

Abstract
Mutations in the gene encoding phospholipase C-γ2 (PLCγ2) have been shown to be associated with resistance to targeted therapy of chronic lymphocytic leukemia (CLL) with the Bruton's tyrosine kinase inhibitor ibrutinib. The fact that two of these mutations, R665W and L845F, imparted upon PLCγ2 an ∼2–3-fold ibrutinib-insensitive increase in the concentration of cytosolic Ca2+ following ligation of the B cell antigen receptor (BCR) led to the assumption that the two mutants exhibit constitutively enhanced intrinsic activity. Here, we show that the two PLCγ2 mutants are strikingly hypersensitive to activation by Rac2 such that even wild-type Rac2 suffices to activate the mutant enzymes upon its introduction into intact cells. Enhanced “basal” activity of PLCγ2 in intact cells is shown using the pharmacologic Rac inhibitor EHT 1864 and the PLCγ2F897Q mutation mediating Rac resistance to be caused by Rac-stimulated rather than by constitutively enhanced PLCγ2 activity. We suggest that R665W and L845F be referred to as allomorphic rather than hypermorphic mutations of PLCG2. Rerouting of the transmembrane signals emanating from BCR and converging on PLCγ2 through Rac in ibrutinib-resistant CLL cells may provide novel drug treatment strategies to overcome ibrutinib resistance mediated by PLCG2 mutations or to prevent its development in ibrutinib-treated CLL patients.
Keywords: leukemia, lymphoma, phosphatidylinositol signaling, phospholipase C, Rac (Rac GTPase), chronic lymphocytic leukemia, ibrutinib resistance, leukemia
Introduction
Inositol-phospholipid-specific phospholipases C (PLCs)3 regulate many fundamental functions of normal and neoplastic B cells (1, 2). They catalyze the formation of inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol and, at the same time, decrease the local or general plasma membrane abundance of their substrate, phosphatidylinositol 4,5-bisphosphate (PtdInsP2) (3). Three members of the six mammalian PLC subfamilies, β, γ, δ, ϵ, ζ, and η, play important roles in B cells as follows: PLCβ2, PLCβ3, and PLCγ2. PLCβ2 and PLCβ3 are important in mediating B cell responses to G-protein-coupled chemokine receptors (4). PLCγ2 serves as a key component of the B cell receptor (BCR) signalosome by interacting with cell surface receptor activation, e.g. by antigens (5), cleavage fragments of the third complement component (6), and bacterial, viral, or autoimmunity host DNA (7), and even certain chemokines (8). PLCγ2 activation results in InsP3-mediated increases in the concentration of free Ca2+, diacylglycerol-mediated activation of protein kinases C, and changes in transmembrane signaling directly mediated by PtdInsP2 (9).
Several lines of evidence point to an important contribution of enhanced BCR signaling in the pathogenesis, progression, and/or maintenance of B cell leukemias and lymphomas. For example, leukemic B cells of patients with chronic lymphocytic leukemia (CLL) specifically express a restricted immunoglobulin heavy variable (IGHV) gene repertoire, suggesting that CLL development represents an antigen-superantigen-driven process (10). Furthermore, the presence or absence of somatic mutations in rearranged IGHV genes determines the clinical course of CLL, with patients carrying mutated IGHV genes generally following a more indolent course (10). In CLL, the BCR repertoire is characterized by subsets of closely homologous (“stereotyped”) immunoglobulin V(D)J sequences, which are directly involved in antigen binding. This, together with the finding that most malignant B cells thrive only poorly in vitro, further supports the notion of a role of antigenic drive in B cell tumorigenicity (11). Recent evidence suggests that, at least in CLL, BCRs also induce cell-autonomous signaling independent of extrinsic antigens that is caused by intra- or inter-BCR interactions (12). Finally, there is evidence for the existence of constitutively activated protein kinases and transcription factors downstream of PLCγ2 in leukemic B cells of certain CLL cells (13–15). The observation that some of the signaling components upstream of PLCγ2, such as the protein-tyrosine kinases Syk and Btk, can promote B cell proliferation and/or survival, either along the pathway of normal B cell development or at specific stages following malignant transformation, is well in line with this concept (16–18).
The orally bioavailable irreversible Btk inhibitor ibrutinib has recently undergone a remarkably successful evolution as a second-line treatment of patients with relapsed or refractory CLL or mantle cell lymphoma and as a first-line treatment of patients with CLL carrying a del(17p) or TP53 mutation (19, 20). Currently, the drug is being evaluated for treatment of other diseases, including other malignancies, autoimmune disease, inflammatory diseases, osteoclast-associated bone diseases, and ischemic stroke (21–26). As is the case for other targeted tumor therapies (27), ibrutinib treatment is characterized, in some cases, by the development of acquired drug resistance (28). Thus, whole-exome sequencing of six CLL patients with late relapses revealed C481S mutations in BTK of five patients and three distinct mutations in PLCG2 of two patients as follows: L845F, R665W, and S707Y in one patient with tumor cells also harboring a BTK C481S mutation and PLCG2 R665W representing the sole mutation in the other patient (29). Although the resistance mechanism conferred by the BTK C481S mutation is immediately apparent from the fact that the thiol group of Cys-481 is the site of covalent linkage of ibrutinib to Btk close to its ATP-binding site, the mechanisms of action of the mutations found in PLCG2 remained less well understood. Whereas S707Y had previously been reported as a constitutively activating mutation in the dominantly inherited human disease APLAID (autoinflammation and PLCγ2-associated antibody deficiency and immune dysregulation) (30), the R665W and L845F mutants of PLCγ2 appeared to be functionally normal in reconstituted DT40 chicken B cells in the absence of BCR stimulation, but to mediate moderately enhanced and markedly prolonged ibrutinib-resistant increases in [Ca2+]i following BCR ligation with anti-IgM (29). Very recent evidence showed Btk-independent activation of the overexpressed R665W PLCγ2 mutant after B cell receptor engagement in Btk-deficient DT40 cells, suggesting Btk independency of this mutant (31). When the same mutant was expressed in PLCγ2-deficient DT40 cells containing endogenous wild-type Btk, BCR-mediated PLCγ2 activation was resistant to ibrutinib, but sensitive to pharmacologic inhibitors of Syk and Lyn. These results suggested the existence of protein-tyrosine kinase mechanisms emanating from BCR and bypassing Btk to activate R665W to mediate ibrutinib resistance even in tumor cells lacking BTK mutations (31).
We have previously shown that PLCγ2 is specifically activated by Rac GTPases by a mechanism independent of PLCγ2 tyrosine phosphorylation, but dependent on the direct interaction of activated Rac with the bipartite split PH domain (spPH) juxtaposed between the two halves, X and Y, of the PLCγ2 catalytic domain (32, 33). Studies using a Rac-resistant mutant of PLCγ2, F897Q, reconstituted into PLCγ2-deficient DT40 B cells recently showed that Rac-mediated stimulation of PLCγ2 amplifies BCR-mediated Ca2+ signaling (34). The fact that failure to proliferate in response to immunoglobulin receptor stimulation had also been observed in mice carrying deletions in all three genes encoding Vav guanine nucleotide exchange factors of Rac GTPases, Vav1, −2, and −3 (35), prompted us to examine, in this work, the effect of the PLCG2 mutations R665W and L845F on the Rac-PLCγ2 interaction in intact cells and in a cell-free system in vitro. The results show that the two mutations take marked stimulatory effects on this interaction. These stimulatory effects may not only contribute to the mechanism(s) of ibrutinib resistance caused by mutations in PLCG2 rather than BTK, but they also provide novel strategies to tackle ibrutinib resistance in CLL and other debilitating human diseases.
Results
The first experiment was designed to determine whether the two PLCγ2 mutants R665W and L845F exhibit constitutive activity in intact cells. To this end, the two mutants were expressed in COS-7 cells to be radiolabeled with [3H]inositol for measurement of [3H]inositol phosphate formation. Wild-type PLCγ2 and PLCγ2 carrying an M28L germ line mutation identified in an ibrutinib-resistant patient were analyzed for comparison. Fig. 1A shows that, in contrast to wild-type PLCγ2 and PLCγ2M28L, the mutants R665W and L845F caused marked, up to 18-fold, increases in basal inositol phosphate formation when expressed in increasing amounts (Fig. 1, A, left panel, and B). Only slight, 1.4-fold, increases were apparent at the highest amounts of wild-type and PLCγ2M28L. There was no difference between wild-type and M28L mutant PLCγ2 in terms of their stimulatory responses to constitutively active Rac2G12V (Fig. 1A, right panel), indicating that PLCγ2M28L did not harbor a defect in enzyme activation.
FIGURE 1.
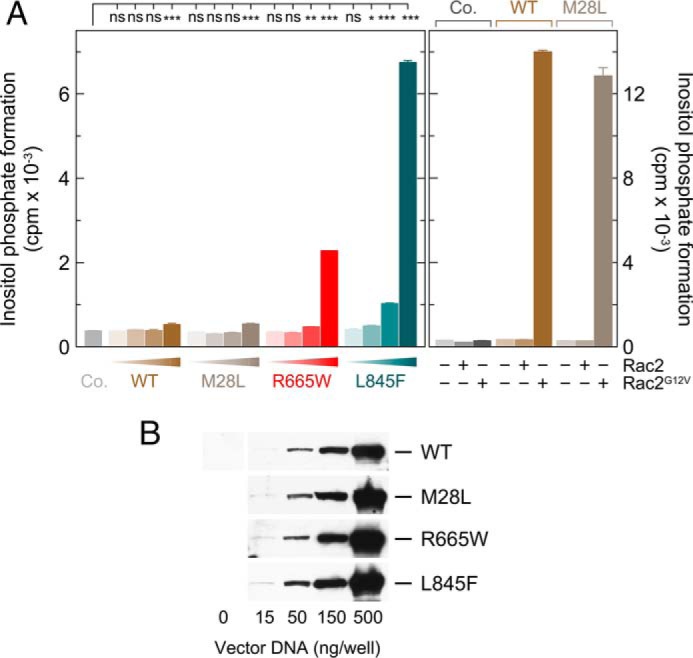
PLCγ2 point mutations identified in CLL patients differ in their ability to confer enhanced basal activity to the enzyme. A, left panel, COS-7 cells were transfected as indicated with 500 ng/well of either empty vector (Co., control) or increasing amounts (15, 50, 150, and 500 ng/well) of vector encoding either wild-type PLCγ2 (WT), PLCγ2M28L (M28L), PLCγ2R665W (R665W), and PLCγ2L845F (L845F). Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was then determined. Right panel, COS-7 cells were cotransfected with 525 ng/well of empty vector (Co.) or 500 ng/well of vector encoding either wild-type PLCγ2 (WT) or PLCγ2M28L (M28L), together with 25 ng/well each of either empty vector or vector encoding wild-type Rac2 (Rac2) or its constitutively active mutant Rac2G12V (Rac2G12V). B, homogenates from cells functionally analyzed in A, left panel, were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope present on wild-type and mutant PLCγ2.
To determine and compare the sensitivity of wild-type PLCγ2 to stimulation by constitutively active Rac2 to the sensitivities of the mutants R665W and L845F, the PLCγ2 isozymes were coexpressed with increasing amounts of Rac2G12V. Fig. 2A shows that there were striking increases in inositol phosphate formation in response to increasing amounts of Rac2G12V. Specifically, the maximal increase in Rac2G12V efficacy was ∼6.7- and 35-fold for PLCγ2R665W and PLCγ2L845F, respectively. In addition, we consistently observed that the two point mutations caused an increase in the potency of Rac2G12V, which was ∼4.5- and 6.5-fold for PLCγ2R665W and PLCγ2L845F, respectively. The increase in Rac2-stimulated PLC activity caused by the PLCγ2 mutations was not caused by changes in PLCγ2 protein production in transfected cells (Fig. 2B).
FIGURE 2.
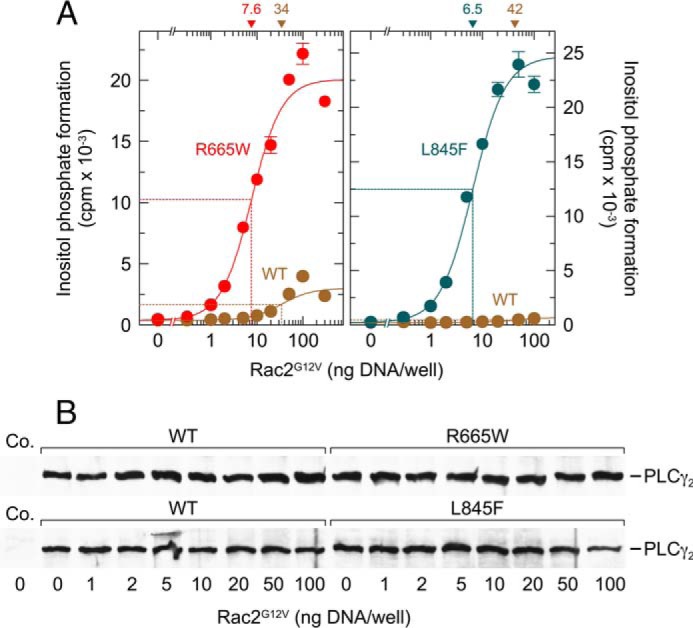
Point mutations R665W and L845F augment the responsiveness of PLCγ2 to activated Rac2. A, COS-7 cells were transfected as indicated with 150 ng/well vector encoding wild-type PLCγ2 (WT) or PLCγ2R665W (R665W) (left panel) or with 50 ng/well vector encoding wild-type PLCγ2 (WT) or PLCγ2L845F (L845F) (right panel) and increasing amounts of vector encoding Rac2G12V. Note that the amounts of vector DNA encoding mutant PLCγ2 was different in the left and right panels to observe full-range stimulation of the two mutants by Rac2G12V without running out of available phospholipid substrate. Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was then determined. The ED50 values of vector encoding Rac2G12V for the stimulation of wild-type or mutant PLCγ2 activity obtained by non-linear curve fitting are shown above the graphs in nanograms/well. B, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope. Co., control.
Most interestingly, enhanced sensitivity of PLCγ2R665W and PLCγ2L845F to Rac2 was not limited to constitutively active Rac2G12V but was also observed for wild-type Rac2 (Fig. 3A). Specifically, although there was no effect of increasing amounts of Rac2 on the activity of wild-type PLCγ2, the mutants R665W and L845F were activated up to 5.1- and 4.2-fold, respectively. There was little, if any, change in the expression of wild-type or mutant PLCγ2 in the presence of increasing amounts of Rac2 (Fig. 3B).
FIGURE 3.
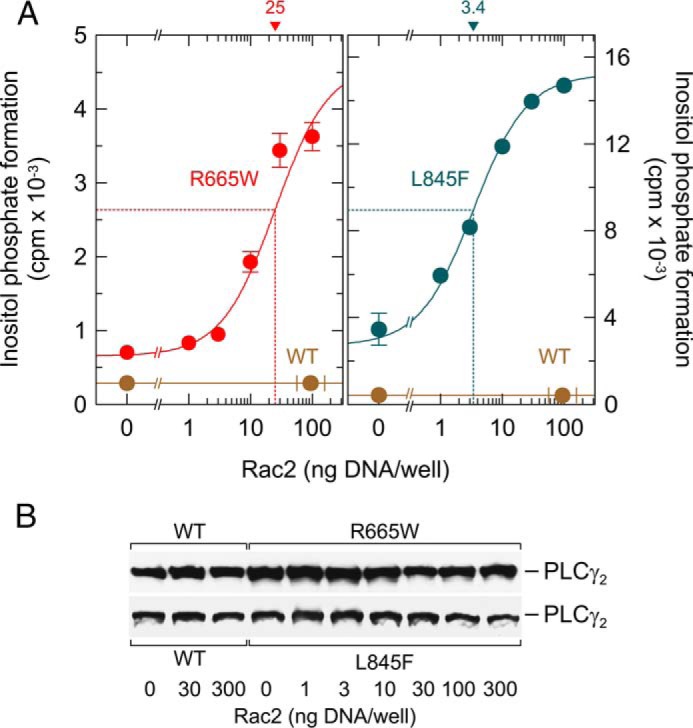
Point mutations R665W and L845F augment the responsiveness of PLCγ2 to exogenous wild-type Rac2. A, COS-7 cells were transfected as indicated with 500 ng/well vector encoding wild-type PLCγ2 (WT) or PLCγ2R665W (R665W) (left panel) or wild-type PLCγ2 (WT) or PLCγ2L845F (L845F) (right panel) and increasing amounts of vector encoding Rac2. Note that the vectors encoding mutant PLCγ2 were used at the same maximal amount (500 ng/well) in the left and right panels to observe the stimulation by wild-type Rac. Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was then determined. The ED50 values of vector encoding Rac2 for the stimulation of mutant PLCγ2 activity obtained by non-linear curve fitting are shown above the graphs in nanograms. B, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope.
We have previously shown that PLCγ2 is sensitive to stimulation by exogenous, constitutively active Vav1, Vav1ΔN, presumably via activation of endogenous Rac GTPases present in COS-7 cells (33). Fig. 4A shows that the two ibrutinib resistance mutations imparted on PLCγ2 a marked increase in its responsiveness to stimulation by Vav1ΔN, which amounted to ∼9.3- and 12-fold for the mutants R665W and L845F, respectively. As observed for Rac2G12V, there was an increase in the apparent potency of Vav1ΔN by the mutations, which was ∼5.3- and 5.7-fold for PLCγ2R665W and PLCγ2L845F, respectively. The results shown in Fig. 4B suggest that the increase in inositol phosphate formation by wild-type PLCγ2, but not by its mutants may have, at least in part, been due to a slight increase in the production of the protein at increasing concentrations of Vav1ΔN. The ability of activated Vav1 to catalyze activation of Rho GTPases (36) is dependent on an intact interaction network between the DH, PH, and cysteine-rich domain regions of the Vav1 protein and is abolished by an Asp to Ala mutation at position 376, D376A (37). Fig. 4C shows that the inactivating Vav1 mutation caused an almost complete (∼85%) and a complete loss in the stimulatory effect of Vav1ΔN on the activity of R665W and L845F, respectively.
FIGURE 4.
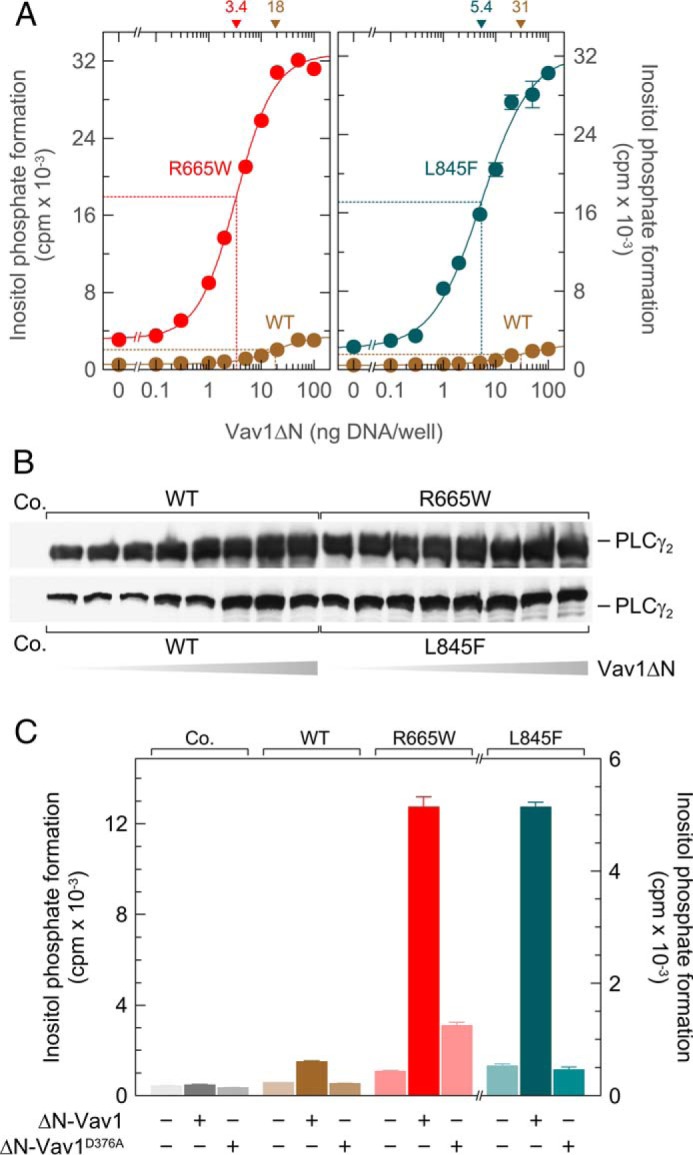
Point mutations R665W and L845F augment the responsiveness of PLCγ2 to exogenous activated Vav1 even in the absence of exogenous Rac. A, COS-7 cells were transfected as indicated with 500 ng/well vector encoding wild-type PLCγ2 (WT) or PLCγ2R665W (R665W) (left panel) or 50 ng/well vector encoding WT PLCγ2 (WT) or PLCγ2L845F (L845F) (right panel) and increasing amounts of vector encoding Vav1ΔN. Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was then determined. The ED50 values of vector encoding Vav1ΔN for the stimulation of wild-type or mutant PLCγ2 activity obtained by non-linear curve fitting are shown above the graphs in nanograms/well. B, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope. C, ability of the point mutations R665W and L845F to enhance stimulation of PLCγ2 by activated Vav1 is dependent on an intact interaction network between the DH, PH, and cysteine-rich domain regions of the Vav1 protein. COS-7 cells were transfected as indicated with 500 ng each per well of either empty vector (Co., Control), vector encoding wild-type PLCγ2 (WT), or vector encoding PLCγ2R665W (R665W), or 50 ng/well vector encoding PLCγ2L845F (L845F) and 100 ng each per well of vector encoding either Vav1ΔN (ΔN-Vav1) or its DH domain mutant Vav1ΔND376A (ΔN-Vav1D376A). Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was then determined.
The abilities of wild-type and constitutively active Rac2 to cause enhanced activation of PLCγ2R665W in comparison with wild-type PLCγ2 were dependent on the C-terminal isoprenylation of the exogenous Rac2 proteins (Fig. 5A). Specifically, their stimulatory effect on PLCγ2R665W activity was completely lost upon replacement of the cysteine residue at position −4 from the Rac2 C terminus, which normally serves as a substrate for geranylgeranylation, by a serine residue, C189S (38). This was despite the fact that the C189S mutants were expressed at similar, if not somewhat enhanced levels, as compared with their respective counterparts (Fig. 5B).
FIGURE 5.
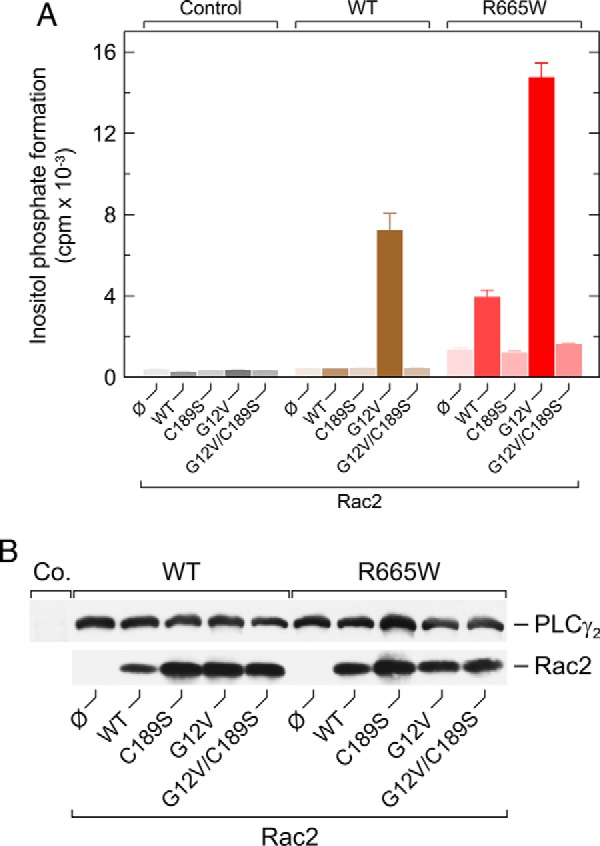
Abilities of exogenous wild-type and constitutively activated Rac2 to activate PLCγ2R665W are dependent on C-terminal isoprenylation of Rac2. A, COS-7 cells were transfected with 500 ng/well empty vector (Co., control) or vector encoding either wild-type PLCγ2 (WT) or PLCγ2R665W (R665W), together with 10 ng each per well of empty vector (Ø) or vector encoding Rac2 (WT), Rac2C189S (C189S), Rac2G12V (G12V), or Rac2G12V C189S (G12V|C189S). Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was then determined. B, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope present on wild-type and mutant PLCγ2 (top) or antiserum reactive against Rac2 (bottom).
Figs. 3A, 4, A and C, and 5A also confirm the increase in basal activity of PLCγ2R665W and PLCγ2L845F even in the absence of exogenous PLCγ2 stimuli such as Rac2 and Vav1ΔN observed in Fig. 1. The fact that COS-7 cells are transformed cells prompted us to investigate the following possibilities: (i) that enhanced basal activity is caused by cell-autonomous activation of the mutant PLCγ2 isoforms, e.g. by spontaneously active cell surface receptors, and (ii) that Rac participates in this activation. To this end, F897Q mutants of wild-type PLCγ2 and its mutants R665W and L845F were generated and functionally characterized (Fig. 6A). We have previously shown that the F897Q substitution blocks activation of PLCγ2 by constitutively active Rac2 and abolishes binding of GTPγS-activated Rac2 to PLCγ2 spPH, while leaving the overall fold of PLCγ2 spPH unaffected (33). Fig. 6A, left panel, shows that the F897Q mutation, as expected, caused a complete or near complete loss of activation of wild-type PLCγ2 by Rac2G12V and of PLCγ2R665W by wild-type and G12V mutant Rac2. More importantly, however, the increased “basal” PLCγ2R665W activity, determined in the absence of exogenous Rac2, was reduced by about 74% (Fig. 6A, left panel). Fig. 6A, right panel, shows that F897Q mutation caused similar reductions of basal activity of the R665W and L845F variants of PLCγ2 (∼68 and ∼78%, respectively). This indicates that the basal activities of the R665W and L845F mutants are strongly dependent on Rac1 endogenously present in COS-7 cells. These reductions in PLCγ2 activity were not related to reduced PLCγ2 protein synthesis in transfected cells (Fig. 6B).
FIGURE 6.
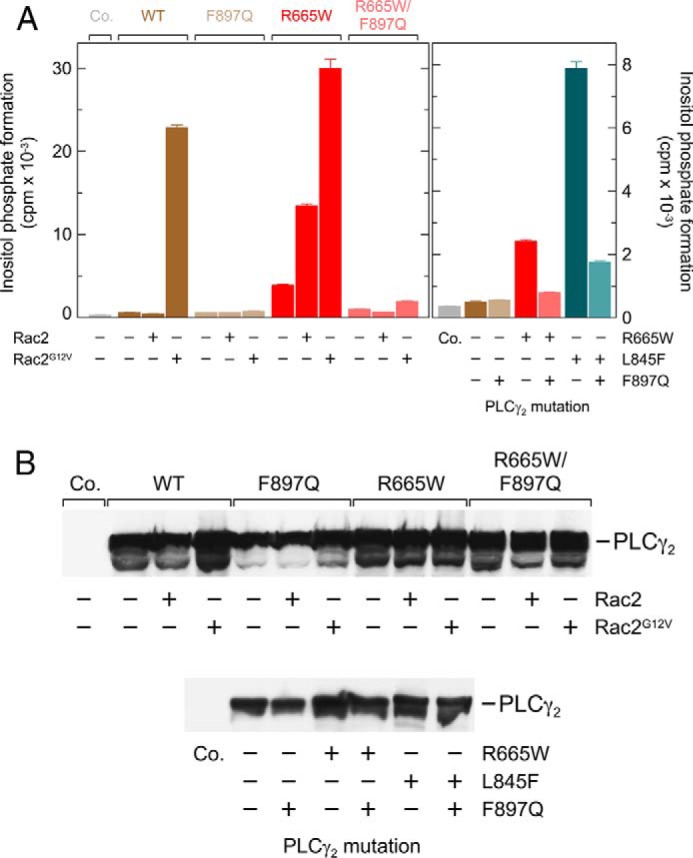
Enhanced Rac2- and Rac2G12V-stimulated activity of PLCγ2R665W as well as enhanced basal activity of PLCγ2R665W and PLCγ2L845F are prevented by a point mutation of PLCγ2, F897Q, mediating resistance of the enzyme to stimulation by activated Rac2. A, COS-7 cells were transfected with 500 ng/well empty vector (Co., Control) or vector encoding either wild-type PLCγ2 (WT), PLCγ2F897Q (F897Q), PLCγ2R665W (R665W), or PLCγ2 R665W/F897Q (R665W|F897Q), together with 25 ng/well empty vector or vector encoding Rac2 or Rac2G12V (left panel). In the right panel, COS-7 cells were transfected with 500 ng/well (from left to right) empty vector (Co.) or vector encoding either wild-type PLCγ2, PLCγ2F897Q, PLCγ2R665W, PLCγ2 R665W/F897Q, PLCγ2L845F, or PLCγ2 L845F/F897Q. Note that the vectors encoding mutant PLCγ2 were used at the same maximal amount (500 ng/well) to observe the stimulation by wild-type Rac2 (left panel) and enhanced basal activity of the PLCγ2 mutants (right panel).Twenty four hours after transfection, the cells were incubated for 20 h with myo-[2-3H] inositol, and inositol phosphate formation was determined. B, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope.
To obtain further independent evidence for an involvement of active Rac in the enhanced “basal” activities of PLCγ2R665W and PLCγ2L845F, we employed the Rac-specific pharmacologic inhibitor EHT 1864 and its inactive analog EHT 4063 (39). EHT 1864 is known to bind with high affinity to Rac1, Rac1b, Rac2, and with somewhat lower affinity to Rac3. The inhibitor has been suggested to place Rac in an inactive state by promoting the loss of bound guanine nucleotide, rather than interfering with RhoGEF-induced Rac activation, as described for other Rac inhibitors such as NSC23766 (40, 41). Fig. 7A, left panel, shows that EHT 1864, but not EHT 4063, caused a clear (∼55%) inhibition of basal inositol phosphate formation by PLCγ2R665Wand PLCγ2L845F. There was a smaller (∼25%) not quite statistically significant (p = 0.0676) inhibitory effect for wild-type PLCγ2. No effect of EHT 1864 was observed in the absence of exogenous PLC isozyme and in the presence of PLCδ1Δ44, a constitutively active variant of PLCδ1. PLCδ1 is an evolutionarily divergent relative to PLCγ2 and insensitive to stimulation by Rac (32, 42). The inhibitory effect of EHT 1864 on basal inositol phosphate formation by PLCγ2L845F was concentration-dependent with an IC50 of about 1 μm (Fig. 7A, right panel), which is slightly lower than, but still in line with, the previously reported value of about 5 μm for modulation of γ-secretase-mediated amyloid precursor protein (APP) processing (39). There was no effect of EHT 1864 and EHT 4063 on the expression of the various recombinant PLC isozymes in transfected cells (Fig. 7B).
FIGURE 7.
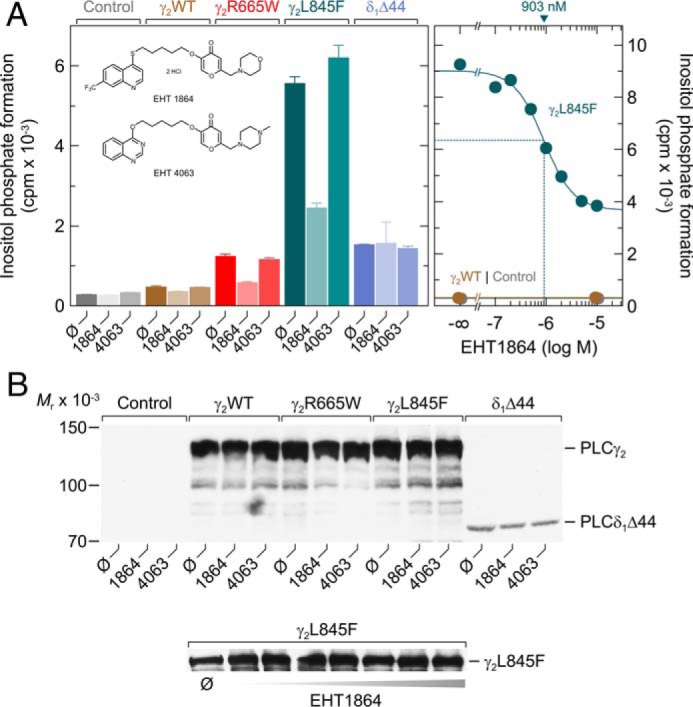
Enhanced basal activities of PLCγ2R665W and PLCγ2L845F are specifically reduced by the Rac inhibitor EHT 1864. A, COS-7 cells were transfected with 500 ng/well empty vector (Control) or vector encoding wild-type PLCγ2 (γ2WT), PLCγ2R665W (γ2R665W), PLCγ2L845F (γ2L845F), or 30 ng/well of vector encoding PLCδ1Δ44 (δ1Δ44). Twenty four hours after transfection, the cells were incubated for 18 h with myo-[2-3H]inositol in the absence (Ø) or presence of 5 μm EHT 1864 or 5 μm of its inactive congener EHT 4063, followed by determination of inositol phosphate formation (left panel). The inset shows the structural formulas of EHT 1864, 5-(5-(7-(trifluoromethyl)quinolin-4-ylthio)pentyloxy)-2-(morpholino-methyl)-4H-pyran-4-one dihydrochloride and EHT 4063, 5-(5-(quinazolin-4-yloxy)pentyl-oxy)-2-((4-methylpiperazin-1-yl)methyl)-4H-pyran-4-one. In the right panel, COS-7 cells were transfected with 500 ng/well empty vector (Control) or vector encoding either wild-type PLCγ2 (γ2WT) or PLCγ2L845F (γ2L845F). Twenty four hours after transfection, the cells were incubated for 18 h with myo-[2-3H]inositol in the absence or presence of EHT 1864 at the concentrations indicated at the abscissa. The IC50 value of EHT 1864 for the inhibition of mutant PLCγ2 activity obtained by non-linear curve fitting is shown above the graph in nanomolar. B, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope present on wild-type and mutant PLCγ2 as well as on PLCδ1Δ44.
The two PLCγ isoforms are distinct in their response to activated Rac, with PLCγ1, in marked contrast to PLCγ2, showing little if any stimulation (32). We therefore set out to examine the functional effects of the PLCγ2 R665W mutation in the PLCγ1 context. The two isozymes are very similar in the overall structure and amino acid sequence in the region close to the point mutation R665W in PLCγ2 and the corresponding residue, Arg-687, in PLCγ1 (cf. Fig. 8A, right panel, inset; PLCγ1, LMRVPR687DGAFL; PLCγ2 LMRIPR665DGAFL, single divergent residue underlined). Fig. 8A, left panel, shows that overexpression of wild-type PLCγ1 and wild-type PLCγ2 led to only minor, if any, changes in basal inositol phosphate formation. Introduction of the point mutations R687W and R665W into PLCγ1 and PLCγ2, respectively, led to no change in activity at lower expression levels. Only at the highest amount of cDNA used for transfection, 500 ng/well, an ∼2-fold increase in activity was evident in either case. Fig. 8A, right panel, shows that neither wild-type nor the R687W mutant PLCγ1 responded to wild-type Rac2, in contrast to the ∼5.8-fold enhancement witnessed for the R665W mutation in PLCγ2. Although PLCγ1R687W exhibited an ∼2.3-fold statistically significant (p < 0.01) stimulatory response to Rac2G12V, this response was by far less prominent than the ∼18.4-fold increase observed for PLCγ2R665W. The enhanced basal activity of PLCγ1R687W was insensitive to the Rac inhibitor EHT 1864 (Fig. 8B), unlike those of the PLCγ2 mutants associated with ibrutinib resistance, PLCγ2R665W and PLCγ2L845F (cf. Fig. 7A), indicating that the basal activity of PLCγ1R687W is independent of Rac activity, despite its low but statistically significant sensitivity to exogenous Rac2G12V. The latter was also evident, as described before (32), for wild-type PLCγ1 (Fig. 8A, right panel). Hypersensitivity to protein tyrosine phosphorylation offers a possible explanation for the increased basal activity of PLCγ1R687W. Fig. 8C shows that although there were noticeable differences in the expression levels of the PLCγ1 versus PLCγ2 isozymes, in particular at low transfection levels, these differences did not explain the marked functional differences observed in Fig. 8A, right panel. There were no effects of the EHT compounds on the expression of wild-type or R687W mutant PLCγ1 or endogenous Rac in the experiment shown in Fig. 8, B and D.
FIGURE 8.

Point mutation in PLCγ1, corresponding to R665W in PLCγ2, R687W, causes enhanced basal activity of PLCγ1 in intact cells that is insensitive to the Rac inhibitor EHT 1864. A, COS-7 cells were transfected with increasing amounts of vector encoding either wild-type PLCγ2 (γ2WT), PLCγ2R665W (γ2R665W), PLCγ1 (γ1WT), or PLCγ1R687W (γ1R687W) (left panel). Right panel, COS-7 cells were transfected with 500 ng/well empty vector (Control) or vector encoding either wild-type PLCγ2 (γ2WT), PLCγ2R665W (γ2R665W), PLCγ1 (γ1WT), or PLCγ1R687W (γ1R687W) together with 25 ng/well empty vector or vector encoding Rac2 or Rac2G12V, as indicated at the abscissa. The inset shows a superimposition of the three-dimensional structures of the C-terminal SH2 domains of PLCγ2R665W (γ2R665W) and PLCγ1R687W (γ1R687W), as predicted by Swiss-Model using the structure of SH2n-SH2c tandem of PLCγ1 as a template (4FBN (67), data not shown) and visualized using the PyMOL Molecular Graphics System. The predicted structures of the wild-type and mutant SH2c domains of PLCγ1 and PLCγ2 and the localizations of the residues in positions 687 (γ1) and 665 (γ2) are virtually identical to each other. Of the two wild-type structures, only the two arginines present in positions 687 (γ1) and 665 (γ2) are shown in gray. B, COS-7 cells were transfected with 500 ng/well empty vector (Co., control) or vector encoding either wild-type PLCγ1 (γ1WT) or PLCγ1R687W (γ1R687W). Twenty four hours after transfection, the cells were incubated for 18 h with myo-[2-3H]inositol in the absence (Ø) or presence of 5 μm EHT 1864 or EHT 4063. Subsequently, inositol phosphate formation was determined. C, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope present on wild-type and mutant PLCγ (top and center panels) or antiserum reactive against Rac2 (bottom panel). D, homogenates from cells functionally analyzed in B were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope present on wild-type and mutant PLCγ1 (top panel) or antibody reactive against Rac1 (bottom panel).
Next, wild-type and R665W mutant PLCγ2 were produced as recombinant polypeptides in baculovirus-infected insect cells and purified to near homogeneity by sequential column chromatography (Fig. 9A). The two purified preparations were adjusted to contain the same amounts of PLCγ2 by label-free quantitative mass spectrometry and then used for cell-free determination of inositol phosphate formation from artificial lipid vesicles containing radiolabeled PtdInsP2 as a substrate. Fig. 9B, left panel, shows that wild-type and R665W mutant PLCγ2 displayed a similar dependence on free Ca2+ for PtdInsP2 hydrolysis under these conditions, with half-maximal and maximal hydrolysis occurring at ∼1 and 20 μm free Ca2+, respectively. Maximal activity was slightly (∼1.4-fold) higher for PLCγ2R665W than for the wild-type enzyme. Upon functional reconstitution of the two PLCγ2 isoforms with isoprenylated Rac2 that had also been produced in and purified to near homogeneity from baculovirus-infected insect cells, the R665W mutant PLCγ2 exhibited a response to increasing concentrations of the poorly hydrolysable Rac2-activating guanine nucleotide analog GTPγS that was clearly different from that of its wild-type counterpart. Specifically, GTPγS showed a higher potency (EC50 ∼155 nm versus ∼830 nm) and a higher efficacy (stimulation by ∼57 versus ∼21 pmol inositol phosphates × min−1) to activate the R665W mutant PLCγ2 in comparison with its wild-type counterpart. Thus, functional differences between wild-type and R665W mutant PLCγ2 were particularly striking at limited activation of Rac2. A maximal, almost 7-fold difference in Rac2-stimulated activity was observed between wild-type and R665W mutant PLCγ2 at about 100 nm GTPγS (cf. dotted lined in Fig. 9B, right panel).
FIGURE 9.
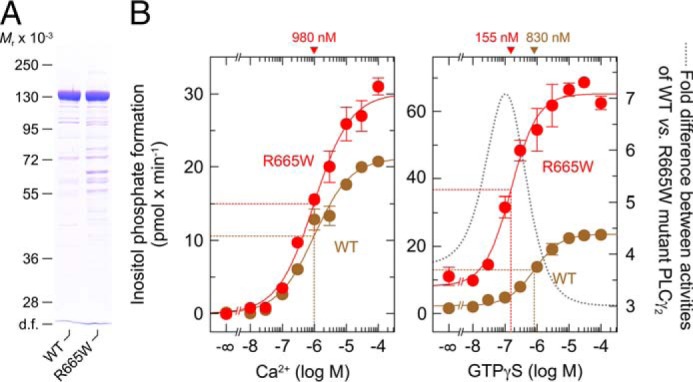
Purified PLCγ2R665W displays a slight enhancement of basal activity and a marked increase in the sensitivity to purified Rac2 in a reconstituted system in vitro. A, recombinant wild-type PLCγ2 and PLCγ2R665W were purified from baculovirus-infected insect cells. Aliquots of the two purified preparations were analyzed by label-free quantitative mass spectrometry to determine the relative abundance of PLCγ2-specific tryptic peptides in the two purified protein preparations. Samples adjusted to contain the same quantities of wild-type and R665W mutant PLCγ2 were subjected to SDS-PAGE and Coomassie Blue staining. B, aliquots of the two samples analyzed in A containing equal quantities of purified recombinant wild-type PLCγ2 and PLCγ2R665W were incubated at increasing concentrations of free Ca2+ and 2.5 mm sodium deoxycholate with phospholipid vesicles containing [3H]PtdInsP2. There was no difference between wild-type and R665W mutant PLCγ2 in the concentrations of free Ca2+ required to observe half-maximal stimulatory effects (left panel). The EC50 value of Ca2+ for the stimulation of wild-type or mutant PLCγ2 activity obtained by non-linear curve fitting is shown above the graphs in nanomolar. In the right panel, equal quantities of purified recombinant wild-type PLCγ2 and PLCγ2R665W were reconstituted with purified Rac2 in the presence of 30 nm free Ca2+ and 1 mm sodium deoxycholate (32) and incubated at increasing concentrations of GTPγS, as indicated at the abscissa, with phospholipid vesicles containing [3H]PtdInsP2. The EC50 values of GTPγS for the stimulation of wild-type or mutant PLCγ2 activity obtained by non-linear curve fitting is shown above the graphs in nanomolar.
COS-7 cells exhibit endogenous expression of EGF receptors, known to be coupled to activation of several intracellular signaling intermediates, including Rac (43). Upon heterologous expression of PLCγ2 in COS-7 cells, the enzyme is phosphorylated at tyrosine residues and translocated to the plasma membrane to mediate enhanced PtdInsP2 hydrolysis (44, 45). These previous findings led us to compare the activation of wild-type and L845F mutant PLCγ2 by endogenously expressed EGF receptors and to examine the relative contribution of tyrosine phosphorylation-mediated and Rac-mediated activation by also studying the two PLCγ2 isoforms carrying either replacements of four tyrosines known to be phosphorylated by upstream tyrosine kinases during enzyme activation by phenylalanines (4F) or the F897Q mutation blocking activation by Rac. Fig. 10A, left panel, shows that there was a concentration-dependent increase of wild-type and L845F mutant PLCγ2 stimulation by EGF, which was half-maximal at ∼13 and 6.9 ng/ml EGF, respectively, and maximal at about 50 ng/ml in both cases. Maximal EGF-stimulated PLCγ2 activity was about 4.5-fold higher in the presence of PLCγ2L845F in comparison with wild-type PLCγ2. The results obtained with the 4F, F897Q, and 4F/F897Q mutants of the two variants suggest that about half of the responses of both wild-type and L845F mutant PLCγ2 were due to tyrosine phosphorylation- and Rac-mediated activation. Similar findings were obtained for PLCγ2R665W and its F897Q variant. Interestingly, PLCγ2L845F was sensitive to activation by EGF even in the additional presence of the 4F and F897Q mutations. Wild-type and mutant PLCγ2 isozymes were present at equal amounts throughout the experiment shown in Fig. 10A, left panel, and B. The ∼12.5-fold enhancement of PLCγ2L845F-mediated inositol phosphate formation by EGF was almost completely blocked (−95%) by the EGF receptor inhibitor cetuximab (Fig. 10A, right panel).
FIGURE 10.
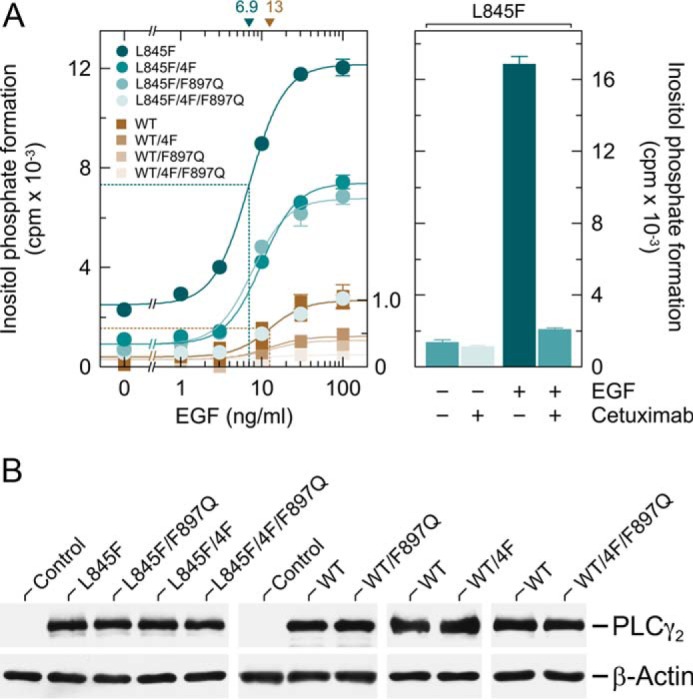
PLCγ2L845F is hypersensitive to activation by EGF receptor(s) endogenously expressed in COS-7 cells by a mechanism dependent on both protein tyrosine phosphorylation and activation by Rac. A, COS-7 cells were transfected with 150 ng/well vectors encoding either PLCγ2L845F (L845F), PLCγ2L845F/4F (L845F/4F), PLCγ2L845F/F897Q (L845F/F897Q), or PLCγ2L845F/4F/F897Q (L845F/4F/F897Q) or wild-type PLCγ2 (WT), PLCγ24F (WT/4F), PLCγ2F897Q (WT/F897Q), or PLCγ24F/F897Q (WT/4F/F897Q) (all constructed in pMT2 vector; Tyr → Phe substitutions at amino acid positions 753, 759, 1197, and 1217). Eighteen hours after transfection, the cells were incubated for a further 24 h with myo-[2-3H]inositol in the absence of serum and then treated for 60 min in the presence of 20 mm LiCl with increasing concentrations of EGF (1, 3, 10, 30, and 100 ng/ml), followed by determination of inositol phosphate formation. Background inositol phosphate formation in response to addition of EGF at increasing concentrations was determined in parallel on cells transfected with empty vector and subtracted from the individual values, with appropriate consideration of error propagation (68). The data on PLCγ2L845F and its mutants are from one experiment; the data on wild-type PLCγ2 and its mutants are from three experiments, each comparing the activity of wild-type PLCγ2 and one of the mutants. The latter activities were normalized to the maximal activity of PLCγ2L845F/4F/F897Q used as an internal control in one of the latter experiments (shown as a fraction of 1.0 on the right y axis). The EC50 values of EGF for the stimulation of wild-type or L845F mutant PLCγ2 activity obtained by non-linear curve fitting are shown above the graphs in nanograms/ml (left panel). In the right panel, COS-7 cells were transfected with 150 ng/well vector encoding PLCγ2L845F (L845F) and then incubated as described above and treated for 60 min in the absence or presence of 20 μg/ml cetuximab without or with 10 ng/ml EGF in medium containing 20 mm LiCl prior to determination of inositol phosphate formation. B, homogenates from cells functionally analyzed in A, left panel, were subjected to SDS-PAGE and immunoblotting using an antiserum reactive against PLCγ2 or antibody reactive against β-actin.
At least two ibrutinib-resistant patients have been described thus far harboring more than one PLCG2 mutations, including one with the coexistence of R665W and L845F (46). This prompted us to determine the effects of a compound R665W/L845F mutation on the functions of PLCγ2. Fig. 11A shows that basal activity of PLCγ2R665W/L845F was much higher than that of either PLCγ2R665W or PLCγ2L845F, despite similar levels of protein expression (cf. Fig. 11D). Specifically, when 150 ng of DNA encoding mutant PLCγ2 was used per well for transfection, the enhancement was ∼1.1-, 2.6-, and 70-fold over the activity observed for mock-transfected control cells for PLCγ2R665W, PLCγ2L845F, and PLCγ2R665W/L845F, respectively. In addition to basal phospholipase C activity, Rac2-, Rac2G12V-, and Vav1ΔN-stimulated activity was also markedly enhanced for PLCγ2R665W/L845F in comparison with the other two mutants (Fig. 11, B and C). For example, inositol phosphate formation in the presence of Vav1ΔN was enhanced ∼1.2-, 3.1-, 9.9-, and 61-fold for wild-type PLCγ2, PLCγ2R665W, PLCγ2L845F, and PLCγ2 R665W/L845F when compared with expression of Vav1ΔN alone (Fig. 11C, right panel). The compound mutation also markedly enhanced the apparent potency of Rac2G12V and Vav1ΔN to activate PLCγ2. Thus, half-maximal activation was observed for PLCγ2R665W/L845F at about 10–20-fold lower transfection levels of Rac2G12V and Vav1ΔN than for either of the single mutants, PLCγ2R665W or PLCγ2L845F (Fig. 11C).
FIGURE 11.

PLCγ2 ibrutinib resistance mutations R665W and L845F synergize to enhance basal enzyme activity and to sensitize the enzyme to stimulation by activated Vav1 and Rac2. A, COS-7 cells were transfected either with 500 ng/well empty vector (Co., control) or increasing amounts (15, 50, 150, and 500 ng/well) of vector encoding either PLCγ2 (WT), PLCγ2R665W (R665W), PLCγ2L845F (L845F), or the compound mutant PLCγ2R665W/L845F (R665W/L845F). B, COS-7 cells were transfected with 15 ng/well either empty vector (Co., Control) or vector encoding either PLCγ2 (WT), PLCγ2R665W (R665W), PLCγ2L845F (L845F), or the compound mutant PLCγ2R665W/L845F (R665W/L845F), together with 25 ng/well empty vector or vector encoding Rac2 or Rac2G12V. C, left panel, COS-7 cells were transfected as indicated with 15 ng/well vector encoding either PLCγ2R665W (R665W), PLCγ2L845F (L845F), or PLCγ2R665W/L845F (R665W/L845F) and increasing amounts of vector encoding Rac2G12V. Right panel, COS-7 cells were transfected with 50 ng/well vector encoding either PLCγ2R665W (R665W), PLCγ2L845F (L845F), or PLCγ2R665W/L845F (R665W/L845F) and increasing amounts of vector encoding Vav1ΔN. The ED50 values of vector encoding Rac2G12V or Vav1ΔN for the stimulation of mutant PLCγ2 activity obtained by non-linear curve fitting are shown above the graphs in nanograms/well. In all panels, the cells were incubated 24 h after transfection for 20 h with myo-[2-3H]inositol, and inositol phosphate formation was determined. D, upper panel, homogenates from cells functionally analyzed in A were subjected to SDS-PAGE and immunoblotting using an antibody reactive against the c-Myc epitope present on wild-type and mutant PLCγ2 or antibody reactive against β-actin. Lower panel, homogenates from cells functionally analyzed in B were subjected to SDS-PAGE and immunoblotting using an antiserum reactive against Rac2.
Discussion
The functions of PLCγ2 mutants mediating ibrutinib resistance in CLL patients have previously mostly been characterized following their reconstitution into PLCγ2-deficient DT40 B cells (29). Even in those cells, expressing only the mutant PLCγ2 rather than a combination of wild-type and mutant PLCγ2 isozymes, there was no evidence of autonomous PLCγ2 signaling. Instead, the PLCγ2 mutants R665W and L845F still relied on BCR activation. The main change was an ∼2–3-fold increase in the level of cytosolic Ca2+ upon BCR ligation, which was insensitive to inhibition by ibrutinib and, interestingly, did not return to baseline within the time frame of the experiment. Subsequent experiments in reconstituted DT40 cells showed that BCR-mediated activation of PLCγ2R665W was enhanced even in Btk−/− cells in comparison with the wild-type enzyme, suggesting that the mutant functionally bypasses Btk upon BCR activation (31). In cells expressing PLCγ2R665W, the BCR-mediated increase in Ca2+ was sensitive to pharmacologic inhibitors of Syk and Lyn, signaling components previously known to be essential for BCR-mediated InsP3 generation and rapid Ca2+ mobilization, respectively, in DT40 cells (47). These results suggested that the R665W mutation renders PLCγ2 independent of Btk and therefore capable of mediating ibrutinib resistance in CLL cells.
Using the same experimental model, reconstituted PLCγ2−/−DT40 cells, we have previously shown that interaction of PLCγ2 with Rac amplifies the BCR-induced Ca2+ signaling by increasing the sensitivity of the cells to BCR ligation, augmenting the BCR-mediated Ca2+ release from intracellular stores, enhancing the Ca2+ entry from the extracellular compartment, and facilitating the nuclear translocation of the Ca2+-regulated nuclear factor of activated T cells (34). Although performed in a different cellular system, the results presented here suggest that the bypass of Btk exploited by the PLCγ2 mutant R665W may also be based on an increased sensitivity of this and another mutant, L845F, to enhanced activation by Rac.
Our results showing marked increases in basal inositol phosphate formation upon expression of the two PLCγ2 mutants R665W and L845F at increasing levels, suggest, at first glance, that the mutants exhibit constitutively enhanced intrinsic activity. Given that constitutive protein activity is a hallmark of hypermorphic mutations (48), this is consistent with the designation of the two PLCγ2 mutations as belonging to the hypermorphic class (31). However, several lines of evidence presented in this work suggest that enhanced constitutive PLC activity, such as that observed for other mutants of several PLC isozymes (42), including PLCγ2 (49), is unlikely to be the main molecular mechanism of ibrutinib resistance. Specifically, both mutants are strikingly hypersensitive to activation by Rac2 and its upstream regulator Vav1, such that even wild-type Rac2 suffices to activate the mutant enzymes, but not their wild-type counterpart, upon its introduction into intact COS-7 cells (Fig. 3A). This view is strongly supported by the fact that enhanced basal activity of the mutant enzymes is markedly reduced by the F897Q mutation of PLCγ2 (Fig. 6A), which has previously been shown to block PLCγ2 activation by Rac but not by loss of SH-mediated autoinhibition or Ca2+ (34). Further support stems from the observations that this activity is subject to specific inhibition by the small molecule Rac inhibitor EHT 1864 (Fig. 7A) and that the purified PLCγ2R665W displays only a subtle increase of its basal activity (Fig. 9B). Hence, it appears likely that the mutants are hypersensitive to activated Rac2 rather than simply constitutively active.
Several lines of evidence have been presented suggesting that Rac is activated by BCR ligation. Thus, several elements of the canonical BCR signaling cascade, e.g. Syk (50), Btk (51), and BLNK (52, 53), are known to physically interact with and activate the Rac activator Vav, by processes not necessarily involving the protein kinase activity of Btk. BCR cross-linking caused activation of both Rac1 and Rac2 within minutes (54). Total internal reflection fluorescence microscopy has shown that PLCγ2, Vav, BLNK, and Btk synergize to form highly coordinated microsignalosomes. Very interestingly, efficient assembly of the latter is absolutely dependent on Lyn and Syk (55). Therefore, it appears likely that one of the major functional consequences of the two PLCγ2 mutations conferring ibrutinib resistance to intact cells, including B lymphocytes, is hypersensitivity of PLCγ2 to activated Rac. The finding that the enhanced PLCγ2 stimulation by ligation of endogenous EGF receptors requires the replacement by phenylalanines of the tyrosine residues involved in enzyme activation in addition to a F897Q mutation to be maximally reduced (Fig. 10A, left panel) suggests that tyrosine phosphorylation may be involved, in addition to direct PLCγ2-Rac interaction in mediating hypersensitivity of PLCγ2 to Rac. In intact B cells, this hypersensitivity is likely to be the molecular basis of a relatively focused rewiring of the signaling pathways immediately downstream of the BCR, such that PLCγ2 loses its dependence on activated Btk (56) and gains sensitivity to the pathway made up of Lyn, Syk, Vav, and Rac. Hence, in our opinion, the two PLCγ2 ibrutinib resistance mutations, R665W and L845F, are not simply and solely hypermorphic. We suggest that they would be better termed allomorphic, according to the Greek word αλλοσ for other, different.
In B cells, activation of Rac is not limited to BCR activation, but also occurs upon activation of BCR coreceptors, such as CD19/CD21, integrins, as well as certain G-protein-coupled chemokine and Toll-like receptors (cf. discussion and references in Ref. 34). It is thus possible that the rewiring process induced by the ibrutinib resistance mutations also enhances the sensitivity of PLCγ2 to extracellular ligands of these cell surface proteins, such as cleavage fragments of the third complement component, pathogen-derived molecules, extracellular matrix proteins, and chemokines. This may ultimately allow costimulatory signals to become stimulatory in their own right and as such to alter the interactions of the ibrutinib-resistant tumor cells with their protective microenvironments, for example (57). Interestingly, integrin-mediated adhesion and migration in response to the chemokines CXCL12 or CXCL13, as well as in vivo homing to lymphoid organs, was impaired in Btk-deficient (pre-)B cells, whereas CXCL12-mediated activation of Rac was intact. Deficiency of PLCγ2 also curtailed the CXCL12-mediated migratory response (8). Independence of Btk and increased sensitivity of the PLCγ2 mutants to Rac by rewiring may provide CLL cells with the ability to home to and remain in protective microenvironments for survival and expansion even in the presence of ibrutinib-mediated Btk inhibition.
Enhanced sensitivity of PLCγ2 by signaling mechanisms emanating from BCR or other B cell surface receptors bypassing Btk may provide novel mechanisms for targeted treatment of CLL and, possibly, B cell lymphomas. Thus, inhibitors of Syk and Lyn have been shown to oppose ibrutinib resistance mediated by PLCG2 mutations (31). Although the exact position of PI3Kδ relative to other components within the BCR signalosome is still controversial, some evidence puts class IA PI3Ks, at least in part, upstream of Vav and Rac (58). Hence, inhibition of PI3Kδ with idelalisib may also interfere with Rac-mediated activation of wild-type and, even more so, R665W or L845F mutant PLCγ2 isozymes. Pharmacologic interventions at the level of Rac itself or other upstream activators also appear to be a viable option. These include inhibition of Rac C-terminal modification or of Rac protein-protein interaction, e.g. by small molecules like EHT 1864, or prevention of integrin, CD19, or chemokine receptor activation (59–61). Very recent results suggest that ibrutinib therapy of CLL patients favors selection and expansion of rare subclones already present before ibrutinib treatment, including subclones containing mutations in PLCG2 (62). Hence, it may be worthwhile to investigate combining ibrutinib with adjuvant drugs of this type ab initio to suppress this selection and expansion.
Experimental Procedures
Materials
The mouse monoclonal antibody 9B11 reactive against the c-Myc epitope (EQKLISEEDL) and the rabbit polyclonal antiserum reactive against human PLCγ2 (sc-407) were obtained from Cell Signaling Technology and Santa Cruz Biotechnology, respectively. The rabbit polyclonal antiserum reactive against human Rac2 (sc-96) was purchased from Santa Cruz Biotechnology. The anti-β-actin antibody (clone AC-15) and the anti-Rac1 antibody (clone 23A8) were obtained from Sigma and Merck Millipore, respectively. The Rac inhibitor EHT 1864 and its inactive analog EHT 4063 were synthesized as described previously (63). Human epidermal growth factor (EGF) (E9644) was from Sigma. ProGreen baculovirus vector DNA (A1) was purchased from AB Vector. GTPγS (catalog no. 10220647001) was purchased from Roche Applied Bioscience. Cetuximab is marketed by Merck.
Construction of Vectors
The construction of complementary DNAs encoding c-Myc epitope-tagged human PLCγ1 (1291 amino acids, accession number ABB84466), human PLCγ2 (1265 amino acids, accession number NP_002652), and F897Q mutant of PLCγ2 was described previously (34). The construction of all other vectors and of the baculoviruses was outlined in Refs. 32, 33. Complementary DNAs encoding mutants of PLCγ1 and PLCγ2 were constructed by in vitro mutagenesis using the QuikChange II XL site-directed mutagenesis kit (200521, Agilent Technologies). The primer sequences and PCR protocols are available from the authors upon request. A vector encoding c-Myc epitope-tagged human PLCδ1Δ44 was kindly supplied by J. Sondek (42).
Cell Culture and Transfection
COS-7 cells were maintained at 37 °C in a humidified atmosphere of 90% air and 10% CO2 in Dulbecco's modified Eagle's medium (DMEM) (catalog no. 41965-039, Gibco) supplemented with 10% (v/v) fetal calf serum (catalog no. 10270-106, Gibco), 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from PAA Laboratories). Prior to transfection, COS-7 cells were seeded into 24-well plates at a density of 0.75 × 105 cells/well and grown for 24 h in 0.5 ml of medium/well. For transfection, plasmid DNA (500–800 ng/well) was diluted in 50 μl of jetPRIME® buffer, and 1–1.6 μl of jetPRIME® was added according to the manufacturer's instructions. The total amount of DNA was maintained constant by adding empty vector. Four hours after the addition of the DNA-jetPRIME® complexes to the dishes, the medium was replaced by fresh medium, and the cells were incubated for a further 20 h at 37 °C and 10% CO2.
Radiolabeling of Inositol Phospholipids and Analysis of Inositol Phosphate Formation
Twenty four hours after transfection, COS-7 cells were washed once with 0.3 ml/well Dulbecco's PBS (PAA Laboratories) and then incubated for 18 h in 0.2 ml/well DMEM containing supplements as described above, supplemented with 2.5 μCi/ml myo-[2-3H]inositol (NET1156005MC, PerkinElmer Life Sciences) and 10 mm LiCl. The cells were then washed once with 0.2 ml/well of Dulbecco's PBS and lysed by addition of 0.2 ml/well 10 mm ice-cold formic acid. The analysis of inositol phosphate formation was performed as described previously (33).
To examine EGF-mediated PLCγ2 stimulation, COS-7 cells were radiolabeled for 24 h in serum-free DMEM as described previously (45). Briefly, cells were washed twice with 0.3 ml/well DMEM containing the above supplements except serum and then incubated for 24 h in 0.2 ml/well of the same medium supplemented with 0.25% fatty-acid-free bovine serum albumin (catalog no. A8806, Sigma) and 2.5 μCi/ml myo-[2-3H]inositol. The cells were then washed with 0.3 ml/well Dulbecco's PBS and incubated for 1 h in 0.2 ml/well DMEM without serum containing the above supplements, 20 mm LiCl, and increasing concentrations of EGF. After removal of the medium, the cells were lysed by addition of 0.2 ml/well of 10 mm ice-cold formic acid for analysis of inositol phosphate formation.
Expression and Purification of Proteins
Post-translationally modified Rac2 was expressed as a glutathione S-transferase fusion protein in baculovirus-infected insect cells and solubilized from the particulate fraction. The Rac2 portion of the fusion protein was purified as detailed previously (33). c-Myc epitope-tagged PLCγ2 and PLCγ2R665W were purified from soluble fractions of baculovirus-infected High FiveTM insect cells grown in suspension culture by sequential chromatography on HiTrapTM Heparin HP and Resource Q (GE Healthcare) as described before for PLCβ2Δ (64). Our attempts at purifying PLCγ2L845F for functional analysis have thus far been unsuccessful, mostly due to the lower expression of the enzyme in baculovirus-infected insect cells and to its lower stability during purification.
Label-free Quantitative Mass Spectrometry
Equal volumes (5 μl each) of purified wild-type or R665W mutant PLCγ2 were mixed with 300 fmol of the PierceTM peptide retention time calibration mixture (PRTC, catalog no. 88320, ThermoFisher) containing 15 known synthetic tryptic peptides. Samples were reduced for 20 min at room temperature with 5 mm DTT and subsequently alkylated for 20 min at 37 °C with 50 mm iodoacetamide. The samples were then subjected to tryptic digestion overnight at 37 °C. The resulting peptides were identified by LC/MS analysis. Using the SEQUEST search engine within the Proteome DiscovererTM software suite (1.4.1.14, Thermo Scientific), mass spectra were correlated with a database containing the sequences of wild-type and mutant PLCγ2, a concatenation of the PRTC peptide sequences, and sequences of common contaminants commonly encountered in proteomic experiments, as used in the MaxQuant software (65). For relative quantitation, the precursor ions area detector node within Proteome DiscovererTM was used; preceding event detection was set to 4 ppm.
Measurement of PLC Activity in Vitro
Phospholipase C activity was determined as described (64, 66) with minor modifications. In brief, aliquots (10 μl) of purified PLCγ2 proteins appropriately diluted in buffer containing 60 mm Tris/maleate, pH 7.3, 84 mm KCl, 3.6 mm EGTA, 2.4 mm dithiothreitol, 2 mg/ml bovine serum albumin were incubated for 45 min at 30 °C in a volume of 60 μl containing 50 mm Tris/maleate, pH 7.3, 70 mm KCl, 3 mm EGTA, 2 mm dithiothreitol, 536 μm phosphatidylethanolamine, 33.4 μm [3H]PtdInsP2 (185 GBq/mmol), 0.33 mg/ml bovine serum albumin, and the concentrations of sodium deoxycholate and free Ca2+ specified in the figure legends. For reconstitution of wild-type and mutant PLCγ2 with Rac2, purified PLCγ2 was reconstituted with 5 μl of purified isoprenylated Rac2 and incubated with the phospholipid substrate as described above. The concentration of CaCl2 required to adjust the concentration of free Ca2+ to the desired value was calculated using the program EqCal for Windows (Biosoft, Ferguson, MO). The reaction was terminated, and the samples were analyzed for inositol phosphates, as described (64).
Miscellaneous
SDS-PAGE and immunoblotting were performed according to standard protocols using antibodies reactive against the c-Myc epitope for wild-type and mutant PLCγ2. Immunoreactive proteins were visualized using the ECL Western blotting detection system (GE Healthcare). All experiments were performed at least three times. Similar results and identical trends were obtained each time. Data from representative experiments are shown as means ± S.E. of triplicate determinations. In Figs. 2A, 3A, 4A, 7A, 9B, 10A, and 11C, the data were fitted by nonlinear least squares curve fitting to three- or four-parameter dose-response equations using GraphPad Prism®, version 5.04. In certain cases, the global curve fitting procedure contained in Prism® was used to determine whether the best fit values of selected parameters differed between data sets. The simpler model was selected unless the extra sum of squares F-test had a p value of less than 0.05. Repeated measures analysis of variance with Tukey's post test contained in the GraphPad InStat software package (version 3.10; GraphPad Software, La Jolla, CA) was used for the statistical analysis of the data shown in Figs. 1A and 8A. Statistically significant effects are denoted by ***, p < 0.001; **, p > 0.001 and p < 0.01; and *, p > 0.01 and p < 0.05. Non-significant changes are denoted by ns, p < 0.05.
Author Contributions
C. W., E. H., A. S., S. W., J. D., and M. Z. performed the experiments and analyzed the data. P. G. provided overall direction and wrote the manuscript with input from L. D., D. M., S. S., and the other authors.
Acknowledgments
The expert technical assistance of Susanne Gierschik and Norbert Zanker is greatly appreciated.
This work was supported by Grant SFB 1074, TP A8 from the Deutsche Forschungsgemeinschaft. The authors declare that they have no conflicts of interest with the contents of this article.
- PLC
- inositol-phospholipid-specific phospholipase C
- SH2
- Src homology 2
- PH
- pleckstrin homology
- spPH
- split PH domain
- Rac
- Ras-related C3 botulinum toxin substrate
- BCR
- B cell receptor
- InsP3
- inositol 1,4,5-trisphosphate
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- CLL
- chronic lymphocytic leukemia
- DH
- diffuse B-cell lymphoma (Dbl) homology.
References
- 1. Hikida M., and Kurosaki T. (2005) Regulation of phospholipase C-γ2 networks in B lymphocytes. Adv. Immunol. 88, 73–96 [DOI] [PubMed] [Google Scholar]
- 2. Jumaa H., Hendriks R. W., and Reth M. (2005) B cell signaling and tumorigenesis. Annu. Rev. Immunol. 23, 415–445 [DOI] [PubMed] [Google Scholar]
- 3. Kadamur G., and Ross E. M. (2013) Mammalian phospholipase C. Annu. Rev. Physiol 75, 127–154 [DOI] [PubMed] [Google Scholar]
- 4. Shirakawa A. K., Liao F., Zhang H. H., Hedrick M. N., Singh S. P., Wu D., and Farber J. M. (2010) Pathway-selective suppression of chemokine receptor signaling in B cells by LPS through downregulation of PLC-β2. Cell. Mol. Immunol. 7, 428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Packard T. A., and Cambier J. C. (2013) B lymphocyte antigen receptor signaling: initiation, amplification, and regulation. F1000Prime Rep. 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tedder T. F. (2010) Innate and adaptive receptors interact to balance humoral immunity. J. Immunol. 184, 2231–2232 [DOI] [PubMed] [Google Scholar]
- 7. Rawlings D. J., Schwartz M. A., Jackson S. W., and Meyer-Bahlburg A. (2012) Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol. 12, 282–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Gorter D. J., Beuling E. A., Kersseboom R., Middendorp S., van Gils J. M., Hendriks R. W., Pals S. T., and Spaargaren M. (2007) Bruton's tyrosine kinase and phospholipase Cγ2 mediate chemokine-controlled B cell migration and homing. Immunity 26, 93–104 [DOI] [PubMed] [Google Scholar]
- 9. Zheng H., Nam J. H., Pang B., Shin D. H., Kim J. S., Chun Y. S., Park J. W., Bang H., Kim W. K., Earm Y. E., and Kim S. J. (2009) Identification of the large-conductance background K+ channel in mouse B cells as TREK-2. Am. J. Physiol. Cell Physiol. 297, C188–C197 [DOI] [PubMed] [Google Scholar]
- 10. Ghia P., Chiorazzi N., and Stamatopoulos K. (2008) Microenvironmental influences in chronic lymphocytic leukaemia: the role of antigen stimulation. J. Intern. Med. 264, 549–562 [DOI] [PubMed] [Google Scholar]
- 11. Stamatopoulos K., Belessi C., Moreno C., Boudjograh M., Guida G., Smilevska T., Belhoul L., Stella S., Stavroyianni N., Crespo M., Hadzidimitriou A., Sutton L., Bosch F., Laoutaris N., Anagnostopoulos A., et al. (2007) Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood 109, 259–270 [DOI] [PubMed] [Google Scholar]
- 12. Dühren-von Minden M., Übelhart R., Schneider D., Wossning T., Bach M. P., Buchner M., Hofmann D., Surova E., Follo M., Köhler F., Wardemann H., Zirlik K., Veelken H., and Jumaa H. (2012) Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 489, 309–312 [DOI] [PubMed] [Google Scholar]
- 13. Muzio M., Apollonio B., Scielzo C., Frenquelli M., Vandoni I., Boussiotis V., Caligaris-Cappio F., and Ghia P. (2008) Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood 112, 188–195 [DOI] [PubMed] [Google Scholar]
- 14. Hewamana S., Alghazal S., Lin T. T., Clement M., Jenkins C., Guzman M. L., Jordan C. T., Neelakantan S., Crooks P. A., Burnett A. K., Pratt G., Fegan C., Rowntree C., Brennan P., and Pepper C. (2008) The NF-κB subunit Rel A is associated with in vitro survival and clinical disease progression in chronic lymphocytic leukemia and represents a promising therapeutic target. Blood 111, 4681–4689 [DOI] [PubMed] [Google Scholar]
- 15. Stevenson F. K., Krysov S., Davies A. J., Steele A. J., and Packham G. (2011) B-cell receptor signaling in chronic lymphocytic leukemia. Blood 118, 4313–4320 [DOI] [PubMed] [Google Scholar]
- 16. Kulathu Y., Grothe G., and Reth M. (2009) Autoinhibition and adapter function of Syk. Immunol. Rev. 232, 286–299 [DOI] [PubMed] [Google Scholar]
- 17. Hendriks R. W., and Kersseboom R. (2006) Involvement of SLP-65 and Btk in tumor suppression and malignant transformation of pre-B cells. Semin. Immunol. 18, 67–76 [DOI] [PubMed] [Google Scholar]
- 18. Davis R. E., Ngo V. N., Lenz G., Tolar P., Young R. M., Romesser P. B., Kohlhammer H., Lamy L., Zhao H., Yang Y., Xu W., Shaffer A. L., Wright G., Xiao W., Powell J., et al. (2010) Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cramer P., Langerbeins P., Eichhorst B., and Hallek M. (2016) Advances in first-line treatment of chronic lymphocytic leukemia current recommendations on management and first-line treatment by the German CLL Study Group (GCLLSG). Eur. J. Haematol. 96, 9–18 [DOI] [PubMed] [Google Scholar]
- 20. de Claro R. A., McGinn K. M., Verdun N., Lee S. L., Chiu H. J., Saber H., Brower M. E., Chang C. J., Pfuma E., Habtemariam B., Bullock J., Wang Y., Nie L., Chen X. H., Lu D. R., et al. (2015) FDA approval: ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin. Cancer Res. 21, 3586–3590 [DOI] [PubMed] [Google Scholar]
- 21. Wilson W. H., Young R. M., Schmitz R., Yang Y., Pittaluga S., Wright G., Lih C. J., Williams P. M., Shaffer A. L., Gerecitano J., de Vos S., Goy A., Kenkre V. P., Barr P. M., Blum K. A., et al. (2015) Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 21, 922–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chakraborty R., Kapoor P., Ansell S. M., and Gertz M. A. (2015) Ibrutinib for the treatment of Waldenstrom macroglobulinemia. Expert Rev. Hematol. 8, 569–579 [DOI] [PubMed] [Google Scholar]
- 23. Kokhaei P., Jadidi-Niaragh F., Sotoodeh J. A., Osterborg A., Mellstedt H., and Hojjat-Farsangi M. (2016) Ibrutinib-A double-edge sword in cancer and autoimmune disorders. J. Drug Target 24, 1–13 [DOI] [PubMed] [Google Scholar]
- 24. Honigberg L. A., Smith A. M., Sirisawad M., Verner E., Loury D., Chang B., Li S., Pan Z., Thamm D. H., Miller R. A., and Buggy J. J. (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U.S.A. 107, 13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shinohara M., Chang B. Y., Buggy J. J., Nagai Y., Kodama T., Asahara H., and Takayanagi H. (2014) The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone 60, 8–15 [DOI] [PubMed] [Google Scholar]
- 26. Ito M., Shichita T., Okada M., Komine R., Noguchi Y., Yoshimura A., and Morita R. (2015) Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6, 7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mancini M., and Yarden Y. (2016) Mutational and network level mechanisms underlying resistance to anti-cancer kinase inhibitors. Semin. Cell Dev. Biol. 50, 164–176 [DOI] [PubMed] [Google Scholar]
- 28. Komarova N. L., Burger J. A., and Wodarz D. (2014) Evolution of ibrutinib resistance in chronic lymphocytic leukemia (CLL). Proc. Natl. Acad. Sci. U.S.A. 111, 13906–13911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woyach J. A., Furman R. R., Liu T. M., Ozer H. G., Zapatka M., Ruppert A. S., Xue L., Li D. H., Steggerda S. M., Versele M., Dave S. S., Zhang J., Yilmaz A. S., Jaglowski S. M., Blum K. A., et al. (2014) Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 370, 2286–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou Q., Lee G. S., Brady J., Datta S., Katan M., Sheikh A., Martins M. S., Bunney T. D., Santich B. H., Moir S., Kuhns D. B., Long Priel D. A., Ombrello A., Stone D., Ombrello M. J., et al. (2012) A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am. J. Hum. Genet. 91, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu T. M., Woyach J. A., Zhong Y., Lozanski A., Lozanski G., Dong S., Strattan E., Lehman A., Zhang X., Jones J. A., Flynn J., Andritsos L. A., Maddocks K., Jaglowski S. M., Blum K. A., et al. (2015) Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood 126, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Piechulek T., Rehlen T., Walliser C., Vatter P., Moepps B., and Gierschik P. (2005) Isozyme-specific stimulation of phospholipase C-γ2 by Rac GTPases. J. Biol. Chem. 280, 38923–38931 [DOI] [PubMed] [Google Scholar]
- 33. Walliser C., Retlich M., Harris R., Everett K. L., Josephs M. B., Vatter P., Esposito D., Driscoll P. C., Katan M., Gierschik P., and Bunney T. D. (2008) Rac regulates its effector phospholipase Cγ2 through interaction with a split pleckstrin homology domain. J. Biol. Chem. 283, 30351–30362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walliser C., Tron K., Clauss K., Gutman O., Kobitski A. Y., Retlich M., Schade A., Röcker C., Henis Y. I., Nienhaus G. U., and Gierschik P. (2015) Rac-mediated stimulation of phospholipase Cγ2 amplifies B cell receptor-induced calcium signaling. J. Biol. Chem. 290, 17056–17072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujikawa K., Miletic A. V., Alt F. W., Faccio R., Brown T., Hoog J., Fredericks J., Nishi S., Mildiner S., Moores S. L., Brugge J., Rosen F. S., and Swat W. (2003) Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J. Exp. Med. 198, 1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cook D. R., Rossman K. L., and Der C. J. (2014) Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33, 4021–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chrencik J. E., Brooun A., Zhang H., Mathews I. I., Hura G. L., Foster S. A., Perry J. J., Streiff M., Ramage P., Widmer H., Bokoch G. M., Tainer J. A., Weckbecker G., and Kuhn P. (2008) Structural basis of guanine nucleotide exchange mediated by the T-cell essential Vav1. J. Mol. Biol. 380, 828–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gutierrez L., Magee A. I., Marshall C. J., and Hancock J. F. (1989) Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. EMBO J. 8, 1093–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Désiré L., Bourdin J., Loiseau N., Peillon H., Picard V., De Oliveira C., Bachelot F., Leblond B., Taverne T., Beausoleil E., Lacombe S., Drouin D., and Schweighoffer F. (2005) RAC1 inhibition targets amyloid precursor protein processing by γ-secretase and decreases Aβ production in vitro and in vivo. J. Biol. Chem. 280, 37516–37525 [DOI] [PubMed] [Google Scholar]
- 40. Shutes A., Onesto C., Picard V., Leblond B., Schweighoffer F., and Der C. J. (2007) Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 282, 35666–35678 [DOI] [PubMed] [Google Scholar]
- 41. Onesto C., Shutes A., Picard V., Schweighoffer F., and Der C. J. (2008) Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 439, 111–129 [DOI] [PubMed] [Google Scholar]
- 42. Hicks S. N., Jezyk M. R., Gershburg S., Seifert J. P., Harden T. K., and Sondek J. (2008) General and versatile autoinhibition of PLC isozymes. Mol. Cell 31, 383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coso O. A., Chiariello M., Yu J. C., Teramoto H., Crespo P., Xu N., Miki T., and Gutkind J. S. (1995) The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 81, 1137–1146 [DOI] [PubMed] [Google Scholar]
- 44. Matsuda M., Paterson H. F., Rodriguez R., Fensome A. C., Ellis M. V., Swann K., and Katan M. (2001) Real time fluorescence imaging of PLCγ translocation and its interaction with the epidermal growth factor receptor. J. Cell Biol. 153, 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Everett K. L., Bunney T. D., Yoon Y., Rodrigues-Lima F., Harris R., Driscoll P. C., Abe K., Fuchs H., de Angelis M. H., Yu P., Cho W., and Katan M. (2009) Characterization of phospholipase Cγ enzymes with gain-of-function mutations. J. Biol. Chem. 284, 23083–23093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maddocks K. J., Ruppert A. S., Lozanski G., Heerema N. A., Zhao W., Abruzzo L., Lozanski A., Davis M., Gordon A., Smith L. L., Mantel R., Jones J. A., Flynn J. M., Jaglowski S. M., Andritsos L. A., et al. (2015) Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 1, 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takata M., Sabe H., Hata A., Inazu T., Homma Y., Nukada T., Yamamura H., and Kurosaki T. (1994) Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 13, 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilkie A. O. (1994) The molecular basis of genetic dominance. J. Med. Genet. 31, 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Everett K. L., Buehler A., Bunney T. D., Margineanu A., Baxendale R. W., Vatter P., Retlich M., Walliser C., Manning H. B., Neil M. A., Dunsby C., French P. M., Gierschik P., and Katan M. (2011) Membrane environment exerts an important influence on Rac-mediated activation of phospholipase Cγ2. Mol. Cell. Biol. 31, 1240–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deckert M., Tartare-Deckert S., Couture C., Mustelin T., and Altman A. (1996) Functional and physical interactions of Syk family kinases with the Vav proto-oncogene product. Immunity 5, 591–604 [DOI] [PubMed] [Google Scholar]
- 51. Guinamard R., Fougereau M., and Seckinger P. (1997) The SH3 domain of Bruton's tyrosine kinase interacts with Vav, Sam68 and EWS. Scand. J. Immunol. 45, 587–595 [DOI] [PubMed] [Google Scholar]
- 52. Fu C., Turck C. W., Kurosaki T., and Chan A. C. (1998) BLNK: a central linker protein in B cell activation. Immunity 9, 93–103 [DOI] [PubMed] [Google Scholar]
- 53. Wienands J., Schweikert J., Wollscheid B., Jumaa H., Nielsen P. J., and Reth M. (1998) SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J. Exp. Med. 188, 791–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arana E., Vehlow A., Harwood N. E., Vigorito E., Henderson R., Turner M., Tybulewicz V. L., and Batista F. D. (2008) Activation of the small GTPase Rac2 via the B cell receptor regulates B cell adhesion and immunological-synapse formation. Immunity 28, 88–99 [DOI] [PubMed] [Google Scholar]
- 55. Weber M., Treanor B., Depoil D., Shinohara H., Harwood N. E., Hikida M., Kurosaki T., and Batista F. D. (2008) Phospholipase C-γ2 and Vav cooperate within signaling microclusters to propagate B cell spreading in response to membrane-bound antigen. J. Exp. Med. 205, 853–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Takata M., and Kurosaki T. (1996) A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-γ2. J. Exp. Med. 184, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ten Hacken E., and Burger J. A. (2016) Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 1863, 401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hawkins P. T., Anderson K. E., Davidson K., and Stephens L. R. (2006) Signalling through Class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 34, 647–662 [DOI] [PubMed] [Google Scholar]
- 59. Montresor A., Toffali L., Mirenda M., Rigo A., Vinante F., and Laudanna C. (2015) JAK2 tyrosine kinase mediates integrin activation induced by CXCL12 in B-cell chronic lymphocytic leukemia. Oncotarget 6, 34245–34257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Rourke L. M., Tooze R., Turner M., Sandoval D. M., Carter R. H., Tybulewicz V. L., and Fearon D. T. (1998) CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity 8, 635–645 [DOI] [PubMed] [Google Scholar]
- 61. Stamatopoulos B., Meuleman N., De Bruyn C., Pieters K., Mineur P., Le Roy C., Saint-Georges S., Varin-Blank N., Cymbalista F., Bron D., and Lagneaux L. (2012) AMD3100 disrupts the cross-talk between chronic lymphocytic leukemia cells and a mesenchymal stromal or nurse-like cell-based microenvironment: pre-clinical evidence for its association with chronic lymphocytic leukemia treatments. Haematologica 97, 608–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Burger J. A., Landau D. A., Taylor-Weiner A., Bozic I., Zhang H., Sarosiek K., Wang L., Stewart C., Fan J., Hoellenriegel J., Sivina M., Dubuc A. M., Fraser C., Han Y., Li S., et al. (2016) Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat. Commun. 7, 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leblond B., Petit S., Picard V., Taverne T. Schweighoffer F. (September 10, 2004) World Patent WO2004076445
- 64. Illenberger D., Stephan I., Gierschik P., and Schwald F. (2000) Stimulation of phospholipase C-β2 by Rho GTPases. Methods Enzymol. 325, 167–177 [DOI] [PubMed] [Google Scholar]
- 65. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 66. Camps M., Hou C. F., Jakobs K. H., and Gierschik P. (1990) Guanosine 5′-[γ-thio]triphosphate-stimulated hydrolysis of phosphatidylinositol 4,5-bisphosphate in HL-60 granulocytes: evidence that the guanine nucleotide acts by relieving phospholipase C from an inhibitory constraint. Biochem. J. 271, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bunney T. D., Esposito D., Mas-Droux C., Lamber E., Baxendale R. W., Martins M., Cole A., Svergun D., Driscoll P. C., and Katan M. (2012) Structural and functional integration of the PLCγ interaction domains critical for regulatory mechanisms and signaling deregulation. Structure 20, 2062–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Meyer S. L. (1975) Data Analysis for Scientists and Engineers, pp. 39–48, Peer Management Consultants, Evanston, IL [Google Scholar]