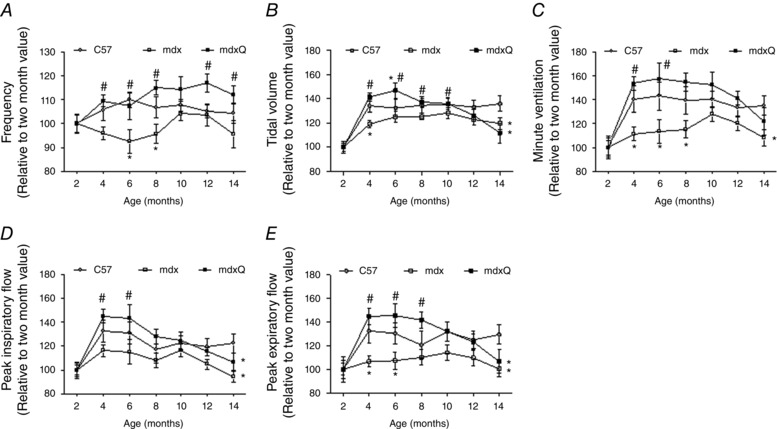
Respiratory function was measured using whole‐body unrestrained plethysmography on alternating months throughout a 12 month study. A, respiratory frequency was increased throughout the testing period compared to mdx and was similar to C57. B–E, oral quercetin administration transiently improved tidal volume (B), minute ventilation (C), peak inspiratory flow (D) and peak expiratory flow (E). *Significantly different from C57; #significantly different from mdx. C57 (n = 7), mdx (n = 7), mdxQ (n = 6).
After completion of the final in vivo functional measure at 14 months of age we also measured in vitro muscle function in diaphragms taken from these mice. We found that specific tension was decreased approximately 50% in mdx and mdxQ compared to C57 (Fig. 3 A). During the test to measure resistance to contraction‐induced injury the mdx group produced 7% less force than C57 during the fourth contraction and in the fifth contraction the mdx and mdxQ groups produced 11 and 8% less force compared to the C57 group, respectively. Resistance to contraction‐induced injury was similar between the mdx and mdxQ groups throughout the testing procedure (Fig. 3 B).
Figure 3. In vitro muscle function following 14 months of dietary quercetin enrichment .
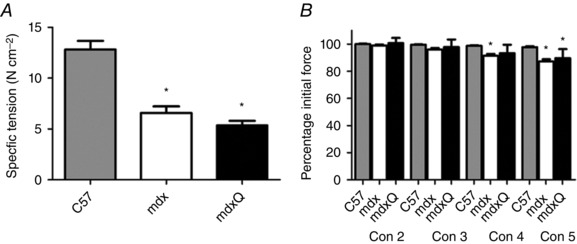
A, the average of the specific tension measured in diaphragm strips used for measurement of resistance to fatigue or contraction‐induced injury was calculated and found to be similar in mdx and mdxQ groups and both were significantly less than C57. B, dietary quercetin enrichment did not improve resistance to contraction‐induced injury during five eccentric contractions. Force produced during contractions 2–5 (Con 2–5) was normalized to maximal force produced during the first contraction. *Significantly different from C57; #significantly different from mdx. C57 (n = 7), mdx (n = 6), mdxQ (n = 3).
Histopathology
Dystrophin‐deficient and healthy muscle had a similar number of muscle fibres per area at the conclusion of the investigation (Fig. 4 A–D). At 14 months of age diaphragms from the mdx group had a 7‐fold increase in the number of infiltrating immune cells (Fig. 4 E) and a 25‐fold increase in centralized nuclei (Fig. 4 F) compared to C57 that were not corrected by dietary quercetin enrichment. At 14 months of age the > 30‐fold increase in fibrosis was also unaffected by the addition of quercetin to the diet (Fig. 5). The distribution of fibre area was measured and found to be similar between mdx and mdxQ groups but did differ between healthy and dystrophic muscle such that at 14 months of age there was a larger frequency of small fibres (< 50–100 μm2) and a smaller frequency of middle size fibres (200–500 μm2) in dystrophic muscle compared to healthy (Fig. 6 A–E). This apparent shift in fibre size distribution translated to a mean fibre area reduction of nearly 50% and a doubling of the variance coefficient in mdx and mdxQ compared to C57 (Fig. 6 F). Of interest, the percentage of fibres with an area greater than the mean was decreased by 13% in mdx compared to C57 (Fig. 6 G). Dietary quercetin enrichment preserved this measure of histological injury such that it was greater in mdxQ compared to mdx and similar to C57 and may be indicative of previous therapeutic efficacy.
Figure 4. Histopathological injury was not corrected by dietary quercetin enrichment at the conclusion of the 12 month treatment period .
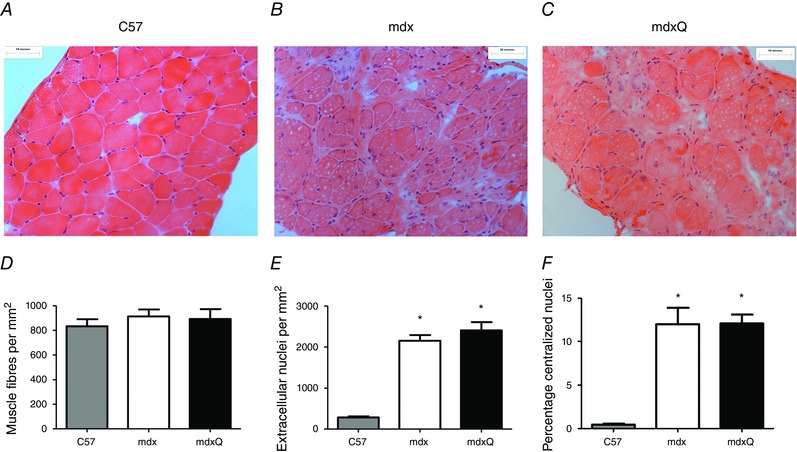
A–C, representative images of H&E‐stained diaphragm from 14 month old C57, mdx, and mdxQ animals. D. The number of muscle fibres/area were similar between groups. E. Extracellular nuclei and fibres with a F) centralized nucleus were increase in mdx and mdxQ compared to C57 and were not corrected by dietary quercetin enrichment. * indicates significantly different from C57. C57 (n = 7), mdx (n = 6), mdxQ (n = 6). [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 5. Fibrosis is not reversed by dietary quercetin enrichment .
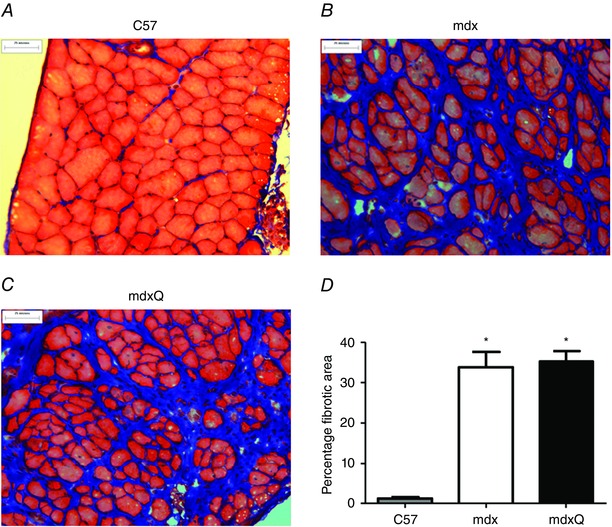
A–C, representative images following trichrome staining of 14 month old diaphragm for C57, mdx and mdxQ, respectively. D, the area of fibrosis was quantified and expressed relative to the total area. *Significantly different from C57. C57 (n = 7), mdx (n = 6), mdxQ (n = 6). [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 6. Fibre area distribution is significantly altered by dystrophin deficiency .
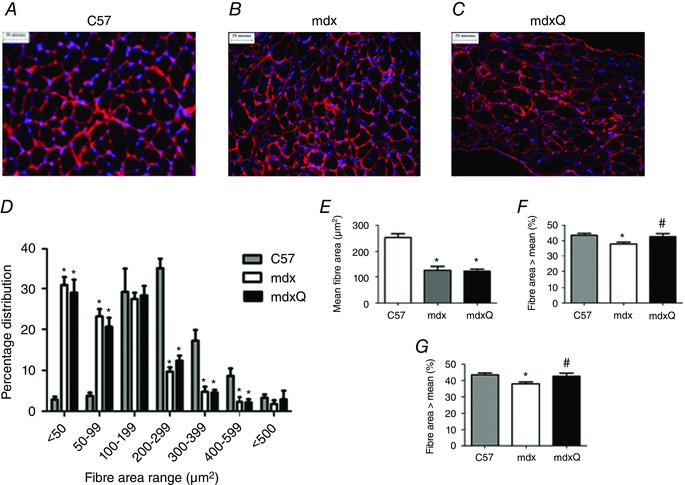
A–C, representative images of laminin‐probed diaphragm sections from C57, mdx and mdxQ animals at 14 months of age. D, fibre area distribution was altered by dystrophin deficiency such that there was a greater frequency of smaller fibres at the expense of larger fibres. This outcome resulted in (E) mean fibre size to be decreased in diseased animals independent of quercetin treatment. F, the variability of the fibre area distribution was increased in mdx and mdxQ compared to C57. G, the percentage of fibres with an area higher than the mean was decreased in mdx compared to C57 and was corrected by dietary quercetin enrichment. *Significantly different from C57; #significantly different from mdx. C57 (n = 7), mdx (n = 6), mdxQ (n = 6). [Colour figure can be viewed at wileyonlinelibrary.com]
Biochemistry
Our in vivo functional measures demonstrated some degree of protection conferred via the quercetin treatment that had abated by the conclusion of the 12 month study despite continued and sustained quercetin exposure. This outcome suggests a failure of quercetin to maintain PGC‐1α pathway activation. To assess this possibility we measured expression of genes driven either by PGC‐1α pathway activity or by quercetin supplementation (Table 2). Most metabolic genes were reduced either statistically or numerically in mdx compared to C57 and the addition of quercetin to the diet generally exaggerated these reductions. Several genes related to the Pgc‐1α pathway were also decreased by the quercetin treatment including Errα and Pgc‐1α. In addition, gene expression related to muscle repair, inflammation and apoptosis, oxidative stress and force transmission were largely decreased in mdx and many were further decreased in mdxQ.
Table 2.
Dystrophin deficiency alters gene expression
| C57 | mdx | mdxQ | |
|---|---|---|---|
| Mitochondrial biogenesis (PGC‐1α pathway genes) | |||
| Sirt1 | 1.00 ± 0.24 | −1.69 ± 0.14 | −2.52 ± 0.07 |
| Ppargc1a (Pgc‐1α) | 1.00 ± 0.17 | −2.77 ± 0.04* | −3.23 ± 0.02*, # |
| Esrra (Errα) | 1.00 ± 0.10 | −1.96 ± 0.13 | −5.38 ± 0.05*, # |
| Nrf1 | 1.00 ± 0.11 | −1.02 ± 0.22 | −1.51 ± 0.12 |
| Nrip1 | 1.00 ± 0.14 | 1.12 ± 0.25 | −1.16 ± 0.08 |
| Tfam | 1.00 ± 0.15 | 1.44 ± 0.26 | 1.23 ± 0.30 |
| Metabolic genes | |||
| Fnip1 | 1.00 ± 0.10 | −1.88 ± 0.06* | −2.60 ± 0.04* |
| Mtor | 1.00 ± 0.13 | −1.09 ± 0.10 | −1.54 ± 0.14 |
| Prkaa1 | 1.00 ± 0.25 | −1.03 ± 0.19 | −1.15 ± 0.11 |
| Gapdh | 1.00 ± 0.16 | −2.31 ± 0.10* | −3.59 ± 0.04* |
| Pfkm | 1.00 ± 0.07 | −2.20 ± 0.03* | −3.38 ± 0.03*, # |
| Pparg | 1.00 ± 0.17 | −1.73 ± 0.15 | −1.49 ± 0.12 |
| Cs | 1.00 ± 0.11 | −2.22 ± 0.08* | −4.67 ± 0.02*, # |
| Mdh1 | 1.00 ± 0.17 | −2.54 ± 0.13* | −6.03 ± 0.04*, # |
| Atp1a2 | 1.00 ± 0.08 | −1.74 ± 0.06* | −2.73 ± 0.05*, # |
| Cybb | 1.00 ± 0.19 | 1.93 ± 0.18* | 1.62 ± 0.29* |
| Cycs | 1.00 ± 0.16 | −2.24 ± 0.06 | −2.59 ± 0.10* |
| Mb | 1.00 ± 0.18 | −2.95 ± 0.09* | −7.52 ± 0.03*, # |
| Mt‐atp6 | 1.00 ± 0.17 | −2.39 ± 0.03* | −3.68 ± 0.01*, # |
| Mt‐co1 | 1.00 ± 0.17 | −2.34 ± 0.03* | −3.25 ± 0.01* |
| Mt‐co2 | 1.00 ± 0.15 | −2.21 ± 0.10* | −4.29 ± 0.04*, # |
| Mt‐cyb | 1.00 ± 0.19 | −2.39 ± 0.03* | −3.50 ± 0.02*, # |
| Mt‐nd1 | 1.00 ± 0.15 | −2.40 ± 0.02* | −4.02 ± 0.02*, # |
| Mt‐nd4 | 1.00 ± 0.18 | −2.44 ± 0.02* | −3.51 ± 0.02*, # |
| Uqcrc1 | 1.00 ± 0.17 | −2.61 ± 0.11* | −5.70 ± 0.04*, # |
| Tfb1m | 1.00 ± 0.35 | −1.68 ± 0.09 | −2.27 ± 0.23 |
| Tfb2m | 1.00 ± 0.15 | −1.70 ± 0.08* | −2.69 ± 0.05* |
| Ucp1 | 1.00 ± 0.23 | −1.62 ± 0.26 | −1.72 ± 0.28 |
| Ucp3 | 1.00 ± 0.19 | −3.23 ± 0.13* | −5.01 ± 0.05* |
| Ak1 | 1.00 ± 0.24 | −4.53 ± 0.10* | −3.44 ± 0.12* |
| Akt1 | 1.00 ± 0.41 | −2.17 ± 0.10 | −1.94 ± 0.12 |
| Ckm | 1.00 ± 0.16 | −3.57 ± 0.07* | −7.79 ± 0.02*, # |
| Inflammation/antioxidant genes | |||
| Nfκb1 | 1.00 ± 0.16 | −1.06 ± 0.21 | −1.83 ± 0.08 |
| Il1b | 1.00 ± 0.32 | −1.69 ± 0.19 | 1.26 ± 0.47 |
| Il5 | 1.00 ± 0.54 | −1.01 ± 0.63 | −2.06 ± 0.17 |
| Il6 | 1.00 ± 0.50 | −1.48 ± 0.20 | −2.85 ± 0.07 |
| Tlr4 | 1.00 ± 0.17 | 1.41 ± 0.48 | −1.41 ± 0.14 |
| Tnf | 1.00 ± 0.37 | −1.03 ± 0.25 | −1.77 ± 0.06 |
| Traf1 | 1.00 ± 0.25 | 3.12 ± 0.69* | 1.22 ± 0.38 |
| Traf2 | 1.00 ± 0.14 | −1.18 ± 0.22 | −2.27 ± 0.05* |
| Cat | 1.00 ± 0.12 | −1.30 ± 0.11 | −1.88 ± 0.06* |
| Gpx1 | 1.00 ± 0.26 | −1.33 ± 0.09 | −1.89 ± 0.10 |
| Gpx4 | 1.00 ± 0.18 | −1.42 ± 0.17 | −2.49 ± 0.08* |
| Gsr | 1.00 ± 0.21 | −1.35 ± 0.13 | −1.45 ± 0.07 |
| Nfe2l2 | 1.00 ± 0.11 | −1.5 ± 0.07 | −2.32 ± 0.04* |
| Nos2 | 1.00 ± 0.10 | −2.44 ± 0.14* | −1.19 ± 0.20# |
| Prdx2 | 1.00 ± 0.08 | −1.45 ± 0.06* | −3.19 ± 0.02* |
| Sod1 | 1.00 ± 0.16 | −1.14 ± 0.19 | −2.07 ± 0.13* |
| Sod2 | 1.00 ± 0.09 | −2.49 ± 0.06* | −4.58 ± 0.03*, # |
| Apoptosis genes | |||
| Bax | 1.00 ± 0.16 | 1.19 ± 0.31 | −1.61 ± 0.14 |
| Bcl2 | 1.00 ± 0.20 | 1.23 ± 0.21 | 1.17 ± 0.20 |
| Bnip2 | 1.00 ± 0.19 | −1.61 ± 0.14 | −2.88 ± 0.05*, # |
| Casp3 | 1.00 ± 0.55 | 2.63 ± 0.35 | 1.08 ± 0.29 |
| Trp53 | 1.00 ± 0.15 | 1.24 ± 0.11 | −1.63 ± 0.05*, # |
| Xiap | 1.00 ± 0.10 | −1.12 ± 0.10 | −1.56 ± 0.05* |
| Muscle repair and protein turnover genes | |||
| Fbl | 1.00 ± 0.15 | 1.43 ± 0.51 | −1.74 ± 0.06# |
| Gata2 | 1.00 ± 0.11 | −1.10 ± 0.19 | −2.00 ± 0.17*, # |
| Hspa1a | 1.00 ± 0.20 | 2.17 ± 0.75 | 5.58 ± 4.04 |
| Hspa5 | 1.00 ± 0.08 | −1.04 ± 0.15 | −1.86 ± 0.09*, # |
| Hspb1 | 1.00 ± 0.19 | 1.16 ± 0.37 | −1.85 ± 0.15 |
| Mef2c | 1.00 ± 0.13 | −1.25 ± 0.09 | −1.64 ± 0.08 |
| Mstn | 1.00 ± 0.16 | −8.02 ± 0.07* | −7.02 ± 0.11* |
| Myf5 | 1.00 ± 0.18 | −1.84 ± 0.05 | −2.86 ± 0.09* |
| Myocd | 1.00 ± 0.26 | 1.25 ± 0.59 | 1.08 ± 0.45 |
| Myod1 | 1.00 ± 0.38 | −1.10 ± 0.14 | −1.40 ± 0.12 |
| Myof | 1.00 ± 0.47 | −1.58 ± 0.04 | −1.47 ± 0.07 |
| Myog | 1.00 ± 0.13 | 4.21 ± 1.01* | 1.91 ± 0.31*, # |
| Poldip2 | 1.00 ± 0.14 | −1.82 ± 0.15* | −2.87 ± 0.06* |
| Tgfb1 | 1.00 ± 0.13 | 1.17 ± 0.19 | −1.30 ± 0.10 |
| Structural and sarcomeric genes | |||
| Dag1 | 1.00 ± 0.13 | −1.19 ± 0.11 | −1.54 ± 0.07 |
| Dtna | 1.00 ± 0.17 | −1.82 ± 0.09* | −2.38 ± 0.07* |
| Dysf | 1.00 ± 0.27 | −1.54 ± 0.12 | −1.43 ± 0.13 |
| Myh1 | 1.00 ± 0.17 | −4.74 ± 0.07* | −9.20 ± 0.02* |
| Myh2 | 1.00 ± 0.12 | −1.44 ± 0.08 | −2.11 ± 0.07* |
| Myh7 | 1.00 ± 0.10 | 1.36 ± 0.04 | −1.29 ± 0.08*, # |
| Sgca | 1.00 ± 0.15 | −2.36 ± 0.10* | −3.32 ± 0.06* |
| Utrn 3’ | 1.00 ± 0.17 | −1.30 ± 0.17 | −2.35 ± 0.05*, # |
| Utrn 5’ | 1.00 ± 0.12 | 1.36 ± 0.42 | −1.81 ± 0.07*, # |
Expression of genes involved in inflammation, apoptosis, muscle repair, muscle structure, metabolism and the Pgc‐1α pathway were largely decreased in dystrophic muscle compared to healthy. Diaphragms from 14 month old animals are represented in this table. Data are shown as fold change (mean ± SEM) relative to C57. *Significantly different from C57; #significantly different from mdx. C57 (n = 5), mdx (n = 4), mdxQ (n = 5).
Given these changes in gene expression and the transient nature of functional improvement with the quercetin diet we also measured relative protein abundance of components of the PGC‐1α pathway (Fig. 7). Quercetin is demonstrated to increase PGC‐1α pathway activity via the deacetylase, SIRT1 (Davis et al. 2009; Dong et al. 2014; Zhao et al. 2014). We found that SIRT1 protein abundance was increased 2.5‐ and 2.0‐fold in mdx and mdxQ, respectively, compared to C57. We also measured abundance of histone 3 acetylation of lysine 9, an indicator of SIRT1 activity, and found that it was also increased 3.0‐fold in mdx and mdxQ compared to C57. As SIRT1 is a deacetylase, this elevation is reflective of impaired SIRT1 activity despite the increase in SIRT1 abundance. As there is also evidence of a quercetin‐mediated increase in AMPK activation (Ahn et al. 2008; Suchankova et al. 2009; Dong et al. 2014) we measured the relative abundance of p‐AMPKαT172 and the downstream protein p‐ACCS79. We found that dystrophin deficiency caused a 7.0‐fold reduction of both proteins. Dietary quercetin enrichment caused a 3.0‐fold increase in mdxQ compared to mdx in p‐AMPKαT172 and p‐ACCS79, although the change in p‐ACCS79 failed to reach statistical significance. PGC‐1α protein abundance was similar to C57 with quercetin treatment while PGC‐1α in mdx increased 1.75‐fold in mdx compared to C57.
Figure 7. PGC‐1α pathway is altered by dystrophin deficiency .
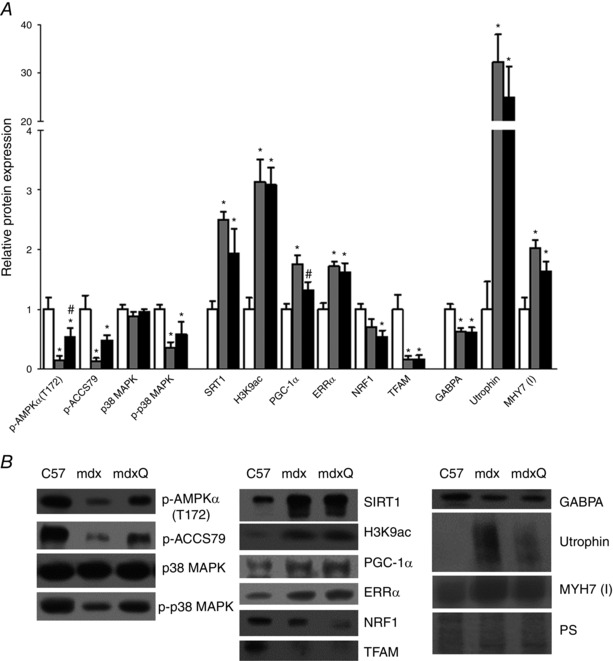
A, proteins along the PGC‐1α pathway are altered in 14 month old dystrophic diaphragm muscle but are similar following 12 months of quercetin administration. B, representative Western blots for proteins evaluated in A. *Significantly different from C57; #significantly different from mdx. White bar = C57 (n = 6), grey bar = mdx (n = 5), black bar = mdxQ (n = 5).
The PGC‐1α pathway is also regulated by p38 MAPK via phosphorylation (Fan et al. 2004; Pogozelski et al. 2009). We found that total p38 MAPK was similar between groups but p‐p38 MAPK was decreased similarly in the mdx and mdxQ groups (Fig. 7).
The PGC‐1α pathway can lead to mitochondrial biogenesis via an ERRα/NRF1/TFAM pathway (Fig. 7). We found that ERRα was increased 1.7‐ and 1.6‐fold in mdx and mdxQ compared to C57, respectively. NRF1 was decreased by 1.9‐fold in mdxQ compared to C57, but mdx was similar to both. TFAM was decreased 6.0‐fold in both mdx and mdxQ groups compared to C57. To assess mitochondrial content we measured the abundance of cytochrome c, COXIV and SDHA (Fig. 8). Dystrophin deficiency caused a 1.7‐fold reduction in cytochrome c and a 1.7‐fold reduction in SDHA relative abundance compared to C57; COX IV was not readily detectable in dystrophic skeletal muscle compared to C57.
Figure 8. Mitochondrial content was decreased in 14 month old dystrophic diaphragm tissue .
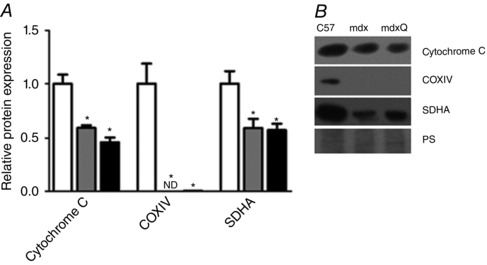
A, dystrophin deficiency caused a reduction in the relative abundance of several markers of mitochondrial content that were not corrected following 12 months of dietary quercetin enrichment. B, representative Western blots for proteins evaluated in A. *Significantly different from C57; #significantly different from mdx; ND = not detectable. White bar = C57 (n = 6), grey bar = mdx (n = 5), black bar = mdxQ (n = 5).
A second pathway driven by PGC‐1α results in increased utrophin expression via GABPA. Relative GABPA protein abundance was decreased 1.6‐fold in mdx and mdxQ compared to C57. Despite this, utrophin protein abundance was increased more than 20‐fold in mdx and mdxQ compared to C57. In addition, type I myosin heavy chain was increased 2.0‐ and 1.6‐fold in mdx and mdxQ compared to C57.
Discussion
DMD remains the most common fatal X‐linked disease worldwide. While there are a number of therapies poised for regulatory approval there are still hurdles to overcome and these therapies are not appropriate for all mutations. Hence, there remains a critical need to develop novel therapies for this disease that could be used by a large percentage of DMD patients. We and others found that activation of the PGC‐1α pathway provides therapeutic relief to dystrophic muscle using prevention (Handschin et al. 2007; Selsby et al. 2012) and rescue paradigms (Godin et al. 2012; Hollinger et al. 2013; Hollinger & Selsby, 2015). We found that oral delivery of the PGC‐1α activator, quercetin, decreased histological injury in the diaphragm and heart following a 6 month dosing regimen (Ballmann et al. 2015; Hollinger et al. 2015), an outcome that makes this work easily translatable to human patients. Based on these compelling preliminary studies the purpose of this investigation was to determine the extent to which long‐term quercetin administration would protect respiratory and diaphragmatic function and decrease histological injury in dystrophin‐deficient mice. To complete these experiments, a series of in vivo measures of respiratory function were assesses throughout the lifespan and in vitro physiological measures of diaphragmatic muscle function an assessment of histopathological injury were made in senescence.
We found that a quercetin‐enriched diet led to transient protection of respiratory function that abated by the conclusion of our life long study period. For example, peak inspiratory flow, indicative of diaphragmatic function, was increased for the first 4 months of treatment and peak expiratory flow, indicative of elastic recoil, was increased for the first 6 months of treatment in mdxQ compared to mdx. Consistent with our previous work, measures of injury were decreased after 6 months of quercetin treatment (Hollinger et al. 2015), but following 12 months of quercetin treatment protection of respiratory function subsided. In vitro muscle function and histological analyses performed at the conclusion of the study were similar between treated and untreated groups. One exception to this was that larger fibres may be spared, which when extrapolated into the human setting means that long‐term quercetin may have a positive effect on muscle function.
To investigate the possibility of a developed PGC‐1α pathway insensitivity, we measured protein expression of pathway components and effector proteins. Moreover, the expression of genes shown or thought to be driven by the PGC‐1α pathway were examined. In dystrophic muscle, we found that relative protein abundance of pathway components, SIRT1/PGC‐1α/ERRα, were increased compared to C57 but downstream pathway components (NRF1/TFAM) were decreased as was relative abundance of oxidative metabolism genes and proteins. Despite the elevation in SIRT1 content, activity of SIRT1, a deacetylase, was decreased in dystrophic muscle, which limits the capacity of SIRT1 to inhibit repression of PGC‐1α via deacetylation (Nemoto et al. 2005; Gerhart‐Hines et al. 2007). The addition of quercetin to the diet had little impact on pathway activation at the conclusion of the study period. Given the clear functional benefits found earlier during treatment we interpret these data to result from developed quercetin insensitivity whereby the continued administration of quercetin failed to drive PGC‐1α pathway activity in our ageing mice. It seems likely that this failure is related to a disease‐related blunting of SIRT1 activity and a limited capacity for subsequent PGC‐1α activation. Indeed, SIRT1 activation may be regulated by protein kinase A (PKA)‐mediated phosphorylation driven by upstream cAMP (Gerhart‐Hines et al. 2011; Chao & Tontonoz, 2012). Hence, depressed ATP content in dystrophic muscle (Cole et al. 2002) may decrease cAMP concentrations limiting PKA‐mediated activation of SIRT1.
Also of importance, at the conclusion of the study period AMPK activation (p‐AMPKαT172) and activity (p‐ACCS79) were increased in mdxQ compared to mdx. AMPK may be activated by quercetin (Ahn et al. 2008) or decreased food intake (Chang et al. 2015), among other factors. While mice assigned to mdxQ occasionally consumed less food than mdx, food consumption at the conclusion of the study period (12 months) and when considered in aggregate (average) was similar between groups. Given this outcome, it seems likely that AMPK activation was quercetin dependent. As AMPK signalling was increased and there was no attenuation in disease severity, this strongly suggests that this therapeutic target is insufficient to provide long‐term protection to dystrophic skeletal muscle.
The role of decreased transcript expression of PGC‐1α pathway components and oxidative genes in mdxQ compared to mdx despite similar protein content is unclear but may point toward persistent subclinical improvement in the intracellular environment, which improved transcript or mitochondrial stability. Also unclear was discordant transcript expression and protein abundance of PGC‐1α pathway components in dystrophic muscle where transcript expression was suppressed but protein abundance was increased compared to healthy muscle. The cytoskeletal protein, utrophin, also exhibited a similar pattern in which transcript expression was decreased in dystrophic muscle compared to healthy but protein abundance was increased more than 20‐fold. This observation, and that of PGC‐1α pathway components, may be explained in part by the type I shift caused by the preferential loss of type II fibres in dystrophic muscle (Petrof et al. 1993 b). Over‐representation of type I fibres is accompanied by increased transcript expression of utrophin A, which is more stable than utrophin splice variants expressed in type II muscle (Chakkalakal et al. 2008). Hence, transcript abundance is decreased in dystrophic muscle, not because of an improvement in intracellular environment, but because healthy muscle is able to maintain its full complement of type II fibre and produces a utrophin transcript that is more unstable. Utrophin protein abundance is enhanced by internal ribosomal entry site as well as the 3′ untranslated region, which help to improve translational efficiency (Gramolini et al. 2001; Miura et al. 2008). Similar findings of decreased transcript expression coupled with increased protein abundance at the time of tissue harvest have been reported (Gramolini et al. 1999; Miura et al. 2005, 2008).
Genes related to inflammation including nuclear factor of kappa light polypeptide gene enhancer in B cells (NF‐κB), tumour necrosis factor (TNF) and two TNF receptors were numerically decreased in mdxQ compared to mdx. These data are in good agreement with a previous study focused on sarcoidosis (Boots et al. 2011), an inflammatory disease with components of oxidative stress and inflammation, and is additional evidence of persistent subclinical benefits.
While there are numerous reports of short‐term success using Pgc‐1α pathway activation, long‐term studies are uncommon. Six months following neonatal Pgc‐1α gene transfer, dystrophic diaphragms were more resistant to contraction‐induced injury; however, specific tension and fatigue resistance were similar between groups (Selsby et al. 2012). In addition, only partial protection was found in 15 month old mice 3 months following Pgc‐1α gene transfer (Hollinger & Selsby, 2015). The overwhelming success of short‐term studies in young animals contrasted by the limited success in long‐term studies in older mice raises the possibility of inhibited pathway activation associated with either prolonged pathway activation, an effect of ageing, accumulated disease‐related injury or some combination of these variables. Supporting this notion, 9 month old animals in our previous work with quercetin had improved histopathological markers following 6 months of quercetin feeding but we were unable to detect robust pathway activation in tissues collected at the study endpoint (Hollinger et al. 2015). In contrast, in hearts from these animals pathway activation was more apparent (Ballmann et al. 2015). These disparate outcomes point toward a tissue‐specific effect and makes disease‐related injury a more likely possibility as the diaphragm suffers greater disease than the heart at this time point.
In conclusion, 12 months of dietary quercetin enrichment increased respiratory function for the first 4–6 months of treatment. By the conclusion of the study period at 14 months of age respiratory function, histopathology and a biochemical evaluation of these tissues were largely similar between quercetin‐treated and untreated mdx mice. Our current data combined with our previous findings (Hollinger et al. 2015) suggest that quercetin provides powerful and broad therapeutic relief, but becomes less effective between 6 and 8 months. We suggest that this quercetin insensitivity results from a failure to maintain PGC‐1α pathway activation due to suppressed SIRT1 activity. Despite this, this pathway remains a viable avenue for effective therapy, but additional strategies to sustain PGC‐1α pathway activity should be considered. It is clear that oral quercetin delivery provides a simple and effective means to protect dystrophic muscle, and hence determining the mechanism underlying pathway silencing is critical.
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
This research was designed by J.S., J.Q. and C.B., with data collection, analysis and interpretation by J.S., J.Q., C.B., J.R. and H.S. Manuscript development and editing was shared between all authors. All authors approve and agree to be accountable for this work. Finally, all authors qualify for authorship.
Funding
This work was supported by the Duchenne Alliance and its member foundations (Ryan's Quest, Hope for Gus, Team Joseph, Michael's Cause, Duchenne Now, Zack Heger Foundation, Pietro's Fight, RaceMD, JB's Keys, Romito Foundation, Harrison's Fund, Alex's Wish, and Two Smiles One Hope Foundation) grants 100065 and 100071 to J.S. and J.Q.
References
- Ahn J, Lee H, Kim S, Park J & Ha T (2008). The anti‐obesity effect of quercetin is mediated by the AMPK and MAPK signalling pathways. Biochem Biophys Res Commun 373, 545–549. [DOI] [PubMed] [Google Scholar]
- Ballmann C, Hollinger K, Selsby JT, Amin R & Quindry JC (2015). Histological and biochemical outcomes of cardiac pathology in mdx mice with dietary quercetin enrichment. Exp Physiol 100, 12–22. [DOI] [PubMed] [Google Scholar]
- Barton ER, Morris L, Kawana M, Bish LT & Toursel T (2005). Systemic administration of l‐arginine benefits mdx skeletal muscle function. Muscle Nerve 32, 751–760. [DOI] [PubMed] [Google Scholar]
- Boots AW, Drent M, de Boer VC, Bast A & Haenen GR (2011). Quercetin reduces markers of oxidative stress and inflammation in sarcoidosis. Clin Nutr 30, 506–512. [DOI] [PubMed] [Google Scholar]
- Brooks SV & Faulkner JA (1988). Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404, 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussee V, Tardif F & Tremblay JP (1997). Muscle fibres of mdx mice are more vulnerable to exercise than those of normal mice. Neuromuscul Disord 7, 487–492. [DOI] [PubMed] [Google Scholar]
- Carter GT, Wineinger MA, Walsh SA, Horasek SJ, Abresch RT & Fowler WM, Jr (1995). Effect of voluntary wheel‐running exercise on muscles of the mdx mouse. Neuromuscul Disord 5, 323–332. [DOI] [PubMed] [Google Scholar]
- Cerletti M, Negri T, Cozzi F, Colpo R, Andreetta F, Croci D, Davies KE, Cornelio F, Pozza O, Karpati G, Gilbert R & Mora M (2003). Dystrophic phenotype of canine X‐linked muscular dystrophy is mitigated by adenovirus‐mediated utrophin gene transfer. Gene Ther 10, 750–757. [DOI] [PubMed] [Google Scholar]
- Chakkalakal JV, Miura P, Belanger G, Michel RN & Jasmin BJ (2008). Modulation of utrophin A mRNA stability in fast versus slow muscles via an AU‐rich element and calcineurin signalling. Nucleic Acids Res 36, 826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Su H, Zhang D, Wang Y, Shen Q, Liu B, Huang R, Zhou T, Peng C, Wong CC, Shen HM, Lippincott‐Schwartz J & Liu W (2015). AMPK‐dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell 60, 930–940. [DOI] [PubMed] [Google Scholar]
- Chao LC & Tontonoz P (2012). SIRT1 regulation – it ain't all NAD. Mol Cell 45, 9–11. [DOI] [PubMed] [Google Scholar]
- Cole MA, Rafael JA, Taylor DJ, Lodi R, Davies KE & Styles P (2002). A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul Disord 12, 247–257. [DOI] [PubMed] [Google Scholar]
- Davis JM, Murphy EA, Carmichael MD & Davis B (2009). Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am J Physiol Regul Integr Comp Physiol 296, R1071–1077. [DOI] [PubMed] [Google Scholar]
- Dong J, Zhang X, Zhang L, Bian HX, Xu N, Bao B & Liu J (2014). Quercetin reduces obesity‐associated ATM infiltration and inflammation in mice: a mechanism including AMPKα1/SIRT1. J Lipid Res 55, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duance VC, Stephens HR, Dunn M, Bailey AJ & Dubowitz V (1980). A role for collagen in the pathogenesis of muscular dystrophy? Nature 284, 470–472. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG & Campbell KP (1990). Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315–319. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Roberds SL, Anderson RD, Sharp NJ, Kornegay JN & Campbell KP (1994). Alpha‐dystroglycan deficiency correlates with elevated serum creatine kinase and decreased muscle contraction tension in golden retriever muscular dystrophy. FEBS Lett 350, 173–176. [DOI] [PubMed] [Google Scholar]
- Fan M, Rhee J, St‐Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument‐Bromage H, Tempst P & Spiegelman BM (2004). Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC‐1α: modulation by p38 MAPK. Genes Dev 18, 278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart‐Hines Z, Dominy JE, Jr , Blattler SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP & Puigserver P (2011). The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD+ . Mol Cell 44, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart‐Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z & Puigserver P (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC‐1α. EMBO J 26, 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin R, Daussin F, Matecki S, Li T, Petrof BJ & Burelle Y (2012). Peroxisome proliferator‐activated receptor γ coactivator 1‐α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post‐necrotic mdx mouse skeletal muscle. J Physiol 590, 5487–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BS, Delgado Diaz DC & Kostek MC (2013). Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin Nutr 32, 104–111. [DOI] [PubMed] [Google Scholar]
- Gramolini AO, Belanger G & Jasmin BJ (2001). Distinct regions in the 3′ untranslated region are responsible for targeting and stabilizing utrophin transcripts in skeletal muscle cells. J Cell Biol 154, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramolini AO, Karpati G & Jasmin BJ (1999). Discordant expression of utrophin and its transcript in human and mouse skeletal muscles. J Neuropathol Exp Neurol 58, 235–244. [DOI] [PubMed] [Google Scholar]
- Guedes‐Dias P & Oliveira JM (2013). Lysine deacetylases and mitochondrial dynamics in neurodegeneration. Biochim Biophys Acta 1832, 1345–1359. [DOI] [PubMed] [Google Scholar]
- Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP & Spiegelman BM (2007). PGC‐1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 21, 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinger K, Gardan‐Salmon D, Santana C, Rice D, Snella E & Selsby JT (2013). Rescue of dystrophic skeletal muscle by PGC‐1α involves restored expression of dystrophin‐associated protein complex components and satellite cell signalling. Am J Physiol Regul Integr Comp Physiol 305, R13–23. [DOI] [PubMed] [Google Scholar]
- Hollinger K & Selsby JT (2015). PGC‐1 α gene transfer improves muscle function in dystrophic muscle following prolonged disease progress. Exp Physiol 100, 1145–1158. [DOI] [PubMed] [Google Scholar]
- Hollinger K, Shanely RA, Quindry JC & Selsby JT (2015). Long‐term quercetin dietary enrichment decreases muscle injury in mdx mice. Clin Nutr 34, 515–522. [DOI] [PubMed] [Google Scholar]
- Hollinger K, Yang CX, Montz RE, Nonneman D, Ross JW & Selsby JT (2014). Dystrophin insufficiency causes selective muscle histopathology and loss of dystrophin–glycoprotein complex assembly in pig skeletal muscle. FASEB J 28, 1600–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Cheng G, Lu H, Aronica M, Ransohoff RM & Zhou L (2011). Impaired respiratory function in mdx and mdx/utrn +/– mice. Muscle Nerve 43, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Handschin C, St‐Pierre J & Spiegelman BM (2007). AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1α. Proc Natl Acad Sci USA 104, 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V, Burt M, Lunde JA & Jasmin BJ (2014). Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1‐PGC‐1α axis. Am J Physiol Cell Physiol 307, C66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V, Miura P, Burt M, Boudreault L, Khogali S, Lunde JA, Renaud JM & Jasmin BJ (2011). Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet 20, 3478–3493. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV & Faulkner JA (2001). Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol 535, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Andrews M, Holcik M & Jasmin BJ (2008). IRES‐mediated translation of utrophin A is enhanced by glucocorticoid treatment in skeletal muscle cells. PLoS One 3, e2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Thompson J, Chakkalakal JV, Holcik M & Jasmin BJ (2005). The utrophin A 5′‐untranslated region confers internal ribosome entry site‐mediated translational control during regeneration of skeletal muscle fibres. J Biol Chem 280, 32997–33005. [DOI] [PubMed] [Google Scholar]
- Morris CA, Selsby JT, Morris LD, Pendrak K & Sweeney HL (2010). Bowman–Birk inhibitor attenuates dystrophic pathology in mdx mice. J Appl Physiol 109, 1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM & Finkel T (2005). SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC‐1α. J Biol Chem 280, 16456–16460. [DOI] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM & Sweeney HL (1993. a). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Stedman HH, Shrager JB, Eby J, Sweeney HL & Kelly AM (1993. b). Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am J Physiol 265, C834–841. [DOI] [PubMed] [Google Scholar]
- Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT & Yan Z (2009). p38γ Mitogen‐activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One 4, e7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quindry JC, Ballmann CG, Epstein EE & Selsby JT (2016). Plethysmography measurements of respiratory function in conscious unrestrained mice. J Physiol Sci 66, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby J, Pendrak K, Zadel M, Tian Z, Pham J, Carver T, Acosta P, Barton E & Sweeney HL (2010). Leupeptin‐based inhibitors do not improve the mdx phenotype. Am J Physiol Regul Integr Comp Physiol 299, R1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby JT ( 2011). Increased catalase expression improves muscle function in mdx mice. Exp Physiol 96, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby JT, Acosta P, Sleeper MM, Barton ER & Sweeney HL (2013). Long‐term wheel running compromises diaphragm function but improves cardiac and plantarflexor function in the mdx mouse. J Appl Physiol (1985) 115, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby JT, Morine KJ, Pendrak K, Barton ER & Sweeney HL (2012). Rescue of dystrophic skeletal muscle by PGC‐1α involves a fast to slow fibre type shift in the mdx mouse. PloS One 7, e30063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT & Kelly AM (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539. [DOI] [PubMed] [Google Scholar]
- Suchankova G, Nelson LE, Gerhart‐Hines Z, Kelly M, Gauthier MS, Saha AK, Ido Y, Puigserver P & Ruderman NB (2009). Concurrent regulation of AMP‐activated protein kinase and SIRT1 in mammalian cells. Biochem Biophys Res Commun 378, 836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield PM, Tinsley JM, Wood MJ, Gilbert R, Karpati G & Davies KE (2000). Prevention of the dystrophic phenotype in dystrophin/utrophin‐deficient muscle following adenovirus‐mediated transfer of a utrophin minigene. Gene Ther 7, 201–204. [DOI] [PubMed] [Google Scholar]
- Zhao LR, Du YJ, Chen L, Liu ZG, Pan YH, Liu JF & Liu B (2014). Quercetin protects against high glucose‐induced damage in bone marrow‐derived endothelial progenitor cells. Int J Mol Med 34, 1025–1031. [DOI] [PubMed] [Google Scholar]