

Abstract
The overlapping network of kinase-substrate interactions provides exquisite specificity in cell signaling pathways, but also presents challenges to our ability to understand the mechanistic basis of biological processes. Efforts to dissect kinase-substrate interactions have been particularly limited by their inherently transient nature. Here, we use a library of FRET sensors to monitor these transient complexes, specifically examining weak interactions between the catalytic domain of protein kinase Cα and 14 substrate peptides. Combining results from this assay platform with those from standard kinase activity assays yields four novel insights into the kinase-substrate interaction. First, preferential binding of non-phosphorylated versus phosphorylated substrates leads to enhanced kinase-specific activity. Second, kinase-specific activity is inversely correlated with substrate binding affinity. Third, high affinity substrates can suppress phosphorylation of their low affinity counterparts. Finally, the substrate-competitive inhibitor bisindolylmaleimide I displaces low affinity substrates more potently leading to substrate selective inhibition of kinase activity. Overall, our approach complements existing structural and biophysical approaches to provide generalizable insights into the regulation of kinase activity.
Keywords: fluorescence resonance energy transfer (FRET), inhibitor, phosphorylation, protein kinase C (PKC), substrate specificity
Introduction
The canonical role of a protein kinase is to recognize, bind, and phosphorylate specific serine, threonine, or tyrosine residues on a substrate protein (1, 2). Given that kinases are one of the largest gene families in eukaryotes and are involved in nearly every cellular function (3), significant work has focused on understanding the mechanisms that dictate substrate-kinase pairings (4). Although the catalytic domains of eukaryotic kinases are structurally conserved (5, 6), the local environment around the substrate binding pocket of the kinase catalytic domain varies between kinases (7). This has led to the view that phosphorylation site recognition occurs through conserved residues flanking the phospho-residue on the substrate. Linear sequence motifs have now been identified for most kinases through a combination of structural analysis and peptide libraries (4, 8). Additionally, these phospho-sites are frequently found in unstructured regions of the substrate protein that may allow for more flexible accommodation in the kinase active site (9, 10). After recognizing the substrate, the catalytic domain then transfers a phosphate group from ATP to the phospho-residue on the substrate. Phospho-transfer is followed by release of ADP and the phosphorylated substrate to prime the kinase for another catalytic cycle (11).
Crystallographic and NMR approaches have provided detailed structural information on the catalytic domains of individual kinases in complex with a variety of nucleotide analogs and inhibitors (6, 12, 13). However, the lack of defined secondary structure surrounding the phospho-motif and the inherent transient nature of the interaction has limited efforts to dissect the different states of the kinase-substrate interaction in the catalytic cycle. Consequently, only a few crystal structures have thus far been reported for the kinase catalytic domain bound to substrate (4). Here, we instead use a recently developed approach to compare weak (μm) interactions between any two proteins or protein domains (14, 15). We apply this approach to directly compare the binding affinities of a kinase catalytic domain for a variety of weak binding substrate peptides. As a model system, we focus on the nodal kinase protein kinase Cα (PKCα). PKCα is a serine/threonine kinase of the AGC superfamily that phosphorylates a range of substrates implicated in diverse cellular functions including cell growth, muscle contraction, and immune response (16, 17).
In this study, we combine quantitative FRET measurements with traditional kinase activity assays to understand different catalytic states of the PKC-substrate interaction. Specifically, we explore four questions in substrate-kinase regulation using pairwise comparison of different substrate peptides bound to the PKCα catalytic domain in the presence of different nucleotides or inhibitors. 1) How does phosphorylation alter substrate affinity and kinase-specific activity? 2) How does substrate binding affinity correlate with kinase specific activity? 3) How does affinity alter catalytic activity in the presence of multiple substrates? 4) How does substrate affinity affect potency of substrate-competitive inhibitors?
Results
In this study, the PKCα peptide sensors are based around an ER/K linker, which separates the catalytic domain of PKCα and a variety of short peptides ≤14 residues in length (Fig. 1). These peptides are based off of the phosphorylation sites of known PKC substrates or predicted optimal PKC substrate peptides (18). To prevent phosphorylation, an alanine residue is substituted for the serine or threonine phospho-residue (Fig. 2). The linker itself contains an ER/K α-helix, ∼10 nm in length, flanked by a FRET donor (mCerulean or GFP) and a FRET acceptor (mCitrine or mCherry, respectively) (14). The ER/K FRET linker also contains an N-terminal tobacco etch virus (TEV)3 protease cleavage site, and each discrete unit of this ER/K linker is separated by (Gly-Ser-Gly)2–4 linkers (GSG) to provide rotational freedom. Figure 2 lists the sequences along with the basal FRET ratios for each peptide sensor.
FIGURE 1.
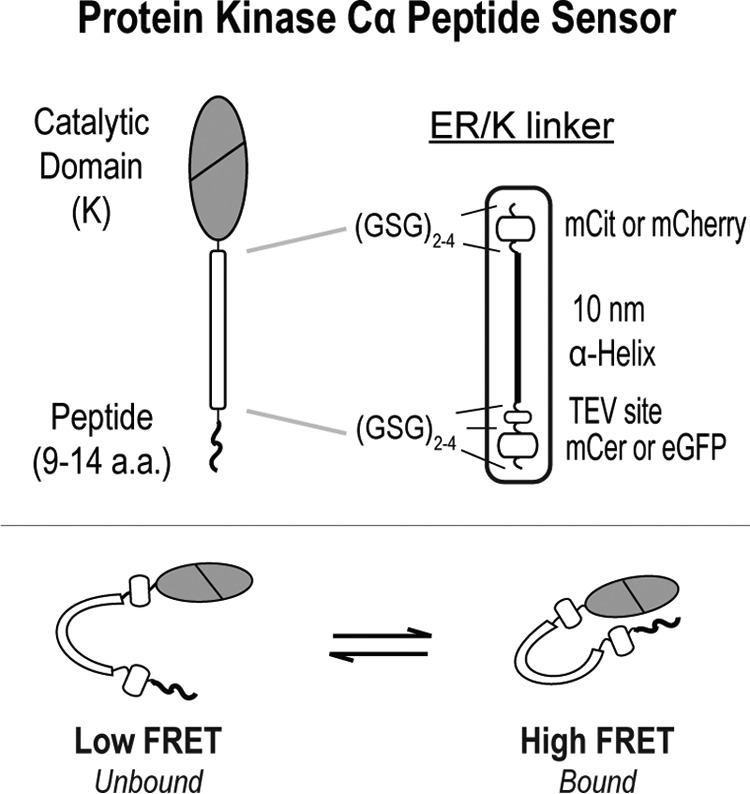
Schematic of PKCα peptide FRET sensors. PKCα peptide sensors are based around an ER/K FRET linker (14, 15), which separates the catalytic domain of PKCα and a variety of short peptides ≤14 residues in length (Fig. 2). In the majority of the experiments, the fluorophores used in the ER/K linker are monomeric Cerulean (mCer, FRET donor) and monomeric Citrine (mCit, FRET acceptor). For the time-resolved measurements in Fig. 4A, monomeric eGFP and monomeric Cherry (mCherry) were used instead.
FIGURE 2.

PKCα FRET sensors display a range of basal FRET interactions. Peptides used in activity assays (Ser- or Thr-containing) or FRET-based sensors (Ala- or Asp-containing) are shown. In the amino acid sequence, the phosphorylation site is highlighted in yellow. Blue (hydrophobic) or purple (basic) shading indicates consensus site residues important for phosphorylation (18). The basal FRET ratio is shown for each alanine-containing sensor and are derived from at least three protein preparations (n ≥ 3 preparations).
Loss of Affinity for Phosphorylated Substrate Enhances Kinase-specific Activity
During catalysis, a kinase must bind both non-phosphorylated substrate and ATP in the active site of its catalytic domain. To be efficient, it is also important that the kinase preferentially selects for non-phosphorylated substrate. To test these transient interactions, we examined three PKCα peptide sensors. We selected sensors that displayed a range of basal FRET ratios (Fig. 2). Upon addition of 100 μm ATP, there is a significant increase in the peptide interaction with the catalytic domain by steady-state FRET (p < 0.0001; Fig. 3A). Unsurprisingly, this ATP-dependent substrate interaction appears to be a wider kinase mechanism as it also exists for two strong-binding pseudo-peptides for PKA (19) (Fig. 3B). Next, we tested the sensor containing substrate number 14, which is derived from the pseudosubstrate sequence of PKCα (20), with additional nucleotide-based compounds. In addition to ATP, the peptide-catalytic domain interaction also occurs in the presence of 100 μm ADP and 100 μm of the non-hydrolysable nucleoside analog sangivamycin (Fig. 3C, Sang).
FIGURE 3.

Loss of affinity for phosphorylated substrate enhances kinase specific activity. A–C, increase in the FRET ratio (ΔFRET) upon addition of 100 μm ATP, 100 μm ADP, or 100 μm sangivamycin (Sang) for sensors containing short alanine-substituted substrate peptides and either (A and C) the catalytic domain of PKCα or (B) the catalytic domain of PKA. For PKC peptide sequence information see Fig. 2. D, FRET ratios for PKCα peptide number 14 sensors containing an alanine (S14A), serine (S14S), or aspartic acid (S14D, phosphomimetic) with 100 μm ATP. E, schematic of a subset of steps in the catalytic cycle of PKCα. Coloring denotes steady-state FRET ratios of PKCα peptide number 14 sensors containing either an aspartic acid (S14D) or an alanine residue (S14A) with 100 μm ATP or 100 μm ADP. States are arranged based on progression of FRET through the catalytic cycle and do not indicate the relative prevalence of all possible states. F, ATP consumption of the catalytic domain of PKCα for serine-containing peptide number 14 upon addition of equal concentrations of either aspartic acid-substituted (S14D) or alanine-substituted (S14A) peptide. For all FRET and activity readings, data are derived from at least three independent protein preparations with at least two replicated measurements for each condition per experiment (mean ± S.E., n ≥ 3 experiments).
To examine the effects of phosphorylation, we mutated the alanine in peptide number 14 to a serine. Given that this peptide is known to be phosphorylated by PKC (20), it is assumed that serine-containing peptide number 14 in the FRET sensor will be phosphorylated in the presence of ATP. Aspartic acid has been successfully used with other PKC substrates to mimic phosphorylation (21–23). Hence, a phosphomimetic version of peptide number 14 was also created by mutating the alanine in the peptide to aspartic acid. In the presence of ATP, both the serine- and the aspartic acid-containing FRET sensors have a significantly lower FRET ratio compared with the alanine-containing peptide (p < 0.0001, Fig. 3D). Given the potential heterogeneity in the phosphorylation state of the serine residue, in subsequent experiments the aspartic acid-substituted peptide was used to mimic the phosphorylation state. With the alanine and aspartic acid-containing substrate number 14 sensors, we measured the FRET ratio of the peptide-catalytic domain interaction during different states of the catalytic cycle (Fig. 3E). As compared with alanine-substituted substrate, the catalytic domain has decreased affinity for the phosphomimetic substrate suggesting a simple mechanism for selecting non-phosphorylated substrate and thereby increasing productive phosphoryl-transfer. To test this preferential substrate selection, we monitored ATP turnover of the catalytic domain for serine-containing peptide number 14 in the presence of either equimolar alanine-substituted peptide or the aspartic acid-containing phosphomimetic peptide. Indeed, the alanine-substituted peptide is significantly better at inhibiting catalytic activity (Fig. 3F).
Inverse Correlation between Substrate Affinity and Kinase Activity
To test whether changes in steady-state FRET reflect changes in fractional binding of the peptide to the catalytic domain (Fig. 1), we turned to time-correlated single photon counting (TCSPC). TCSPC is a method for measuring time-resolved fluorescence that provides information on complex decay kinetics and population states (24). Using this method we measured the relative population of interacting to non-interacting species for four peptide sensors displaying a range of steady-state FRET ratios (Fig. 2). As illustrated in Fig. 4A, increases in steady-state FRET strongly correlate with higher fractional binding (also see Table 1). Interestingly, this data also suggests that the substrate-kinase interaction can occur in the absence of nucleotide. Next, we compared the affinity of peptide-kinase interaction to the activity of the catalytic domain for that same peptide. In Fig. 4B, the steady-state FRET ratio was measured for 14 peptide sensors in the absence of nucleotide and plotted versus activity of the isolated catalytic domain in the presence of excess substrate peptide. All activity measurements were taken under the same conditions. We observed a trend where catalytic activity decreases with increasing basal FRET ratios (i.e. higher peptide binding). This trend was not complete (R2 = 0.62) as a few peptides displayed similar affinity but significantly different activity (e.g. peptide 1 versus 6, Fig. 4B). This suggests that in addition to affinity, other mechanisms are likely involved in dictating catalytic activity. The data also indicates that the peptide based on the pseudosubtrate of PKCα (number 14) binds more strongly than peptides derived from cellular substrates (Figs. 2 and 4B, numbers 1–8). This finding is consistent with the dominant role of the pseudosubstrate in PKC autoinhibition (20).
FIGURE 4.

Inverse correlation between substrate affinity and kinase activity. A, comparison between steady-state FRET and fractional binding for four alanine-containing peptide sensors. For fractional binding, fluorescence decay single photon counting data were fit to I(t) = A1e−t/τ1 + A2e−t/τ2, where τ1 was set to the lifetime of a 10-nm ER/K linker control (see Table 1). Molecular fraction of the bound state(s) was calculated as A2/(A1 + A2). B, steady-state FRET of PKCα catalytic domain sensors with 14 short peptides (Fig. 2) compared with activity of the catalytic domain with corresponding substrate peptides. Activity assays were performed at 21–22 °C with equimolar peptide concentrations (500 μm) and kinase (50 nm). Specific activity is reported as mole of ATP consumed per mol of kinase per s (s−1). In all experiments, data are derived from at least three protein preparations (mean ± S.E., n ≥ 3 experiments).
TABLE 1.
Lifetimes and fractional binding for peptide-catalytic sensors
Mean ± S.E. n = 3 (number of protein preparations). Data were fit to I(t) = A1e−t/τ1 + A2e−t/τ2, where τ1 is fixed to the lifetime of a donor linker control (2.56 ± 0.01 ns).
| Peptide | Mol fraction unbound | Mol fraction bound | τ2, ns |
|---|---|---|---|
| 11 | 0.85 ± 0.01 | 0.15 ± 0.01 | 0.88 ± 0.06 |
| 10 | 0.84 ± 0.01 | 0.16 ± 0.01 | 0.83 ± 0.06 |
| 8 | 0.89 ± 0.01 | 0.11 ± 0.01 | 0.66 ± 0.06 |
| 6 | 0.92 ± 0.01 | 0.08 ± 0.01 | 0.49 ± 0.05 |
High Affinity Substrates Suppresses Phosphorylation of Low Affinity Counterparts
To further examine this trade-off between peptide affinity and activity, we compared activity of two different sets of peptide sensors for the same substrate. In one set, we made a sensor where peptide number 14 was separated from the catalytic domain by either a 10- or 30-nm ER/K linker. By increasing the linker length, we are changing the effective concentration of the peptide for the catalytic domain from ∼10 μm to 100 nm (14). Reducing the effective concentration should weaken the apparent peptide affinity, as observed by a decrease in the FRET ratio, and result in a higher basal activity of this sensor. Indeed, the sensor with the 30-nm linker has significantly higher activity than the 10-nm linker for the same substrate peptide (Fig. 5A, red). For our second set of sensors, we compared the activity of two different 10-nm peptide FRET sensors for the same substrate. Sensors contained either a high affinity alanine-peptide (number 11) or a lower affinity alanine-peptide (number 8). In agreement with the previous set, the higher affinity peptide sensor (number 11) has a much lower activity than the lower affinity peptide sensor (number 8) (Fig. 5A, black). Finally, we tested this trade-off between affinity and activity by examining the effect of a high affinity-low activity peptide (number 12) on catalytic turnover with a low affinity-high activity peptide (number 6). Concentrations of both substrates were held constant, and indeed when both peptides are present the activity is essentially inhibited to levels matching the high affinity-low activity peptide (Fig. 5B). This suggests a new model of substrate selectivity where high-affinity interactions may out-compete low affinity, high activity substrates.
FIGURE 5.

High affinity substrates suppress phosphorylation of low affinity counterparts. A, relative activity of sensors containing either the alanine-substituted peptide number 14 with different linker distances (10 versus 30 nm; 1 versus 2; red) or containing different alanine-substituted peptides (peptide number 8 versus number 11; 3 versus 4; black) with the same 10-nm linker. Activity data are plotted versus the steady-state FRET ratio of the peptide sensors. B, activity of the isolated catalytic domain for peptide number 6, peptide number 12, and equimolar concentrations of peptide numbers 6 and 12. For both FRET and activity experiments, data are derived from at least three protein preparations with at least two replicates for each condition per independent experiment (mean ± S.E., n ≥ 3 experiments).
Kinase-substrate Binding Affinity Impacts Inhibitor Potency
As PKC appears to play a role in numerous diseases, one of the goals of therapeutic drug discovery is to develop selective modulators of PKC activity (25). These include ATP-competitive inhibitors like sangivamyicin (Fig. 3C) and substrate-competitive inhibitors like bisindolylmaleimide I (BimI) (26). Using a high affinity (number 11) and a low affinity (number 8) peptide sensor, we examined the effect of BimI on the ATP-dependent peptide-kinase interaction. We selected these peptides as they display significantly different basal FRET ratios but also provide a dynamic FRET range for the BimI titration. In the presence of BimI, the substrate-kinase interaction is indeed abrogated as measured by steady-state FRET (Fig. 6A). Higher concentrations of BimI, however, are required to disrupt the higher affinity peptide interaction (Fig. 6A, black). This is further reflected when comparing the percent inhibition of catalytic activity for three different substrate peptides (order of affinity: 11 > 8 > 6) (Fig. 6, B and C). Overall, this demonstrates that the potency of a substrate-competitive inhibitor is strongly influenced by the strength of the substrate-kinase interaction (Fig. 6D).
FIGURE 6.

Kinase-substrate binding affinity impacts inhibitor potency. A, titration of peptide numbers 8 and 11 FRET sensors in the presence of 100 μm ATP by the substrate competitive inhibitor BimI. The FRET ratio for each sensor was normalized to the FRET ratio of that sensor in the absence of any inhibitor. B, inhibition of PKCα catalytic domain activity by 0.1 μm BimI for three different substrate peptides (order of peptide affinity: 11 > 8 > 6). Activity assays were performed with equimolar peptide concentrations (500 μm) and kinase (50 nm) and specific activity is reported as mole of ATP consumed per mol of kinase per s (s−1). C, percent inhibition of PKCα catalytic domain activity by 0.1 μm BimI for substrate peptides in B. Percent inhibition is graphed versus the steady-state FRET ratio for the corresponding alanine-substituted FRET sensors. For all FRET and activity readings, data are derived from at least three protein preparations with at least two replicates per independent experiment (mean ± S.E., n ≥ 3 experiments). D, for PKCα, varying substrate affinity reduces the potency of substrate-competitive inhibitors like BimI.
Discussion
To selectively perturb pathophysiological signaling, an important avenue to realizing the potential of nodal kinases as therapeutic targets is achieving substrate-specific kinase inhibition. In this study, we use a novel set of FRET-based sensors to uncover an inverse correlation between substrate-binding affinity and kinase-specific activity for PKCα (Fig. 4). We find that substrate-competitive inhibitors such as BimI displace the kinase-substrate interactions in inverse proportion to the binding affinity of the interaction. Consequently, these inhibitors display varying potency for different substrates (Fig. 6, B and C), suggesting a simple mechanism to achieve substrate-selective kinase inhibition. Although the generality of these mechanisms in other kinases need further exploration, the FRET sensors developed for this study are easily adaptable to other catalytic domain-substrate interactions (Fig. 3B). Regardless, our sensors demonstrate that substrate affinity, which influences the apparent potency of inhibitors, needs to be an important consideration in designing small molecule screens. Hence, our sensors provide a novel assay platform to probe the mechanisms of substrate-selective kinase inhibition. For instance, we were able to detect instances of both positive and negative allostery between kinase and substrate in the presence of sangivamycin (Fig. 3C) and BimI (Fig. 6A), respectively.
Specific activity was measured in the presence of saturating substrate concentrations (500 μm; Fig. 4) such that the variations in specific activity likely reflect changes in maximal catalytic turnover or kcat of PKCα. For a multistep enzymatic process, such as substrate phosphorylation, kcat is determined by the rate-limiting step(s) of the reaction. Although we cannot distinguish what step(s) are rate-limiting from these experiments, our data do suggest that substrates differentially influence catalytic activity, potentially through substrate release or associated conformational changes. There is some precedence for this with PKA, which is kinetically the best characterized AGC kinase (11, 27, 28). Although the overall trend in our measurements suggest an inverse correlation between substrate-kinase affinity and specific activity, some peptides do display similar affinity but significantly different activity (Fig. 4B; e.g. substrates 10, 13, and 14). We speculate that these substrates differentially stabilize conformational states in the catalytic domain of PKCα and thus alter overall catalytic turnover through mechanisms other than binding affinity. A more detailed analysis of this discrepancy requires approaches that can resolve the conformational states of the kinase and therefore are outside the scope of this study.
The inverse correlation between substrate affinity and kinase-specific activity of select substrates identified here impacts the interpretation of kinase-specific activity as measured in standard in vitro kinase assays. First, high specific activity is implicitly equated with higher selectivity for the substrate used in the kinase assay (29, 30). However, high affinity substrates inherently have low specific activities and can outcompete their low affinity counterparts. Hence, kinase-substrate affinity is an important consideration when identifying the target kinase by in vitro kinase screens. Second, the inverse correlation between substrate affinity and kinase activity may increase productive phosphoryl-transfer events. Upon phosphoryl-transfer, the kinase catalytic domain loses affinity for the substrate, preferentially binding non-phosphorylated substrate over phosphorylated substrate (Fig. 3, D and E). This coordination between nucleotide and substrate binding leads to rapid phosphorylation of a pool of non-phosphorylated substrate (Fig. 3F). Enhancing activity through such mechanisms gains added significance when considering both the transient nature of the kinase active state and the constant competition with neighboring phosphatases (7, 31–33).
In this study, we observed that a higher affinity substrate is able to suppress phosphorylation of its lower affinity counterpart (Fig. 5B). This presents an intriguing biochemical mechanism for cellular substrate selection. For example, affinity-based competition between substrates could impact the ordered phosphorylation of a multisite substrate or when the kinase is localized to a region containing multiple substrates (34, 35). Affinity-based selection between substrates is also important to consider in diseases such as cancer where kinase substrates are frequently overexpressed (e.g. Refs. 36 and 37). There are numerous factors, however, that complicate experimentally testing this phenomenon in vivo, including varied expression and subcellular localization of both the substrate and the kinase.
Nonetheless, the differential phosphorylation of substrates based on kinase-binding affinities provides a novel framework to examine several observations in the kinase field. First, our measurements are consistent with studies that used mixed peptide libraries phosphorylated by individual kinases to identify substrate recognition motifs (8, 18, 38, 39). Inherent in this screening methodology is the enrichment of high affinity substrates that get phosphorylated at the expense of others. Second, early work from Dekker et al. (40, 41) showed that swapping the pseudosubstrate peptide of PKCη with that of PKCα changed effector-dependent substrate activity and selectivity. If substrates compete with the pseudosubstrate for binding to the active site, then altering the affinity of the pseudosubstrate-kinase interaction would influence the phosphorylation profile of the kinase. Third, competition between phosphorylation sites on Wee1 and other CDK1 targets influences the sensitivity of phosphorylation levels to activating stimulus (42). The non-linear relationship between substrate phosphorylation and stimulus mirrors the non-additive effects of multiple substrates with varying affinity interacting with the same kinase.
There are over 500 kinases within the human protein kinome. A clear extension from this work will be to explore whether the mechanisms identified here are observed for other AGC kinases as well as non-AGC kinases. For example, in agreement with previous studies, we observed that both PKC and PKA show positive cooperativity between substrate and ATP binding (43). We posit that it is likely not a universal kinase mechanism as a recent paper (44) found that negative cooperativity is observed in the tyrosine kinases Src, Abl, and Hck. Although the negative cooperativity could stem from differences between kinase families, an important autoinhibitory mechanism for both PKC and PKA is pseudosubstrate binding. Thus, positive cooperativity between ATP and substrate may instead reflect a need to achieve strong basal autoinhibition. Overall, the FRET-based system presented in this study provides a novel approach for exploring the impact of substrates on kinase regulation.
Experimental Procedures
Reagents and Constructs
Sangivamycin (Sigma) and BimI (EMD) were solubilized in DMSO, ATP (EMD) in 20 mm imidazole (pH 7.5), and ADP (Sigma) in 50 mm HEPES (pH 7.5). Peptides 1–14 (Fig. 2) were custom-synthesized by GenScript and solubilized in water. All compounds and peptides were aliquoted and stored at either −20 or −80 °C to prevent multiple freeze-thaw cycles. Human protein kinase A (PKA) α cDNA was purchased from DNASU. PKA sensors contained either the pseudosubstrate peptide from the type Iα regulatory subunit of protein kinase A (RIαPep; KGRRRRGAISAEV) or a peptide derived from protein kinase inhibitor protein (PKIPep; TTYADFIASGRTGRRNAIHD) (Fig. 3B) (19). Human PKCα cDNA was purchased from Open Biosystems as described previously (15). PKA and PKCα constructs were cloned using PCR (Expand High Fidelity PCR System, Sigma) or site-directed mutagenesis (Pfu-Turbo, Agilent) into pBiex1 (Novagen) Sf9 expression plasmid vector. For activity and steady-state FRET experiments, sensors contained a monomeric Cerulean (mCer, donor) and monomeric Citrine (mCit, acceptor) FRET pair separated by a 10-nm ER/K single α-helix, as previously described (Fig. 1) (15). For time-resolved TCSPC experiments, the ER/K linker instead contained monomeric eGFP (donor) (45) and monomeric Cherry (mCherry, acceptor).
Insect Cell Expression and Protein Purification
pBiex1 vectors were transiently transfected into Sf9 insect cells cultured in Sf900-II media (Invitrogen) using Escort IV transfection reagent (Sigma) and Opti-MEM I (Life Technologies). Cells were lysed 72 h post-transfection in 20 mm HEPES (pH 7.5), 200 mm NaCl, 4 mm MgCl2, 0.5% sucrose, 0.5% IGEPAL, 2 mm DTT, 50 μg/ml of PMSF, 5 μg/ml of aprotinin, and 5 μg/ml of leupeptin. Lysate was clarified via high-speed centrifugation and incubated with anti-FLAG M2 affinity resin (Sigma) for 1–2 h. Resin was washed three times with 20 mm HEPES (pH 7.5), 150 mm KCl, 5 mm MgCl2, 2 mm DTT, 50 μg/ml of PMSF, 5 μg/ml of aprotinin, and 5 μg/ml of leupeptin. Protein was eluted using FLAG peptide (Sigma), and buffer exchanged into PKC buffer (20 mm HEPES (pH 7.5), 5 mm MgCl2, 0.5 mm EGTA, and 2 mm DTT) using 40-kDa cutoff Zeba Spin Desalting Columns (Pierce). Before each experiment, protein samples were centrifuged at ∼220,000 × g at 4 °C to remove any insoluble protein. Protein concentration was determined for centrifuged protein from the fluorescent emission of mCit (excitation 490, emission 525 nm) compared with a standard on a FluoroMax-4 fluorimeter (Horiba, Scientific) or by eGFP absorbance on a NanoDrop One (Thermo). For experiments requiring isolated catalytic domain, resin-bound sensors were incubated with TEV protease, before being washed, eluted, and desalted. TEV cleavage was ≥95% as assessed by mCer and mCit fluorescence.
FRET Measurements
All experiments were performed with 30–50 nm protein in PKC buffer at 21–22 °C. Samples were prepared in tubes pre-coated with 0.1 mg/ml of BSA to limit protein sticking to tube walls. Protein samples were excited at 430 nm (mCer) with an 8-nm band pass, and emission monitored from 450 to 650 nm. The FRET ratio was calculated from the ratio of the emission for mCit (525 nm) to mCer (475 nm). Final concentrations of 100 μm ATP, 100 μm ADP, 100 μm sangivamycin, or BimI were added as indicated. For each experimental condition, ≥3 independent protein batches were used and at least two replicates were measured for each independent experiment (n ≥ 3).
Time-resolved Measurements
Time-resolved fluorescence decay measurements were taken by single photon counting (SPC-130-EM, Becker and Hickl) using a 485-nm subnanosecond pulse diode laser (PicoQuant), a 519-bandpass filter, and a PMH-100 photomultiplier module (Photonics Solutions) (46). Sensors contained the eGFP and mCherry FRET pair, and all experiments were performed with 50–100 nm protein in PKC buffer containing 100 μm ATP and 0.1 mg/ml of BSA. Data were fit to the second exponential decay, I(t) = A1e−t/τ1 + A2e−t/τ2, were τ1 was set to the lifetime of a 10 nm ER/K linker control. Each experimental condition was collected from three independent protein preparations (n = 3).
Kinase Activity Assay
All activity assays were performed with 50 nm isolated catalytic domain in PKC buffer at 21–22 °C for 2–6 min. Experiments comparing the basal activity of different FRET sensors (Fig. 5A) were performed with 50 μm MBP peptide from GenScript. For all other activity experiments (Figs. 3F, 4B, 5B, and 6B), the final peptide concentration was 500 μm. Activity was measured by monitoring ATP consumption with the Kinase-Glo Max Luminescence Assay Kit (Promega). End-point luminescence was measured in white, 96-well plates using a M5e Spectramax spectrophotometer (Molecular Devices). For each experimental condition, ≥2 independent experiments were performed for ≥3 protein preparations (n ≥ 6).
Author Contributions
R. F. S. and S. S. planned and designed experiments; R. F. S. performed and analyzed experiments; R. F. S. and S. S. wrote the manuscript.
Acknowledgment
Time-resolved fluorescence decay measurements were taken at the Biophysical Technology Center at the University of Minnesota.
This work was supported in part by the American Heart Association Scientist Development Grant 13SDG14270009 and National Institutes of Health Grants 1DP2 CA186752-01 and 1-R01-GM-105646-01-A1 (to S. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TEV
- tobacco etch virus
- TCSPC
- time-correlated single photon counting
- BimI
- bisindolylmaleimide I
- eGFP
- enhanced green fluorescent protein
- Sang
- sangivamycin.
References
- 1. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pearce L. R., Komander D., and Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 3. Manning G., Whyte D. B., Martinez R., Hunter T., and Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 4. de Oliveira P. S., Ferraz F. A., Pena D. A., Pramio D. T., Morais F. A., and Schechtman D. (2016) Revisiting protein kinase-substrate interactions: toward therapeutic development. Sci. Signal. 9, re3. [DOI] [PubMed] [Google Scholar]
- 5. Hanks S. K., and Hunter T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596 [PubMed] [Google Scholar]
- 6. Taylor S. S., and Kornev A. P. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ubersax J. A., and Ferrell J. E. Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541 [DOI] [PubMed] [Google Scholar]
- 8. Songyang Z., Blechner S., Hoagland N., Hoekstra M. F., Piwnica-Worms H., and Cantley L. C. (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 4, 973–982 [DOI] [PubMed] [Google Scholar]
- 9. Fuxreiter M., Tompa P., and Simon I. (2007) Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23, 950–956 [DOI] [PubMed] [Google Scholar]
- 10. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., and Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adams J. A. (2001) Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 101, 2271–2290 [DOI] [PubMed] [Google Scholar]
- 12. Taylor S. S., Ilouz R., Zhang P., and Kornev A. P. (2012) Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Endicott J. A., Noble M. E., and Johnson L. N. (2012) The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 81, 587–613 [DOI] [PubMed] [Google Scholar]
- 14. Sivaramakrishnan S., and Spudich J. A. (2011) Systematic control of protein interaction using a modular ER/K α-helix linker. Proc. Natl. Acad. Sci. U.S.A. 108, 20467–20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Swanson C. J., Ritt M., Wang W., Lang M. J., Narayan A., Tesmer J. J., Westfall M., and Sivaramakrishnan S. (2014) Conserved modular domains team up to latch-open active protein kinase Cα. J. Biol. Chem. 289, 17812–17829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Newton A. C. (1995) Protein kinase C: structure, function, and regulation. J. Biol. Chem. 270, 28495–28498 [DOI] [PubMed] [Google Scholar]
- 17. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishikawa K., Toker A., Johannes F. J., Songyang Z., and Cantley L. C. (1997) Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272, 952–960 [DOI] [PubMed] [Google Scholar]
- 19. Taylor S. S., Kim C., Vigil D., Haste N. M., Yang J., Wu J., and Anand G. S. (2005) Dynamics of signaling by PKA. Biochim. Biophys. Acta 1754, 25–37 [DOI] [PubMed] [Google Scholar]
- 20. House C., and Kemp B. E. (1987) Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science 238, 1726–1728 [DOI] [PubMed] [Google Scholar]
- 21. Schulz S., Doller A., Pendini N. R., Wilce J. A., Pfeilschifter J., and Eberhardt W. (2013) Domain-specific phosphomimetic mutation allows dissection of different protein kinase C (PKC) isotype-triggered activities of the RNA binding protein HuR. Cell. Signal. 25, 2485–2495 [DOI] [PubMed] [Google Scholar]
- 22. López-Sánchez I., Garcia-Marcos M., Mittal Y., Aznar N., Farquhar M. G., and Ghosh P. (2013) Protein kinase C-theta (PKCθ) phosphorylates and inhibits the guanine exchange factor, GIV/Girdin. Proc. Natl. Acad. Sci. U.S.A. 110, 5510–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verma S. K., Ganesan T. S., and Parker P. J. (2008) The tumour suppressor RASSF1A is a novel substrate of PKC. FEBS Lett. 582, 2270–2276 [DOI] [PubMed] [Google Scholar]
- 24. Lakowicz J. R. (2006) Principles of fluorescence spectroscopy, 3rd Ed., Springer, New York [Google Scholar]
- 25. Mochly-Rosen D., Das K., and Grimes K. V. (2012) Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 11, 937–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith I. M., and Hoshi N. (2011) ATP competitive protein kinase C inhibitors demonstrate distinct state-dependent inhibition. PloS One 6, e26338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou J., and Adams J. A. (1997) Participation of ADP dissociation in the rate-determining step in cAMP-dependent protein kinase. Biochemistry 36, 15733–15738 [DOI] [PubMed] [Google Scholar]
- 28. Shaffer J., and Adams J. A. (1999) Detection of conformational changes along the kinetic pathway of protein kinase A using a catalytic trapping technique. Biochemistry 38, 12072–12079 [DOI] [PubMed] [Google Scholar]
- 29. Johnson S. A., and Hunter T. (2005) Kinomics: methods for deciphering the kinome. Nat. Methods 2, 17–25 [DOI] [PubMed] [Google Scholar]
- 30. Meng Z., Moroishi T., Mottier-Pavie V., Plouffe S. W., Hansen C. G., Hong A. W., Park H. W., Mo J. S., Lu W., Lu S., Flores F., Yu F. X., Halder G., and Guan K. L. (2015) MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 6, 8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sim A. T., and Scott J. D. (1999) Targeting of PKA, PKC and protein phosphatases to cellular microdomains. Cell Calcium 26, 209–217 [DOI] [PubMed] [Google Scholar]
- 32. Bauman A. L., and Scott J. D. (2002) Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nat. Cell Biol. 4, E203–206 [DOI] [PubMed] [Google Scholar]
- 33. Hirschi A., Cecchini M., Steinhardt R. C., Schamber M. R., Dick F. A., and Rubin S. M. (2010) An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 17, 1051–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amess B., Manjarrez-Hernandez H. A., Howell S. A., Learmonth M., and Aitken A. (1992) Multisite phosphorylation of the 80 kDa (MARCKS) protein kinase C substrate in C3H/10T1/2 fibroblasts: quantitative analysis of individual sites by solid-phase microsequencing. FEBS Lett. 297, 285–291 [DOI] [PubMed] [Google Scholar]
- 35. Woods A., and Couchman J. R. (1992) Protein kinase C involvement in focal adhesion formation. J. Cell Sci. 101, 277–290 [DOI] [PubMed] [Google Scholar]
- 36. Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M. R., Carotenuto A., De Feo G., Caponigro F., and Salomon D. S. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 [DOI] [PubMed] [Google Scholar]
- 37. Chen C. H., Cheng C. T., Yuan Y., Zhai J., Arif M., Fong L. W., Wu R., and Ann D. K. (2015) Elevated MARCKS phosphorylation contributes to unresponsiveness of breast cancer to paclitaxel treatment. Oncotarget 6, 15194–15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hutti J. E., Jarrell E. T., Chang J. D., Abbott D. W., Storz P., Toker A., Cantley L. C., and Turk B. E. (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29 [DOI] [PubMed] [Google Scholar]
- 39. Obata T., Yaffe M. B., Leparc G. G., Piro E. T., Maegawa H., Kashiwagi A., Kikkawa R., and Cantley L. C. (2000) Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J. Biol. Chem. 275, 36108–36115 [DOI] [PubMed] [Google Scholar]
- 40. Dekker L. V., McIntyre P., and Parker P. J. (1993) Altered substrate selectivity of PKC-η pseudosubstrate site mutants. FEBS Lett. 329, 129–133 [DOI] [PubMed] [Google Scholar]
- 41. Dekker L. V., McIntyre P., and Parker P. J. (1993) Mutagenesis of the regulatory domain of rat protein kinase C-η: a molecular basis for restricted histone kinase activity. J. Biol. Chem. 268, 19498–19504 [PubMed] [Google Scholar]
- 42. Kim S. Y., and Ferrell J. E. Jr. (2007) Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell 128, 1133–1145 [DOI] [PubMed] [Google Scholar]
- 43. Kim J., Li G., Walters M. A., Taylor S. S., and Veglia G. (2016) Uncoupling catalytic and binding functions in the cyclic AMP-dependent protein kinase A. Structure 24, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foda Z. H., Shan Y., Kim E. T., Shaw D. E., and Seeliger M. A. (2015) A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat. Commun. 6, 5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Altman D., Goswami D., Hasson T., Spudich J. A., and Mayor S. (2007) Precise positioning of myosin VI on endocytic vesicles in vivo. PLoS Biol. 5, e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Autry J. M., Rubin J. E., Svensson B., Li J., and Thomas D. D. (2012) Nucleotide activation of the Ca-ATPase. J. Biol. Chem. 287, 39070–39082 [DOI] [PMC free article] [PubMed] [Google Scholar]