

Abstract
The biochemical mechanisms through which eosinophils contribute to asthma pathogenesis are unclear. Here we show eosinophil peroxidase (EPO), an abundant granule protein released by activated eosinophils, contributes to characteristic asthma-related phenotypes through oxidative posttranslational modification (PTM) of proteins in asthmatic airways through a process called carbamylation. Using a combination of studies we now show EPO uses plasma levels of the pseudohalide thiocyanate (SCN−) as substrate to catalyze protein carbamylation, as monitored by PTM of protein lysine residues into Nϵ-carbamyllysine (homocitrulline), and contributes to the pathophysiological sequelae of eosinophil activation. Studies using EPO-deficient mice confirm EPO serves as a major enzymatic source for protein carbamylation during eosinophilic inflammatory models, including aeroallergen challenge. Clinical studies similarly revealed significant enrichment in carbamylation of airway proteins recovered from atopic asthmatics versus healthy controls in response to segmental allergen challenge. Protein-bound homocitrulline is shown to be co-localized with EPO within human asthmatic airways. Moreover, pathophysiologically relevant levels of carbamylated protein either incubated with cultured human airway epithelial cells in vitro, or provided as an aerosolized exposure in non-sensitized mice, induced multiple asthma-associated phenotypes including induction of mucin, Th2 cytokines, IFNγ, TGFβ, and epithelial cell apoptosis. Studies with scavenger receptor-A1 null mice reveal reduced IL-13 generation following exposure to aerosolized carbamylated protein, but no changes in other asthma-related phenotypes. In summary, EPO-mediated protein carbamylation is promoted during allergen-induced asthma exacerbation, and can both modulate immune responses and trigger a cascade of many of the inflammatory signals present in asthma.
Keywords: asthma, eosinophil, inflammation, peroxidase, post-translational modification (PTM)
Introduction
Asthma is a complex inflammatory disorder of the airways, typically characterized by eosinophilia, mucus hypersecretion, epithelial apoptosis, and airway reactivity. Inflammatory cells such as eosinophils and neutrophils participate in tissue injury through the release of cytotoxic products and formation of reactive oxidant species (1–6). Some of the most abundant proteins in them are their respective heme-dependent peroxidases, eosinophil peroxidase (EPO),3 and myeloperoxidase (MPO). The normal function of these enzymes is to produce reactive oxidants and free radical species that inflict oxidative damage upon invading parasites and pathogens as part of the innate host defense system. However, the reactive species EPO and MPO form can also oxidatively modify host tissues. Under physiological conditions, both EPO and MPO use hydrogen peroxide (H2O2) as one substrate and an assortment of co-substrates to generate an array of reactive oxidant species that can damage both proteins and lipids at sites of inflammation (7–12, 13). For example, we have shown that eosinophils use EPO-generated oxidants to promote modification of lung and airway proteins during allergen challenge in mild asthma, and in severe asthma (9, 14–16). We have identified and structurally characterized specific posttranslational modifications (PTMs) formed on proteins, and used detection of specific brominated and chlorinated residues as tools for monitoring molecular signatures for reactive halogenating species formed by EPO- and MPO-dependent oxidative injury in asthma and other inflammatory disorders (14, 17–24). Specifically, eosinophil activation results in EPO release and catalytic generation of a unique oxidant species, hypobromous acid (HOBr), a reactive brominating oxidant that can form the molecular footprints, 3-bromotyrosine and 3,5-dibromotyrosine (9, 14, 17). We have shown that increased levels of 3-bromotyrosine are observed in the airways of subjects with atopic asthma, and can be monitored as a non-invasive marker in the urine of asthmatic adults and children alike, with levels of urinary 3-bromotyrosine being indicative of asthma control and predictive of asthmatic exacerbations (14, 15, 17, 25). Alternatively, we also first reported that activated eosinophils use EPO as a catalytic sink for consuming nitric oxide (NO), both forming NO-derived oxidants capable of producing the PTM protein bound 3-nitrotyrosine, and inhibiting NO-dependent bronchodilation (9, 19, 20, 26, 27). Thus, leukocytes in general and activated eosinophils in particular, have the potential for inflicting oxidative modifications on proteins and lipids in asthmatic airways via numerous pathways.
Both EPO and MPO use the pseudo-halide thiocyante (SCN−) as a preferred co-substrate with H2O2 (28, 29). SCN− in vivo originates primarily from diet and environmental exposure. The normal plasma levels of SCN− typically found in non-smokers range from 20 to 100 μm depending upon dietary intake (30), whereas levels in smokers can be substantially higher (31–33). By comparison, physiological levels of the cosubstrate H2O2 vary widely with levels in extracellular spaces and in the plasma estimated to range between 1 and 18 μm under typical conditions (34, 35), but under conditions of leukocyte degranulation and respiratory burst at sites of inflammation, local concentrations as high as 1000 μm H2O2 have been suggested (reviewed in Ref. 36). Under physiological levels of H2O2 and cosubstrates, EPO and MPO utilize SCN− as a preferred substrate to primarily form hypothiocyanous acid (HOSCN), a weak oxidant with bacteriostatic activity. Using multinuclear NMR, mass spectrometry, and other analytical approaches, we first reported that in addition to HOSCN, a minor product formed following SCN− oxidation by both MPO and EPO in vitro is cyanate (OCN−), with EPO being more efficient at generating OCN− at plasma levels of halides (i.e. 100 mm Cl−, and 100 μm Br−) and SCN− (28). Cyanate is in equilibrium with urea under physiological conditions. In end stage renal disease elevated levels of urea lead to enhanced levels of OCN−, and consequently, marked increases in protein carbamylation (37). Nucleophilic groups on proteins such as amino, hydroxyl, or thiol groups may become covalently modified with a carbamyl (NH2CO) group upon exposure to the electrophile OCN−, altering protein primary structure, charge and function, often leading to enzymatic loss of function (38–41). These alterations in structure and charge were first investigated by biochemists studying the reversible denaturation-renaturation of proteins with urea (38–41). The reaction with nucleophilic ϵ-amino groups of lysine residues produces Nϵ-carbamyl-lysine, also known as homocitrulline (HCit) (40, 42). The abundance of lysine compared with alternative susceptible amino acids, and the enhanced nucleophilicity of the ϵ-amino lysine moiety compared with that of alternative targets like arginine or histidine, makes lysine side chains a major target for carbamylation in vivo.
In prior studies (43) we demonstrated MPO-catalyzed oxidation of SCN− serves as an alternative pathway to uremia for promotion of protein carbamylation in vivo at sites of inflammation and vascular disease. We further showed that carbamylated proteins were themselves pro-inflammatory and triggered T cell activation (44). Moreover, recent studies by others have shown that either direct OCN− exposure to endothelial cells in culture, or via infusion in vivo, promotes protein carbamylation, is pro-inflammatory and inhibits NO-dependent functions (45, 46). Airway lining fluid has high levels (millimolar) of SCN−, where it exerts bacteriostatic activity (47). Because our prior in vitro studies revealed EPO preferentially utilizes SCN− as substrate, and is even more efficient at producing OCN− than MPO (28), we hypothesized that under eosinophilic inflammatory conditions, such as occurs within asthmatic airways, EPO might promote protein carbamylation and accompanying adverse proinflammatory effects. Herein we demonstrate for the first time that EPO-catalyzed protein carbamylation occurs during allergen-triggered asthma exacerbation and contributes to altered immune responses, triggering a cascade of inflammatory processes present in asthma.
Results
EPO Utilizes SCN− and H2O2 at Physiological Levels as Substrates to Carbamylate Proteins
Because we previously showed EPO catalyzes production of OCN− from SCN− and H2O2 (28), we first tested whether isolated human EPO could catalyze protein carbamylation, as monitored by protein-bound HCit, under physiological conditions of enzyme and reactants. Catalytic levels of EPO (60 nm) were incubated with albumin (1 mg/ml) as protein target, H2O2 (100 μm final), and plasma concentrations of Cl− (100 mm), Br− (100 μm), and SCN− (100 μm). Protein in the reaction mixture was recovered, hydrolyzed to its constituent amino acids, and protein-bound HCit was examined by HPLC with on-line electrospray ionization tandem mass spectrometry (LC/MS/MS) as described (43). In protein hydrolysates recovered from reaction mixtures of the complete EPO/H2O2/SCN− system, a new analyte was readily observed that possessed the same retention time as authentic HCit standard, as monitored using multiple parent to daughter ion transitions (e.g. m/z 190 → 173, 190 → 127, and 190 → 84) characteristic of HCit (Fig. 1A). To further confirm HCit was formed by the complete EPO/H2O2/SCN− system, the full scan collision-induced dissociation mass spectrum of the new analyte was obtained and found to be identical to authentic HCit (Fig. 1B).
FIGURE 1.

Protein carbamylation is catalyzed by the EPO/SCN−/H2O2 system. A, extracted ion chromatograms in positive multiple reaction monitoring mode of HCit standard and the hydrolysate of BSA after reaction with EPO/SCN−/H2O2 with parent-to-daughter transitions, m/z 190 → 173, 190 → 127, and 190 → 84, respectively. B, MS/MS spectra in positive mode of HCit standard, and the HCit isotoplogue in the albumin hydrolysate after reaction with SCN−, [13C]SCN−, or [13C,15N]SCN− and H2O2 in the presence of EPO. The mass spectra were acquired by Triple TOF with positive information-dependent acquisition mode. C, reaction requirements and quantification of protein (BSA, 1 mg/ml) carbamylation by the EPO/SCN−/H2O2 system under physiological concentrations of halides (100 mm Cl−, 100 μm Br−, 100 μm SCN−), 100 μm H2O2, and 60 nm EPO with catalase (500 nm) and methionine (500 μm) as indicated. D and E, protein carbamylation occurs across physiological concentration ranges of SCN− and H2O2. The concentrations of H2O2 in D and SCN− in E are 300 and 100 μm, respectively. Data are presented as mean ± S.D. for two (C) independent experiments or scatter plots for individual experiments with mean ± S.D. indicated (D and E).
In additional studies, we used different isotope-labeled SCN− (i.e. natural abundance versus [13C]SCN− versus [13C,15N]SCN−) as co-substrate for EPO to trace at the atomic level the incorporation of atoms from SCN− substrate into the posttranslational modification of protein lysine residues forming protein-bound HCit. Notably, with incorporation of native and each heavy isotope-labeled SCN−, we observed generation of the corresponding anticipated HCit isotopologue (i.e. natural abundance HCit versus [13C]HCit and [13C,15N]HCit), eluting at the same retention time as HCit. Importantly, high resolution mass spectra of each isotopologue confirmed both parent ions and fragments anticipated for the isotopologues based on predicted elemental composition of the compounds, with assigned daughter ions based upon a neutral loss of NH3, formic acid, and isocyanic acid in that order (Fig. 1B). These studies thus further confirm at the atomic level the source of SCN− in generation of protein-bound HCit during EPO catalyzed protein carbamylation.
To further explore the mechanism of EPO-catalyzed protein carbamylation, we explored the reaction requirements by quantifying the level of protein-bound HCit formed using stable isotope dilution LC/MS/MS analyses of proteins recovered from reaction mixtures of the complete EPO/H2O2/SCN− system, or various controls (Fig. 1C). Formation of HCit required each component of the complete system, and occurred in the absence or presence of plasma levels of Cl− and Br−. The addition of catalase to quench H2O2 blocked the EPO-catalyzed protein carbamylation. On the other hand, addition of halogenating oxidant scavengers like methionine failed to impact EPO-catalyzed protein carbamylation. Quantification of the extent of EPO-catalyzed protein carbamylation showed dose-dependent increases in HCit formation across pathophysiologically relevant ranges of both SCN− and H2O2 (32, 33, 48) (Fig. 1, D and E). Collectively, these results suggest that EPO-mediated protein carbamylation can occur under physiologically-relevant conditions.
EPO Promotes Protein Carbamylation at Sites of Inflammation
To directly test whether EPO can catalyze protein carbamylation in vivo, protein-bound HCit was quantified in tissues recovered from animal models of eosinophilic inflammation using EPO-deficient (EPO-KO) mice. Initially we used a well characterized model where large numbers of eosinophils (>106, 20–30%) are induced into the peritoneal cavity of mice via a sensitization/challenge protocol using a whole-protein extract of the helminth Mesocestoides corti. Subsequent intra-peritoneal injection of zymosan, a yeast cell wall glucan, triggers activation of the recruited peritoneal eosinophils, resulting in eosinophil degranulation and EPO release, coupled with H2O2 generation from the oxidant burst (20, 49). As illustrated in Fig. 2A, peritoneal lavage proteins recovered at baseline in the absence of any provocative challenge already contain protein-bound HCit because of the ubiquitous presence of urea in body tissues and fluids. However, in WT mice, but not EPO-KO mice, nearly a 2-fold enhancement in protein carbamylation is observed following both eosinophil recruitment with M. corti antigen injection, and then leukocyte activation at time of peak eosinophil recruitment (72 h post-M. corti) with zymosan treatment. A contribution of HCit production by MPO from activation of alternatively elicited leukocytes was minimal in this model because EPO-KO mice showed no incremental increase in protein-bound HCit following M. corti and zymosan treatment (Fig. 2A). To further demonstrate EPO-catalyzed protein oxidation in vivo using this inflammatory model, the content of 3-bromotyrosine was simultaneously determined in recovered peritoneal lavage proteins. Again, a significant increase (p < 0.001) was only observed in peritoneal proteins recovered from WT but not in EPO-KO mice following M. corti/zymosan challenge (Fig. 2A).
FIGURE 2.

EPO is a catalytic source for carbamylation at sites of inflammation and within asthma airway specimens. A, abundance of protein-bound HCit recovered from peritoneal lavage supernatants of wild-type (WT) and EPO-deficient (EPO-KO) mice (n = 5 per group) presensitized to M. corti whole protein extract and then injected with normal saline (NS), M. corti antigen, or M. corti antigen with zymosan (M. corti/zymosan) as indicated after cell pellet removal. Abundance of protein-bound bromotyrosine (BrTyr) as an indicator of EPO-specific activity is listed. B, abundance of protein-bound HCit in homogenates of lung tissue after ovalbumin allergic asthma induction. Ovalbumin pre-sensitized wild-type (WT) and EPO-KO mice (n = 6 per group) 24 h after final challenge of either normal saline (NS) or OVA challenge. Data represent the mean ± S.D. for independent replicates (A and B). Protein-bound BrTyr levels as in A are indicated. C, representative fluorescence microscopy of a human asthma airway biopsy specimen identifies EPO and carbamyl protein localization. Sections were immunostained with either monoclonal antibodies to EPO (left panel) or carbamylated proteins (middle panel). The merged image (right panel) reveals co-localization of EPO and carbamyl proteins. Nuclei were stained with DAPI and insets are magnifications of the boxed sections. Protein-bound bromotyrosine level, expressed as BrTyr/Tyr (μmol/mol), was reported previously (20).
We next examined the role of EPO in promoting protein carbamylation in a mouse asthma model, aerosolized ovalbumin (OVA) challenge. WT and EPO-KO mice were initially sensitized to OVA via intraperitoneal injection (i.p.) with the adjuvant alum. Sensitized mice were subsequently challenged with aerosolized OVA or vehicle (normal saline) as control, as described under “Experimental Procedures.” Twenty-four hours following aerosolized challenge, a marked ∼8-fold increase in lung tissue protein-bound HCit content was noted in WT mice relative to normal saline controls. In marked contrast, the EPO-KO group showed no increase in lung protein-bound HCit content following allergen challenge (Fig. 2B). Parallel changes in lung tissue levels of the alternative EPO selective oxidation product, 3-bromotyrosine, were also noted.
To initially explore whether EPO might catalyze protein carbamylation in humans, particularly within asthmatic airways, we investigated the localization of EPO and carbamylated protein using immunofluorescence microscopy of biopsies taken from the human asthmatic airway from 5 distinct subjects (2–3 biopsies/subject) with mild asthma. We found all biopsies from each subject to similarly stain positive for EPO and carbamylated protein co-localization. A representative figure is presented (Fig. 2C) showing EPO immunostaining was co-localized with epitopes recognized by a monoclonal antibody selective for carbamylated proteins. Collectively, these results show that EPO serves as a major pathway for promoting protein carbamylation in vivo where eosinophils are present and activated, including within human asthmatic airways.
Allergen Exposure Triggers Eosinophil Activation and Induction of Protein Carbamylation within Airways of Atopic Asthmatics
Eosinophil recruitment and activation is a cellular hallmark of atopic asthma. We therefore sought to test whether allergen exposure resulted in eosinophil activation and protein carbamylation within airways of subjects with asthma. A segmental bronchoscopic allergen challenge was performed in mild atopic asthmatics to induce a controlled, local acute asthma reaction after which airway fluid proteins were recovered. We have previously used this model to show allergen exposure in atopic asthmatics induces eosinophil activation and EPO-catalyzed oxidative tissue injury through reactive brominating species (14). In this model, human subjects (healthy controls and non-steroid dependent allergic asthmatics; n = 6 each) underwent fiber optic bronchoscopy in a specific segment of one lung that was challenged with a known allergen at a predefined dose and for a set time period as described under “Experimental Procedures.” Comparatively, the corresponding lung segment in the contralateral lung was lavaged with normal saline as a control (t = 0 h). Bronchial airway lavage (BAL) was collected and centrifuged to isolate cells for cell counts and differentials, whereas BAL supernatant was processed to determine levels of protein-bound HCit and 3-bromotyrosine by stable isotope dilution using LC/MS/MS. After 48 h fiber optic bronchoscopy was repeated and the allergen-challenged lung segment was lavaged with normal saline and biopsies taken. Biopsies were fixed in 4% buffered formalin and used for immunofluorescent studies as described under “Experimental Procedures.” BAL was processed for cell counts and differentials, and protein-bound HCit and 3-bromotyrosine levels were quantified by LC/MS/MS. Controls (n = 6) and asthmatics (n = 6) were sex- and age-matched non-smokers. At baseline, BAL cell counts and differentials were nearly identical between the two groups, including the percentage of recovered aveolar macrophages, lymphocytes, neutrophils, and eosinophils (e.g. eosinophil % of 0.2 ± 0.2 versus 0.1 ± 0.2 for non-asthmatic controls versus asthmatics; p = NS) as previously reported (14). However, after allergen challenge, eosinophils were markedly increased among the atopic asthmatics (p = 0.01 for comparison with baseline) but not the non-asthmatic controls (16 ± 6 versus 0.3 ± 0.3%, asthmatics versus non-asthmatic controls; p = 0.03) (14). Examination of endobronchial biopsies from non-asthmatics by immunohistochemistry in this study demonstrated no eosinophilia (<1 eosinophil/high power field) and negligible (<0.5%) eosinophils in BAL irrespective of allergen or normal saline challenge (data not shown). In contrast, after allergen challenge but not normal saline challenge, a robust infiltration of eosinophils in allergic asthmatic subjects was revealed by histological examination of endobronchial biopsy specimens and cytological examination of BAL fluids, as reported in Wu et al. (14). The level of protein-bound HCit present in BAL supernatant proteins recovered at baseline (t = 0 h) and after segmental allergen challenge (48 h) in both non-asthmatic controls and the atopic asthmatic subjects are shown in Fig. 3. At baseline, no significant difference was observed between the mild asthmatics versus non-asthmatic controls. In contrast, a dramatic increase in protein-bound HCit was observed in asthmatic subjects, but not non-asthmatic controls, 48 h after allergen exposure (mean ± S.D.; 2.09 ± 0.78 versus 0.29 ± 0.22 mmol of HCit/mol of Lys; asthmatic versus control, respectively; p = 0.002) (Fig. 3). Concomitantly, levels of protein-bound 3-bromotyrosine, the molecular fingerprint of EPO-catalyzed hypobromous acid oxidation, were 10-fold elevated only in the allergen-challenged asthmatic subjects at 48 h, indicative of eosinophil activation.
FIGURE 3.
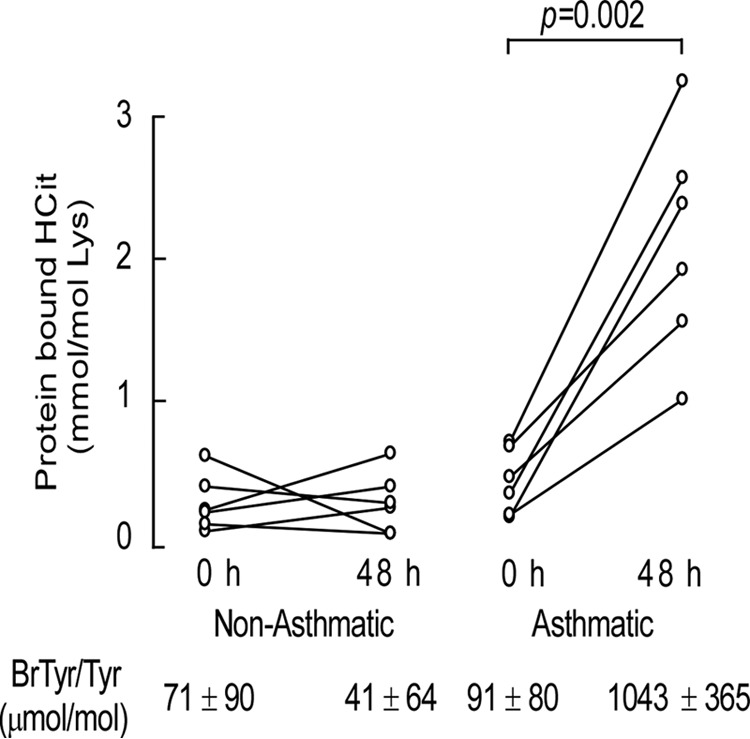
Protein-bound HCit is elevated in asthmatic subjects. Quantification of HCit in proteins recovered from non-asthmatic and asthmatic subjects at baseline and after segmental allergen challenge. Healthy control and allergic mild asthmatic subjects (n = 6 distinct subjects) underwent fiber optic bronchoscopy, and a specific segment of one lung was lavaged with normal saline to obtain a baseline sample (t = 0 h). The same specific segment in the contralateral lung was then exposed to allergen. Forty-eight hours later, fiber optic bronchoscopy was repeated on the allergen-exposed segment and lavaged with normal saline. Cells in the BAL were removed by centrifugation, and the levels of protein-bound HCit recovered in the supernatant at baseline and after segmental allergen challenge were then determined by LC/MS/MS. Abundance of protein-bound bromotyrosine (BrTyr) as an indicator of EPO-specific activity for each group is listed, as previously reported (14).
Carbamylated Proteins Contribute to Asthma Phenotypes
Characteristic features of asthma include increased airway hyperreactivity, increased mucus production, and increased epithelial cell apoptosis leading to denudation of the epithelial cell lining in the asthmatic lung (16). We hypothesized that production of carbamylated protein may participate in these events. We first investigated the effect of carbamylated albumin (c-albumin) exposure on mucus production within lung epithelial cells. Importantly, a pathophysiological level of human albumin carbamylation was used for these studies. Specifically, A549 human airway epithelial cells were incubated with either native human albumin (8.0 μg/ml), which has a comparatively low level of HCit (0.16 mmol/mol of Lys) or comparable concentrations of albumin that had first been carbamylated either chemically with OCN− (1.5 mmol of HCit/mol Lys), or enzymatically with EPO using the complete EPO/SCN−/H2O2 system (2.5 mmol of HCit/mol of Lys) to levels of protein-bound HCit similar to that observed in the asthmatic airway (Fig. 3, 2.09 ± 0.78 mmol of HCit/mol of Lys). Periodic acid-Schiff (PAS)-hematoxylin staining of the A549 cells 24 h following exposure revealed that c-albumin produced by either chemical or enzymatic (EPO-catalyzed) modification, but not native albumin, led to increased polysaccharide content by PAS staining (Fig. 4A, middle and right panels). Based upon increased PAS staining in response to carbamylated protein exposure, we next examined the specific effect that c-albumin exposure had on mucin expression in airway epithelial cells. Mucin 5AC (MUC5AC) is the dominant mucin protein expressed in the asthmatic airway (50). We therefore transfected A549 human alveolar basal epithelial cells with the plasmid pGL3-Basic being driven by the muc5AC promoter inserted upstream of the reporter gene, firefly luciferase. The control plasmid, pRL-TK, and a transfection efficiency indicator plasmid, pCG-GFP, were also used to co-transfect the A549 cells simultaneously to normalize transgene expression levels. Using a Dual-Glow luciferase assay kit we monitored the relative expression of muc5AC-driven luciferase expression. In transfected A549 cells, c-albumin exposure led to a significant increase in muc5AC-driven reporter gene expression, as determined by luciferase activity (Fig. 4B). Increased MUC5AC protein levels, as determined by MUC5AC-specific ELISA, were also observed to be elevated in parallel treated cells (Fig. 4C).
FIGURE 4.

Carbamyl protein exposure increases mucin expression. A, PAS-hematoxylin staining of A549 cells 24 h after exposure to albumin or carbamyl-albumin (c-albumin). B, expression from the muc5AC promoter in A549 cells monitored by Dual-Glow luciferase activity assay after transfection with pmuc5AC-Luc3-basic and pTK-RL. Cells were starved for 1 day and then incubated in serum-free medium with either albumin, EPO/SCN−/H2O2 catalyzed c-Alb (EPO) or OCN− carbamylated albumin (c-Alb (OCN−)), as described under “Experimental Procedures,” and muc5AC promoter-driven luciferase expression was determined. C, MUC5AC accumulation in A549 cells incubated with carbamylated protein as in B was determined by MUC5AC-specific ELISA. D, secretion of MUC5AC into the cell growth media from NCI-H292 monitored by MUC5AC-specific ELISA after incubation with the indicated albumin, EPO-carbamylated albumin or OCN−-carbamylated albumin proteins as in B. The results were expressed as the relative concentration of MUC5AC normalized to control cells having a value of 1 arbitrary unit (AU). E, MUC5AC accumulation in A549 cells incubated for 1 day with 20 μg/ml of carbamylated ovalbumin either by reaction with OCN− or the EPO/SCN−/H2O2 complete system, or brominated ovalbumin either by reaction with HOBr or EPO/Br−/H2O2, with modified ovalbumin labeled as c-Ova (OCN−), c-Ova (EPO), Br-Ova (HOBr), and Br-Ova (EPO), respectively. The concentration of MUC5AC was determined by ELISA and the results are expressed as the relative concentration to the cells incubated with ovalbumin. F, RT-PCR determination of MUC5AC expression in mice after OVA or c-OVA challenge. Cumulative n values for animal numbers from several replicate studies are listed where RT-PCR were analyzed for MUC5AC gene expression. G, immunohistochemical staining of mouse lung tissues with anti-MUC 5AC antibody from C57BL/6J WT mice challenged with c-OVA versus OVA. H, ELISA quantitation of MUC5AC expression in lung tissues. Scatter plots indicate individual replicates and the mean ± S.E. is indicated (B–F and H). Cumulative n values for animal numbers are reported (F and H).
To independently confirm carbamylated protein exposure-induced mucin production, another human airway epithelial cell line, NCI-H292, was exposed to control versus two different conditions that foster protein carbamylation and potentially mimic the setting as might occur in the asthmatic lung. Specifically, NCI-H292 cells were incubated in serum-containing media either alone (control), in the same media supplemented with OCN− (100 μm) to chemically promote protein carbamylation, or the same media supplemented with EPO, SCN−, and an H2O2-generating system (glucose/glucose oxidase; i.e. EPO/SCN−/Glc/Gox) (14). Twenty-four hours later the concentrations of MUC5AC secreted into the medium was quantified using a MUC5AC-specific ELISA, as described under “Experimental Procedures.” Remarkably, the airway epithelial cells significantly enhanced MUC5AC secretion under both conditions that promote protein carbamylation (Fig. 4D).
EPO can also catalyze protein bromination (17). We therefore sought to test whether protein exposed to EPO-generated brominating oxidants can increase mucin expression in cultured airway epithelial cells similar to carbamylated protein. We investigated this possibility by incubating A549 cells with ovalbumin modified by reaction with OCN−, EPO/SCN−/H2O2, HOBr, or EPO/Br−/H2O2, and quantified the MUC5AC expression present in cell lysates by ELISA. Notably, we found that only the chemical carbamylation (OCN−) and enzymatic carbamylation (EPO/SCN−/H2O2)-modified ovalbumin triggered enhanced MUC5AC expression in the airway epithelial cells (Fig. 4E).
We next sought to test whether carbamylated protein exposure to airway lining epithelial cells in vivo might similarly lead to increased mucin production. Importantly, for these studies we used naive mice (i.e. non-sensitized), and exposed them for the first time with nebulized protein solution (40 mg/ml) of either carbamyl-ovalbumin (c-OVA, 1.1 mmol of HCit/mol of Lys) or native ovalbumin (OVA, 0.15 mmol of HCit/mol of Lys), as described under “Experimental Procedures.” (Note, ovalbumin was selected as the target protein for aerosolized exposure testing because of its low background level of PB-HCit, and because commercial sources of albumin were found to have higher carbamylation levels at baseline, possibly because of urea used during albumin isolation.) Animals were sacrificed 24 h after treatment and lung tissues were harvested with identical lobes from each treatment group collected for immunohistochemistry, RNA expression analysis, or protein and mass spectrometry studies. Real time (RT)-PCR quantitation of naive mouse lung Muc5ac mRNA expression level after exposure to nebulized OVA versus c-OVA is shown in Fig. 4F. As can be seen, Muc5ac mRNA levels were significantly higher (p = 0.002) in mice exposed to aerosolized c-OVA relative to native OVA (Fig. 4F). In addition, mouse airway tissues showed increased MUC5AC-specific immunofluorescent staining in response to aerosolized c-OVA (Fig. 4G, right), and MUC5AC-specific ELISA quantification showed significantly higher (p = 0.002) protein content in the airway lumen of c-OVA exposed mice compared with native OVA-aerosolized control mice (Fig. 4H).
In parallel studies we incubated primary human airway epithelial cells with native and individually oxidatively modified ovalbumin (chemically versus enzymatically brominated, or carbamylated) and found that only carbamyl-ovalbumin enhance glycoprotein expression, which was reflected by the enhanced PAS red staining area (Fig. 5). Thus, exposure of human airway epithelial cells to OVA incubated with either OCN− or the EPO/SCN−/H2O2 system resulted in enhanced PAS positive staining, whereas exposure to OVA previously exposed to either HOBr or the EPO/Br−/H2O2 system failed to alter glycoprotein expression and PAS staining.
FIGURE 5.

Carbamyl-ovalbumin rather than brominated ovalbumin induces mucin expression. A–E, representative PAS-hematoxylin staining of primary human airway epithelial cells after incubation with 80 μg/ml of modified versus non-modified ovalbumin for 3 days. Ovalbumin modifications include carbamylated ovalbumin generated either by reaction with reagent OCN− or exposure to the EPO/SCN−/H2O2 complete system as described under “Experimental Procedures,” or brominated ovalbumin generated either by reaction with reagent HOBr or by exposure to the complete EPO/Br−/H2O2 system as described under “Experimental Procedures.” F, computer-assisted quantification of mean PAS staining area in cells cultured in a 24-well plate after exposure to different modified ovalbumin for 3 days. Each treatment was performed in biological duplicates with 4 pictures taken under microscopy for each condition. The PAS positive staining is shown in red and cumulative random area of 216.5 × 165.0 μm for each treatment was integrated using ImagePro software. Scatter plots indicate individual replicates and the mean ± S.E. is indicated.
Carbamylated Proteins Enhance Asthma-associated Cytokine Expression and Increased Airway Epithelial Cell Apoptosis
Cytokines underlie the inflammation of asthma and cytokine antagonists are an effective strategy to treat asthma (reviewed in Ref. 51). Many of the cytokines involved in the pathophysiology of asthma are secreted from T cells of the Th2 type, such as IL-4, IL-5, IL-9, and IL-13 (52). Th1 cells secrete IFNγ, which has pleiotropic effects on Th2 cells, and can counteract, enhance, or prolong Th2 responses (53). TGFβ, is secreted from T-regulatory cells, is associated with tissue remodeling in asthma, and is detected at higher levels during asthma exacerbation (54). To test whether carbamylated proteins (HCit-containing) could trigger or enhance immune responses that contribute to asthma, we examined expression levels of cytokines known to contribute to asthma exacerbation and airway remodeling in lungs recovered from the naive (non-sensitized) mice exposed to aerosolized native ovalbumin (OVA, 0.15 mmol of HCit/mol of Lys) versus c-OVA (1.1 mmol of HCit/mol of Lys) (Fig. 6, A–E). All cytokines monitored (IFNγ, TGFβ, IL-4, IL-5, and IL-13 mRNA) showed significantly higher levels of expression in mice exposed to aerosolized c-OVA compared with OVA. However, despite the significant increases in cytokine expression levels induced by c-OVA, within this short time period (72 h), no observed changes in airway elastance or hyperreactivity in the naive animals were observed (Fig. 6, F and G).
FIGURE 6.

c-OVA enhances cytokine expression and carbamylation generating systems induce apoptosis of airway epithelial cells. A–E, RT-PCR determination of expression levels of IFNγ, TGFβ, IL-4, IL-5, and IL-13 mRNA in mice 72 h after OVA or c-OVA challenge. p values were calculated by Student's t test. Cumulative n values for animal numbers from several replicate studies are reported. F and G, pulmonary function measured as resistance and elastance of lung airways in C57BL/6J wild-type (WT) mice exposed to OVA and c-OVA. F and G, airway hyperresponsiveness (F) and lung mechanics (G) were measured in mice (n = 19 per group) in response to increasing doses of inhaled methacholine following exposure to nebulized OVA or c-OVA as described under “Experimental Procedures.” p values were found not to be significantly different between the treatment groups. H, TUNEL assay showing A549 cell apoptosis in response to incubation in medium containing carbamylating agents EPO/SCN−/Glc/Gox or OCN− as indicated. Control reactions consisted of similar levels of Glc/Gox and SCN− in the absence of added EPO. Nucleus staining by DAPI in the same cells is shown in the top panel. I, graphic representation of TUNEL-positive cells counted in H, p values were calculated by Wilcoxon rank sum test. Scatter plots indicate individual replicates with mean ± S.E. indicated (A–E and I) or data were presented as mean ± S.E. (F and G). p values were calculated by Wilcoxon rank sum test.
Apoptosis of airway epithelial cells is known to contribute to airway remodeling and plays an important role in the pathogenesis of asthma (2). To determine the potential impact of carbamylated proteins on airway epithelial cell apoptosis we examined A549 human alveolar basal epithelial cells following 24 h exposure to two distinct carbamylating systems, either EPO/SCN−/Glc/Gox or OCN−. Serving as a control, cells were similarly exposed in the same media supplemented with comparable levels of the H2O2 generating the Glc/Gox system and SCN− but in the absence of any added EPO. Apoptosis induced in response to HCit-generating treatments was monitored by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and quantified as described under “Experimental Procedures.” As can be seen, exposure of cells to either condition for generating carbamylated proteins led to significant increases in the level of TUNEL-positive cells (Fig. 6, H and I).
Effects of Scavenger Receptor SR-A1 on Carbamylated Protein-enhanced Asthma-associated Phenotypes and Cytokine Expression
Scavenger receptor A1 (SR-A1) has previously been shown to be involved in recognition of carbamylated lipoproteins (43). To examine whether SR-A1 might also recognize carbamylated proteins in airway epithelial cells and play a role in asthma-associated pathophysiological responses, we first tested whether SR-A1 is expressed in airway epithelial cells. Western analyses with antibody specific for SR-A1 revealed the expression of SR-A1 in A549 and NCI-H292 cells is clearly visible. SR-A1 expression was also seen in HCAEC, HUVEC, BASMC, and Raw 264.7 cells and in mouse liver homogenate (Fig. 7A). Other scavenger receptors previously suggested to serve as receptor for alternative carbamylated proteins (55) were also examined, including LOX-1 and CD36, but no detectable expression of these proteins was observed in airway epithelial cells by Western blot analyses (data not shown).
FIGURE 7.

Identification of scavenger receptor, SR-A1, in human airway epithelial cells and carbamyl ovalbumin-enhanced muc5AC expression is not mediated by SR-A1. A, Western blot for SR-A1 protein expression in HCAEC, HUVEC, A549, NCI-H292, BASMC, and Raw264.7 whole cell extracts and mouse liver homogenate as indicated. B-G, RT-PCR quantitation (for mRNA) of relative MUC5AC, IFNγ, TGFβ, IL-4, IL-5, and IL-13 expression in lung tissue from C57BL/6J WT versus SR-A1−/− mice 72 h after OVA or c-OVA challenge. Relative MUC5AC, IFNγ, TGFβ, IL-4, IL-5, and IL-13 mRNA expression levels were normalized to Arp mRNA for each mouse and the mean value of WT mice challenged with OVA for each gene was normalized to a value of 1 arbitrary unit (AU). Scatter plots indicate individual replicate animals and the mean ± S.E. is indicated. p values were calculated by Wilcoxon rank sum test.
To explore the role of SR-A1 in recognition of carbamylated proteins in airway epithelial cells in vivo, we examined whether carbamylated protein exposure (via nebulization as before) similarly leads to increased mucin production and asthma-associated cytokine expression in wild type versus SR-A1−/− mice. Importantly, for these studies we used wild type C57Bl/6J and SR-A1−/− naive mice (i.e. non-sensitized), and exposed them with nebulized protein solution (40 mg/ml) of either c-OVA or OVA as described under “Experimental Procedures.” Animals were tested for pulmonary function/airway reactivity in response to methocholine challenge, and lung tissues were collected from mice to quantify various asthma-associated cytokine and MUC5AC expression levels by RT-PCR. As can be seen, c-OVA exposure, but not native OVA exposure, led to increased mucin and asthma-associated cytokine expression in both WT and SR-A1−/− mice (Fig. 7B). However, lack of SR-A1 did not significantly affect c-OVA-induced muc5AC expression or asthma-associated cytokine genes (Fig. 7, B-G) except for IL-4 and IL-13. Interestingly, lack of SR-A1 leads to an increased IL-4 response to both OVA and c-OVA compared with WT animals (p = 0.05 and p = 0.03, respectively) (Fig. 7E), whereas absence of SR-A1 significantly decreased IL-13 expression in response to c-OVA (p = 0.01); however, IL-13 induced changes were still significantly increased (albeit attenuated) from c-OVA-exposed SRA−/− animals (p = 0.006) (Fig. 7G). Given the possible role of SR-A1 as a receptor for carbamylated protein in the airways we examined pulmonary function in parallel to the mice treated as described above. Although SR-A1 is involved in carbamylated protein-enhanced IL-13 expression to some extent, we did not see any differences in mice pulmonary function by monitoring airway elastance or hyperreactivity in naive mice during methocholine challenge testing (Fig. 8).
FIGURE 8.

Resistance and elastance of lung airways in C57BL/6J wild-type (WT) versus SR-A1−/− mice challenged with OVA and c-OVA. A and B, pulmonary function measured as airway hyperresponsiveness (A) and lung mechanics (B) were measured in mice in response to increasing doses of inhaled methacholine following exposure to nebulized OVA or c-OVA as described under “Experimental Procedures.” Results are from a single experiment. Treatment regime and genotype are indicated with n > 10 mice per group. Data were expressed per group as mean ± S.E.
Discussion
Both the prevalence and severity of asthma continue to increase, with a significant healthcare burden (56). The recruitment and activation of eosinophils in the airways is a characteristic feature of asthma, and the role of eosinophils in the pathogenesis of this disorder has long been a focus of intense investigation. Classically, eosinophils are recognized for their oxidative and antimicrobial activities, but more recently, eosinophils have been recognized for directly regulating T cell function and immune responses, contributing to allergic respiratory inflammation (57). Accordingly, an improved understanding of the basic chemical pathways available to eosinophils for generating specific reactive oxidants and diffusible radical species in vivo, and for eliciting changes in inflammatory signaling is of interest. Herein we show that eosinophil activation within asthmatic airways, such as following exposure to allergen, results in EPO-catalyzed protein carbamylation, with accompanying induction of pro-inflammatory and immune modulatory effects. A scheme illustrating the chemistry involved and the impact of this pathway on asthma-promoting features is presented in Fig. 9.
FIGURE 9.
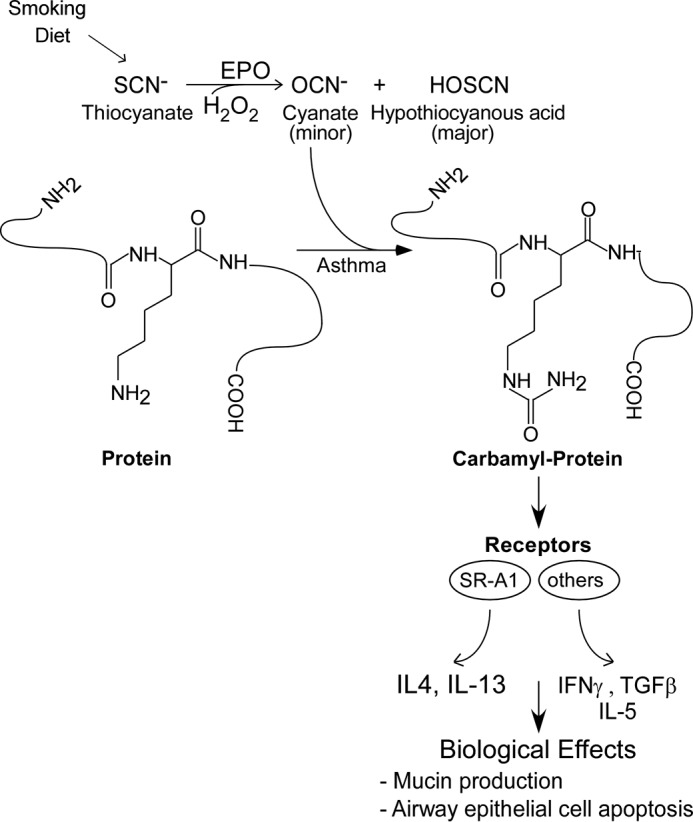
Proposed pathway for the promotion of EPO of protein carbamylation and enhancement of asthma pathophysiology. Diet and tobacco smoke exposure are major determinants of plasma SCN− concentration. Leukocyte EPO uses H2O2 and SCN− as co-substrates to generate OCN− and promote protein carbamylation at sites of eosinophilic inflammation. Carbamylated protein participates in multiple asthma-associated phenotypes including induction of mucin, cytokine expression, and epithelial cell apoptosis. Whether the induction is mediated by scavenger receptors such as SR-A1 and others need further investigation.
Posttranslational modification of lysine residues by carbamylation results in formation of protein-bound homocitrulline (Nϵ-carbamyllysine), a modification thought until recently to result from reaction of urea driven OCN− formation, especially in end stage renal disease. In renal disease, protein carbamylation is markedly increased and has been suggested to serve as an important contributor to the “toxemia of uremia” (37). Recently we identified another pathway to carbamylate proteins at sites of inflammation that involves the mammalian heme-peroxidase MPO and production of OCN− from the pseudohalide substrate thiocyante (SCN−) and H2O2 (43). SCN− is present at abundant levels in vivo and its levels are dependent upon environmental exposures including both diet and tobacco smoke (30, 58, 59). Hydrogen peroxide is ubiquitously made via the electron transport chain in response to oxidative phosphorylation during normal cellular respiration (60). High levels of H2O2 are also produced by activated phagocytes during their respiratory burst, and play an important role in host defense (61, 62). We previously showed EPO, the heme-dependent peroxidase found in eosinophils, preferentially uses SCN− and H2O2 as co-substrates to produce not only hypothiocyanous acid (HOSCN) as the major product, but also OCN− as a minor product (28). In those initial studies, we did not examine whether EPO can catalyze protein carbamylation, nor did we explore potential physiological effects of this potential reaction. Our studies clearly show isolated human EPO catalyzes protein carbamylation using SCN− and H2O2 as co-substrates under physiological conditions. Furthermore, use of EPO-KO mice and examination of human asthmatic airway biopsies confirm a dominant role for EPO in protein carbamylation during eosinophil activation in vivo in both animal models of asthma and within human asthmatic airways. Moreover, provocative segmental allergen challenge studies in atopic asthmatics revealed robust protein carbamylation in response to allergen within asthmatic airways. Finally, functional studies reveal either chemical (KOCN) or EPO-catalyzed carbamylation of target proteins confers a gain of function activity to the modified proteins, enabling them to trigger a cascade of asthma-related phenotypes including mucus and cytokine production and airway epithelial cell apoptosis. The present studies thus show that protein carbamylation, a PTM observed in other proinflammatory and chronic conditions, similarly occurs in asthma. These studies suggest PTM of proteins via carbamylation serves as a common underlying proinflammatory biochemical process in a variety of conditions including asthma, and may thus serve as a potential pharmacological target for asthma.
In the airways of both humans and mice, SCN− levels are quite high (millimolar range), averaging 10–100-fold higher than plasma, due to an active transport process involving the basolateral Na+ symporter and efflux via the cystic fibrosis transporter (63, 64). Such high levels are thought to exist due to bacteriostatic activity of SCN−, a substrate for lactoperoxidase, which similarly is high in concentration in saliva, airway lining fluid, tears, milk, and other fluids that line mucus membranes (65). Although the HOSCN formed by SCN− oxidation by mammalian heme peroxidases (EPO, MPO, and lactoperoxidase) is bacteriostatic, it is remarkably nontoxic to host cells (66). Moreover, because it can serve as a preferred substrate to mammalian heme peroxidases, SCN− can modulate the production of more potent and cytotoxic oxidants by EPO and MPO (HOBr and HOCl, respectively), and thus has the potential to limit the extent of some oxidative reactions mediated by activated leukocytes (28, 66–68). The combination of bacteriostatic activity and potential for reduced collateral oxidative tissue damage to host has led to investigation in recent animal model studies of the potential efficacy of aerosolized SCN− as an adjuvant therapy for cystic fibrosis (69). However, despite these presumed beneficial effects of SCN−, the present studies suggest that in asthma, SCN− oxidation by EPO leads to carbamylation of proteins, as monitored by increased protein-bound HCit levels, and the potential to propagate more potent asthma-promoting responses. It is thus of interest that a national population based study utilizing data from the United States National Health and Nutrition Examination Surveys (NHANES) recently showed that urinary SCN− concentrations are strikingly associated with multiple lung problems including increased risk of wheezing, coughing, chronic bronchitis, emphysema, and sleep complaints (70). In addition, SCN− levels are increased not only in smokers, but also in subjects exposed to second hand smoke (31, 71), with second hand environmental tobacco smoke exposure being a major known trigger for asthma exacerbations and severity (72–74). It is tempting to speculate that protein carbamylation within asthmatic airways in part could account for the association between second hand smoke exposure and worse asthma severity. Further studies in this area seem warranted.
The present studies add to a growing body of work suggesting that the spatiotemporal generation of carbamylated proteins in distinct sites in chronic diseases can induce distinct proinflammatory cascades. For example, in prior studies we showed that HCit-containing lipoproteins within the atherosclerotic lesion can be generated by MPO-catalyzed carbamylation, which endows them with multiple pro-atherogenic biological properties including increased cholesterol accumulation and foam cell formation, enhanced endothelial cell apoptosis, and induction of vascular smooth muscle cell proliferation (43). Other investigators have more recently shown that carbamylated lipoproteins can also promote endothelial dysfunction and additional proinflammatory and proatherogenic activities (75). We have also shown that within inflamed joint space fluid from subjects with rheumatoid arthritis protein-bound HCit is increased, and direct injection of HCit containing peptides into the inter-articular joint space has been shown to selectively activate T cells and induce a proinflammatory erosive arthropathy in mice (44). Whereas these alternative studies involve MPO-driven inflammatory conditions characterized by monocytes/macrophages or neutrophils, results shown herein reveal that eosinophils, via secretion of EPO, similarly promote protein carbamylation within the asthmatic lung and airways. Either endogenous production or delivery (as aerosolized exposure) of HCit containing proteins was shown to elicit multiple asthma-associated phenotypes, including increased mucin expression, asthma-promoting cytokines (IL-4, IL-5, IL-13, and INFγ), TGFβ expression, and airway epithelial cell apoptosis. Because carbamylated protein exposure failed to impact airway reactivity, the immediate implication for the present findings is that airway hyperreactivity and loss of elastance develop via distinct processes in asthma and not as an immediate response to mucin production or expression/exposure to Th2 cytokines triggered by exposure to carbamylated proteins. It is interesting to note that recent asthma model studies with mice lacking eosinophils similarly indicate a role of eosinophils in mucin production, Th2 cytokines, and cytokines conductive to tissue remodeling, and less or no significant effects on airway elastance and hyperreactivity (76).
One question the present studies suggest is the identification of the molecular receptor(s) within asthmatic airways that recognizes carbamylated proteins and triggers the pro-asthma-related phenotypes fostered. We previously suggested that one potential receptor involved in recognition of carbamylated low density lipoprotein is SR-A1 (43). Our studies with SR-A1−/− mice revealed significant reduction in c-OVA-induced IL-13 expression, although on the whole, the majority of cytokines induced by c-OVA remained unchanged, as did changes in airway responsiveness and mucus production. Thus, our studies suggest an alternative receptor(s) also contributes to asthma-related phenotypes provoked by exposure to carbamylated proteins (Fig. 9). It is notable that multiple alternative receptors exist for interaction with carbamylated proteins. For example, it has recently been reported that HCit on apolipoprotein A1 of carbamylated high density lipoprotein confers recognition by scavenger receptor class B, type 1 (SRARB1), and macrophage foam cell formation (77). Yet additional recent studies have suggested that the class E scavenger receptor lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in human vein endothelial cells can also recognize and bind carbamylated low density lipoprotein, leading to increased reactive oxygen species generation and increased cellular apoptosis (78). Carbamylated erythropoietin has been shown to be recognized not by the erythropoietin receptor, but by a heterodimer of the erythropoietin receptor and IL-3 receptor β (CD131), with carbamylated erythropoietin signaling through this heterodimeric receptor, providing protection from ischemia/reperfusion injury (79). Thus, many candidate receptors exist for recognition of various carbamylated proteins, and future studies are needed to identify the receptor(s) responsible for recognition of carbamylated proteins and the downstream immune modulatory and asthma-associated phenotypes induced within asthmatic airways.
Experimental Procedures
Materials and General Procedures
All chemicals, unless otherwise noted, were obtained from Sigma, and all solvents were HPLC grade. Stable isotope-labeled compounds were purchased from Cambridge Isotope Lab (Tewksbury, MA), unless otherwise stated.
Protein Carbamylation in Vitro
Reactions were carried out at 37 °C for 8 h in 50 mm sodium phosphate buffer (pH 7.0) supplemented with 1 mg of protein/ml of bovine albumin (BSA) or ovalbumin (OVA) wherever indicated. EPO (60 nm), thiocyanate, H2O2, and other components were added at different concentrations as indicated. Typical reactions used either the complete EPO system (60 nm EPO, 100 μm SCN−, 100 μm H2O2) or reagent OCN− (100 μm, as the potassium salt) under the incubation conditions listed and reactions were terminated with 50 nm catalase, 40 μm butylated hydroxytoluene, and 100 μm diethylenetriaminepentaacetic acid and dialysis against PBS containing 100 μm diethylenetriaminepentaacetic acid as described (43). Where indicated, a glucose (Glc, 100 μg/ml)/glucose oxidase (Gox, 20 ng/ml) system was used to generate H2O2 (14).
Cell Culture
A549 human adenocarcinoma lung cells (ATCC CCL-185) were grown in Dulbecco's modified Eagle's media (DMEM) with 1 mg/ml of glucose and 10% fetal bovine serum (FBS) and NCI-H292 human mucoepidermal pulmonary carcinoma cells (ATCC CRL-1848) were grown in RPMI 1640 media supplemented with 1 mg/ml of glucose and 10% FBS. Human bronchial airway epithelial cells were isolated from the bronchus dissected from non-asthmatic lungs obtained at explantation using previously described methods (80–82). In brief, human bronchus was digested overnight at 4 °C in protease solution with antibiotic/antimycotic (Sigma). The epithelial side of the bronchus was scraped with a sterile glass slide to remove the remaining adherent cells. Cells were seeded on tissue culture plates precoated with coating medium containing 29 μg/ml of collagen (Vitrogen; Collagen, Palo Alto, CA), 10 μg/ml of BSA (Biofluids), and 10 μg/ml of fibronectin (Calbiochem, La Jolla, CA), in serum-free bronchial epithelial basal medium (Clontech) supplemented with 1% penicillin, streptomycin and fungizone.
For cell culture experiments using the glucose/glucose oxidase system to generate H2O2, the complete EPO system (60 nm EPO, 100 μm SCN−, 20 ng/ml of glucose oxidase) was added to A549 cells in culture (90% confluence), whereas control reactions lacked the addition of SCN−. Glucose levels in the culture medium (DMEM) were adjusted to 1 mg/ml. In cell culture experiments where indicated, albumin, carbamylated albumin, native OVA, or c-OVA was added at 8 μg/ml, unless otherwise stated, and cells were harvested and analyzed as described below.
Chemical Synthesis of Nϵ-[13C,15N]Carbamyl-lysine
Nϵ-[13C,15N]Carbamyl-lysine ([13C,15N]HCit) was synthesized for use as internal standard in mass spectrometry studies. It was produced by reaction of 10 mg/ml of polylysine with 6 m [13C2,15N]urea at 37 °C for 2 days followed by 6 n HCl hydrolysis and then purified by HPLC on a Phenyl column (4.6 × 250 mm, 5 μm Rexchrom Phenyl) (Regis, Morton Grove, IL) as immobile phase and the mobile phase consisting of solvent (A), 0.2% formic acid in water, and solvent (B), 0.2% formic acid in acetonitrile over a linear gradient from 100% A to 10% B in 10 min. The eluent fractions with [13C,15N]HCit were collected and its identity confirmed by high resolution mass spectrometry, and the material was then dried under SpeedVac. The produced [13C,15N]HCit concentration was determined by mass spectrometry with gravimetrically prepared non-labeled HCit serving as standard. The product (purity >99%) was confirmed by high resolution mass spectrometry in positive mode with parent ion m/z = 192.1196, and daughter ions: m/z = 174.0960, 128.0905, and 84.0813, which correspond to neutral loss of 15NH3, formic acid, and [13C]isocyanic acid in that order.
Protein-bound Homocitrulline Quantification by Mass Spectrometry Analysis
Protein carbamylation levels, expressed as protein-bound homocitrulline (mmol/mol of Lys), was determined as previously described (43). Stable isotope-dilution HPLC with online electrospray ionization tandem mass spectrometry (LC/ESI/MS/MS) following delipidation and desalting and overnight hydrolysis with HCl was used to quantify lysine and homocitrulline in protein hydrolysates from BAL fluid or tissues. [13C6,15N2]Lys and [13C,15N]HCit were added to samples before hydrolysis and used as internal standards (83). Calibration curves were prepared using varying lysine and homocitrulline levels and a fixed amount of stable isotope-labeled internal standards undergoing hydrolysis and DSC-SCX column extraction. Sample (10 μl) was injected onto a Phenyl column (4.6 × 250 mm, 5 μm Rexchrom Phenyl) (Regis, Morton Grove, IL) at a flow rate of 0.8 ml/min. Separation was performed employing a gradient starting at 10 mm ammonium formate aqueous solution for 0.5 min, then increasing linearly to 25% methanol containing 0.1% formic acid and 5 mm ammonium formate over 3 min, followed by this solution isocratically for 15 min. The HPLC column effluent was introduced into an API 365 triple quadrupole mass spectrometer with an Ionics EP 10+ upgrade (Concord, Ontario, CA) interfaced to a Cohesive Technologies Aria LX Series HPLC multiplexing system (Franklin, MA). Analyses were performed using electrospray ionization (ESI) in positive-ion mode with multiple reaction monitoring of parent and characteristic daughter ions specific for components monitored. The transitions monitored were mass-to-charge ratio (m/z): m/z 147 → 84 for Lys; m/z 190 → 127 for HCit; m/z 155 → 90 for [13C6,15N2]Lys; and m/z 192 → 128 for [13C,15N]HCit.
High Resolution Mass Determination Using Triple TOF Mass Spectrometry
Protein lysate was injected onto a silica column (2 × 150 mm, 5 μm) (Phenomenx) at a flow rate of 0.2 ml/min. Separation was performed employing a gradient starting at 10 mm ammonium acetate aqueous solution for 2 min, then increasing linearly to 20% B (acetonitrile: methanol: acetic acid, 50:50:0.1, v/v/v) over 8 min. The HPLC column effluent was then introduced into an AB SCIEX TripleTOFTM 5600 mass spectrometer. MS/MS spectra in positive mode on an information-dependent acquisition were recorded during the LC-MS experiment. The TOF-MS was calibrated using APCI positive calibration solution (ABSciex) delivered with a Calibrant Delivery System.
Mouse Models
All studies were performed on wild type (WT or EPO+/+) or EPO knock-out (EPO−/− or EPO-KO) or SR-A1 knock-out (SR-A1−/−) mice backcrossed onto a C57BL/6J background (≫10 generations). EPO-KO mice were obtained from J. Lee, Mayo Clinic. SR-A1 knock-out mice were previously obtained from Dr. T. Kodama (University of Tokyo) and bred in our institute (84). C57BL/6J mice were purchased from Jackson Laboratory. Lavage and cell pellet from WT and EPO-KO mice after intraperitoneal (i.p.) injection of helminth M. corti antigen and yeast spore coat (zymosan) were provided by the Mayo Clinic. Animals receiving M. corti were sensitized by subcutaneous injection of 250 μg of helminth whole protein extract of M. corti and 8 × 109 heat-killed pertussis organisms (Michigan Department of Public Health, Lansing MI). Animals (n = 5 per group) were then challenged on day 21 by i.p. injection with 200 μg of M. corti whole protein extract. At 72 h post-challenge, elicited peritoneal leukocytes (usually >2 × 106 cells, with >25% eosinophils) were activated by i.p. injection of zymosan (250 mg/kg of body weight). Peritoneal lavage was performed 4 h later and cells were harvested by centrifugation. Lavage fluid and separated cells were processed as described to prevent further oxidation (20). Cell counts and differentials were performed as described (20) and lavage fluid was processed for determination of protein-bound HCit as described above.
Ovalbumin Asthma Challenge Model
Mice aged 8–12 weeks were sensitized by 2 i.p. injections of either normal saline or 20 μg of OVA (Sigma, grade IV)/2.25 mg of Imject Alum (Al(OH)3-Mg(OH)2; Pierce) in 100 μl of saline on days 0 and 14. Mice were challenged on days 24, 25, and 26 by 20-min inhalations of an aerosol (nebulized 1% OVA in normal saline; control mice received a 20-min aerosol of nebulized normal saline). Mice on day 28 were assessed for pulmonary histopathologies, tissue harvest, cellular infiltrates, or lung function as described (49). Lungs from EPO+/+ and EPO−/− mice (n = 6 per group), challenged with OVA and normal saline (NS) as a control, were also provided by the Mayo Clinic. The lung tissue was homogenized and the homogenate was used for assay of protein-bound HCit and MUC5AC expression.
Ovalbumin or Carbamyl-ovalbumin Challenge
The indicated genotypes of mice were challenged with 10 ml of aerosol of either nebulized 40 mg/ml of ovalbumin or 40 mg/ml of carbamyl-ovalbumin in a chamber the first day, then repeated 1 more time on day 2, 24 h just prior to pulmonary function measurement and tissue harvesting on day 3. The lung tissue was divided into three parts so that the same area of lung was obtained from each mouse for each specific analysis, which included being fixed in 4% formalin for immunohistochemical staining of MUC5AC, snap frozen in liquid N2 for ELISA analysis of MUC5AC, or covered with 1 ml TRIzol (TRIzol® reagent, Invitrogen, catalog number 15596-026) for RNA isolation, cDNA preparation, and real-time PCR analysis of MUC5AC and cytokines expression. When analyzing RT-PCR for various genes' expression as indicated, the number of successful reactions meeting QC are indicated by n values inserted into the figures.
Airway Hyperresponsiveness and Lung Mechanics
Airway hyperresponsiveness and lung mechanics were measured in mice (n = 9 per group) in response to increasing doses of inhaled methacholine as described (85). Mice were anesthetized with an i.p. injection of pentobarbital sodium (40–50 mg/kg). Following the intubation and placement of a 19-gauge cannula through a tracheotomy incision, the cannula was affixed with a silk suture ligation. Mice were connected to a computer-controlled piston ventilator (flexiVent, SCIREQ, Montreal, Canada) run at a rate of 150 breaths/min, with a tidal volume of 10 ml/kg, and a positive end-expiratory pressure of 3 cm of H2O. A muscle relaxant (pancuronium bromide, 0.8 mg/kg) was administered to the mice and the lungs were expanded twice to total lung capacity at an amplitude pressure of 30 cm of H2O. Methacholine in saline was aerosolized at doses of 0, 2.5, 5, 10, and 25 mg/ml and delivered over 10 s through an in-line nebulizer for each mouse. FlexiVent 5.2 software (SCIREQ) was utilized to obtain resistance measurements through multiple linear regression fitting of measured pressure and volume from each mouse to the model of linear motion of the lung. Only coefficients of determination of >0.90 were used for calculations. Lung resistance and lung compliance were obtained through the forced oscillation technique. Tissue elastance (H) and dampening were obtained by a constant phase model.
Segmental Allergen Challenge Model in Humans
Human subjects (healthy control and mild allergic asthmatic) underwent fiber optic bronchoscopy, and a specific segment of one lung was challenged by administration of a predetermined allergen at a known dose and for a set time interval as described (14). In the same lung segment, in the contralateral lung, the lung segment is lavaged with sterile normal saline and the BAL was collected (t = 0 h). Cells were centrifuged for determination of cell numbers and differentials. BAL supernatant was collected and processed for protein-bound HCit levels by LC/ESI/MS/MS (14). Forty-eight hours later, fiber optic bronchoscopy was repeated, the lung segment was challenged with allergen, lavaged with sterile normal saline, and BAL fluid was collected. Cells were removed by centrifugation and cell numbers and differentials were determined. BAL supernatant was collected and processed for protein-bound HCit levels as described above.
Immunolocalization of EPO and Carbamyl Proteins
Asthma airway specimens were fixed in 3% paraformaldehyde to prepare ultra-thin sections. The sections were incubated with primary antibodies, anti-homocitrulline antibody (43) was prepared as k2-Fab monoclonal antibody with HA tag to carbamyl-LDL for carbamyl proteins and mouse anti-human EPO antibody for EPO, diluted 1:50 in PBS, at room temperature for 2 h, and the corresponding secondary antibody, FITC-labeled mouse anti-HA antibody and anti-mouse IgG (H +L) Texas Red-lined antibody for 30 min. Sections were mounted with VECTASHIELD® mounting medium with DAPI as described (43).
Plasmids, Transfection, and Luciferase Assays
The reporter construct muc5AC contains 3.7-kb 5′-flanking region of the human MUC5AC mucin gene in a luciferase reporter vector pGL3 (86). Transient transfections of A549 cells (6-well dishes) were performed in triplicate using 0.5 μg of muc5AC luciferase reporter gene and 13.5 μg of empty vector (pGL3 basic)/well with Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's instructions. One day after transfection, the cells were starved overnight and then incubated in DMEM with albumin or carbamylated albumin (8 μg/ml) using either the EPO/SCN−/H2O2 system or OCN− carbamylation for an additional 24 h before harvesting for luciferase assays. Relative luciferase expression levels were determined using the Dual-Glow luciferase assay kit per the manufacturer's direction (Promega, Madison, WI). Cells were lysed and the activities of firefly and Renilla luciferase measured using the Dual-Glow system. The luminescence was read under a Wallac 1420 VICTOR3TM illuminator (PerkinElmer Life Sciences). Firefly luciferase activities were normalized to the Renilla luciferase activities.
Mucin Analysis
The cytochemical staining of mucin for cultured cells was carried out using the PAS kit (395B-1KT, Sigma). Cells after removal of medium were fixed with formalin/ethanol (1:9, v/v) fixative solution and followed by PAS reagent staining and hematoxylin counterstain according to protocol of these reagents provided by Sigma. MUC5AC protein was measured using ELISA following the protocol as described previously (87).
RNA Isolation
RNA was isolated from cells or lung tissue by TRIzol® reagent following the protocol as described by the manufacturer (Life Technologies). RNase-free DNase (Promega) was used to remove contaminating DNA in the prepared RNA as per the manufacturer's direction.
Real-time PCR of Target Genes
First strand cDNA was synthesized using the Brillant II SYBR® first-strand cDNA synthesis system (Agilent) with 1 μg of RNA, 100 ng of oligo(dT)20, and 100 ng of random hexamers per reaction (20 μl). Newly made cDNA was amplified by PCR in a final volume of 20 μl including Brillant II SYBR® Green QPCR master mix (Agilent), 1 μl of cDNA, and 7.5 pmol of SYBR probe and 10 pmol of each primer using a Step One Plus Real-time PCR System (Applied Biosystem). Reactions were initiated for 10 min at 95 °C, followed by 40 cycles; 15 s at 95 °C, followed by 1 min at 60 °C. The amount of target cDNA amplified was normalized using the housekeeping gene acidic ribosomal protein (ARP) (88) as reference. All primers were synthesized by information-dependent acquistion and the sequences were as follows: Muc5AC, 5′-ACG ACA CTT TTC AGT ACC AAT GAC-3′ and 5′-GCT TCC TTA CAG ATG CAG TCC T-3′ (89); IFNγ, 5′-GCT CTG AGA CAA TGA ACG CT-3′ and 5′-AAA GAG ATA ATC TGG CTC TGC-3′ (90); TGFβ, 5′-ACC GCA ACA ACG CCA TCT AT-3′ and 5′-GTA ACG CCA GGA ATT GTT GC-3′ (91); IL-4, 5′-TCG GCA TTT TGA ACG AGG TC-3′ and 5′-GAA AAG CCC GAA AGA GTC TC-3′ (92); IL-5, 5′-TCA CCG AGC TCT GTT GAC AA-3′ and 5′-CCA CAC TTC TCT TTT TGG CG-3′ (93); IL-13, 5′-GCC AGC CCA CAG TTC TA CAG C-3′ and 5′-GTG ATG TTG CTC AGC TCC TCA-3′ (94); ARP, 5′-TCA TCC AGC TGT TTG ACA A-3′ and 5′-ATT GCG GAC ACC CTG TAG GAA G-3′. Relative gene expression was determined by the 2−ΔΔCT method (95) using Step One Plus analysis software. RT-PCR was performed on biological duplicates in technical triplicate reactions. Melt curves were run for each primer pair set to confirm that fluorescence was specific under the conditions used. Reaction conditions (primer concentrations and annealing temperature) for each primer pair/template was optimized beforehand. We confirmed that carbamylated OVA treatment did not affect the threshold cycle (CT) of our reference gene ARP when compared with ovalbumin treatment at any given input cDNA concentration. The normalized gene expression data were plotted relative to the OVA-treated samples as the unity measure (RQ = 1 arbitrary unit).
Immunohistochemical Staining of MUC5AC in Mice Airway Tissues
MUC5AC protein was immunostained in paraffin sections of mouse airways with a mouse monoclonal antibody (SPM488, NeoMarkers, Fremont, CA) to mouse MUC5AC. Normal goat serum (50–062Z, Invitrogen) at 10% and goat anti-mouse IgG (H+L) (31160, Thermo Scientific) (diluted 1:50) was used as the blocking solution. Alexa Fluor® 488 goat anti-mouse IgG (H+L) (A11001, Life Technologies) was used as the secondary antibody (diluted at 1:100). Sections were mounted with VECTASHIELD® mounting medium with DAPI.
Apoptotic Cell Detection
Cell apoptosis detection was carried out using DeadEndTM Fluorometric TUNEL System (Promega) exactly as suggested by the manufacturer.
Immunoblotting Identification of Scavenger Receptor, SR-A1 in Airway Epithelial Cells
SDS-PAGE Western blotting analysis was employed to identify scavenger receptor, SR-A1 protein expression in human coronary aortic endothelial cell (HCAEC), human umbilical vein endothelial cells (HUVEC), A549, NCI-H292, bovine aortic smooth muscle cell (BASMC), Raw264.7 cell lysates, and mouse liver homogenate. Protein samples were solubilized in sample buffer (125 mm Tris-HCl, pH 6.8, with 4% (w/v) SDS, 20% (v/v) glycerol, 120 mm β-mercaptoethanol, bromphenol blue), heated to 95 °C, and separated on 12% polyacrylamide gel, each well was loaded with 30 μg of protein. After transfer onto PVDF membrane, proteins were detected using specific primary antibodies (anti-SR-A1, Clone No. SRA-E5, TransGenic Inc.) and secondary antibody, anti-mouse IgG coupled to horseradish peroxidase (NA931V, GE Healthcare) for ECL detection (Amersham Biosciences, GE Healthcare).
Statistical Analysis
Data are presented in scatter plots or as mean ± S.E. Comparisons between groups were performed using unpaired Student's t test or Wilcoxon rank sum test. p < 0.05 was considered significant between groups.
Study Approvals
Animals
All animal experiments were approved by the Cleveland Clinic and Mayo Clinic IACUC and were performed in accordance with NIH, Cleveland Clinic, and Mayo Clinic guidelines.
Human Tissue Samples
All human tissue samples were obtained from the Cleveland Clinic, Department of Pulmonology and Critical Care Medicine as part of a clinical investigation study. All participants gave written informed consent, and the Institutional Review Board of the Cleveland Clinic approved the study protocol. Clinical investigations were conducted in accordance with the Declaration of Helsinki principles.
Author Contributions
Z. W., J. A. D., and S. L. H. wrote the manuscript. S. L. H. conceived the project and Z. W., J. A. D., and S. L. H. designed and supervised the studies. Z. W., J. A. D., and S. L. H. analyzed the data. Z. W., J. B., M. A. A., N. A. L., and J. J. L. performed animal asthma studies and/or provided experimental expertise. M. J. T., M. K., S. A. C., S. C. E., and R. A. D. performed the human asthma challenge studies.
Acknowledgments
We thank S. Hörkkö, University of Oulu, Finland, for the generous gift of the carbamylation-specific single chain antibody. We thank G. Chang and L. Ruple for assistance in measuring pulmonary function and also E. R. Rodriguez for immunohistochemical staining. Mass spectrometry studies were performed on instruments housed in a facility in part supported through an AB SCIEX Center of Innovation award.
This work was supported, in whole or in part, by National Institutes of Health Grants P01 HL103453 (to S. L. H.), P01 HL081064 (to S. E.), P01 HL076491 (to S. L. H.), HL058723 (to N. A. L.), HL124165 (to J. J. L.), and 1R01HL130819 (to Z. W.) and National Center for Advancing Translational Sciences Grant UL1 TR000439. Dr. Wang and Dr. Hazen are named as co-inventors on patents held by the Cleveland Clinic relating to cardiovascular and inflammation diagnostics. Dr. Hazen reports having been paid as a consultant for the following companies: Esperion and P&G. Dr. Hazen reports receiving research funds from Astra Zeneca, P&G, Pfizer Inc., and Takeda. Dr. Wang reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics from Cleveland Heart Lab. Dr. Hazen reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from the companies shown below: Cleveland Heart Lab., Siemens, Esperion, and Frantz Biomarkers, LLC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EPO
- eosinophil peroxidase
- PTM
- posttranslational modification
- MPO
- myeloperoxidase
- HOBr
- hypobromous acid
- HCit
- homocitrulline
- HOSCN
- hypothiocyanous acid
- SCN−
- thiocyanate
- OCN−
- cyanate
- OVA
- ovalbumin
- PAS
- periodic acid-Schiff
- MUC5AC
- mucin 5AC
- SR-A1
- scavenger receptor A1
- Gox
- glucose oxidase
- c-OVA
- carbamylated ovalbumin
- BAL
- bronchial airway lavage
- ARP
- acidic ribosomal protein
- HCAEC
- human coronary aortic endothelial cell
- HUVEC
- human umbilical vein endothelial cell
- BASMC
- bovine aortic smooth muscle cell
- NS
- normal saline.
References
- 1. Andreadis A. A., Hazen S. L., Comhair S. A., and Erzurum S. C. (2003) Oxidative and nitrosative events in asthma. Free Radic. Biol. Med. 35, 213–225 [DOI] [PubMed] [Google Scholar]
- 2. Comhair S. A., Xu W., Ghosh S., Thunnissen F. B., Almasan A., Calhoun W. J., Janocha A. J., Zheng L., Hazen S. L., and Erzurum S. C. (2005) Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am. J. Pathol. 166, 663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Comhair S. A., and Erzurum S. C. (2010) Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 12, 93–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyce J. A., Bochner B., Finkelman F. D., and Rothenberg M. E. (2012) Advances in mechanisms of asthma, allergy, and immunology in 2011. J. Allergy Clin. Immunol. 129, 335–341 [DOI] [PubMed] [Google Scholar]
- 5. Fulkerson P. C., and Rothenberg M. E. (2013) Targeting eosinophils in allergy, inflammation and beyond. Nat. Rev. Drug Discov. 12, 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang L., Diaz P. T., Best T. M., Stimpfl J. N., He F., and Zuo L. (2014) Molecular characterization of redox mechanisms in allergic asthma. Ann. Allergy Asthma Immunol. 113, 137–142 [DOI] [PubMed] [Google Scholar]
- 7. Hazen S. L., Hsu F. F., Duffin K., and Heinecke J. W. (1996) Molecular chlorine generated by the myeloperoxidase-hydrogen peroxide-chloride system of phagocytes converts low density lipoprotein cholesterol into a family of chlorinated sterols. J. Biol. Chem. 271, 23080–23088 [DOI] [PubMed] [Google Scholar]
- 8. Carr A. C., van den Berg J. J., and Winterbourn C. C. (1998) Differential reactivities of hypochlorous and hypobromous acids with purified Escherichia coli phospholipid: formation of haloamines and halohydrins. Biochim. Biophys. Acta 1392, 254–264 [DOI] [PubMed] [Google Scholar]
- 9. MacPherson J. C., Comhair S. A., Erzurum S. C., Klein D. F., Lipscomb M. F., Kavuru M. S., Samoszuk M. K., and Hazen S. L. (2001) Eosinophils are a major source of nitric oxide-derived oxidants in severe asthma: characterization of pathways available to eosinophils for generating reactive nitrogen species. J. Immunol. 166, 5763–5772 [DOI] [PubMed] [Google Scholar]
- 10. Zhang R., Brennan M. L., Shen Z., MacPherson J. C., Schmitt D., Molenda C. E., and Hazen S. L. (2002) Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 277, 46116–46122 [DOI] [PubMed] [Google Scholar]
- 11. Zhang R., Shen Z., Nauseef W. M., and Hazen S. L. (2002) Defects in leukocyte-mediated initiation of lipid peroxidation in plasma as studied in myeloperoxidase-deficient subjects: systematic identification of multiple endogenous diffusible substrates for myeloperoxidase in plasma. Blood 99, 1802–1810 [PubMed] [Google Scholar]
- 12. Albert C. J., Thukkani A. K., Heuertz R. M., Slungaard A., Hazen S. L., and Ford D. A. (2003) Eosinophil peroxidase-derived reactive brominating species target the vinyl ether bond of plasmalogens generating a novel chemoattractant, α-bromo fatty aldehyde. J. Biol. Chem. 278, 8942–8950 [DOI] [PubMed] [Google Scholar]
- 13. Poliakov E., Brennan M. L., Macpherson J., Zhang R., Sha W., Narine L., Salomon R. G., and Hazen S. L. (2003) Isolevuglandins, a novel class of isoprostenoid derivatives, function as integrated sensors of oxidant stress and are generated by myeloperoxidase in vivo. FASEB J. 17, 2209–2220 [DOI] [PubMed] [Google Scholar]
- 14. Wu W., Samoszuk M. K., Comhair S. A., Thomassen M. J., Farver C. F., Dweik R. A., Kavuru M. S., Erzurum S. C., and Hazen S. L. (2000) Eosinophils generate brominating oxidants in allergen-induced asthma. J. Clin. Investig. 105, 1455–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wedes S. H., Khatri S. B., Zhang R., Wu W., Comhair S. A., Wenzel S., Teague W. G., Israel E., Erzurum S. C., and Hazen S. L. (2009) Noninvasive markers of airway inflammation in asthma. Clin. Transl. Sci. 2, 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bousquet J., Mantzouranis E., Cruz A. A., Aït-Khaled N., Baena-Cagnani C. E., Bleecker E. R., Brightling C. E., Burney P., Bush A., Busse W. W., Casale T. B., Chan-Yeung M., Chen R., Chowdhury B., Chung K. F., et al. (2010) Uniform definition of asthma severity, control, and exacerbations: document presented for the World Health Organization Consultation on Severe Asthma. J. Allergy Clin. Immunol. 126, 926–938 [DOI] [PubMed] [Google Scholar]
- 17. Wu W., Chen Y., d'Avignon A., and Hazen S. L. (1999) 3-Bromotyrosine and 3,5-dibromotyrosine are major products of protein oxidation by eosinophil peroxidase: potential markers for eosinophil-dependent tissue injury in vivo. Biochemistry 38, 3538–3548 [DOI] [PubMed] [Google Scholar]
- 18. Mitra S. N., Slungaard A., and Hazen S. L. (2000) Role of eosinophil peroxidase in the origins of protein oxidation in asthma. Redox Rep. 5, 215–224 [DOI] [PubMed] [Google Scholar]
- 19. Podrez E. A., Abu-Soud H. M., and Hazen S. L. (2000) Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic. Biol. Med. 28, 1717–1725 [DOI] [PubMed] [Google Scholar]
- 20. Brennan M. L., Wu W., Fu X., Shen Z., Song W., Frost H., Vadseth C., Narine L., Lenkiewicz E., Borchers M. T., Lusis A. J., Lee J. J., Lee N. A., Abu-Soud H. M., Ischiropoulos H., and Hazen S. L. (2002) A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 277, 17415–17427 [DOI] [PubMed] [Google Scholar]
- 21. Brennan M. L., and Hazen S. L. (2003) Amino acid and protein oxidation in cardiovascular disease. Amino acids 25, 365–374 [DOI] [PubMed] [Google Scholar]
- 22. Cannady S. B., Batra P. S., Leahy R., Citardi M. J., Janocha A., Ricci K., Comhair S. A., Bodine M., Wang Z., Hazen S. L., and Erzurum S. C. (2007) Signal transduction and oxidative processes in sinonasal polyposis. J. Allergy Clin. Immunol. 120, 1346–1353 [DOI] [PubMed] [Google Scholar]
- 23. Citardi M. J., Song W., Batra P. S., Lanza D. C., and Hazen S. L. (2006) Characterization of oxidative pathways in chronic rhinosinusitis and sinonasal polyposis. Am. J. Rhinol. 20, 353–359 [DOI] [PubMed] [Google Scholar]
- 24. Nicholls S. J., and Hazen S. L. (2009) Myeloperoxidase, modified lipoproteins, and atherogenesis. J. Lipid Res. 50, S346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wedes S. H., Wu W., Comhair S. A., McDowell K. M., DiDonato J. A., Erzurum S. C., and Hazen S. L. (2011) Urinary bromotyrosine measures asthma control and predicts asthma exacerbations in children. J. Pediatr. 159, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abu-Soud H. M., Khassawneh M. Y., Sohn J. T., Murray P., Haxhiu M. A., and Hazen S. L. (2001) Peroxidases inhibit nitric oxide (NO) dependent bronchodilation: development of a model describing NO-peroxidase interactions. Biochemistry 40, 11866–11875 [DOI] [PubMed] [Google Scholar]
- 27. Wu W., Chen Y., and Hazen S. L. (1999) Eosinophil peroxidase nitrates protein tyrosyl residues: implications for oxidative damage by nitrating intermediates in eosinophilic inflammatory disorders. J. Biol. Chem. 274, 25933–25944 [DOI] [PubMed] [Google Scholar]
- 28. Arlandson M., Decker T., Roongta V. A., Bonilla L., Mayo K. H., MacPherson J. C., Hazen S. L., and Slungaard A. (2001) Eosinophil peroxidase oxidation of thiocyanate: characterization of major reaction products and a potential sulfhydryl-targeted cytotoxicity system. J. Biol. Chem. 276, 215–224 [DOI] [PubMed] [Google Scholar]
- 29. Wever R., Kast W. M., Kasinoedin J. H., and Boelens R. (1982) The peroxidation of thiocyanate catalysed by myeloperoxidase and lactoperoxidase. Biochim. Biophys. Acta 709, 212–219 [DOI] [PubMed] [Google Scholar]
- 30. Olea F., and Parras P. (1992) Determination of serum levels of dietary thiocyanate. J. Anal. Toxicol. 16, 258–260 [DOI] [PubMed] [Google Scholar]
- 31. Husgafvel-Pursiainen K., Sorsa M., Engström K., and Einistö P. (1987) Passive smoking at work: biochemical and biological measures of exposure to environmental tobacco smoke. Int. Arch. Occup. Environ. Health 59, 337–345 [DOI] [PubMed] [Google Scholar]
- 32. Morgan P. E., Pattison D. I., Talib J., Summers F. A., Harmer J. A., Celermajer D. S., Hawkins C. L., and Davies M. J. (2011) High plasma thiocyanate levels in smokers are a key determinant of thiol oxidation induced by myeloperoxidase. Free Radic. Biol. Med. 51, 1815–1822 [DOI] [PubMed] [Google Scholar]
- 33. Talib J., Kwan J., Suryo Rahmanto A., Witting P. K., and Davies M. J. (2014) The smoking-associated oxidant hypothiocyanous acid induces endothelial nitric oxide synthase dysfunction. Biochem. J. 457, 89–97 [DOI] [PubMed] [Google Scholar]
- 34. Lacy F., Kailasam M. T., O'Connor D. T., Schmid-Schönbein G. W., and Parmer R. J. (2000) Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension 36, 878–884 [DOI] [PubMed] [Google Scholar]
- 35. Lacy F., O'Connor D. T., and Schmid-Schönbein G. W. (1998) Plasma hydrogen peroxide production in hypertensives and normotensive subjects at genetic risk of hypertension. J. Hypertens. 16, 291–303 [DOI] [PubMed] [Google Scholar]
- 36. Dröge W. (2002) Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95 [DOI] [PubMed] [Google Scholar]
- 37. Kraus L. M., and Kraus A. P. Jr. (1991) Tyrosine and N-carbamoyl-tyrosine in end-stage renal disease during continuous ambulatory peritoneal dialysis. J. Lab. Clin. Med. 118, 555–562 [PubMed] [Google Scholar]
- 38. Stark G. R., Stein W. H., and Moore S. (1960) Reactions of the cyanate present in aqueous urea with amino acids and proteins. J. Biol. Chem. 235, 3177–3181 [Google Scholar]
- 39. Stark G. R. (1964) On the reversible reaction of cyanate with sulfhydryl groups and the determination of NH2-terminal cysteine and cystine in proteins. J. Biol. Chem. 239, 1411–1414 [PubMed] [Google Scholar]
- 40. Stark G. R. (1965) Reactions of cyante with functional groups of proteins: II. formation, decomposition and properties of N-carbamylimidazole. Biochemistry 4, 588–595 [DOI] [PubMed] [Google Scholar]
- 41. Bobb D., and Hofstee B. H. (1971) Gel isoelectric focusing for following the successive carbamylations of amino groups in chymotrypsinogen A. Anal. Biochem. 40, 209–217 [DOI] [PubMed] [Google Scholar]
- 42. Stark G. R. (1977) Cleavage at cysteine after cyanylation. Methods Enzymol. 47, 129–132 [DOI] [PubMed] [Google Scholar]
- 43. Wang Z., Nicholls S. J., Rodriguez E. R., Kummu O., Hörkkö S., Barnard J., Reynolds W. F., Topol E. J., DiDonato J. A., and Hazen S. L. (2007) Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 13, 1176–1184 [DOI] [PubMed] [Google Scholar]
- 44. Mydel P., Wang Z., Brisslert M., Hellvard A., Dahlberg L. E., Hazen S. L., and Bokarewa M. (2010) Carbamylation-dependent activation of T cells: a novel mechanism in the pathogenesis of autoimmune arthritis. J. Immunol. 184, 6882–6890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. El-Gamal D., Rao S. P., Holzer M., Hallström S., Haybaeck J., Gauster M., Wadsack C., Kozina A., Frank S., Schicho R., Schuligoi R., Heinemann A., and Marsche G. (2014) The urea decomposition product cyanate promotes endothelial dysfunction. Kidney Int. 86, 923–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gould N. S., Gauthier S., Kariya C. T., Min E., Huang J., and Brian D. J. (2010) Hypertonic saline increases lung epithelial lining fluid glutathione and thiocyanate: two protective CFTR-dependent thiols against oxidative injury. Respir. Res. 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chandler J. D., Min E., Huang J., Nichols D. P., and Day B. J. (2013) Nebulized thiocyanate improves lung infection outcomes in mice. Br. J. Pharmacol. 169, 1166–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jin B. Y., Sartoretto J. L., Gladyshev V. N., and Michel T. (2009) Endothelial nitric-oxide synthase negatively regulates hydrogen peroxide-stimulated AMP-activated protein kinase in endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 106, 17343–17348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Denzler K. L., Borchers M. T., Crosby J. R., Cieslewicz G., Hines E. M., Justice J. P., Cormier S. A., Lindenberger K. A., Song W., Wu W., Hazen S. L., Gleich G. J., Lee J. J., and Lee N. A. (2001) Extensive eosinophil degranulation and peroxidase-mediated oxidation of airway proteins do not occur in a mouse ovalbumin-challenge model of pulmonary inflammation. J. Immunol. 167, 1672–1682 [DOI] [PubMed] [Google Scholar]
- 50. Fahy J. V. (2002) Goblet cell and mucin gene abnormalities in asthma. Chest 122, 320S–326S [DOI] [PubMed] [Google Scholar]
- 51. Corren J. (2011) Cytokine inhibition in severe asthma: current knowledge and future directions. Curr. Opin. Pulm. Med. 17, 29–33 [DOI] [PubMed] [Google Scholar]
- 52. Barnes P. J. (2001) Th2 cytokines and asthma: an introduction. Respir. Res. 2, 64–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu M., Eckart M. R., Morgan A. A., Mukai K., Butte A. J., Tsai M., and Galli S. J. (2011) Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J. Clin. Investig. 121, 3133–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Halwani R., Al-Muhsen S., Al-Jahdali H., and Hamid Q. (2011) Role of transforming growth factor-β in airway remodeling in asthma. Am. J. Respir. Cell Mol. Biol. 44, 127–133 [DOI] [PubMed] [Google Scholar]
- 55. Apostolov E. O., Shah S. V., Ray D., and Basnakian A. G. (2009) Scavenger receptors of endothelial cells mediate the uptake and cellular proatherogenic effects of carbamylated LDL. Arterioscler. Thromb. Vasc. Biol. 29, 1622–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sullivan P. W., Slejko J. F., Ghushchyan V. H., Sucher B., Globe D. R., Lin S. L., and Globe G. (2014) The relationship between asthma, asthma control and economic outcomes in the United States. J. Asthma 51, 769–778 [DOI] [PubMed] [Google Scholar]
- 57. Jacobsen E. A., Helmers R. A., Lee J. J., and Lee N. A. (2012) The expanding role(s) of eosinophils in health and disease. Blood 120, 3882–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scanlon C. E., Berger B., Malcom G., and Wissler R. W. (1996) Evidence for more extensive deposits of epitopes of oxidized low density lipoprotein in aortas of young people with elevated serum thiocyanate levels. PDAY Research Group. Atherosclerosis 121, 23–33 [DOI] [PubMed] [Google Scholar]
- 59. Demkowska I., Polkowska Z., and Namieśnik J. (2011) Non-invasive biological fluid matrices analysed to assess exposure to environmental tobacco smoke. J. Expo. Sci. Environ. Epidemiol. 21, 656–661 [DOI] [PubMed] [Google Scholar]
- 60. Boveris A., Oshino N., and Chance B. (1972) The cellular production of hydrogen peroxide. Biochem. J. 128, 617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Curnutte J. T., Whitten D. M., and Babior B. M. (1974) Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N. Engl. J. Med. 290, 593–597 [DOI] [PubMed] [Google Scholar]
- 62. Babior B. M., Curnutte J. T., and McMurrich B. J. (1976) The particulate superoxide-forming system from human neutrophils: properties of the system and further evidence supporting its participation in the respiratory burst. J. Clin. Investig. 58, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fragoso M. A., Fernandez V., Forteza R., Randell S. H., Salathe M., and Conner G. E. (2004) Transcellular thiocyanate transport by human airway epithelia. J. Physiol. 561, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wijkstrom-Frei C., El-Chemaly S., Ali-Rachedi R., Gerson C., Cobas M. A., Forteza R., Salathe M., and Conner G. E. (2003) Lactoperoxidase and human airway host defense. Am. J. Respir. Cell Mol. Biol. 29, 206–212 [DOI] [PubMed] [Google Scholar]
- 65. Thomas E. L., Milligan T. W., Joyner R. E., and Jefferson M. M. (1994) Antibacterial activity of hydrogen peroxide and the lactoperoxidase-hydrogen peroxide-thiocyanate system against oral streptococci. Infect. Immun. 62, 529–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Slungaard A., and Mahoney J. R. Jr. (1991) Thiocyanate is the major substrate for eosinophil peroxidase in physiologic fluids: implications for cytotoxicity. J. Biol. Chem. 266, 4903–4910 [PubMed] [Google Scholar]
- 67. Tahboub Y. R., Galijasevic S., Diamond M. P., and Abu-Soud H. M. (2005) Thiocyanate modulates the catalytic activity of mammalian peroxidases. J. Biol. Chem. 280, 26129–26136 [DOI] [PubMed] [Google Scholar]
- 68. Galijasevic S., Saed G. M., Hazen S. L., and Abu-Soud H. M. (2006) Myeloperoxidase metabolizes thiocyanate in a reaction driven by nitric oxide. Biochemistry 45, 1255–1262 [DOI] [PubMed] [Google Scholar]
- 69. Chandler J. D., Min E., Huang J., McElroy C. S., Dickerhof N., Mocatta T., Fletcher A. A., Evans C. M., Liang L., Patel M., Kettle A. J., Nichols D. P., and Day B. J. (2015) Anti-inflammatory and anti-microbial effects of thiocyanate in a cystic fibrosis mouse model. Am. J. Respir. Cell Mol. Biol. 53, 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shiue I. (2015) Urinary thiocyanate concentrations are associated with adult cancer and lung problems: US NHANES, 2009–2012. Environ. Sci. Pollut. Res. Int. 22, 5952–5960 [DOI] [PubMed] [Google Scholar]
- 71. Chen Y., Pederson L. L., and Lefcoe N. M. (1990) Exposure to environmental tobacco smoke (ETS) and serum thiocyanate level in infants. Arch. Environ. Health 45, 163–167 [DOI] [PubMed] [Google Scholar]
- 72. Chilmonczyk B. A., Salmun L. M., Megathlin K. N., Neveux L. M., Palomaki G. E., Knight G. J., Pulkkinen A. J., and Haddow J. E. (1993) Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. N. Engl. J. Med. 328, 1665–1669 [DOI] [PubMed] [Google Scholar]
- 73. Menzies D., Nair A., Williamson P. A., Schembri S., Al-Khairalla M. Z., Barnes M., Fardon T. C., McFarlane L., Magee G. J., and Lipworth B. J. (2006) Respiratory symptoms, pulmonary function, and markers of inflammation among bar workers before and after a legislative ban on smoking in public places. JAMA 296, 1742–1748 [DOI] [PubMed] [Google Scholar]
- 74. Morgan W. J., Crain E. F., Gruchalla R. S., O'Connor G. T., Kattan M., Evans R. 3rd, Stout J., Malindzak G., Smartt E., Plaut M., Walter M., Vaughn B., Mitchell H., and Inner-City Asthma Study Group. (2004) Results of a home-based environmental intervention among urban children with asthma. N. Engl. J. Med. 351, 1068–1080 [DOI] [PubMed] [Google Scholar]
- 75. Speer T., Owala F. O., Holy E. W., Zewinger S., Frenzel F. L., Stähli B. E., Razavi M., Triem S., Cvija H., Rohrer L., Seiler S., Heine G. H., Jankowski V., Jankowski J., Camici G. G., et al. (2014) Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur. Heart J. 35, 3021–3032 [DOI] [PubMed] [Google Scholar]
- 76. Jacobsen E. A., Lee N. A., and Lee J. J. (2014) Re-defining the unique roles for eosinophils in allergic respiratory inflammation. Clin. Exp. Allergy 44, 1119–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Holzer M., Gauster M., Pfeifer T., Wadsack C., Fauler G., Stiegler P., Koefeler H., Beubler E., Schuligoi R., Heinemann A., and Marsche G. (2011) Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox Signal. 14, 2337–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Son J. N., Lho Y., Shin S., Kwon S. H., Moon K. C., and Ha E. (2011) Carbamylated low-density lipoprotein increases reactive oxygen species (ROS) and apoptosis via lectin-like oxidized LDL receptor (LOX-1) mediated pathway in human umbilical vein endothelial cells. Int. J. Cardiol. 146, 428–430 [DOI] [PubMed] [Google Scholar]
- 79. Xu X., Cao Z., Cao B., Li J., Guo L., Que L., Ha T., Chen Q., Li C., and Li Y. (2009) Carbamylated erythropoietin protects the myocardium from acute ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Surgery 146, 506–514 [DOI] [PubMed] [Google Scholar]
- 80. Zheng S., De B. P., Choudhary S., Comhair S. A., Goggans T., Slee R., Williams B. R., Pilewski J., Haque S. J., and Erzurum S. C. (2003) Impaired innate host defense causes susceptibility to respiratory virus infections in cystic fibrosis. Immunity 18, 619–630 [DOI] [PubMed] [Google Scholar]
- 81. Xu W., Janocha A. J., Leahy R. A., Klatte R., Dudzinski D., Mavrakis L. A., Comhair S. A., Lauer M. E., Cotton C. U., and Erzurum S. C. (2014) A novel method for pulmonary research: assessment of bioenergetic function at the air-liquid interface. Redox Biol. 2, 513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lauer M. E., Fulop C., Mukhopadhyay D., Comhair S., Erzurum S. C., and Hascall V. C. (2009) Airway smooth muscle cells synthesize hyaluronan cable structures independent of inter-alpha-inhibitor heavy chain attachment. J. Biol. Chem. 284, 5313–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koeth R. A., Kalantar-Zadeh K., Wang Z., Fu X., Tang W. H., and Hazen S. L. (2013) Protein carbamylation predicts mortality in ESRD. J. Am. Soc. Nephrol. 24, 853–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kunjathoor V. V., Febbraio M., Podrez E. A., Moore K. J., Andersson L., Koehn S., Rhee J. S., Silverstein R., Hoff H. F., and Freeman M. W. (2002) Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277, 49982–49988 [DOI] [PubMed] [Google Scholar]
- 85. Swaidani S., Cheng G., Lauer M. E., Sharma M., Mikecz K., Hascall V. C., and Aronica M. A. (2013) TSG-6 protein is crucial for the development of pulmonary hyaluronan deposition, eosinophilia, and airway hyperresponsiveness in a murine model of asthma. J. Biol. Chem. 288, 412–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li D., Gallup M., Fan N., Szymkowski D. E., and Basbaum C. B. (1998) Cloning of the amino-terminal and 5′-flanking region of the human MUC5AC mucin gene and transcriptional up-regulation by bacterial exoproducts. J. Biol. Chem. 273, 6812–6820 [DOI] [PubMed] [Google Scholar]
- 87. Takeyama K., Dabbagh K., Lee H. M., Agustí C., Lausier J. A., Ueki I. F., Grattan K. M., and Nadel J. A. (1999) Epidermal growth factor system regulates mucin production in airways. Proc. Natl. Acad. Sci. U.S.A. 96, 3081–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sanders P. N., Koval O. M., Jaffer O. A., Prasad A. M., Businga T. R., Scott J. A., Hayden P. J., Luczak E. D., Dickey D. D., Allamargot C., Olivier A. K., Meyerholz D. K., Robison A. J., Winder D. G., Blackwell T. S., et al. (2013) CaMKII is essential for the proasthmatic effects of oxidation. Sci. Transl. Med. 5, 195ra197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lankford S. M., Macchione M., Crews A. L., McKane S. A., Akley N. J., and Martin L. D. (2005) Modeling the airway epithelium in allergic asthma: interleukin-13-induced effects in differentiated murine tracheal epithelial cells. In Vitro Cell. Dev. Biol. Anim. 41, 217–224 [DOI] [PubMed] [Google Scholar]
- 90. Guilloteau L. A., Dornand J., Gross A., Olivier M., Cortade F., Vern Y. L., and Kerboeuf D. (2003) Nramp1 is not a major determinant in the control of Brucella melitensis infection in mice. Infect. Immun. 71, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hufnagl K., Wagner B., Winkler B., Baier K., Hochreiter R., Thalhamer J., Kraft D., Scheiner O., Breiteneder H., and Wiedermann U. (2003) Induction of mucosal tolerance with recombinant Hev b 1 and recombinant Hev b 3 for prevention of latex allergy in BALB/c mice. Clin. Exp. Immunol. 133, 170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kim D. Y., Park B. S., Hong G. U., Lee B. J., Park J. W., Kim S. Y., and Ro J. Y. (2011) Anti-inflammatory effects of the R2 peptide, an inhibitor of transglutaminase 2, in a mouse model of allergic asthma, induced by ovalbumin. Br. J. Pharmacol. 162, 210–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bae C. J., Lee J. W., Bae H. S., Shim S. B., Jee S. W., Lee S. H., Lee C. K., Hong J. T., and Hwang D. Y. (2011) Detection of allergenic compounds using an IL-4/luciferase/CNS-1 transgenic mice model. Toxicol. Sci. 120, 349–359 [DOI] [PubMed] [Google Scholar]
- 94. Lee K. S., Park S. J., Hwang P. H., Yi H. K., Song C. H., Chai O. H., Kim J. S., Lee M. K., and Lee Y. C. (2005) PPAR-γ modulates allergic inflammation through up-regulation of PTEN. FASEB J. 19, 1033–1035 [DOI] [PubMed] [Google Scholar]
- 95. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−δδ CT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]