

Abstract
Multifunctional proteins play a variety of roles in metabolism. Here, we examine the catalytic function of the combined 3-deoxy-d-arabino heptulosonate-7-phosphate synthase (DAH7PS) and chorismate mutase (CM) from Geobacillus sp. DAH7PS operates at the start of the biosynthetic pathway for aromatic metabolites, whereas CM operates in a dedicated branch of the pathway for the biosynthesis of amino acids tyrosine and phenylalanine. In line with sequence predictions, the two catalytic functions are located in distinct domains, and these two activities can be separated and retain functionality. For the full-length protein, prephenate, the product of the CM reaction, acts as an allosteric inhibitor for the DAH7PS. The crystal structure of the full-length protein with prephenate bound and the accompanying small angle x-ray scattering data reveal the molecular mechanism of the allostery. Prephenate binding results in the tighter association between the dimeric CM domains and the tetrameric DAH7PS, occluding the active site and therefore disrupting DAH7PS function. Acquisition of a physical gating mechanism to control catalytic function through gene fusion appears to be a general mechanism for providing allostery for this enzyme.
Keywords: allosteric regulation, Bacillus, crystal structure, inhibition mechanism, multifunctional enzyme, X-ray scattering, DAHPS, aromatic amino acid, chorismate mutase, shikimate
Introduction
Allostery is the biochemical process whereby binding a remote ligand triggers protein functional changes. Allosteric regulation of key metabolic enzymes is an important control system for many metabolic pathways. The shikimate pathway, which is found in plants and microorganisms, is the biosynthetic route for the formation of the aromatic amino acids and other important aromatic metabolites (1). The first committed step of the pathway, catalyzed by 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase (DAH7PS),2 is generally controlled by allosteric inhibition arising from the remote binding of end products or intermediates of the pathway (2).
DAH7PS catalyzes the metal ion-dependent condensation reaction of phosphoenolpyruvate (PEP) and erythrose 4-phosphate (E4P) to generate 3-deoxy-d-arabino-heptulosonate-7-phosphate (DAH7P, Fig. 1). The pathway has six further steps to the common intermediate chorismate. Chorismate is substrate for a variety of enzymes, including those that catalyze steps that ultimately lead to the aromatic amino acids Phe, Tyr, or Trp. Allosteric regulation is observed both at the pathway entry via DAH7PS and at the branch points from chorismate. All known DAH7PS enzymes share a (β/α)8 barrel catalytic domain, which houses the active site. However, there are distinct variations between DAH7PS enzymes from different organisms in the structural extensions to the barrel that support allostery.
FIGURE 1.

Shikimate pathway for aromatic amino acid biosynthesis. The first step of the pathway is catalyzed by the enzyme DAH7PS. The pathway branches at chorismate, and chorismate mutase catalyzes the formation of prephenate from chorismate, which in turn will form Tyr and Phe.
DAH7PS enzymes are divided into two distinct types, which differ in sequence, subunit size, and quaternary structure. Type I enzymes are generally smaller than their type II counterparts and have been shown to display allosteric control from a single allosteric effector or to display no allostery. Type I enzymes are further divided into two subtypes. The type Iα subgroup contains the well characterized Escherichia coli, Saccharomyces cerevisiae, and Neisseria meningitidis enzymes (3–5). These enzymes have an allosteric site formed by an N-terminal and loop extension and are sensitive to one of the aromatic amino acids. Type Iβ enzymes are more varied and either comprise just the core catalytic barrel and show no allosteric regulation or have a discrete domain found at either the N or C termini of the barrel (6). This addition can be either an ACT domain (7, 8) (named after the enzymes in which it was originally identified, aspartokinase, chorismate mutase, and TyrA) or a chorismate mutase (CM) enzyme (9), which catalyzes a branch point step of the pathway (Fig. 1). For the latter, previously characterized DAH7PS enzymes have dual catalytic functionality (both DAH7PS and CM activity), and the DAH7PS activity is inhibited by the binding of CM substrate chorismate or its product prephenate. Type II enzymes are the most complex, and the only fully characterized example is from Mycobacterium tuberculosis (10). M. tuberculosis DAH7PS has multiple extensions to the core barrel providing three allosteric binding sites that work synergistically (11). In addition, this protein can form a non-covalent complex with the M. tuberculosis CM allowing for complex allosteric control of both CM and DAH7PS activities (12).
In accordance with the different extensions to the core catalytic barrel, the molecular mechanisms by which allostery is achieved vary between the DAH7PS types. Whereas the type Iβ DAH7PS from Thermotoga maritima undergoes significant ACT domain movement associated with the binding of inhibitor Tyr (7), for other enzymes, such as the type II M. tuberculosis DAH7PS (13) and the type Iα N. meningitidis DAH7PS (14), allosteric inhibition is conferred by more subtle structural changes and alteration in protein dynamics. The variation in allosteric machinery and mechanism for this enzyme is intriguing both from a mechanistic and evolutionary point of view.
Here, we examine in detail the mechanism of allostery for the DAH7PS from Geobacillus sp. (strain Y412MC61), a thermophilic soil bacterium. From sequence inspection, this DAH7PS is predicted to have an N-terminal CM domain and be a member of the type Iβ group. The independence of the individual catalytic activities of the DAH7PS and CM domains has already been demonstrated for the Bacillus subtilis enzyme (15), and a structure of an uninhibited form of an enzyme of this type has been previously determined for the DAH7PS from Listeria monocytogenes (LmoDAH7PS) (16). Here, we explore the functional properties of this dual activity protein and demonstrate directly the structural changes associated with the allosteric response induced by prephenate binding. Based on our new x-ray crystal structure and small angle x-ray scattering experiments, we reveal that prephenate binding to the CM domain stabilizes a more compact conformation of the protein, limiting the catalytic functionality of the DAH7PS domain active site.
Results
DAH7PS and CM Catalytic Activities of Wild-type GspDAH7PS Can Be Separated into Two Functional Enzymes
The DAH7PS protein complex from Geobacillus sp. putatively includes two functional units, represented by a DAH7PS and a CM domain. To test this prediction and to investigate the dependence of the catalytic activity of each functional unit of the GspDAH7PS complex, the full-length protein (GspDAH7PSWT) was expressed, and two truncated forms were generated consisting of either only the predicted DAH7PS (GspDAH7PSD) or CM (GspDAH7PSC) domains (Fig. 2). The full-length protein indeed displayed both DAH7PS and CM activity (Table 1), and as for other characterized DAH7PS enzymes, the DAH7PS activity of GspDAH7PSWT was found to be dependent on the presence of a divalent metal ion (Fig. 3A). Consistent with the thermophilic nature of Geobacillus sp., maximum DAH7PS and CM activity was observed at 70 °C (Fig. 3, B and C). The DAH7PS from Geobacillus sp. is therefore a bifunctional thermophilic enzyme, which possesses both DAH7PS and CM activity.
FIGURE 2.

A, sequence alignment of GspDAH7PS, LmoDAH7PS unregulated PfuDAH7PS. and EcoCM. B, SDS-polyacrylamide gel of purified GspDAH7PSWT (lane 1), GspDAH7PSD (lane 2), and GspDAH7PSC (lane 3) after purification. Lane M, molecular mass markers.
TABLE 1.
Kinetic parameters for GspDAH7PSWT, GspDAH7PSD, and GspDAH7PSC
| Protein | DAH7PS activitya |
CM activity |
||||||
|---|---|---|---|---|---|---|---|---|
| KmPEP | KmE4P | kcat | kcat/KmPEP | kcat/KmE4P | Km | kcat | kcat/Km | |
| μm | μm | s−1 | mm−1s−1 | mm−1s−1 | μm | s−1 | mm−1s−1 | |
| GspDAH7PSWT | 87 ± 9 | 95 ± 8 | 45 ± 4 | 0.52 | 0.47 | 88 ± 6 | 1.9 ± 0.1 | 2.1 × 10−2 |
| GspDAH7PSD | 98 ± 11 | 62 ± 5 | 63 ± 5 | 0.64 | 1.0 | |||
| GspDAH7PSC | 450 ± 40 | 5.5 ± 0.5 | 1.2 × 10−2 | |||||
a The Michaelis constants for the DAH7PS activity are apparent values determined with a fixed concentration of the invariant substrate (as described under “Experimental Procedures”).
FIGURE 3.
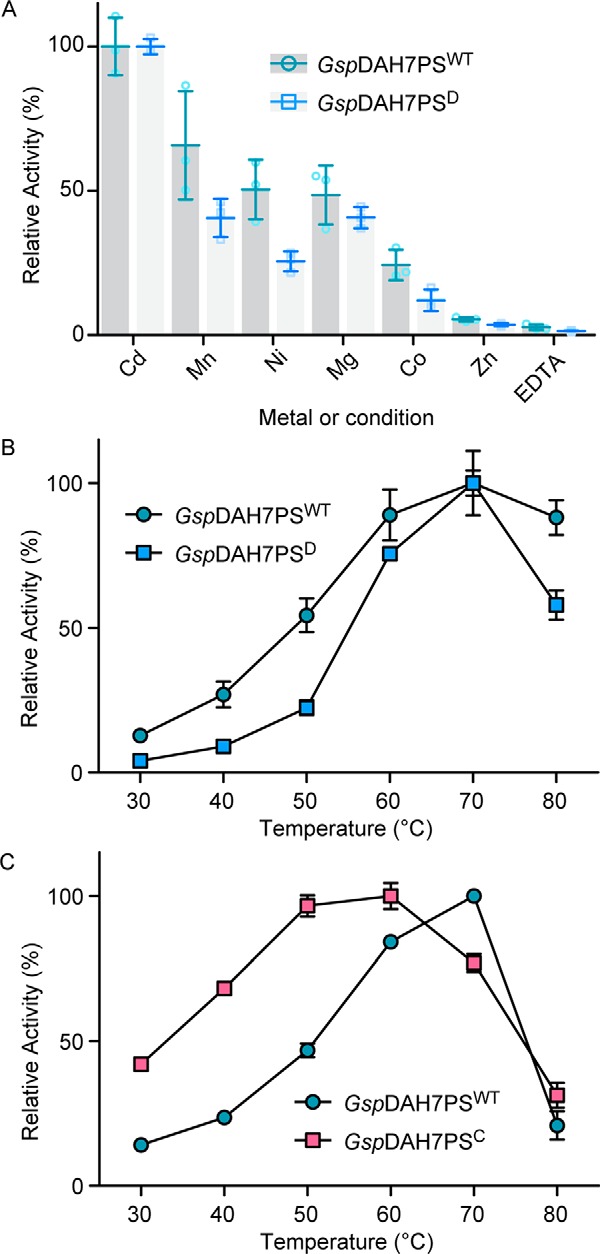
A, activation of the DAH7PS activity of GspDAH7PSWT (green) and GspDAH7PSD (blue) by different divalent metal ions (100 μm) in comparison with the activity with Cd2+. Also shown is the activity of both enzymes in the presence of the metal chelator EDTA. B, dependence of reaction rates of DAH7PS activity (B) and CM activity (C) on temperature. GspDAH7PSWT activity is shown in green; GspDAH7PSD activity is shown in blue, and GspDAH7PSC activity is shown in pink. Error bars represent the standard deviation from triplicate measurements.
Consistent with compartmentalization of activities into the respective domains, in the truncated variants only metal-dependent DAH7PS activity was observed for GspDAH7PSD, and only CM activity was detected for GspDAH7PSC. Intriguingly, whereas the DAH7PS activity of the separated DAH7PS domain was greater than that of the wild-type protein, the efficiency of catalysis of the CM reaction was reduced for the isolated CM domain (Table 1). This change was principally caused by a 5-fold increase in the chorismate Km value for the shorter GspDAH7PSC protein, yet the catalytic turnover, represented by the kcat, was 2-fold higher. The activities of both separated domains were also found to be slightly more sensitive to temperature than was found for the full-length protein (Fig. 3). Thus, the properties of the two functional domains comprising the full DAH7PS-CM protein are affected by its formation, suggesting the possibility of inter-domain communication.
Inhibition of DAH7PS Activity by Prephenate
Structural extensions to DAH7PS core catalytic barrels is a common mechanism for elaborating feedback inhibition on DAH7PS activity (4, 7, 17). The combination of DAH7PS and CM domains may represent another case of this phenomenon. To test this, we assessed whether the product of CM, prephenate, was an inhibitor of the DAH7PS activity, as this would be consistent with the fusion of the CM domain to DAH7PS providing feedback regulation to the pathway. Notably, prephenate inhibited the DAH7PS activity of the full-length GspDAH7PSWT protein but not of separated GspDAH7PSD (Fig. 4). The DAH7PS activity of the full-length enzyme was reduced to ∼50% with 20 μm prephenate and to less than 2% at higher prephenate concentrations compared with the activity in the absence of prephenate. This observation suggests that the inhibitory mechanism directly involves the CM domain, which may provide an allosteric binding site for prephenate. Due to the high CM activity of GspDAH7PSWT, the same inhibition response was observed with chorismate rather than prephenate.
FIGURE 4.
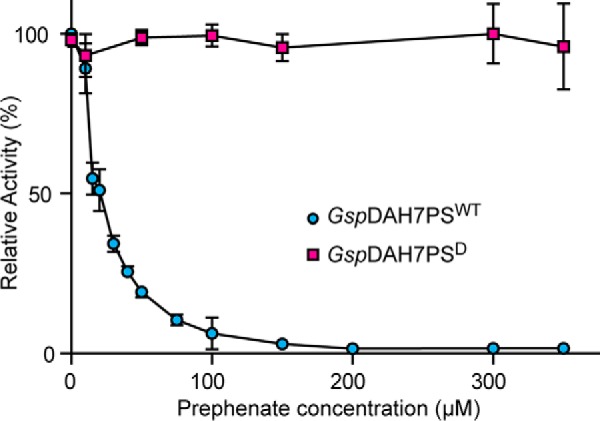
Response of GspDAH7PSWT (blue) and GspDAH7PSD (red) DAH7PS activity to increasing concentrations of prephenate at 60 °C. Error bars represent the standard deviation from triplicate measurements.
Small Angle X-ray Scattering Shows a Conformational Change in the Presence of Prephenate
To examine the effect on the structure of the complex in the presence of prephenate, experimental SAXS data were obtained for GspDAH7PS in the absence and presence of prephenate to shed light on the conformational changes associated with inhibition (Fig. 5). Comparison of the SAXS profiles of GspDAH7PS in the absence and presence of prephenate shows clear differences, consistent with a conformational change resulting from the binding of prephenate (Fig. 5A). This change is associated with an increased compactness of the enzyme in the presence of prephenate by the smaller radius of gyration, derived from the Guinier plot (Rg = 34.8 ± 0.6 Å with prephenate compared with Rg = 36.5 ± 0.4 Å without) (Fig. 5A).
FIGURE 5.
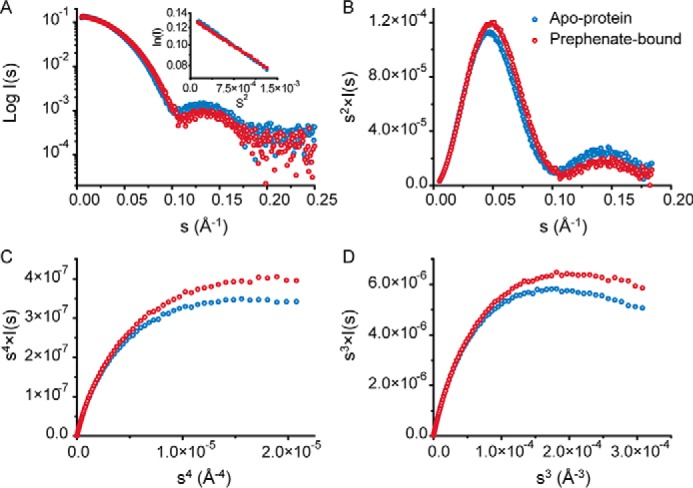
Analysis of the SAXS data for GspDAH7PS in the absence (blue) and presence (red) of prephenate. A, SAXS profiles (log I(q) versus q) with Guinier plots inset. B, Kratky plot (q2·I(q) versus q). C, Porod-Debye plot (q4·I(q) versus q4), and D, SIBYLS plot (q3·I(q) versus q3), limited to the range of the SAXS data for which the Guinier linearity is observed.
Notably, the bell-shaped curve observed in the Kratky plot (Fig. 5B) shows that in both the presence and absence of prephenate the protein exhibits a typical folded shape, without random coils. This verifies that both domains remain folded even in the absence of prephenate, although the slightly more flattened curve for the prephenate-free sample may indicate an increased flexibility of the protein. The Porod-Debye plots (Fig. 5C) present two different plateaus in these two cases, suggesting that the protein experiences a discrete conformational change upon ligand binding without any radical change in its intrinsic flexibility (18, 19). This conclusion is supported by the q3·I(q) versus q3 plot (Fig. 5D) that displays a distinct decrease, rather than a plateau as one would expect in the case of a highly flexible enzyme.
Prephenate Binds to the CM Domain at the Active Site Located within a Helical Bundle
To further probe the intriguing conformational changes in the presence of prephenate and to identify the prephenate-binding site, we determined the crystal structure of GspDAH7PS in complex with the CM product and DAH7PS inhibitor prephenate. GspDAH7PS was crystallized in the presence of Mn2+ and chorismate, and its structure was determined at a resolution of 2.75 Å in the space group P64. The asymmetric unit contains two adjacent chains, with each comprising a catalytic (β/α)8 barrel common to all DAH7PS enzymes (residues 106–362) and a CM domain (residues 1–86) connected by a short linker region (residues 86–105) (Fig. 6A). The two catalytic barrels from each chain in the asymmetric unit are highly similar (Cα r.m.s.d. = 0.11 Å), whereas the CM domains are slightly less similar (Cα r.m.s.d. = 0.23 Å), with one of the chains having more unresolved residues than the other in the linker region connecting the CM domain with the catalytic barrel. However, when taken as a whole, the two chains present a Cα r.m.s.d. of 3.93 Å. This large difference arises from the difference in location of the two CM domains relative to the barrels in each chain (Fig. 6A).
FIGURE 6.

Crystal structure of GspDAH7PS. A, two chains of the asymmetric unit, chain A (left) and chain B (right). The catalytic TIM barrel is shown in blue, the CM domain in green, and the linker region in purple. Allosteric inhibitor prephenate (Pre) is represented with gray sticks, and the active site is identified with the metal ion, represented with an orange sphere. B, tetrameric assembly with the components of one asymmetric unit colored (TIM barrel is shown in blue, the CM domain in green, and the linker region in purple) and the symmetry-related dimer is colored gray. C and D, binding sites of prephenate in the two unique subunits of the asymmetric unit. Residues important for the binding of prephenate and the catalytic activity of the CM domain are shown with sticks. One CM domain is shown in green, and the adjacent chain forming the catalytically active CM is shown in blue. Prephenate is represented with gray sticks. The σA-weighted Fo − Fc omit maps for prephenate are represented with a blue mesh and were generated by deleting the prephenate molecules from the n-1 round of refinement. Electron density is contoured at 3σ (0.1457e/Å3).
A 2-fold symmetry operation reveals the homotetrameric biological unit of GspDAH7PS (Fig. 6B). The DAH7PS barrel subunits are arranged in a similar fashion to the tetrameric assemblies observed for other structurally characterized type Iβ DAH7PS enzymes (6), with neighboring protomers placed so that adjacent barrels face in opposite directions around the tetramer plane. A manganese ion is modeled at the C-terminal end of each of the barrels and is coordinated to residues Cys-126, His-296, Glu-322, and Asp-333, identifying the locality of the DAH7PS active site. These metal-binding residues are conserved in all structurally characterized DAH7PS enzymes. The helical CM domains of two diagonally opposed chains associate in an interlaced dimer that resembles a type II or AroQ helical CM (20).
Difference electron density was observed in the area corresponding to the catalytic site of the CM domain. Because of the observed CM activity of GspDAH7PS, prephenate is generated from the added chorismate, and prephenate presence was confirmed by difference density at the CM domain active site (Fig. 6, C and D). The prephenate-binding site reveals that the residues involved in its binding correspond to a subset of the residues responsible for the catalytic activity of the CM domain (Fig. 6, C and D). More specifically, the position of Arg-10, Arg-27, Lys-38, Asp-47, and Glu-51 suggests they form hydrogen bonds and other electrostatic interactions with prephenate, whereas Val-31, Ile-34, Met-54, Phe-79, and Leu-83 create a hydrophobic shell surrounding the ligand and the aliphatic side chain of Arg-50 contributing to these hydrophobic interactions.
Despite low sequence identity (35%, Fig. 2) the CM domains of GspDAH7PS align well with the structure of E. coli AroQ CM (20) (EcoCM, r.m.s.d. = 2.23 Å) (PDB code 1ECM; Fig. 7A). The GspDAH7PS CM helices show more curvature than is observed in the EcoCM structure, especially noticeable at the end of the first helix (Fig. 7A), which may be the result of the interactions between the CM domains and the DAH7PS barrels. Moreover, the second and third helices of the GspCM domains are shorter than their EcoCM counterparts, with the end of the third α-helix of EcoCM corresponding to the beginning of the linker region, which connects the GspDAH7PS CM domain to the catalytic barrel. The well characterized catalytic site of EcoCM (20–23) exhibits an excellent structural alignment with the active site of the CM domain of GspDAH7PS (Fig. 7B), with the catalytically important charged and polar residues shared by both proteins. Hydrophobic residues Ala-32, Val-35, Ile-81, and Val-85 of EcoCM are not conserved in GspDAH7PS but instead are replaced by alternative hydrophobic residues Val-31, Ile-34, Phe-79, and Leu-83, respectively. In addition, Ser-84, which had been postulated as being important in EcoCM (21), is not conserved and replaced by Ala in GspDAH7PS. These alterations may account for the relatively low CM activity of the GspDAH7PSWT and the truncated GspDAH7PSC protein.
FIGURE 7.
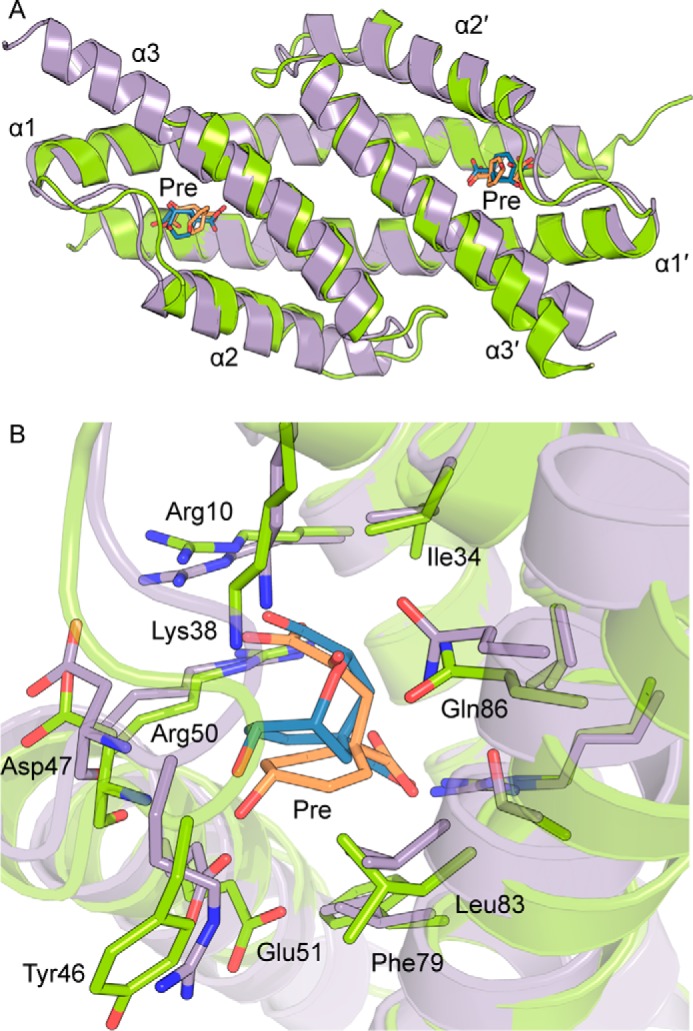
A, structural alignment of the CM domains of GspDAH7PS (residues 1–94) and EcoCM (PDB code 1ECM). GspDAH7PS is shown in green with prephenate in orange sticks, and EcoCM is shown in purple with the transition state intermediate in blue sticks. B, superimposition of the CM active site. Residues of GspDAH7PS are labeled.
For the structures of LmoDAH7PS, which have no ligand bound in the CM active site, the CM domains are found further from the catalytic DAH7PS barrels in an “open” position when compared with the “closed” position they adopt in the inhibited prephenate-bound GspDAH7PS structure. LmoDAH7PS shares sequence identity of 75.1% over 361 residues (91.1% similarity, Fig. 2) with GspDAH7PS, and structural alignment of tetrameric GspDAH7PS with LmoDAH7PS yields r.m.s.d. values of 0.82 Å (PDB code 3NVT) and 1.08 Å (PDB code 3TFC).
Prephenate Binding Is Associated with Closer Association between the Catalytic and Regulatory Domains
To estimate the likely structural changes of GspDAH7PS on the binding of prephenate, a full model of GspDAH7PS in its active open form, referred to hereafter as the “open model,” based on the two known prephenate-free LmoDAH7PS structures was constructed by homology using MODELLER (24). In addition, missing residues and side chains in the crystal structure of GspDAH7PS were reconstructed, and their position optimized, with MODELLER (referred to as the “closed model”).
Comparison of these open and closed models sheds light on the structural changes likely associated with the binding of prephenate and mechanism of the allosteric inhibition of GspDAH7PS (Fig. 8). In the absence of prephenate, there is little contact between the CM domain and the DAH7PS barrels (7.6% solvent-accessible surface area (SASA) of the tetrameric barrels buried by the CM domains), and the structure is extended (Fig. 8A), with the entrance to the active sites of the DAH7PS domains accessible for substrate binding (Fig. 8C). In comparison, the closed model with prephenate bound is more compact (Fig. 8B), with the CM domains slightly bent to partly follow the shape of the DAH7PS barrels (15.6% of the SASA of the tetrameric DAH7PS barrels buried by the CM domains). In addition, a number of hydrogen bonds are formed between the DAH7PS and CM domains, in particular between Asp-326 and Arg-52, Tyr-46 and Glu-129, and Arg-175 and Gly-42, occluding in part the entrance to the active sites, suggesting an explanation for the inhibitory effect of prephenate against the DAH7PS activity (Fig. 8C).
FIGURE 8.

Open (A) and closed (B) models of GspDAH7PS correspond, respectively, to the apo and allosterically inhibited states of the enzyme. The open model consists of a homology model based on the deposited structures of LmoDAH7PS, and the closed model corresponds to the crystal structure of GspDAH7PS for which the missing atoms have been modeled. The CM domains are colored in green, the linker regions in purple, and the main DAH7PS barrels in blue. The substrate PEP and the allosteric inhibitor prephenate are shown in spheres in A and B, respectively, whereas the metal ions are represented with orange spheres. For each model, the inset represents a zoom on a particular region of the CM domain that is disorganized in the open form model and reorganized to accommodate prephenate in the closed form model. In these insets, the residues important for the binding of prephenate are displayed with sticks, and prephenate is shown with gray spheres. C, molecular surface representation of the closed (top) and open (bottom) models of GspDAH7PS. The active site of the DAH7PS domain is visible in the open form model and is identified with a red arrow which points to PEP (shown in sticks).
Portions of CM domains that are disordered in the open model, namely the inter-helical loops, the N-terminal region, and the beginning of the linker region, are found to be ordered in the closed model and lead to extended CM helices (Fig. 8, A and B). These parts of the CM domains include residues identified as being responsible for the catalytic activity of the CM domains and for the binding of prephenate, in particular, Ile-34, Lys-38, Asp-47, Arg-50, and Glu-51. We can therefore postulate that the binding of chorismate or prephenate is accompanied by a reorganization of the secondary structure of the CM domains, viz. the formation of longer helices. This reorganization, together with the binding of the allosteric ligand itself, significantly rigidifies the CM domains, favoring a conformation in which these domains lean on the DAH7PS catalytic barrels.
SAXS Data Confirm That the Conformational Compaction upon Prephenate Binding Observed Crystallographically Also Occurs in Solution
To confirm the conformational changes upon prephenate binding observed in the crystal structure of GspDAH7PS and to gain insights into the possible allosteric regulation mechanism adopted by this enzyme, the SAXS data recorded in the absence and presence of prephenate were compared with the open and closed (prephenate-bound) models of the protein (Fig. 9). The theoretical scattering curves calculated for each model present a good fit with the corresponding experimental curves (χ2 = 0.70 and 0.37 for the closed and open forms, respectively). In contrast, the comparisons of the theoretical curve of the open structure to the data for prephenate-bound GspDAH7PS, and of the closed form to the apoenzyme data, give poor fits (χ2 = 8.02 and 3.13, respectively).
FIGURE 9.
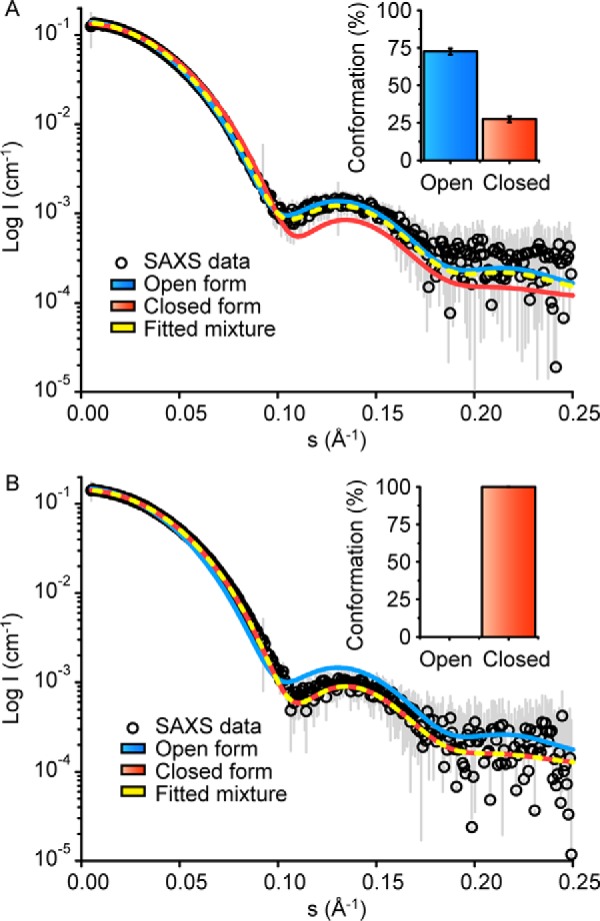
Distribution of open and closed conformations in GspDAH7PS with or without prephenate in solution. Theoretical scattering profiles were generated from models of both the prephenate-free open form (blue line) and the prephenate-bound closed form (red line) using OLIGOMER. These theoretical profiles were compared with the experimental GspDAH7PS SAXS data (open circles) in both the absence (A) and the presence (B) of prephenate. The experimental SAXS data were fitted to a distribution of both open and closed forms using OLIGOMER (yellow dashed line), and column charts show the percentages of open and closed forms GspDAH7PS adopted in solution with and without prephenate.
The experimental SAXS data were further fitted using a mixture of open and closed models of GspDAH7PS. In the absence of prephenate, the analysis shows a better fit of the theoretical scattering from a combination of both open and closed GspDAH7PS models (χ2 = 0.22) to the experimental data obtained in the absence of prephenate. This combination predicts that ≈73% of the protein adopts the open conformation, and ≈27% of the protein adopts the closed form conformation (Fig. 9A). On this basis, a conformational equilibrium may be established between the open and closed forms in the absence of ligand binding to the CM domains, and intermediate states between the open and closed forms may be present. Nevertheless, the open conformation or open-like structures are predominant in the absence of prephenate, suggesting that the closed form is less stable or more difficult to access. In the presence of prephenate, the best fit to the experimental profile (χ2 = 0.37) is obtained with that of the closed GspDAH7PS model alone, predicting that the closed structure, or a set of conformationally similar structures, is the sole form adopted by the prephenate-bound protein (Fig. 9B). These data are consistent with the full loss of DAH7PS activity of GspDAH7PS by prephenate binding observed experimentally.
Discussion
Multifunctional enzymes are observed in many living systems, and they play important roles in substrate channeling, enzyme compartmentalization, and metabolic control (25). In the thermophilic soil bacterium Geobacillus sp., a CM domain is found fused to the N terminus of the first enzyme dedicated to aromatic amino acid biosynthesis, DAH7PS. We have shown here that this fusion gives rise to a bifunctional enzyme presenting both DAH7PS and CM catalytic activity. Additionally, we have shown that the smaller enzyme, CM, can bind its product prephenate, and this inhibits the activity of the fusion partner DAH7PS.
The helical AroQ form of CM is frequently observed as a fusion partner to enzymes involved in Tyr and Phe biosynthesis. For example, not only is it attached to the DAH7PS enzyme studied herein, it is found fused to both prephenate dehydratase and prephenate dehydrogenase enzymes (26–30). Geobacillus sp. has both the AroQ form (fused) and the other form of CM, AroH, encoded by its genome. Sequence inspection reveals that this AroH variant does not seem to be fused to another protein. For the B. subtilis DAH7PS enzyme, the CM catalytic activity of the AroQ in the fusion protein was shown to be extremely low, and it was predicted that the AroH protein was the major contributor to CM activity, and the primary role of the CM domain of the equivalent CM-DAH7PS fusion was for allosteric control (15). In contrast, the AroQ CM domain of the hyperthermophilic Geobacillus sp. fusion protein has moderate activity, suggesting it may play a dual role in both providing catalytic function in tandem with the AroH protein as well as providing a feedback mechanism to control entry into the pathway. Notably, the CM catalytic efficiency is enhanced by its association with DAH7PS, principally due to its significantly lower Michaelis constant.
Through a combination of enzyme kinetics, x-ray crystallography, SAXS, and modeling, we can now build a picture of the molecular details of how the feedback inhibition of DAH7PS is achieved in this class of DAH7PS enzymes. In the absence of the allosteric ligand prephenate, the CM domains remain at a distance from the DAH7PS catalytic barrels, permitting the substrates to enter the DAH7PS active site. The binding of prephenate to the CM domain is then accompanied by a conformational change, evident from SAXS data and comparison of our prephenate-bound crystal structure with the homologous unbound structure of LmoDAH7PS. The latter structure shows a far more extended structure in the absence of ligands within the CM domain binding site. In the presence of prephenate, the protein adopts a more compact and more ordered form, with the extension of helices in the CM domain, which bear residues forming binding contacts with prephenate. Furthermore, the SAXS data suggest that the CM domains remain close to the DAH7PS catalytic barrels in the presence of prephenate, because of the very good fit obtained for the data in the presence of inhibitor, using the closed form model. The compactness of the structure impacts on substrate access to the DAH7PS active site and thus inhibits the enzyme. Conformational transitions and the occasional sampling of conformations resembling the closed form may account for the observed slightly higher activity of the truncated DAH7PS mutant lacking the CM domain.
This mechanism of allostery involving physical gating of the active site has been employed for other DAH7PS enzymes, albeit with structurally distinct regulatory domains. The T. maritima DAH7PS bears an ACT regulatory domain, which on inhibitor Tyr binding also restricts access to the active site (7). These two quite distinct allosteric solutions suggest that adopting this structural approach for the control DAH7PS represents a general mechanism, albeit that both of these allosteric control mechanisms require the same basic tetrameric structure for the DAH7PS modules to operate. Other DAH7PS enzymes have been shown to adopt allosteric regulation mechanisms based on changes in protein dynamics (13, 14) rather than using a physical gating mechanism, which reinforces the versatility of both the protein fold and the approaches to allosteric control.
As noted above, the acquisition of the CM domain by DAH7PS comes with a slight burden to its catalytic function, as evidenced by the moderate increase in DAH7PS catalytic efficiency observed for the truncated protein of GspDAH7PSD. In contrast, the CM activity benefits from its association with its DAH7PS partner, with its independent activity following excision being reduced. This reduction suggests that the transient contacts the CM dimer forms with its DAH7PS partner in the full-length protein are important for its catalytic function as well as for the delivery of allostery. Although it is difficult to unravel the individual contributions to binding and catalysis from the steady-state kinetic parameters, it is notable that the CM activity of the full-length protein has a lower chorismate Km value (by a factor of almost 5) and a lower kcat value (almost a 3-fold decrease). These changes reveal the impact domain interaction observed in the x-ray structure has on CM catalytic function. The decrease in Km value for the fused CM may reflect the contribution that conformational restriction affords by the interaction with DAH7PS, whereas the lower kcat value may reflect the tailoring of the CM active site for prephenate binding and the provision of allostery for the DAH7PS activity, rather than for catalyzing the CM reaction.
The connected and overlapping functions between enzymatic activities demonstrate the complexity of cellular mechanisms for both providing allosteric control and delivering cellular biosynthetic functionality. DAH7PS, through its associations and varied mechanisms for providing allosteric control, provides insight into the evolutionary processes that provide solutions to biosynthetic control.
Experimental Procedures
Construction of Plasmids
The gene sequence encoding GspDAH7PSWT (strain Y412MC61) was codon-optimized for expression in E. coli, synthesized by Geneart (Life Technologies, Inc.), and ligated into the expression vector pET-28b between the NcoI and XhoI sites to give a protein with a non-cleavable C-terminal His6 tag. This plasmid was used as the template for the preparation of constructs for expressing GspDAH7PSD (the DAH7PS portion of GspDAH7PSWT) and GspDAH7PSC (the CM portion of GspDAH7PSWT) variants, created by truncating between Lys-92 and Ala-93. Genes encoding the truncated proteins were amplified using gene-specific primers (Table 2) for cloning using the Gateway system (Life Technologies, Inc.). The products from the first round of PCR were extended using primers that incorporated the attb1 site and attb2 site, and the PCR products were gel-purified. The purified products were cloned into the donor vector pDONR-221 using BP clonase enzyme mix (Life Technologies, Inc.), and the product was subsequently transformed into E. coli One Shot® TOP10 cells (Life Technologies, Inc.). Purified plasmids from transformants were sequence-verified. LR clonase enzyme mix (Life Technologies, Inc.) was used to sub-clone into the destination vector pDEST-17 (for the construct encoding GspDAH7PSD), which encodes an N-terminal His tag and TEV protease cleavage site, or pDEST-15 (for the construct encoding GspDAH7PSC), which encodes an N-terminal GST tag, a His tag, and a TEV protease cleavage site.
TABLE 2.
Primers for generation of constructs
| GspDAH7PSC primers (round 1) | |
| Forward primer | 5′-GGCAGCGGCGCGATGGGTAATGAACGTCTG |
| Reverse primer | 5′-GAAAGCTGGGTGTTATTTACGATGATCATCTTC |
| GspDAH7PSD primers (round 1) | |
| Forward primer | 5′-GGCAGCGGCGCGATGGCACTGCTGGTTAGC |
| Reverse primer | 5′-GAAAGCTGGGTGTTACTCGAGTGCACGAAC |
| Generic primers (round 2) | |
| Forward primer | 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAAA CCTGTATTTTCAGGGCAGCGGCGCG |
| Reverse primer | 5′-GGGGACCACTTTGTACAAGAAAGCTGGT |
Expression and Purification of GspDAH7PSWT, GspDAH7PSD, and GspDAH7PSC
The vectors bearing the genes of GspDAH7PSWT and GspDAH7PSC were transformed into E. coli BL21 (DE3) cells, whereas the vector bearing the gene coding for GspDAH7PSD was transformed into E. coli BL21 (DE3) pBB540/pBB542 cells for expression. Cultures were grown at 37 °C in 1 liter of Luria-Bertani (LB) medium supplemented with 100 μg/ml kanamycin for GspDAH7PSWT, 100 μg/ml ampicillin for GspDAH7PSC, and a combination of 100 μg/ml ampicillin, 25 μg/ml chloramphenicol, and 10 μg/ml spectinomycin for GspDAH7PSD. Expression of target protein was induced through the addition of 0.5 mm isopropyl β-d-thiogalactopyranoside, and the cultures were grown for a further 4 h at 37 °C (for expression of GspDAH7PSWT) or overnight at 23 °C (for the expression of GspDAH7PSD and GspDAH7PSC). Cells were harvested by centrifugation and resuspended in lysis buffer (50 mm 1,3-bis(tris(hydroxymethyl) methylamino) propane (BTP), 200 mm KCl, 2 mm dithiothreitol (DTT), 200 μm PEP, pH 7.2, EDTA-free protease inhibitor) prior to lysis by sonication.
Purification of GspDAH7PSWT and GspDAH7PSD
The crude lysate was heat-treated (30 min at 70 °C for GspDAH7PSWT and 55 °C for GspDAH7PSD) and then clarified by centrifugation at 21,000 × g for 35 min. The supernatant was recovered and filtered, and after addition of 50 mm imidazole, it was loaded onto a HisTrap HP column (GE Healthcare) that had been pre-equilibrated with the binding buffer (20 mm sodium phosphate, 500 mm NaCl, 50 mm imidazole, pH 7.4). The column was washed with binding buffer, and protein was eluted with elution buffer (containing 500 mm imidazole). Fractions containing enzyme were pooled together and run through a desalting column eluting with binding buffer to lower the imidazole concentration, before overnight treatment with TEV protease at a 1% mol/mol concentration (for GspDAH7PSD only). TEV-treated enzyme was subsequently run through a second HisTrap HP column to separate protease and His tag from cleaved enzyme using the same elution conditions as used previously. Untagged enzyme was pooled together for size-exclusion chromatography (SEC).
Purification of GspDAH7PSC
The crude lysate was centrifuged at 21,000 × g for 35 min, and the supernatant was recovered, filtered, and loaded onto a GSTrap HP column (GE Healthcare) that had been pre-equilibrated with the binding buffer (140 mm NaCl, 2.7 mm KCl, 10 mm sodium phosphate buffer, pH 7.3). The column was washed with binding buffer, and protein was eluted with elution buffer (50 mm Tris-HCl containing 10 mm reduced glutathione, pH 8.0). Fractions containing enzyme were pooled together and run through a desalting column eluting with binding buffer, before overnight treatment with TEV protease at a 1% mol/mol concentration. TEV-treated enzyme was subsequently run through a second GSTrap HP column to separate protease and GST tag from cleaved enzyme. Untagged enzyme eluted in the flow-through of the second column, and fractions were pooled for SEC.
All proteins were purified by SEC (Superdex 200 26/60) into buffer containing BTP (10 mm, pH 7.4), KCl (40 mm), PEP (200 μm), and EDTA (10 μm). Fractions containing the desired protein (as determined by gel electrophoresis) were concentrated by ultrafiltration, and aliquots were flash-frozen with liquid N2 and stored at −80 °C.
Determination of Enzymatic Activity
Assays were performed in 50 mm BTP buffer, pH 6.8, at the stated temperature in a total volume of 1 ml. Initial rates were fitted to the Michaelis-Menten equation to determine kinetic parameters using GraFit (Erithacus software).
DAH7PS Assay
A modified continuous assay of Schoner and Herrmann (31) was used as described previously by Schofield et al. (32) for use at elevated temperatures. Enzyme activity was monitored by following the loss of absorbance at 232 nm due to consumption of PEP at 60 °C in the presence of CdCl2 (final concentration of 100 μm) following the addition of E4P to the assay solution containing PEP and enzyme (2.8–3.5 × 10−2 μm). For the determination of kinetic parameters, initial rates of reaction were determined by keeping the concentration of one substrate fixed (PEP concentration was fixed at 338 μm and E4P concentration was fixed at 329 μm) while varying the concentration of the other substrate. The metal activation profiles of GspDAH7PSWT and GspDAH7PSD were determined using standard assays with 195 μm PEP and 197 μm E4P at 60 °C in the presence of 100 μm CdCl2, MnSO4, MgSO4, Zn(CO2CH3)2, NiCl2, CoCl2, EDTA, dipicolinic acid, or no chelator or metal ion (control) and enzyme (2.8 × 10−2 μm).
CM Assay
Steady-state kinetics were measured using a continuous assay monitoring the disappearance of chorismate at 274 nm at 50 °C (extinction coefficient of 2,630 m−1 cm−1 at 50 °C and pH 6.8). Reactions were initiated with the addition of chorismate (Sigma) at a desired concentration to a buffer solution containing enzyme (0.94 μm).
Temperature Dependence of Enzyme Activity
DAH7PS activity was measured at 325 μm PEP, 475 μm E4P for GspDAH7PSWT and 337 μm PEP, 356 μm E4P for GspDAH7PSD and 100 μm Cd2+ in 50 mm BTP, pH 6.8. Enzyme (2.5 × 10−2 μm for GspDAH7PSWT and 10−2 μm for GspDAH7PSD) was added for 5 min prior to the initiation of activity by the addition of E4P. CM activity was measured at 70 μm chorismate for GspDAH7PSWT and 80 μm chorismate for GspDAH7PSC in 50 mm BTP, pH 6.8. Enzyme (0.9 μm for GspDAH7PSWT and 1.25 μm for GspDAH7PSC) was added 5 min prior to initiation of the reaction with chorismate.
Inhibition by Prephenate
The inhibition profiles of DAH7PS activity for GspDAH7PSWT and GspDAH7PSD were determined by varying the concentration of prephenate (Sigma) in buffer with CdCl2 (100 μm), PEP (121 μm), and enzyme (2.5 × 10−2 μm) incubated at 60 °C for 10 min. The reactions were initiated by the addition of E4P (123 μm).
Crystallization and Structure Solution
Crystals of GspDAH7PSWT were grown by hanging-drop vapor diffusion. A protein solution (11 mg ml−1 in 20 mm BTP, 40 mm KCl, 200 μm PEP, pH 7.4) was mixed 1:1 (v/v) with a reservoir solution containing 0.2 m sodium citrate, 0.1 m BTP, pH 6.5, 20% w/v polyethylene glycol (PEG) 3350, and 0.2 mm chorismic acid. The drop sizes were 2 μl, and the reservoir volume was 500 μl. The crystallization trays were incubated at 20 °C until immediately before loading the crystals into the loop for flash cooling prior to data collection. The reservoir solution containing 20% PEG 400 was used as a cryoprotectant. Intensity data were collected at the Australian Synchrotron using the MX2 beamline with an ADSC Quantum 315r detector (Area Detector Systems Corp.) at a temperature of 110 K. The collection and refinement data are provided in Table 3.
TABLE 3.
X-ray data collection and refinement statistics
| PDB 5J6F | |
|---|---|
| Data collection | |
| Space group | P64 |
| Cell dimensions | |
| a, b, c (Å) | 95.44, 95.44, 167.45 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 82.66–2.75 (2.88–2.75)a |
| Unique reflections | 22,482 |
| Rmeas | 0.085 (1.443) |
| Mn(I/σ(I)) | 18.0 (1.9) |
| Completeness (%) | 99.9 (100) |
| Redundancy | 7.6 (7.7) |
| CC1/2 | 0.818 |
| Wilson B-factor (Å2) | 69.0 |
| Refinement | |
| Resolution (Å) | 82.66–2.75 |
| No. of reflections | 21,178 |
| Rwork/Rfree | 0.1822/0.2352 |
| No. of atoms | |
| Protein | 5,366 |
| Ligand (prephenate) | 32 |
| Water | |
| Other (Mn, SO4) | 22 |
| Mean B-factor (Å2) | |
| Protein | 78.54 |
| Ligand (prephenate) | 88.82 |
| Water | |
| Other (Mn, SO4) | 84.78 |
| r.m.s.d from target values | |
| Bond lengths (Å) | 0.019 |
| Bond angles (°) | 2.120 |
| Ramachandran analysisb | |
| Favored regions (%) | 98.1 |
| Allowed regions (%) | 1.9 |
| Outliers (%) | 0.0 |
a Values shown in parentheses are for the highest resolution shell.
b Data are defined by Coot.
Phasing and Model Refinement
Diffraction data sets were processed and scaled using XDS (33) and Aimless (34). Initial phase estimates were calculated by molecular replacement using PHASER (35) with the monomeric core barrel (with CM domains removed) from the previously determined structure of LmoDAH7PS (PDB code 3TFC) as the search model. Two core barrels were placed in the asymmetric unit, and after rigid body refinement using REFMAC5 (36), this solution was further used to place the CM domains in the difference Fourier map using phased molecular replacement in MOLREP (37). Further structural refinement was performed using REFMAC5, and electron density maps were analyzed using Coot (38). Prephenate was modeled after the penultimate round of refinement. Structures were validated using the MolProbity server (39) and the tools of Coot.
Reconstruction of the Missing Residues and Side Chains of GspDAH7PS
Residues 1–2 and 355–362 in chain A as well as residues 88–101 and 355–362 in chain B are absent in the crystal structure model of GspDAH7PS obtained after refinement due to the weak electron density in these regions. These residues, as well as some unmodeled side chains, were reconstructed using MODELLER (24) version 9.13. First, the tetrameric form was generated from the asymmetric dimeric unit in PyMOL (40), and the topologies and parameters for prephenate and Mn2+, obtained from the SwissParam server (41) and the MM3 force field (42–44), respectively, were added to the MODELLER library. Second, 100 models of GspDAH7PS were generated using the “automodel” routine of MODELLER tuned to improve model accuracy. More specifically, the variable target function method was set to slow, and the maximum number of conjugate gradient iterations was set to 300, although the level of molecular dynamics refinement was adjusted to slow and the complete optimization-refinement process was repeated twice. The “env.io.hetatm” instruction was defined as true to include the metal ion and prephenate in the model. During the model generation, only the missing residues and residues with missing atoms were allowed to move, and the chorismate mutase domains were restrained to keep their secondary structure intact, and a symmetry restraint was applied on the α-carbons of the symmetric chains of the tetramer. The best model was selected among the models presenting the lowest MODELLER objective function value and the lowest global DOPE score, following visual inspection and residue-by-residue DOPE score analysis. The C-terminal tail of this model was further refined using the “loopmodel” class of MODELLER (45) using the same protocol as described above, and the best of the 100 models generated was assessed using the same evaluation procedure.
Homology Model Construction of GspDAH7PS in Its Open Form
Homology modeling of GspDAH7PS in its open, active form was performed using the two previously determined structures of DAH7PS from L. monocytogenes EGD-e (PDB codes 3NVT and 3TFC) as templates. For each structure of LmoDAH7PS, the tetrameric form was generated in PyMOL, and the topology and parameters of PEP obtained with SwissParam as well as those of Mn2+ were included into MODELLER libraries. The missing residues and side chains were then added with MODELLER using automodel to create 25 models using the same parameters and the same assessment procedure as described above. The best model for each of the structures was aligned, and their sequences were aligned against the full sequence of GspDAH7PS using T-COFFEE (46). The homology model of GspDAH7PS in its open form was performed with MODELLER using the automodel routine as before with the env.io.hetatm instruction set to true to include PEP and Mn2+ in the model. Secondary structure restraints were applied to the chorismate mutase domains, and symmetry restraints were used for the symmetric chains of the tetramer. A total of 100 models were generated, as above, and the best model was selected among the models presenting the lowest MODELLER objective function value and the lowest global DOPE score, following visual inspection and residue-by-residue DOPE score analysis.
SAXS
Measurements were performed at the Australian Synchrotron SAXS/WAXS beamline equipped with a Pilatus detector (1M, 170 × 170 mm, effective pixel size, 172 × 172 μm). The wavelength of the x-rays was 1.0332 Å. The sample detector distance was 2.6 m, which provided an s range of 0.01–0.35 Å−1 (where q is the magnitude of the scattering vector, which is related to the scattering angle (2θ) and the wavelength (λ) as follows: s = (4π/λ)sin(θ). Data were collected from samples in a 1.5-mm thin-walled glass capillary at 25 °C at 2-s intervals.
Scattering data were collected from GspDAH7PSWT following elution from an SEC column (Superdex 200 5/150), pre-equilibrated with SEC buffer (10 mm BTP, pH 7.4, 150 mm NaCl, 200 μm PEP) or SEC buffer with prephenate (10 mm BTP, pH 7.4, 150 mm NaCl, 200 μm PEP, 400 μm prephenate).
Raw scattering data were processed with Scatterbrain developed at the Australian Synchrotron. Scattered intensity (I) was plotted versus s using Primus (47) and Origin 8. All samples were devoid of an increase in intensity at low s (indicative of aggregation). Guinier plots were linear for sRg <1.3. The data sets for structural analysis were recorded with 447 data points over the range 0.005 ≤ s ≤ 0.35 Å−1. Theoretical scattering curves were generated from the models of open form (active) and closed form (inactive) GspDAH7PS generated as described above and compared with experimental scattering curves using OLIGOMER (47). Theoretical scattering data of two distinct GspDAH7PS forms were further mixed and fitted to the individual experimental scattering profile in the presence or absence of prephenate with OLIGOMER to investigate the volume fraction of each form in the solution with and without ligand (47).
Additional Computational Methods
SASA for the open and closed models were calculated in VMD (48) using the “measure sasa” command and using a solvent probe radius of 1.4 Å. Sequence identities were determined using LALIGN (49) using the BLOSUM50 scoring matrix with an opening gap penalty of −12 and an extending gap penalty of −2. Sequence alignments were generated with T-Coffee (50) and rendered with ESPript 3 (51). Structural figures were generated with PyMOL.
Author Contributions
A. R. N., M. H. O., and Y. B. carried out the experiments. E. J. M. L., Y. B., and M. H. O. analyzed the data. S. P., V. L. M., and T. M. A. assisted with the x-ray structure solution. E. J. P. conceived and coordinated the study. A. R. N., E. J. M. L., T. M. A., Y. B., and E. J. P. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Data were collected at the MX and SAXS beamlines of the Australian Synchrotron with access provided by the New Zealand Synchrotron Group.
This work was supported by New Zealand Marsden Fund Grant UOC1105, the Biomolecular Interaction Centre, and the University of Canterbury High Performance Computing. The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (code 5J6F) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DAH7PS
- 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase
- BTP
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- CM
- chorismate mutase
- E4P
- d-erythrose 4-phosphate
- GspDAH7PS
- Geobacillus sp. DAH7PS
- GspDAH7PSC
- Geobacillus sp. CM domain only
- GspDAH7PSD
- Geobacillus sp. DAH7PS domain only
- PDB
- Protein Data Bank
- PEP
- phosphoenolpyruvate
- SEC
- size-exclusion chromatography
- TEV
- tobacco etch virus
- r.m.s.d.
- root mean square deviation
- SAXS
- small angle x-ray scattering
- SASA
- solvent-accessible surface area.
References
- 1. Knaggs A. R. (2003) The biosynthesis of shikimate metabolites. Nat. Prod. Rep. 20, 119–136 [DOI] [PubMed] [Google Scholar]
- 2. Light S. H., and Anderson W. F. (2013) The diversity of allosteric controls at the gateway to aromatic amino acid biosynthesis. Protein Sci. 22, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. König V., Pfeil A., Braus G. H., and Schneider T. R. (2004) Substrate and metal complexes of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from Saccharomyces cerevisiae provide new insights into the catalytic mechanism. J. Mol. Biol. 337, 675–690 [DOI] [PubMed] [Google Scholar]
- 4. Shumilin I. A., Zhao C., Bauerle R., and Kretsinger R. H. (2002) Allosteric inhibition of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase alters the coordination of both substrates. J. Mol. Biol. 320, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 5. Cross P. J., Pietersma A. L., Allison T. M., Wilson-Coutts S. M., Cochrane F. C., and Parker E. J. (2013) Neisseria meningitidis expresses a single 3-deoxy-d-arabino heptulosonate 7-phosphate synthase that is inhibited primarily by phenylalanine. Protein Sci. 22, 1087–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schofield L. R., Anderson B. F., Patchett M. L., Norris G. E., Jameson G. B., and Parker E. J. (2005) Substrate ambiguity and crystal structure of Pyrococcus furiosus 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase: an ancestral 3-deoxyald-2-ulosonate-phosphate synthase? Biochemistry 44, 11950–11962 [DOI] [PubMed] [Google Scholar]
- 7. Cross P. J., Dobson R. C., Patchett M. L., and Parker E. J. (2011) Tyrosine latching of a regulatory gate affords allosteric control of aromatic amino acid biosynthesis. J. Biol. Chem. 286, 10216–10224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lang E. J., Cross P. J., Mittelstädt G., Jameson G. B., and Parker E. J. (2014) Allosteric ACTion: the varied ACT domains regulating enzymes of amino acid metabolism. Curr. Opin. Struct. Biol. 29, 102–111 [DOI] [PubMed] [Google Scholar]
- 9. Wu J., Sheflyan G. Y., and Woodard R. W. (2005) Bacillus subtilis 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase revisited: resolution of two long-standing enigmas. Biochem. J. 390, 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Webby C. J., Baker H. M., Lott J. S., Baker E. N., and Parker E. J. (2005) The structure of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes. J. Mol. Biol. 354, 927–939 [DOI] [PubMed] [Google Scholar]
- 11. Blackmore N. J., Reichau S., Jiao W., Hutton R. D., Baker E. N., Jameson G. B., and Parker E. J. (2013) Three sites and you are out: ternary synergistic allostery controls aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J. Mol. Biol. 425, 1582–1592 [DOI] [PubMed] [Google Scholar]
- 12. Sasso S., Okvist M., Roderer K., Gamper M., Codoni G., Krengel U., and Kast P. (2009) Structure and function of a complex between chorismate mutase and DAHP synthase: efficiency boost for the junior partner. EMBO J. 28, 2128–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiao W., Hutton R. D., Cross P. J., Jameson G. B., and Parker E. J. (2012) Dynamic cross-talk among remote binding sites: the molecular basis for unusual synergistic allostery. J. Mol. Biol. 415, 716–726 [DOI] [PubMed] [Google Scholar]
- 14. Lang E. J., Heyes L. C., Jameson G. B., and Parker E. J. (2016) Calculated pKa variations expose dynamic allosteric communication networks. J. Am. Chem. Soc. 138, 2036–2045 [DOI] [PubMed] [Google Scholar]
- 15. Wu J., and Woodard R. W. (2006) New insights into the evolutionary links relating to the 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase subfamilies. J. Biol. Chem. 281, 4042–4048 [DOI] [PubMed] [Google Scholar]
- 16. Light S. H., Halavaty A. S., Minasov G., Shuvalova L., and Anderson W. F. (2012) Structural analysis of a 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase with an N-terminal chorismate mutase-like regulatory domain. Protein Sci. 21, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Webby C. J., Jiao W., Hutton R. D., Blackmore N. J., Baker H. M., Baker E. N., Jameson G. B., and Parker E. J. (2010) Synergistic allostery, a sophisticated regulatory network for the control of aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 285, 30567–30576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hammel M. (2012) Validation of macromolecular flexibility in solution by small-angle x-ray scattering (SAXS). Eur. Biophys. J. 41, 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee A. Y., Karplus P. A., Ganem B., and Clardy J. (1995) Atomic structure of the buried catalytic pocket of Escherichia coli chorismate mutase. J. Am. Chem. Soc. 117, 3627–3628 [Google Scholar]
- 21. Hur S., and Bruice T. C. (2002) The mechanism of catalysis of the chorismate to prephenate reaction by the Escherichia coli mutase enzyme. Proc. Natl. Acad. Sci. U.S.A. 99, 1176–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu D. R., Cload S. T., Pastor R. M., and Schultz P. G. (1996) Analysis of active site residues in Escherichia coli chorismate mutase by site-directed mutagenesis. J. Am. Chem. Soc. 118, 1789–1790 [Google Scholar]
- 23. Lassila J. K., Keeffe J. R., Kast P., and Mayo S. L. (2007) Exhaustive mutagenesis of six secondary active-site residues in Escherichia coli chorismate mutase shows the importance of hydrophobic side chains and a helix N-capping position for stability and catalysis. Biochemistry 46, 6883–6891 [DOI] [PubMed] [Google Scholar]
- 24. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 25. Elleuche S. (2015) Bringing functions together with fusion enzymes-from nature's inventions to biotechnological applications. Appl. Microbiol. Biotechnol. 99, 1545–1556 [DOI] [PubMed] [Google Scholar]
- 26. Tzin V., Malitsky S., Aharoni A., and Galili G. (2009) Expression of a bacterial bi-functional chorismate mutase/prephenate dehydratase modulates primary and secondary metabolism associated with aromatic amino acids in Arabidopsis. Plant J. 60, 156–167 [DOI] [PubMed] [Google Scholar]
- 27. Lütke-Eversloh T., and Stephanopoulos G. (2005) Feedback inhibition of chorismate mutase/prephenate dehydrogenase (TyrA) of Escherichia coli: generation and characterization of tyrosine-insensitive mutants. Appl. Environ. Microbiol. 71, 7224–7228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vincent S., Chen S., Wilson D. B., and Ganem B. (2002) Probing the overlap of chorismate mutase and prephenate dehydrogenase sites in the Escherichia coli T-protein: a dehydrogenase-selective inhibitor. Bioorg. Med. Chem. Lett. 12, 929–931 [DOI] [PubMed] [Google Scholar]
- 29. Zhang S., Pohnert G., Kongsaeree P., Wilson D. B., Clardy J., and Ganem B. (1998) Chorismate mutase-prephenate dehydratase from Escherichia coli–study of catalytic and regulatory domains using genetically engineered proteins. J. Biol. Chem. 273, 6248–6253 [DOI] [PubMed] [Google Scholar]
- 30. Turnbull J., Cleland W. W., and Morrison J. F. (1990) Chorismate mutase prephenate dehydrogenase from Escherichia-coli. 1. Kinetic characterization of the dehydrogenase reaction by use of alternative substrates. Biochemistry 29, 10245–10254 [DOI] [PubMed] [Google Scholar]
- 31. Schoner R., and Herrmann K. M. (1976) 3-Deoxy-d-arabino-heptulosonate 7-phosphate synthase. Purification, properties, and kinetics of the tyrosine-sensitive isoenzyme from Escherichia coli. J. Biol. Chem. 251, 5440–5447 [PubMed] [Google Scholar]
- 32. Schofield L. R., Patchett M. L., and Parker E. J. (2004) Expression, purification, and characterization of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Pyrococcus furiosus. Protein Expr. Purif. 34, 17–27 [DOI] [PubMed] [Google Scholar]
- 33. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 38. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 41. Zoete V., Cuendet M. A., Grosdidier A., and Michielin O. (2011) SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 [DOI] [PubMed] [Google Scholar]
- 42. Allinger N. L., Yuh Y. H., and Lii J. H. (1989) Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 111, 8551–8566 [Google Scholar]
- 43. Lii J. H., and Allinger N. L. (1989) Molecular mechanics. The MM3 force field for hydrocarbons. 2. Vibrational frequencies and thermodynamics. J. Am. Chem. Soc. 111, 8566–8575 [Google Scholar]
- 44. Lii J. H., and Allinger N. L. (1989) Molecular mechanics. The MM3 force field for hydrocarbons. 3. The van der Waals' potentials and crystal data for aliphatic and aromatic hydrocarbons. J. Am. Chem. Soc. 111, 8576–8582 [Google Scholar]
- 45. Fiser A., Do R. K., and Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Notredame C., Higgins D. G., and Heringa J. (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 47. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 48. Humphrey W., Dalke A., and Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 49. Huang X. C., and Miller W. (1991) A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 12, 337–357 [Google Scholar]
- 50. Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J. M., Taly J. F., and Notredame C. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]