

Abstract
A general problem is posed by analysis of transcriptional thresholds governing cell fate decisions in metazoan development. A model is provided by testis determination in therian mammals. Its key step, Sertoli cell differentiation in the embryonic gonadal ridge, is initiated by SRY, a Y-encoded architectural transcription factor. Mutations in human SRY cause gonadal dysgenesis leading to XY female development (Swyer syndrome). Here, we have characterized an inherited mutation compatible with either male or female somatic phenotypes as observed in an XY father and XY daughter, respectively. The mutation (a crevice-forming substitution at a conserved back surface of the SRY high mobility group box) markedly destabilizes the domain but preserves specific DNA affinity and induced DNA bend angle. On transient transfection of diverse human and rodent cell lines, the variant SRY exhibited accelerated proteasomal degradation (relative to wild type) associated with increased ubiquitination; in vitro susceptibility to ubiquitin-independent (“default”) cleavage by the 20S core proteasome was unchanged. The variant's gene regulatory activity (as assessed in a cellular model of the rat embryonic XY gonadal ridge) was reduced by 2-fold relative to wild-type SRY at similar levels of mRNA expression. Chemical proteasome inhibition restored native-like SRY expression and transcriptional activity in association with restored occupancy of a sex-specific enhancer element in principal downstream gene Sox9, demonstrating that the variant SRY exhibits essentially native activity on a per molecule basis. Our findings define a novel mechanism of impaired organogenesis, accelerated ubiquitin-directed proteasomal degradation of a master transcription factor leading to a developmental decision poised at the edge of ambiguity.
Keywords: developmental factor, proteasome, protein degradation, reproduction, transcription, DNA-binding domain, Sywer syndrome, disorders of sexual development, intersex
Introduction
Regulation of alternative genetic programs is central to the logic of metazoan development (1). Broad mechanistic insight has been obtained from studies of prokaryotic toggle switches (2) constructed within synthetic operons (3), based in part on the bistable transcriptional circuitry of lysogenic phages (4, 5). Analogous principles are thought to govern developmental decisions in metazoans (6) wherein organogenesis may be directed by alternative states of gene-regulatory networks (7, 8). Such control systems have been delineated in model organisms, in particular through molecular-genetic analysis of sex determination and sex-specific gene regulation (9, 10). In this and previous studies (11–15), we have sought to characterize biochemical determinants of a binary switch in human gonadogenesis (16).
Male development of therian mammals is (with rare exception (17, 18)) initiated by the stage- and lineage-specific expression of Sry (19), a gene contained within the sex-determining region of the Y chromosome (20). Sry encodes an architectural transcription factor (TF)3 whose expression in the embryonic gonadal ridge activates a developmental program leading to a wave of Sertoli cell differentiation and ultimately to testis formation (Fig. 1A) (21). Assignment of Sry as the testis-determining factor was demonstrated in transgenic mice (20) and extended to human embryogenesis through identification of diverse mutations in SRY associated with a distinct disorder of sex development (DSD) (22), designated Swyer syndrome (Fig. 1B). Such mutations cluster in SRY's high mobility group (HMG) box, a sequence-specific DNA-bending domain shared by a conserved family of TFs (designated SRY-related HMG box, Sox (23)). Most Swyer mutations arise de novo as meiotic errors in paternal spermatogenesis (green arrowheads in Fig. 1B) and are commonly associated with marked defects in specific DNA binding relative to wild-type (WT) SRY (15, 22, 24). The three-dimensional structure of a specific SRY HMG box-DNA complex (Fig. 1C) has provided a foundation for interpretation of many such mutations (25–27).
FIGURE 1.
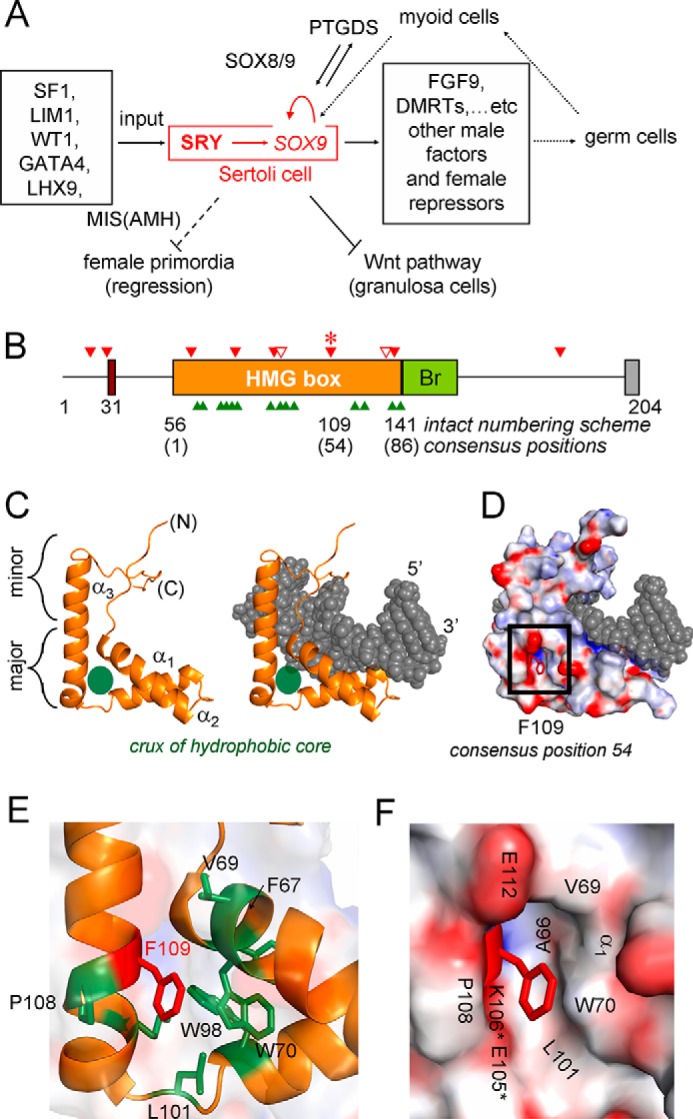
SRY-regulated gene-regulatory network and structure of human SRY HMG box. A, pathway of SRY-dependent testis determination. Red box highlights the central SRY → SOX9 axis. Genetic inputs are shown at left and outputs at right. In the differentiating gonadal ridge, SOX9 functions to activate a male-specific gene-regulatory network, effecting in turn a hormonal pathway of Müllerian-duct regression (dashed ⊥; Müllerian inhibiting substance MIS, also designated anti-Müllerian hormone or AMH) and inhibit granulosa cell fate (solid ⊥; Wnt pathway). B, domain organization; the central HMG box is highlighted in gold. N-terminal serine phosphorylation sites and putative C-terminal PDZ-binding motif are indicated by maroon and gray boxes, respectively; bridge domain is labeled Br. Triangles indicate sites of clinical mutation: green, de novo; solid red, inherited; and open red, mosaic father. C, ribbon model of the SRY HMG box bound to DNA (middle) and with DNA omitted (left). This L-shaped domain consists of major and minor wings; their confluence (green circle) contains a hydrophobic core underlying an angular DNA-bending surface. D, environment of Phe-109 (red stick; consensus residue 54) within space-filling model of SRY domain-DNA complex. The SRY surface is coded by electrostatic potential (negative in red; positive, blue); the DNA surface is shown in gray. Phe-109 (within black box) partially exposed in nonpolar crevice on back surface of the domain. E, expanded view of ribbon model showing side chains near Phe-109 in core: Trp-70, Trp-98, and Leu-101 (consensus positions 15, 43, and 46). Side chains of Phe-67 and Val-69 (consensus positions 12 and 14) buttress the domain's wedge-cantilever motif (Ile-68; see Fig. 9). F, expanded view of Phe-109 side chain (red stick) in crevice boxed in C. Asterisks indicate main-chain borders, whereas neighboring side chains are without asterisks. Coordinates for structural models were obtained from Protein Data Bank (PDB) code 1J46 (27).
This study has focused on a Swyer mutation of complementary structural and biological interest, F109S (consensus box position 54; Fig. 1D) (28). This mutation lies at the back surface of the HMG box; substitution of a large side chain by a smaller one would be predicted to create a destabilizing crevice (Fig. 1, E and F) (25, 26). First identified in a 46-chromosome XY adolescent female presenting with primary amenorrhea and gonadoblastoma in situ (28), the same mutation was found in her father, two brothers, and a paternal uncle. Such differences in phenotype (also known as “variable genetic penetrance”) are likely to reflect either autosomal background (29) and/or stochastic variation in gene expression (30). Analogous background-dependent XY sex reversal has been observed among strains of laboratory mice (31–33), highlighting the tenuous function of murine Sry at the threshold of developmental ambiguity (34).
The multiplicity of inherited Swyer mutations in human SRY (filled red arrowheads in Fig. 1B) presumably reflects the diversity of molecular mechanisms (beyond specific DNA binding and bending) by which a developmental switch may be perturbed in vivo. Patient-directed studies of such representative mutations (e.g., V60L and I90M at respective box positions 5 and 35 (22, 24)) have illuminated, for example, the contribution of SRY's nucleocytoplasmic shuttling (NCS) to the robust specification of human testicular development (11, 14, 35, 36). NCS may be a general requirement of SOX TFs coupled to their post-translational modification (37). In this context the properties of F109S SRY are particularly intriguing. Consensus position 54 is broadly conserved as an aromatic residue (Phe or Tyr (15)) throughout the metazoan superfamily of specific HMG boxes, and yet Phe-109 is not part of any of the following five signature features of human SRY: (i) its angular DNA-binding surface (26); (ii) its basic tail as accessory DNA-binding element (27) and kinetic clamp C-terminal to the HMG box (12); (iii) N-terminal bipartite nuclear localization signals (NLS) (38); (iv) C-terminal monopartite NLS (39); and (v) central nuclear export signal (NES) (40).
What does Phe-109 contribute to the structure and function of SRY, and why does Swyer mutation F109S SRY lead to variable male or female somatic phenotypes? To address these questions, our investigation had two parts. We first undertook biochemical and biophysical studies of the variant HMG box. These experiments demonstrated that the mutation is profoundly destabilizing, and yet its specific DNA-binding and DNA-bending properties are similar to those of the WT SRY domain. We next pursued cell biological studies of the WT and variant SRY (as epitope-tagged intact proteins introduced by transient transfection) in diverse rat and human cell lines. The latter studies employed two rodent and two human cell lines differing in tissue of origin and state of differentiation. Whereas all four lines share the universal ubiquitin-dependent (26S) and default (20S) proteasomal pathway of protein degradation (41–43), only one cell line, an XY lineage derived from the rat embryonic gonadal ridge just prior to onset of Sry expression and morphological differentiation (44), supports SRY-directed transcriptional activation of the male program (Fig. 1A) (21, 45). This ras-immortalized pre-Sertoli cell line (designated CH34 (44)) thus provides a model of the site and stage of endogenous Sry expression (13, 46). A control rat XY cell line (CH15) lacking pre-Sertoli markers (and unresponsive to transfected SRY) was also obtained from the embryonic bipotential ridge and presumably represents a non-Sertoli-related lineage (46). The two SRY-unresponsive human cell lines (HEK 293T and Hs1.TES) were respectively derived from the embryonic kidney (47) and fetal testis (Leydig cell lineage (48)).
Remarkably, F109S SRY underwent, irrespective of particular cellular context, enhanced polyubiquitination and accelerated proteasomal degradation, reducing the mean intracellular SRY concentration at a given level of transfected mRNA expression. In contrast, no significant changes were obtained in the in vitro susceptibility of the variant domain to degradation by the 20S core proteasome. In SRY-responsive CH34 cells, accelerated ubiquitin-associated degradation led to decreased occupancy of a target enhancer element (the testis-specific enhancer of Sox9 (TES) (49)) and in turn with attenuated transcriptional activation of this, the principal endogenous target gene in the program of testicular differentiation (red box in Fig. 1A) (14, 49). Treatment of the cells with chemical proteasome inhibitor MG132 equalized expression of WT and F109S SRY, leading in turn to a striking restoration of native TES enhancer occupancy and rescue of native Sox9 expression. Such subtle effects of the F109S substitution on the structure and function of SRY are in general accordance with past studies of inherited Swyer mutations unrelated in their respective molecular mechanisms of perturbation (13, 14).
Our results demonstrate that a conserved aromatic residue at the back surface of a specific HMG box functions to seal the hydrophobic core and thereby enhances thermodynamic stability and cellular lifetime. F109S and WT SRY differ in efficiency as substrates for polyubiquitination and therefore in proteasomal turnover but exhibit, on a per molecule basis, similar gene-regulatory properties. Together, these findings and their clinical correlation highlight enhanced ubiquitination and accelerated degradation of a master transcription factor as a molecular mechanism of impaired human sex determination. To our knowledge, this is the first observation in a metazoan that such a proteasome-based mechanism may impair the robustness of organogenesis leading to phenotypic variation within a family tree.
Results
Clinical Mutation Destabilizes the HMG Box
Thermal stabilities of the free WT and variant domains were assessed by circular dichroism (CD) (Fig. 2, A and B). The unfolding transition of the variant domain (red in Fig. 2A) was shifted toward lower temperatures (relative to WT) in the range 20–40 °C (black). Estimates of midpoint temperatures were as follows: (variant) 28(±0.5)°C versus (WT) 41(±0.5)°C (vertical arrows in derivative plots, Fig. 2B, and Table 1). Reduced thermal stability was associated with increased Trp fluorescence emission at both 15 and 37 °C (respective filled and open circles in Fig. 2C), suggesting that the core of the variant domain is less stably packed and so more exposed to solvent water. These emission spectra contain contributions from two conserved Trp residues in the core of the major wing (Trp-70 and Trp-98; box positions 15 and 43 as shown in green in Fig. 3E) and an additional non-conserved Trp on the protein surface (Trp-107; box position 53). The similar emission intensities at the two temperatures is likely to reflect offsetting effects of thermal unfolding (enhancing the fluorescence of core Trp side chains) and solvent quenching (more effective at the higher temperature).
FIGURE 2.

Stability of SRY domains. A, thermal unfolding transitions as monitored by CD at 222 nm: black, WT human; red, variant human; and blue, WT murine. B, derivatives of thermal unfolding curves. Arrows indicate midpoint temperatures (Tm in Table 1); the color code is as in A. C, intrinsic Trp fluorescence at 15 °C (filled circles) and 37 °C (open circles): solid black and open black circles, WT human; solid red and open red circles, variant human; and solid blue and open blue circles, WT murine. At a given temperature, decreased emission (due to increased core quenching) indicates more efficient desolvation of the conserved indole rings of Trp-70 and Trp-98 (box positions 15 and 43; Fig. 3E). The spectrum of each domain likewise exhibits a blue shift in emission maximum at the lower temperature (relative to its spectrum at the higher temperature). The human domain contains an additional non-conserved Trp exposed on its surface (Trp-107; box position 52) that is absent in the murine domain. D–F, far-UV CD spectra of the three domains are similar at 4 °C, but their respective α-helical signatures exhibit distinct patterns of attenuation at 25 and 37 °C. Relative to the WT human domain (black), loss of structure is more marked in the variant domain (red) than in the murine domain (blue) in accordance with their respective thermal unfolding transitions (A and B). G–I, guanidine (Gu·HCl)-induced unfolding at 4, 25, and 37 °C as monitored by CD at 222 nm. The variant and murine domains each exhibit greater sensitivity to chemical denaturation than does WT SRY (color code as in D–F). Estimates of Cmid and ΔGu were obtained at 4 °C by application of a two-state model (Table 1). Use of this model at the higher temperatures was limited by possible non-two-state behavior.
TABLE 1.
Biophysical and biochemical characterization of WT and variant SRY
| Domain | 4 °C |
25 °C |
Tmc free | Tm bound | Kdd (15 °C) | Kd (37 °C) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmida | ma,b | ΔGua | Cmida | ma,b | ΔGua | |||||
| WT human | 2.2 | 1.4 | 3.1 | 1.2 | 1.2 | 1.4 | 41 | 51 | 14 ± 2 | 22 ± 6 |
| F109S human | 1.1 | 0.9 | 1.0 | NDe | ND | ND | 28 | 45 | 12 ± 3 | 26 ± 6 |
| WT murine | 1.5 | 1.7 | 2.5 | 0.6 | 1.5 | 0.9 | 33 | 53 | 11 ± 3f | 22 ± 7f |
a Two-state model parameters: ΔGu (kcal/mol), Cmid (M), and m (kcal mol−1 m−1); errors are for each sample ±0.1 kcal/mol (ΔGu), ±0.1 m (Cmid), and ±0.03 kcal mol−1 m−1 (m).
b The m-value (slope d(ΔG)/d(M)) correlates with extent of hydrophobic surfaces exposed on denaturation.
c Tm is the apparent midpoint of thermal unfolding of the free domains or equimolar protein-DNA complex (bound) at 25 μm as monitored by CD (15).
d Dissociation constants (Kd) pertain to the equilibrium between the specific SRY domain and a 15-bp DNA site containing a consensus SRY target site (5′-ATTGTT-3′ and complement): units are in nm as determined by FRET-based titrations (14).
e ND indicates not determined due to apparent non-two state unfolding behavior.
f Values are as reported previously (14).
FIGURE 3.

Temperature dependence of 1H-15N HSQC spectra and DNA-dependent folding. A and B, 1H-15N HSQC spectra of wild-type (A) and F109S variant (B) at 15 °C (blue peaks), 25 °C (gold), and 35 °C (green). For wild-type SRY, three cross-peaks corresponding each Trp side-chain indole NH were observed at 15 and 25 °C, respectively, whereas multiple sets of Trp side-chain cross-peaks were observed at 35 °C, indicating that wild-type Trp residues exist in multiple local conformation at 35 °C. C and D, for F109S mutant, multiple sets of Trp side-chain cross-peaks were observed even at 15 °C, also the main chain amide cross-peaks crowded in the center region (1HN chemical shift range in 7.5–8.5 ppm) at 25 °C (C); and the HSQC spectral pattern is similar as that of WT in 5 m urea (D), suggesting that F109S mutant exists in multiple conformation at lower temperature (15 °C) and partially random-coiled conformation at 35 °C. The results are consistent with the temperature dependence of 1D 1H NMR spectra of wild-type SRY and F109S mutant. E, ribbon model of major wing of the WT human SRY HMG box; helices α1–α3 are as labeled. Invariant Trp residues of the core (Trp-70 and Trp-98; box positions 15 and 43) are shown in green, and Phe-109 (box position 54) is in red. An additional non-conserved Trp shown in gray (Trp-107; box position 52) flexibly projects into solvent from the back surface of helix 3. The minor wing (data not shown) would extend to the top of the page. F and G, 1H-15N HSQC side-chain indole NH resonances of the free WT (F) and F109S (G) domains. Unlike the WT spectrum (which contains three cross-peaks corresponding to the three Trp side chains; assignments as labeled), the variant domain exhibits one cross-peak for Trp-107 but two or more cross-peaks for each of the core Trp side chains (red). H and I, 1H-15N HSQC side-chain indole NH resonances of the DNA-bound WT (H) and F109S (I) domains. The variant spectrum (like the WT spectrum) exhibits three cross-peaks, indicating DNA-dependent stabilization of a unique ground state. Presumptive assignments are as indicated. Black squares indicate positions of the WT resonances. Changes in 1H and 15N chemical shifts (arrows) may reflect the absence of the WT Phe-109 aromatic ring current and/or subtle reorganization of the core due to a residual packing defect around the variant Ser-109 side chain. Spectra were obtained at 25 °C at a proton frequency of 700 MHz; the proteins and their DNA complexes were made 0.5 mm in 50 mm KCl and 10 mm potassium phosphate (pH 7.4). Coordinates were obtained from PDB code 1J46 (27).
Comparison of far-UV CD spectra suggested that the variant domain achieves a native-like fold at 4 °C (Fig. 2D) with progressive loss of α-helical content at 25 and 37 °C (Fig. 2, E and F). Respective thermodynamic stabilities at these three temperatures were probed by chemical denaturation (Fig. 2, G–I). At each temperature, the variant domain exhibited greater sensitivity to guanidine-induced loss of structure than did the WT domain. Application of a two-state model (12, 50) at 4 °C enabled the guanidine concentration at the midpoint of unfolding (Cmid) to be estimated as 1.1(±0.1) m (variant) and 2.2(±0.1) m (WT; Table 1). Inferred thermodynamic stabilities (ΔGu) were 1.0(±0.1) (variant) and 3.1(±0.1) kcal/mol (WT); the variant's loss of stability (ΔΔGu) was thus 2.1(±0.2) kcal/mol. Respective m values (denaturant dependence of free energies of unfolding) were 0.9(±0.03) and 1.4(±0.03) kcal/mol/m; the reduced m value of the variant domain suggested that in the absence of guanidine-HCl its nonpolar surfaces are less efficiently desolvated than those of the WT domain (and so undergo a smaller further change in solvation on denaturation) in accordance with their contrasting Trp fluorescence emission spectra (above).
The reduced stability of the F109S HMG box of human SRY resembled (in exaggerated form) the WT HMG box of murine Sry (shown in blue in Fig. 2). The midpoint unfolding temperature of the murine domain (33(±0.5)°C; Table 1) was thus intermediate between the WT and variant human domains (41 and 28 °C, respectively). Similar intermediate trends were observed in temperature-dependent α-helical CD signatures (Fig. 2, D–F) and in sensitivity to chemical denaturation (Fig. 2, G–I). At 4 °C, the thermodynamic stability of the murine domain (ΔGu) and Cmid guanidine concentration was likewise intermediate between the WT and variant human domains (Table 1). These findings are in general accordance with an analogy proposed between ISS alleles of human SRY and WT murine Sry by Eicher and co-workers (51) (see “Discussion”).
Whereas comparative two-state modeling of protein denaturation was feasible at 4 °C, unfolding of the variant domain at 25 and 37 °C lacked significant cooperativity and so could not be fitted to this model (ND in Table 1). In accordance with these CD studies, 2D 15N-1H “fingerprint” NMR spectra of the variant domain at 15, 25, and 35 °C exhibited less marked chemical shift dispersion than the corresponding spectra of the WT domain (Fig. 3, A–C). Furthermore, the variant's fingerprint NMR spectrum at 35 °C resembles the 15N-1H fingerprint of the WT domain on denaturation in 5 m urea (Fig. 3D). Evidence that the variant domain exhibits two or more conformations at 25 °C was provided by observation of at least five indole 15N-1H cross-peaks for the variant domain (Fig. 3G) rather than the expected three as in the WT spectrum (Fig. 3F). Upon specific DNA binding, the expected three cross-peaks were observed for both WT and variant (Fig. 3, H and I), providing evidence of a single bound conformation.
Clinical Mutation Preserves Native-like Specific DNA-binding and DNA-bending Properties
Despite its marked instability, the variant domain retains specific DNA binding and bending. WT and variant dissociation constants (Kd), determined using a fluorescence resonance energy transfer (FRET)-based equilibrium assay (13), were indistinguishable at 15 and 37 °C (Table 1 and Fig. 4A). Stopped-flow FRET studies of protein-DNA dissociation nonetheless demonstrated that the mutation caused less than a 2-fold increase in rate constant (koff) at each of four temperatures tested (6, 15, 25, and 37 °C) (Table 2 and Fig. 4B). That the WT and variant domains exhibited similar specific protein-DNA affinities in turn implied that the variant domain exhibits a compensating increase in association rate (kon). Conversely, these findings suggest that pre-organized structure within the WT domain imposes a kinetic barrier to specific DNA binding and release.
FIGURE 4.

Specific DNA affinity and kinetic stability of the protein-DNA complexes. A, FRET-based dissociation constant (Kd) graphs at the temperatures reported in Table 1. The variant domains are in red (trace and axis label). B, stopped-flow FRET traces to determine dissociation rate constant (koff) values reported in Table 2; WT traces are in black and Phe-109 in red.
TABLE 2.
Dissociation rates of domain-DNA complexes
| Domain complex | koff (s−1) 37 °C lifetimea | koff (s−1) 25 °C lifetimea | koff (s−1) 15 °C lifetimea | koff (s−1) 6 °C lifetimea |
|---|---|---|---|---|
| ms | ms | ms | ms | |
| WTb | 0.120 ± 0.001 8.3 (±0.1) × 103 | 0.055 ± 0.001 18.2 (±0.3) × 103 | 0.037 ± 0.001 27.0 (±0.8) × 103 | 0.033 ± 0.001 30.3 (±0.9) × 103 |
| F109c | 0.140 ± 0.001 7.1 (±0.1) × 103 | 0.077 ± 0.001 13.0 (±0.02) × 103 | 0.050 ± 0.001 20.0 (±0.4) × 103 | 0.045 ± 0.001 22.2 (±0.1) × 103 |
a Respective dissociation rate constants (koff) were measured by stopped-flow FRET; lifetimes were determined according to (1/koff).
b The WT domain consistent of residues 56–141 of human SRY, spanning the HMG box inclusive of the basic tail.
The WT and variant domains exhibited indistinguishable specific DNA-bending properties. Preservation of native specific DNA bending was demonstrated by a FRET probe of end-to-end distances in a 15-bp DNA duplex at 15 °C (inset in Fig. 5A) and corroborated by permutation gel electrophoresis at 4 °C (Fig. 5, B and C). The decrease in donor emission at 520 nm (fluorescein) corresponded to an enhancement in FRET efficiency (tetramethylrhodamine). Previous studies of the WT SRY domain have shown that this enhancement corresponds to a reduction in mean end-to-end distance from 61 Å (free DNA) to 54 Å (bound DNA) (12, 13) in broad agreement with the permutation gel electrophoresis estimated bend angle of 80 °C under these conditions (Fig. 5D).
FIGURE 5.
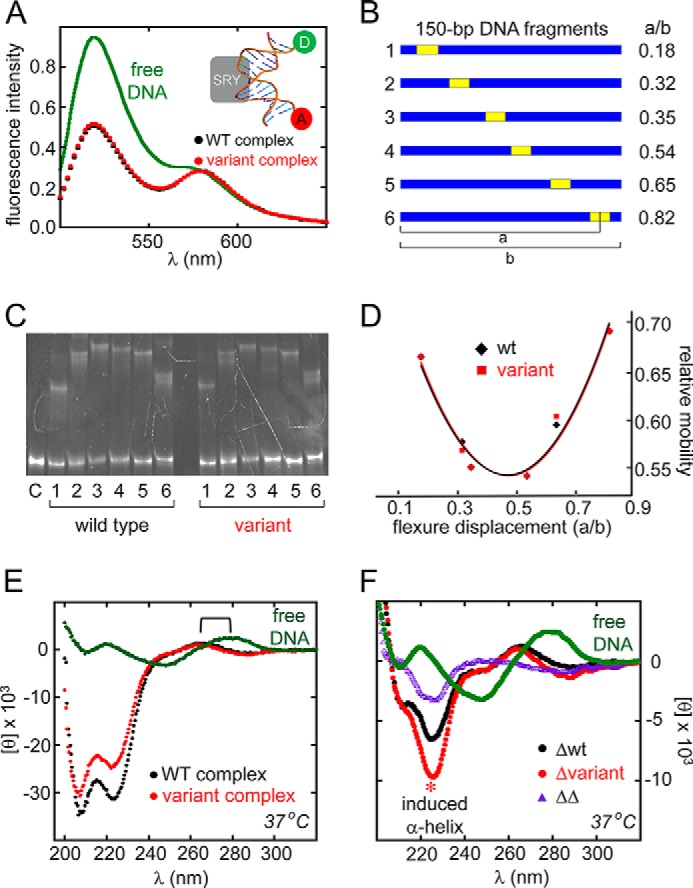
Specific DNA bending and mutual induced fit. A, FRET assay of protein-directed DNA bending. Inset, schematic model of 15-bp DNA site containing a 5′-fluoroscein label at one end (donor; D) and 5′-tetramethylrhodamine label at the other (acceptor; A). Relative to free (A, D)-labeled DNA, binding of the WT (black) or variant (red) SRY domains led to similar enhancements of FRET efficiency as indicated by decreased donor emission at 520 nm. B–D, permutation gel electrophoresis at 4 °C. B, 150-bp duplex DNA fragments, each containing an SRY target site (5′-ATTGTT-3′ and complement) at indicated position. Flexure displacements are at right. C, gel showing dependence of electrophoretic mobility of WT or variant protein-DNA complexes on position of DNA target site within probe. Lanes 1–6 in each set refer to DNA probes in B. Lane C at far left indicates free DNA probe. D, plot of electrophoretic mobilities (vertical axis) versus flexure displacement (horizontal axis). The similar patterns imply indistinguishable DNA bend angles in WT and variant complexes (∠Δθ∠ <1°). E and F, CD studies of mutual induced fit in SRY-DNA complex at 37 °C. E, CD spectra of the free DNA (green), WT complex (black), and variant complex (red). Bracket indicates blue-shift of DNA band reminiscent of classical B → A transition. F, CD difference spectra relative to spectrum of free DNA (green). Black and red spectra represent respective difference WT and variant spectra, obtained by subtracting the spectra of the free domain and free DNA from the spectrum of the complex (a buffer control was also added). Purple spectrum represents the difference between the spectra of the WT complex and variant complex (or equivalently, between the red and black curves). Asterisk highlights marked stabilization of α-helical structure in variant on specific DNA binding.
Variant Domain-DNA Complex Exhibits Native-like Structure
Far-UV CD spectra of the variant domain-DNA complex (containing a 15-bp DNA duplex with central 5′-ATTGTT target site) at low temperatures closely resembled spectra of the WT complex. At 37 °C, restoration of DNA-dependent α-helix content of the variant complex was substantial but incomplete (red and black circles in Fig. 5E). Calculated difference spectra indicated that the extent of DNA-induced α-helical structure in the variant complex was greater than in the WT complex (red asterisk in Fig. 5F), reflecting the more complete thermal unfolding of the unbound variant domain at 37 °C (see above) and its partial stabilization on specific DNA binding. The greater extent of α-helical induction at 37 °C in the variant complex is highlighted by a double-difference spectrum (purple triangles in Fig. 5F).
In the near-UV region of the CD spectra (wherein the DNA contribution predominates) binding of the WT or variant domain was associated with a blue-shift in spectral maximum (from 282 (±0.5) nm (free DNA) to 269 (±0.5) nm (bound DNA)). This 13-nm shift (bracket in Fig. 5E) is reminiscent of a classical B→A double-helical transition (52) in accordance with the A-like DNA conformation observed in the solution structure of the complex (26, 27). A bp-specific view of the bound DNA conformation was provided by 1H NMR spectroscopy (Fig. 6). Relative to the spectrum of the free DNA site (15 bp; Fig. 6A), the chemical shifts of the DNA imino protons (guanosine N1-H and thymidine N3-H; mediating Watson-Crick base pairing) exhibited marked changes on specific DNA binding (both upfield and downfield; Fig. 6, B–E). The WT and variant domain-DNA complexes are each in slow exchange on the NMR time scale (arrows in Fig. 6B) in accordance with the stopped-flow FRET studies above. The pattern of complexation shifts was in each case similar, indicating a general correspondence of bound DNA structures. Subtle differences between the complexes were observed at bp 6–8 (green box at top in Fig. 6), which adjoin the major wing of the WT HMG box-DNA complex (25, 27) and hence presumed site of mutational perturbation in the core. By contrast, the large complexation shifts at positions 10 and 11 (adjoining the minor wing of the HMG box) were essentially identical in the two complexes (violet in Fig. 6).
FIGURE 6.

1H NMR protein-DNA titration. A, imino spectrum of free 15-bp DNA site (downfield guanosine N1H and thymidine N3H resonances). Assignments are as indicated (numbering scheme; top). Base pairs contacted by SRY major wing are labeled in green; contacted by its minor wing (including tail) are labeled in violet (boxes at top). B–D, spectra obtained on addition of successive aliquots of the variant SRY domain; protein-DNA stoichiometries are given at right. Arrows in B indicate complex-specific imino resonances in slow exchange on NMR time scale. E, WT imino spectrum. Vertical segments between E and D indicate small differences in chemical shifts of thymidine N3H resonances at positions 6–8; imino 1H NMR chemical shifts are otherwise similar. Horizontal bracket at top site of side-chain insertion between bp 8 and 9 by “cantilever” residue Ile-68 (triangle within DNA sequence at top) as demonstrated by intermolecular NOE contacts in the WT and variant domain-DNA complexes (see Fig. 7D).
The 1H NMR signature of DNA-dependent minor wing (Fig. 7A) stabilization was essentially identical in the 2D NOESY spectra of the WT and variant complexes (Fig. 7B). This signature is provided by the upfield ring-current shift of the γ1-methyl resonance of the valine (near 0.0 ppm) due to its inter-residue NOE-associated packing within an “aromatic box” comprising His-120, Tyr-124, and Tyr-127 and consensus positions 65, 69, and 72 (arrow and horizontal brackets in Fig. 7B). Although Val-60 does not contact the DNA (violet side chain in Fig. 7A), Tyr-127 projects near a solvated DNA interface.
FIGURE 7.

1H NMR studies of the WT and variant SRY domain-DNA complexes. A, ribbon model of DNA-stabilized mini-core of the minor wing (upper box; Val-60 is shown in violet and Tyr-124 and Tyr-127 in gray) and key structural relationships in the major wing (bottom; Phe-109 in red and Trp-98 and Leu-101 in green). The trajectory of the DNA main chain is shown in blue. B, NMR features of the DNA-stabilized minor wing, complexation shifts and NOE contacts reflecting packing of the γ1-methyl group of Val-60 within the aromatic rings of His-120, Tyr-124, and Tyr-127, are essentially identical in the two complexes. A corresponding NMR signature of the DNA-stabilized major wing, upfield shift of the Leu-101 methyl resonances and NOEs to the Trp-98, is also similar. Box indicates NOEs between δ-methyl resonance of Ile-68 and adenine H2 protons of bp 8 and 9 in an expanded DNA minor groove. C, model of cantilever side chain Ile-68 (box position 13) inserting between successive AT bps in DNA target site (5′-ATTGTT-3 and complement; insertion site in bold). Portions of helices α1, α2, and α3 are shown as gold ribbons. DNA bp 8 and 9 are shown as sticks with following color code: gray, carbon; orange, phosphorus; red, oxygen; and blue, nitrogen. D, NOEs between the upfield-shifted δ-methyl resonance of Ile-68 and flanking imino protons of thymidines at bp 8 and 9. E, 1D 1H NMR aliphatic spectra of free domains (left) and specific DNA complexes (right). Overlay of 2D 1H-NOESY spectra of the WT domain-DNA complex (black) and variant complex (red). Coordinates for structural models were obtained from PDB code 1J46 (27).
Despite their subtle differences in DNA imino 1H NMR chemical shifts adjoining the major wing of the HMG box, the WT and variant domain-DNA complexes exhibited similar NMR signatures of partial side chain intercalation by “cantilever” residue Ile-68 (consensus box position 13). This aliphatic side chain inserts between successive AT base pairs at positions 8 and 9 (Fig. 7, C and D) (26, 44). Such insertion leads to a large upfield ring-current shift in the δ-methyl resonance of Ile-68 (53), which is identical in the 1H NMR spectra of the WT and variant complexes (Fig. 7, D and E). This upfield methyl resonance exhibits prominent intermolecular nuclear Overhauser effects (NOEs) with the flanking imino resonances of thymidines 8 and 9 (Fig. 7D) and to the C2-H resonances of adenines 8 and 9 in the expanded DNA minor groove (box in Fig. 7B). Evidence for the restoration of a native-like major-wing core adjoining the site of mutation was provided by the upfield 1H NMR chemical shift of the δ1-methyl resonance of Leu-101 (box position 46) near 0.1 ppm (Fig. 7E, right) and its preserved NOE to the C2-H proton of Trp-98 (box position 43; green-labeled cross-peaks in Fig. 7B and green side chains in Fig. 7A).
In contrast to the above similarities in the 1H NMR NOESY spectra of the DNA-bound domains, subtle but illuminating differences were observed in the respective 1H NMR spectra of the free domains. Whereas the respective δ1-methyl resonances of Leu-101 in the WT and variant free domains exhibit an attenuated upfield shift (Fig. 7E, left), reflecting greater conformational flexibility than in the DNA-bound domains, this methyl resonance is broader and closer to its random-coil value in the spectrum of the variant (upper panel of Fig. 7E, left) than in the spectrum of the WT domain (lower panel of Fig. 7E, left), presumably due to intermediate exchange between folded and unfolded states at 25 °C in accordance with the above CD studies.
Variant SRY Undergoes Accelerated Proteasomal Degradation in Diverse Cell Lines
To test whether the instability of the F109S HMG box (as an isolated domain) and its marked loss of structure at 37 °C (as probed by CD) affected the mean intracellular protein concentration of full-length F109S SRY relative to WT SRY, respective steady-state levels of expression 24 h following transient transfection were probed by SDS-PAGE followed by anti-HA WB (Fig. 8A). In each of the four cell lines employed (CH15, CH34, HEK 293T, and Hs1.TES), expression of the variant was reduced. The extent of reduction (relative to WT) was 3-fold in the rat cell lines and 6-fold in the human cell lines. In these studies 1× transient transfection was employed (i.e. 1 μg of SRY-encoded plasmid per 106 cells without addition of the parent plasmid; see under “Experimental Procedures”).
FIGURE 8.

F109S SRY exhibits accelerated proteasomal degradation irrespective of cellular context. A, upper box, expression of epitope-tagged WT SRY or variant SRY (v) in four cell lines as indicated. In each case expression of the variant is reduced. Gel represents a WB with anti-HA antiserum in single-well assays. A loading control was provided by α-tubulin (lower box). B, upper box, addition of proteasome inhibitor MG132 in each cell line rescue expression of epitope-tagged F109S SRY to a level similar to that of WT SRY in CH34 cells in the absence of MG132 (left-hand lane). WB employed anti-HA antiserum; loading-control α-tubulin is shown in lower box. C, cycloheximide assays in four cell lines as indicated. WBs exhibited similar time courses relative to α-tubulin. D, semilog plot of SRY expression as a function of time following cycloheximide arrest of translation: upper group, WT SRY; and lower group, F109S SRY (vertical brackets at right). Symbols are as defined at lower left: green ■, CH34 rat cells; ●, CH15 rat cells; ♦, Hs1.TES human cells; and ▴, HEK 293T human cells. Green line highlights data derived from CH34 cells as SRY-responsive model of the pre-Sertoli cell lineage (46). E, histogram showing half-lives of WT SRY (left) and F109S SRY (right) in each cell line. Statistical comparisons: *, Wilcox p values <0.05. Relative to α-tubulin, no significant differences were observed in the intracellular half-lives of WT SRY protein among the four cell lines and likewise for F109S SRY in the same cell lines. n.s., not significant.
To test whether the decreased level of F109S SRY at the protein level was due to proteasomal degradation, chemical proteasome inhibitor MG132 was added 12 h after transient transfection, and the cells harvested 12 h later. In each cell line addition of MG132 resulted in enhancement of the F109S SRY band (Fig. 8B) to a level similar to that of WT SRY in the absence of MG132 (at left in Fig. 8B); addition of MG132 did not enhance WT protein expression. Control studies of subcellular localization (see below) indicated that the differential rates of proteasomal degradation were not due to mislocalization of the variant SRY in the cytoplasm as had been observed in unrelated proteins (54). Control studies of the mRNAs encoding HA-tagged F109S or WT SRY indicated in each cell line that the mutation did not lead to reduced levels of mRNA (which could in principle have resulted from decreased efficiency of transcription or accelerated mRNA decay in the presence of the mutant codon (55)).
That similar decrements in cellular accumulation of F109S SRY (relative to WT) were observed in the four unrelated cellular contexts provided evidence that its mechanism was not dependent on species (rat or human), cell lineage (pre-Sertoli, embryonic gonadal stroma, fetal kidney, or fetal testis of Leydig origin), or stage of development (rat embryonic day E14.5 (46), embryo or human mid-trimester fetus). This conclusion was corroborated through analysis of respective cellular lifetimes following general translational arrest by addition of cycloheximide (56). In each cell line, the apparent lifetime of F109S SRY was shorter than that of WT SRY (Fig. 8C). Quantitation of these bands (relative to α-tubulin loading controls) provided a time course of degradation (semilog plot in Fig. 8D) yielding estimates of apparent cellular lifetimes (half-lives in Fig. 8E). The relative half-life of F109S SRY in CH34 cells observed here (2.8 ± 0.1 h) was reduced relative to WT SRY (5.6 ± 0.4 h) but greater than that implied by a previous study of a clinical SRY variant (de novo mutation W70L; half-life of 2.1 ± 0.1 h) whose HMG box was found to be almost completely unfolded at 37 °C (15).
F109S SRY Exhibited Enhanced Polyubiquitination
To probe the cellular mechanism of accelerated proteasomal degradation of F109S SRY (Fig. 9A), we investigated the extent of its ubiquitination (relative to WT SRY) in each of the above four cell lines. This post-translational modification provides a general signal targeting protein to the 26S proteasome in eukaryotic cells (57). Our protocol exploited the HA tag to immunoprecipitate the transfected proteins. I68A SRY was included to test whether loss of specific DNA binding might influence the extent of ubiquitination. The immunoprecipitated proteins were resolved by SDS-PAGE and probed by an anti-Ub monoclonal antiserum (Fig. 9, B and C). Whereas only trace ubiquitination could be observed in the absence of MG132 (lanes 1–4 in Fig. 9, B and C), as expected treatment with MG132 led to accumulation of multiple ubiquitinated species (lanes 5–8). In each cell line, F109S SRY exhibited enhanced mono- and polyubiquitination (respective lanes 7 in Fig. 9, B and C). Extent of ubiquitination of I68A SRY was similar to that of WT SRY. Quantitation of these results (using the bands contained within the dashed red boxes in the shortest film exposures; Fig. 9, B and C) is shown by the histogram in Fig. 9D (see also Table 3). The fold-increase of ubiquitination of F109S SRY depended on the cell line within the approximate range 2–3 (Table 3). In these assays input- and general loading controls were, respectively, provided by total accumulation of SRY variants (as detected with a polyclonal anti-HA antiserum) and monoclonal blotting of α-tubulin (lower two panels in Fig. 9, B and C). These findings suggest that the decreased cellular half-lives of epitope-tagged F109S SRY and W70L SRY (15) (relative to WT SRY) reflect enhanced modification of the variant proteins by one or more E3 ligases (58) in association with thermodynamic destabilization and segmental unfolding.
FIGURE 9.

F109S promotes ubiquitination but not default 20S proteolysis. A, pathway of ubiquitin-dependent 26S proteasomal degradation (42, 80, 82). Color code: azure, Ub; multicolor complex, 19S particle; blue and yellow, subunits of the 20S core proteasome. The protein substrate and its peptide fragments are shown in brown. B and C, representative anti-Ub Western blots pertaining to rat cell lines CH34 and CH15 (B) and human cell lines HEK 293T and Hs1.TES (C). Each panel contains the following: lanes 1 and 5, empty control; lanes 2 and 6, transient transfection of WT SRY; lanes 3 and 7, transient transfection of F109S SRY; lanes 4 and 8, transient transfection of control I68A SRY. WBs in lanes 1–4 were obtained in the absence of MG132; lanes 5–8 were obtained in the presence of MG132. In each case initial immunoprecipitation (IP) was effected by anti-HA beads. Three exposures are shown from top; below are shown anti-HA WBs as input controls (labeled IB: HA) and α-tubulin loading controls (bottom). Molecular weight markers are as shown at upper and lower left. Signal intensities were measured within red dashed boxes. D, histogram depicting relative extent of ubiquitination on MG132 treatment in each cell line: open bars, WT HA-tagged SRY; black bars, F109S HA-tagged SRY; and gray bars, I68A HA-tagged SRY. At bottom is given the number of biological replicates in each set. Statistical comparisons: * and **, Wilcox p values <0.05 and <0.01. E, pathway of default 20S proteasomal degradation (ubiquitin- and ATP-independent) (59, 80). F, representative Coomassie-stained SDS-polyacrylamide gels monitoring 20S-mediated core proteasomal degradation of isolated SRY HMG boxes (arrow at right; 86 residues) as a function of time: upper panel, WT domain; lower panel, F109S domain. G, quantitation of SDS-PAGE band intensity demonstrated no significant difference in initial rates of degradation. SDS-PAGE assays were conducted in triplicate. A and E were adapted from Ref. 80.
TABLE 3.
Fold-differences in extent of ubiquitination as unmasked by MG132 treatment
| Cell model | Fold difference of ubiquitinationa | S.D. | p valueb |
|---|---|---|---|
| CH34 | 2.35 | ±0.19 | 0.02 |
| CH15 | 2.47 | ±0.23 | 0.04 |
| HEK 293T | 3.19 | ±0.56 | 0.007 |
| Hs1.TES | 1.83 | ±0.09 | 0.03 |
a Ubiquitinated WT SRY in presence of MG132 was defined as base line (1.00) and the fold difference was between band intensity of ubiquitinated F109S and ubiquitinated WT SRY.
b p values were tested by band intensity data sets from WT and F109S SRY with MG132 treatment.
Destabilization of the SRY HMG Box by F109S Did Not Affect 20S Proteasomal Degradation
Short-lived proteins in eukaryotic cells may be degraded by a ubiquitin-independent default pathway through the 20S core proteasome (Fig. 9E) (59). Because F109S perturbs the structure and stability of the free SRY HMG box, we sought to test whether the variant domain was a more efficient substrate for ATP-independent 20S proteasomal degradation in vitro. Assays were conducted at 37 °C (to capture the differential thermal unfolding of the SRY domains under physiological conditions; see Fig. 2 above) at a SRY/core proteasome molar stoichiometry of 3000:1. Under these conditions no significant differences were observed in the rate of 20S degradation between the F109S and WT domains (Fig. 9, F and G). It is possible that their similar susceptibilities to default 20S degradation in vitro reflect the baseline partial unfolding of even the WT domain at 37 °C as demonstrated by the above CD studies (Fig. 2).
Variant SRY Exhibits Reduced Transcriptional Activity in Proportion to the Reduced Mean Intracellular Concentration of the TF
Gene regulatory activity of the WT or variant SRY proteins were evaluated by quantitative reverse-transcriptase polymerase chain reaction (qPCR)-based measurement of endogenous Sox9 mRNA accumulation following their transient transfection (14). The studies were performed at a series of expression-plasmid dilutions, designed to explore a broad range of intracellular SRY concentrations. Whereas standard transient transfection with the undiluted WT plasmid (“1×”) gave rise to a mean protein expression level of 7 × 105 molecules per cell, transient transfection of 50× dilution with the empty parent plasmid (yielding the same total DNA dose per cell) gave rise to 2 × 103 mean molecules per cell. By contrast, 1× transfection of the F109S SRY construct led to mean expression of only 2 × 103 molecules per cell. Systematic characterization of the relationship between plasmid dilution and SRY expression level is provided in Table 4. This experimental maneuver thus enabled adjustment of the transfected TF concentration either within the physiological range (102–104 molecules per cell; Ref 60) or under conditions of marked overexpression (105–106 molecules per cell).
TABLE 4.
Effect of plasmid dose on epitope-tagged SRY expression in rat CH34 cells
| SRY-encoded plasmid dilution | WT SRY |
F109S SRY |
F109S w/MG132a |
||||||
|---|---|---|---|---|---|---|---|---|---|
| 1× | 10× | 50× | 1× | 10× | 50× | 1× | 10× | 50× | |
| Relative SRY signal (on gel) | 6.7 | 2.6 | 1.7 | 2.1 | 1 | 6.1 | 2.8 | 1.9 | |
| Loading protein dilution | 100× | 100× | 1× | 1× | 1× | 1× | 100× | 100× | 1× |
| Relative SRY signal (normalized) | 670 | 260 | 1.7 | 2.1 | 1 | 0.15 | 610 | 280 | 1.9 |
| Molecular abundance of SRYb | 7 × 105 | 3 × 105 | ∼1900 | ∼2200 | ∼1100 | ∼170 | 6 × 105 | 3 × 105 | ∼2000 |
Transcriptional analysis of transfected SRY in the four cell lines required prior characterization of the chromatin state pertaining to its principal endogenous autosomal target gene, Sox9 in rat (chromosome 10) and ortholog SOX9 in human (chromosome 17). To this end, representative histone marks were analyzed in the TESCO element based on side-chain methylation of conserved lysine residues in the N-terminal arm of histone H3 (61). H3 modifications associated with active chromatin (or chromatin accessible to transcriptional activation) were provided by mono-, di-, and tri-methylation of Lys-4 (left-hand panel of Fig. 10A; designated me1 (red), me2 (blue), and me3 (green)), mono-methylation of Lys-9 (middle panel of Fig. 10A), and mono-methylation of Lys-27 (right-hand panel of Fig. 10A). H3 modifications associated with inactive chromatin or chromatin inaccessible to transcriptional activation were provided by di- and tri-methylation of Lys-9 (middle panel of Fig. 10A), and tri-methylation of Lys-27 (right-hand panel of Fig. 10A). Such characterization of orthologous TESCO elements indicated that CH34 cells exhibited activating H3 marks whereas the other three cell lines exhibited repressive H3 marks.
FIGURE 10.

Epigenetic features of the Sox9 core enhancer (TESCO) correlate with SRY occupancy and specific transcriptional activity. A, histone marks in N-terminal arm of histone 3 (61): left, mono-, di-, and tri-methylation of Lys-4 (modifications me1, me2, or me3 in H3K4; shown in red, blue, and green, respectively). Fold-enrichment in TESCO (ChIP primer set a; see “Experimental Procedures”) was evaluated in untransfected cell lines CH34, CH15, Hs1.TES, and HEK 293T; middle, corresponding ChIP analysis of mono-, di-, and tri-methylation of Lys-9; right, ChIP analysis of mono- and tri-methylation of Lys-27. CH34 cells exhibited activating marks at H3K and H3K9 and reduction in repressive mark me3 at H3K9; the other three cell lines exhibited attenuated activating marks and repressive marks at H3K27. B, TESCO occupancy by WT HA-tagged SRY was selectively observed in CH34 cells (left-hand lane of gel with relative quantitation in histogram at right). Transfection conditions were “1×.” Primer sets a, b, and c were as defined by Sekido and Lovell-Badge (49) as homologous sets 4, 6, and 8 in mouse TES. Set b provided a negative control due to absence of specific SRY-binding sites. C, comparison of the relative TESCO occupancies of WT SRY and F109S SRY (variant, v) in CH34 cells. The variant exhibited 2-fold reduction in enhancer occupancy in the absence of MG132 but native occupancy on rescue of protein accumulation by MG132. Transfection conditions were 1×. Extent of attenuation in TESCO binding by F109S SRY is less marked under these conditions than its fold reduction in protein accumulation, reflecting baseline overexpression WT SRY under 1× transfection conditions. A negative control was provided by I68A SRY, which contains a substitution that blocks specific DNA binding (62). D, SRY-dependent transcriptional activation of Sox9 in CH34 cells. F109S SRY exhibited a 2-fold attenuation of Sox9 mRNA accumulation under both 1× and 50× transfection conditions (left- and right-hand histograms). This defect was mitigated by MG132 (blue labels). I68A SRY provided a negative control. E, analysis of SRY-stimulated Sox9 transcriptional activation at equal levels of expression of WT SRY and three variants: F109S, I68A, and G95R. Left, WB documenting equalization of WT and variant SRY accumulation by adjustment of plasmid dilution (see “Experimental Procedures”). α-Tubulin (middle panel) provided a loading control; GFP (bottom panel) mirrored extent of plasmid dilution and hence is strongest under 1× conditions (F109S), intermediate under 25× plasmid dilution, and weakest under 30× plasmid dilution. Right, histogram showing extent of Sox9 mRNA accumulation as stimulated by WT or variant SRY constructs, each at mean level of 105 protein molecules per transfected cell. I68A and G95R SRY variants provided negative controls, in each case due to impaired specific DNA binding (22, 62). Statistical comparisons: * and ** Wilcox p values <0.05 and <0.01, respectively, whereas “ns” indicates p values >0.05.
The above H3 epigenetic codes in the untransfected cell lines were associated with TESCO occupancy by WT SRY on its transient transfection (under 1× conditions). Although such transfection led to similar overexpression of WT SRY in each cell line (Fig. 8A, above), no SRY-specific chromatin immunoprecipitation (ChIP)-based signal was observed in the CH15, HEK 293T, or Hs1.TES cells (Fig. 10B). The ChIP pattern in CH34 cells with primer sets a, b, and c was as described previously (14). Primer sets a and c were designed based on the results of Sekido and Lovell-Badge (49) to enable amplification of a DNA segment containing multiple SRY target sites, whereas primer set b provided a negative control within TESCO. In light of these results, comparative studies of the transcriptional activity of F109S SRY and WT SRY were restricted to CH34 cells.
Comparative ChIP studies of F109S SRY and WT SRY in CH34 cells demonstrated 2-fold reduction in TESCO occupancy in the absence of MG132 and equal occupancy in the presence of MG132 (Fig. 10C). Treatment of the cells with MG132 did not affect TESCO occupancy by WT SRY. Such enhancer binding required DNA recognition by the HMG box as indicated by negative control I68A SRY, a variant that is well folded but without detectable specific DNA binding activity in vitro (62). The 2-fold reduction in TESCO occupancy by F109S SRY was associated with a 2-fold reduction in SRY-directed transcriptional activation of Sox9 under conditions of overexpression (1× transient transfection; left-hand side of Fig. 10D) or reduced expression (50× transient transfection; right-hand side of Fig. 10D). In each case treatment of the cells with MG132 rescued transcriptional activation of Sox9 (blue labels in Fig. 10D). MG132 treatment did not enhance the apparent potency of WT SRY as shown previously (14).
To evaluate the relative transcriptional potencies of F109S SRY and WT SRY on a per molecule basis, plasmid dilution ratios were carefully adjusted to give similar intracellular protein concentrations (Fig. 10E, left). Anti-HA WB band intensities of WT and F109S SRY were similar (top panel in Fig. 10E, left) relative to the α-tubulin loading controls (middle panel) following respective 1× and 25× plasmid dilutions as monitored by GFP co-transfection (bottom panel). Negative controls were provided in this experiment by SRY variants with negligible specific DNA binding activity (I68A and G95R (22, 62)). Results of qPCR-based studies of Sox9 mRNA abundance are shown at right in Fig. 10E. Whereas the negative controls lead, as expected, to no change in the baseline abundance of the Sox9 mRNA, the per molecule potencies of WT and F109S SRY were similar. Although not statistically significant, the small attenuation of the F109S-associated signal was in accordance with the small attenuation of the F109S WB signal (Fig. 10E) due to technical limitations of this protocol.
The above functional analysis was supported by two additional sets of control experiments. The first employed gene-specific qPCR to assess the specificity of SRY-directed transcriptional activation of Sox9 and in particular to exclude induction of a general state of enhanced transcriptional activity. To this end, other members of the Sox family uninvolved in testis determination were probed (Sox2, -3, -4 and -17); none of these genes exhibited SRY-dependent transcriptional activation (left-hand side of Fig. 11A). Similarly, transient transfection of WT SRY did not alter the expression of housekeeping genes (right-hand side of Fig. 11A). Finally, to demonstrate that the relative transcriptional activities of WT and F109S SRY were unaffected by a differences in subcellular localization, an immunocytochemical assay was employed to evaluate the extent of nuclear localization (Fig. 11B). Quantitative analysis (∼500 cells per SRY variant; see Table 5) revealed indistinguishable patterns of nuclear or pancellular protein distributions for WT and F109S SRY (left-hand side of Fig. 11C), whereas control mutations I90M (impairing nuclear export and hence NCS) and R62G (impairing nuclear import) gave rise to the expected perturbations in accordance with past studies (right-hand side of Fig. 11C) (14, 35).
FIGURE 11.

Control studies of off-target genes and nuclear localization in CH34 cells. A, transcriptional assays of selected genes activated by SRY variants in rat embryonic gonadal cell line. RT-Q-rt-PCR was employed to probe mRNA abundances of Sox family members uninvolved in testis development (left) and sex-unrelated housekeeping genes (right). qPCR was analyzed following transfection of SRY variant plasmids, empty vector, or control plasmid expressing a stable but inactive SRY variant (I68A); these genes were not affected by expression of transfected SRY. B and C, F109S does not perturb the nuclear localization of SRY in CH34 cells. B, subcellular localization of HA-tagged SRY variants: DAPI staining (blue, upper left), wild type or variants SRY staining (green, upper right), phalloidin staining for cytoskeletal (red, lower left), and the overly image (lower right). SRY variants: WT (panel a), F109S (panel b), I90M (positive control; panel c), and R62G (negative control; panel d). C, histograms describing the nuclear (light gray) and pan-cellular (white) distribution of SRY variants compared with that of wild type in the presence of proteasomal inhibitor, MG132. Statistical comparisons: **, Wilcox p values <0.01, whereas ns indicates p values > 0.05.
TABLE 5.
Nuclear localization of SRY variants in CH34 cells
Discussion
This study has focused on an inherited mutation in the HMG box of SRY associated in the proband with somatic XY sex reversal, primary amenorrhea, and gonadoblastoma in situ (63). The same mutation was found in the Y chromosomes of the proband's father, fertile paternal uncle, and two brothers (Fig. 12A). Such a DSD pedigree (unusual among Swyer families) suggests that the variant SRY allele is compatible with either male or female somatic phenotypes depending on genetic background or stochastic gene expression (64). Despite the marked biophysical perturbation of the variant HMG box, four members of this pedigree bearing this mutation are male, and only the proband is female. Although such family trees are rare in human populations (65), similar variable DSD phenotypes have commonly been observed among laboratory strains of mice due to Y chromosome-autosome incompatibility (31–33, 51).
FIGURE 12.

Patient pedigree and structural and sequence conservation of the aromatic at HMG consensus position 54. A, family trees pertaining to F109S SRY. Symbols are defined at right. The patient is one of five siblings. Two brothers and the parent uncle with F109S SRY gene are apparently normal. The patient was examined due to the primary amenorrhea. No signs of virilization. Streak gonads were detected, and the phenotypic phenomena of right streak gonad suggests the presence of gonadoblastoma. Cytogenetic studies suggest no evidence for mosaicism of the sex chromosomes. B, alignment of three different HMG domains; representative HMG boxes from architecture-specific (human HMB3 box 2, PDB code 2EQZ, magenta), mouse LEF-1 (PDB code 2LEF (111), dark gray), and human SRY (PDB code 1J46 (27), orange). Chain termini and α-helices are as labeled; the aromatic residue at consensus position 54 is shown as sticks. C, enlarged view of the crux of the angular DNA bending surface the consensus position 54 shown as sticks. The consensus positions of the remaining aromatic residues of the major hydrophobic core motif are labeled in green, which convene at the confluence of the three helices. D, sequence alignments of representative members of the HMG superfamily. Residues of the major core motif are highlighted by corresponding colored box, and consensus numbers are listed below alignments. Consensus positions 15 and 43 are boxed in green and the position 54 in red.
Clinical Mutation F109S Provides Biophysical Insight into General Architectural Rules
Our goal was to decipher possible molecular mechanisms underlying the incomplete genetic penetrance of this mutation. Why was the proband female when the same variant Sry allele was found in male members of her family, in two individuals with evidence of fertility? The Ser substitution at residue 109 alters a core Phe residue (Fig. 12, B and C). This position in the HMG box is (a) invariant among Sry and Sox domains (consensus position 54; lower two panels of Fig. 12D) and (b) conserved as an aromatic residue (Phe or Tyr) among other families of HMG boxes (upper two panels of Fig. 12D) (66–68). This substitution results in loss of aromaticity, hydrophobicity, and side-chain volume, and this was predicted by Clore and co-workers (26) (based on the three-dimensional structure of the SRY HMG box) to be destabilizing. The present biochemical and biophysical studies of the WT and variant box have verified this prediction. These findings are in accordance with mutational studies of diverse globular proteins whose respective hydrophobic cores seldom tolerate such non-conservative substitutions (69, 70).
Although the marked instability of the F109S SRY domain readily rationalizes the phenotype of the proband, what may account for the divergent phenotype of her father and uncle (as fertile males)? The F109S SRY domain was found to retain native-like specific DNA-binding affinity with unperturbed DNA bend angle in a consensus DNA-domain complex. In striking contrast to the temperature-dependent unfolding of the free domain, CD and 1H NMR studies indicated that the native-like structure was largely intact in the specific DNA complex. Such structure was either regained on specific DNA binding (induced fit) or captured by the specific DNA site to predominate in the equilibrium complex (conformational selection) (71). These biophysical mechanisms are of evolutionary interest as the near-native functional properties of unstable or unfolded polypeptides may rationalize how the exquisite structural organization of modern proteins emerged through stepwise stabilization of nascent partial folds (72).
Despite the native-like structure of the variant protein-DNA complex, a subtle increase in rate of protein-DNA dissociation (and hence a corresponding increase in rate of association) was observed in stopped-flow FRET studies. This finding suggests that pre-organized structure in the major wing of the WT domain imposes, at least to this extent, a kinetic barrier to specific DNA binding and release. Analogous compensation between on- and off-rates (likewise associated with specific DNA-dependent protein folding with native-like DNA bending) was observed in studies of the adjoining aromatic substitution W70F in the SRY HMG box (consensus box position 15; Fig. 12C) (15). Although the latter substitution has not to date been observed in a patient, a cavity-associated mutation at the same site (W70L) was identified as a de novo Swyer mutation (63). This mutation (occurring at a site less exposed than is Phe-109) caused a more severe decrement in stability with marked reduction in specific DNA affinity (15). Whereas the present proband had gonadoblastoma in situ, the W70L patient presented with gonadoblastoma as a discrete tumor (63).
Comparative studies of clinical variants promise to uncover general architecture rules governing the structure and function of the SOX HMG box. The failure of kinetic compensation in the case of W70L, for example, was ascribed in unresolvable structural frustration in the bound state due to steric clash imposed by the tetrahedral configuration of the Leu γ-carbon and hence non-planarity of the mutant side chain (15). It would be of future interest to investigate F109L and other non-planar substitutions to compare and contrast these two sites of conserved aromatic residues (box positions 15 and 54; Fig. 12, C and D). We speculate that box position 54, due to its partial exposure to solvent and less constrained packing environment, may tolerate diverse substitutions with respect to maintenance of high affinity specific DNA binding, although we have shown that that Trp-70 (box position 15) can only be substituted by other aromatic amino acids (15).
Clinical mutations have also been found at box position 43 (W98R (46)), at which an invariant Trp adjoins box position 54 (Fig. 12C). The indole ring contributes both to core packing and to specific DNA binding (as part of the cantilever-associated “hydrophobic wedge” in the minor groove and via a hydrogen bond from the indole NH to the DNA backbone (26, 27)). Because this Trp (unlike that at box position 15) is invariant even among distant structure-specific HMG boxes (top panel of Fig. 12D), representing an evolutionary depth of more than 500 million years (66), we imagine that no other natural side chain can fulfill both roles. It is noteworthy that substitution of the homologous Trp by Arg has also been observed in the homologous HMG boxes of SOX9 (in a patient with campomelic dysplasia with XY sex reversal (73)) and SOX10 (in a patient with Kallmann syndrome (74)). Such corresponding mutations highlight how general architectural rules shared by a family of lineage-specific TFs may underlie diverse clinical presentations.
Similarities and Differences between the F109S Human HMG Box and the HMG Box of Mouse Sry
The HMG box of mouse Sry (like the orthologous boxes of muroid rodents in general) is divergent relative to other mammalian taxa (75). Such divergence has been ascribed to the evolutionary dynamics of a muroid-specific C-terminal Gln-rich domain (76, 77), which functions as a transcriptional activation domain (78). This domain is the result of an ancestral invasion of the muroid Y chromosome by a DNA microsatellite and its fortuitous location in-frame within the Sry gene (76). The encoded Gln-rich domain provides intragenic complementation for deleterious mutations in the HMG (76) and also protects the protein from proteasomal degradation (77). Such complementation at the protein level has been proposed to enable the DNA microsatellite to function as a genetic capacitor, permitting the accumulation of cryptic genetic variation in the HMG box and its discharge as a source of evolutionary novelty (76). We speculate that the Gln-rich domains of rodent Sry orthologs either (i) protect these divergent and unstable HMG boxes from polyubiquitination or (ii) hinder degradation of these TFs (once ubiquitinated) from 26S proteasomal degradation.
The instability of the murine Sry HMG box is broadly consistent with an analogy proposed by Eicher and co-workers (51) between Swyer syndrome and the widely observed phenomenon of murine Y/autosome strain incompatibility leading to intersexual phenotypes (33, 79). Our CD studies indicate, however, that the inherited Swyer mutation renders the human SRY HMG box even less stable than the murine HMG box with respect to both thermal unfolding and chemical denaturation. Furthermore, studies of intrinsic Trp fluorescence suggest that the hydrophobic core of the F109S domain is less well protected from solvation than is the core of the murine domain. That the majority of XY humans bearing the F109S mutation develop as males (and in at least two cases as fertile males (28)) suggests that human testis determination is more robust that murine testis determination. It would be of future interest to develop quantitative methods to assess input-output relationships among orthologous developmental switches. Such studies would in principle be akin to those pioneered by Ptashne and co-workers (81) in studies of the phage λ cI and Cro repressors. Quantitative differences in the molecular robustness of a developmental switch may in turn correlate with macro-evolutionary differences the pace of evolutionary change in a taxon, an overarching goal in the emerging “evo-devo” synthesis (10, 18, 29).
Clinical Mutation F109S Provides Biological Insight into Transcriptional Regulation
The biophysical instability of the F109S HMG box and its near-complete unfolding at 37 °C are associated with accelerated proteasomal turnover (relative to WT) of epitope-tagged full-length F109S SRY following transient transfection in four cell lines. These cell lines share the universal eukaryotic proteasomal pathway (82) but otherwise differ in species of origin (rat or human), developmental stage, lineage, and degree of transformation. Despite these marked differences, cellular studies of protein degradation following translational arrest by cycloheximide are remarkable for the uniformity of results.
To delineate mechanisms contributing to the decreased half-life of the mutant SRY, we sought to characterize the ubiquitination state of the variant and WT SRY under conditions of chemical proteasome inhibition. Protein turnover is regulated by various mechanisms, including proteasomal degradation (82, 83). Proteins that misfold due to mutation, chemical damage, or improper maturation are often targeted and hydrolyzed by this process. Two routes of proteasomal degradation have been characterized (57, 58, 82) as follows: (i) ubiquitin-independent hydrolysis by the 20S core proteasome, and (ii) ubiquitin-dependent pathway and hydrolysis by the 26S proteasome complex. Our results have implicated the latter as F109S SRY exhibited increased polyubiquitination (relative to WT SRY and I68A SRY). Although structural analysis of F109S SRY domain suggests that the variant TF resembles, an unfolded protein within the cell, its in vitro ubiquitin-independent degradation by the 20S core proteasome was similar to that of the WT SRY domain. Together, these findings provided evidence that the accelerated degradation of epitope-tagged F109S SRY is due to the ubiquitin-dependent 26S proteasomal pathway.
Of the four mammalian cell lines employed in this study, only CH34 cells exhibit histone marks in the embryonic TESCO (49) associated with an open chromatin structure amenable to transcriptional activation (84). Such a chromatin structure presumably reflects the site and stage of the cell line's origin in the bipotential gonadal ridge just prior to Sry expression (46). Similarly, it was only in CH34 cells that we could observe the following: (a) TESCO occupancy by WT SRY on its transient transfection and (b) SRY-directed transcriptional activation of Sox9. Evidence that such activation was gene- or pathway-specific was provided by control studies of other members of the Sox family not implicated in testis determination and by control studies of housekeeping genes. Together with the above cell line specificity, the absence of SRY-directed transcriptional activation of the latter two sets of endogenous genes implies that SRY-directed activation of Sox9 in CH34 cells provided a mechanistic model of a developmental switch rather than a general state of enhanced transcriptional activity in a transfected cell line.
In our studies enhanced polyubiquitination and accelerated proteasomal degradation of F109S SRY led to a 2-fold decrease in its TESCO enhancer occupancy (relative to WT SRY) and in turn to a 2-fold decrease in extent of SRY-directed Sox9 expression. The relative impairment of target gene regulation was similar in magnitude under standard transfection conditions (1×, corresponding on average to ∼7 × 105 WT SRY molecules per cell and 2 × 103 variant SRY molecules per cell) and upon marked plasmid dilution with the parent plasmid (50×, leading on average to expression of ∼2 × 103 WT SRY molecules per cell and 102 variant SRY molecules per cell). Two lines of evidence indicated that the functional defect of F109S SRY is solely due to its instability and accelerated turnover as follows: (i) chemical proteasome inhibition by MG132 fully rescued TESCO occupancy leading to native Sox9 activation and (ii) equalization of protein expression levels (at a mean value of 103 SRY molecules per cell) by individual adjustment of plasmid dilution ratio (25× for WT SRY; 1× for F109S SRY) likewise led to essentially equal levels of Sox9 mRNA accumulation.
To date, the following three inherited Swyer mutations in SRY have been characterized in biochemical detail (65): V60L (leading to a partial impairment of nuclear import via the N-terminal NLS), I90M (leading to a partial impairment of nuclear export and hence NCS-coupled phosphorylation), and F109S (leading as above to instability). Each of these impairments is associated, independently of molecular mechanism, with a 2-fold decrease in SRY-dependent Sox9 activation under conditions of physiological TF expression (generally 102–104 protein molecules per cell (60)). These findings thus reflect a general feature of human male sex determination, the slim margin by which the output of the SRY-Sox9 regulatory axis functions as a developmental switch. The 2-fold threshold of Sox9 expression is consistent with the pathophysiology of campomelic dysplasia as a syndrome of TF haploinsufficiency (85). In none of these pedigrees has the influence of autosomal genetic variation been distinguished from effects of stochastic gene expression as a mechanism of phenotypic variation.
The remaining four inherited Swyer mutations (Table 6) are each likely to perturb fundamental mechanisms underlying the structure or function of SRY. R30I, for example, adjoins potential sites of serine phosphorylation (bold in sequence LRRSSSFLC; residues 31–33 in human SRY) recognized by PKA (86). Although SRY has not been shown to be phosphorylated in vivo and the adjoining one or more of these three serines are only conserved within primates (76), and not more broadly among therian mammals (76), evidence has been obtained in cell culture that such phosphorylation enhances transcriptional activity (86) and is coupled to NCS (14). We speculate that R30I impairs PKA recognition of this motif and hence recapitulates the functional consequences of impaired nuclear import or export, a 2-fold reduction in SRY-directed transcriptional activation of SOX9 (14) as a general feature of the inherited Swyer syndrome (65). It would be of future interest to test this prediction and more broadly to investigate the relationship (if any) between PKA site selectivity in SRY and transcriptional potency.
TABLE 6.
Inherited Swyer mutations in human SRY
| Mutation | Clinical featuresa |
Locationc activity mechanism |
Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CGD/PGDb | Ovotestis | Gonadal blastoma | Not mosaic father | Sister-brother | Structural location | Relative endogenous Sox9 activation | Mechanism | ||
| % | |||||||||
| S18N | PGD | ■ | ■ | ■ | ■ | N-terminal non-box | 22, 105 | ||
| R30I | CGD | ■ | N-terminal non-box | Impaired phosphorylationd | 22, 106 | ||||
| V60L | CGD | ■ | Minor wing | 46 ± 7 | Impaired nuclear localization | 13, 14, 24 | |||
| R76S | CGD | Major wing α1 | 107 | ||||||
| I90M | CGD | ■ | ■ | Major wing α2 | 52 ± 6 | Impaired nuclear export | 14, 22, 108 | ||
| F109S | CGD | ■ | ■ | Major wing α3 | 51 ± 8 | Accelerated protein degradation | 28 | ||
| Y127F | CGD | ■ | Minor wing | NDe | 109 | ||||
| L163X | CGD | C-terminal non-box | ND | 110 | |||||
a For each inherited mutation, results of prior studies are indicated by ■. Open spaces signify an absence of reported data.
b CGD/PGD, complete gonadal dysgenesis/partial gonadal dysgenesis.
c The three α-helices of the HMG box are designated α1, α2, and α3 (see Fig. 1B).
d This mechanism is presumed based on the proximity of potential sites of serine phosphorylation and presumed targeting by protein kinase A as described in Ref. (86); see under “Discussion.”
e ND indicates not determined.
F109S SRY as a Quantitative Tool
The biophysical instability of a variant SRY HMG box coupled with its retention of native-like specific DNA-binding and -bending properties both underlies the sex-reversed phenotype of the proband and enables the male development of her father, uncle, and brothers. We imagine that such developmental variation is purely a “numbers game,” and in the proband the number of variant SRY molecules per cell in the gonadal ridge fell below the threshold to trigger implementation of the Sertoli cell program.
Beyond the intrinsic interest of these results, we envisage that the fortuitous molecular properties of F109S SRY may enable this variant allele to function as a tool in future quantitative studies of testis determination as a model developmental switch in therian mammals. Just as in the pioneering decades of prokaryotic molecular biology, wherein a small molecule (isopropyl β-d-1-thiogalactopyranoside) was employed to regulate the activity of Lac repressor (87) and in turn the expression of λ cI repressor in engineered operons (2), we imagine that MG132-regulated expression of F109S SRY may provide an analogous chemical “rheostat” by which to measure transcriptional input-output relationships. If so, the binary switch between male and female programs of mammalian gonadogenesis may permit quantitative modeling akin to that of the celebrated genetic switch between the lytic and lysogenic programs of lambdoid bacteriophages (4). Such future directions would highlight the prescience of these prokaryotic model studies (88).
Concluding Remarks
The present results have demonstrated a novel mechanism of impaired human organogenesis, the heightened susceptibility of a master lineage-specific TF to polyubiquitination and proteasomal degradation. This mechanism has historical resonance in studies of simpler eukaryotes: precise control of the cellular lifetimes of mating type-specific TFs was first investigated in Saccharomyces cerevisiae (MATα2 and MATa1 (Ref. 89 and references therein)). Although the latter TFs contain homeodomains as modules of specific DNA binding (90), and so are unrelated to SRY, the corresponding mating-type gene matA in fungus Aspergillus nidulans encodes an HMG-box TF (91). Remarkably, matA may functionally be replaced by human SRY to regulate both early and late stages of sexual development, including fungal gametogenesis (92). Such complementation suggests that SRY-related HMG boxes have deep evolutionary roots in Eukarya. Furthermore, the fundamental role of ubiquitin-directed proteasomal degradation in cell fate decisions (as exemplified in both the yeast mating-type switch (89) and mammalian gonadogenesis) is independent of molecular motif of DNA recognition.
The present results have further shown that the specific DNA-binding and DNA-bending activities of the SRY HMG box are robust to its structural destabilization. Yet, the cell-biological properties of F109S SRY support the notion that the WT TF functions at the edge of developmental ambiguity (13, 14). That a tenuous genetic switch (i.e. a mere 2-fold decrease in SRY-directed transcriptional activation of SOX9) underlies inherited XY sex reversal may seem surprising but is in accordance with the genetics of human DSD as a downstream syndrome of TF haploinsufficiency (93, 94). These clinical correlations highlight the narrow margins of decision-making by opposing sex-specific gene-regulatory networks in the bipotential gonadal ridge (34, 95). In such decisions ubiquitin-directed protein degradation in principle provides a mechanism to control both TF abundance and its time course in relation to morphogenesis. Indeed, exquisite control of the timing of Sry expression in mice has been shown to be critical to the proper initiation and maintenance of testis formation (96–98). We envisage that systematic studies of human SRY variants, as identified in Swyer patients (22, 60) and as tested in model organisms (72), may decipher molecular mechanisms of an archetypical yet enigmatic genetic switch (99). Like the F109S “molecular rheostat” characterized here, the diverse human mutations associated with inherited XY sex reversal promise to provide tools to probe mechanisms of mammalian organogenesis at the edge of ambiguity.
Experimental Procedures
Protein Purification
WT and variant SRY domains of human SRY were expressed in Escherichia coli strain TLYS (New England Biolabs, Inc., Ipswich, MA) and purified as described (100). Purity was determined in each case to be >98% by SDS-PAGE. Results of matrix-assisted laser-desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) were in agreement with expected values.
Circular Dichroism
Far- and near-ultraviolet (UV) CD spectra were obtained at 4, 25, and 37 °C in a 1-mm path length quartz cuvette using an Aviv spectropolarimeter equipped with titrating unit (Aviv Biomedical, Inc., Lakewood, NJ). The domains were made 25 μm in 140 mm KCl and 10 mm potassium phosphate ((pH 7.4) “standard buffer”). Thermal unfolding was monitored at helix-sensitive wavelength 222 nm. CD difference spectra were calculated as the buffer-corrected difference between the observed spectrum of a protein-DNA complex and the sum of the spectra of the free protein and free DNA site.
Protein Stability
Fractional protein unfolding was monitored as a function of guanidine hydrochloride concentration by CD ellipticity at 222 nm. The domains were made 5 μm in standard buffer in a titrating cuvette. The same protein concentration was used in a titrant reservoir containing 7.8 m guanidine-HCl in the same buffer. Thermal unfolding of the free domains and equimolar protein-DNA complexes (25 μm in standard buffer) was monitored using a 12-bp consensus DNA duplex (5′-GTGATTGTTCAG-3′ and complement; core target sequence underlined); CD spectra (200–320 nm) were acquired from 4–90 °C at 2.5 °C increments.
Tryptophan Fluorescence Spectroscopy
Intrinsic Trp fluorescence spectra of the free HMG boxes and their specific DNA complexes were observed in standard buffer at 15 and 37 °C at a concentration of 5 μm. Emission spectra were acquired from 300 to 500 nm following excitation at 295 nm. Extent of the DNA-dependent inner filter effect under these conditions was estimated through control studies as described (15).
Fluorescence Resonance Energy Transfer
Protein-directed DNA bending was probed by steady-state FRET as described (12). This approach employed a 15-bp DNA duplex (5′-TCGGTGATTGTTCAG-3′ (“upper strand”) and complement (“lower strand”); consensus target site underlined). Use of a 15-bp DNA site restricted protein binding to the 1:1 high affinity complex (13). To provide a fluorescent donor, the upper strand was extended at its 5′ terminus by 6-carboxyfluorescein; the dye was flexibly linked to the DNA through a hexanyl linker. To provide a compatible acceptor, the lower strand was extended at its 5′-end by tertamethylrhodamine, also via a hexanyl linker. The labeled DNA strands were purchased from Oligos, Etc., Inc. (Wilsonville, OR). Photophysical control studies verified the mobilities of the probes and excluded non-FRET-related mechanisms of donor quenching (12). FRET studies employed a DNA concentration of 3 μm in 10 mm potassium phosphate, 10 mm Tris-HCl, 140 mm KCl, 1 mm EDTA, and 1 mm dithiothreitol with final pH 8.0 (“FRET buffer”).
FRET-based DNA Binding Assays
Steady-state FRET was employed to determine protein-DNA dissociation constants (Kd); the DNA site was as above. Measurements were made in FRET buffer at 15 and 37 °C. Varying concentrations of the WT or variant SRY domain were titrated at a constant DNA concentration of 25 nm. Emission spectra were recorded from 500–650 nm following excitation at 490 nm. Estimates of Kd were determined by plotting change in fluorescence intensity at 520 nm against total protein concentration. Data were fit to a single-site ligand-binding model (Equation 1) as described (101) using Origin 8.0 software (OriginLab Corp., Northampton, MA).
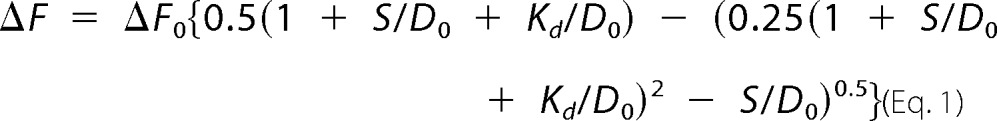 |
In this formalism, ΔF is the change in donor fluorescence observed on addition of the SRY domain relative to the baseline DNA fluorescence; ΔF0 is the maximum fluorescence change obtained in a 1:1 protein-DNA complex; Kd is the dissociation constant; D0 is the concentration of DNA (25 nm); and S is the concentration of SRY domain.
Stopped-flow Kinetic FRET Assay
Rates of protein-DNA dissociation were measured with an Aviv double-mixing stopped-flow apparatus at a fixed temperature (6, 15, 25, or 37 °C); the instrument contained a thermo-electric temperature controller. Fluorescence emission was monitored at 520 nm following excitation at 490 nm using an Aviv ATF 105 spectrofluorometer (12). In brief, a 20-fold molar excess (final stoichiometries) of unlabeled DNA in FRET buffer was employed to sequester the WT or variant SRY domain on rapid mixing. Estimates of dissociation rate constants (koff) were obtained by fitting the traces to a mono-exponential equation; values represent the mean and standard error of four replicates. Control studies of the WT domain indicated that similar time-dependent recovery of donor emission was observed irrespective of the concentration of the unlabeled DNA in the molar excess range 10–50-fold relative to initial concentration of the specific FRET-labeled DNA-protein complex.
Permutation Gel Electrophoresis
Six DNA fragments (150 bp each) containing an SRY-binding site (5′-ATTGTT-3′ and complement) were PCR-amplified from a plasmid previously described (12) such that the binding site was at varying distances from 5′-end (leading to variation in “flexure displacement”). Each fragment (10 nm) was complexed with the WT or variant HMG box (20 nm). Gels were purchased from Bio-Rad, equilibrated, and resolved in 0.5× Tris borate/EDTA buffer (TBE). Protein-DNA complexes were visualized using SYBR Green stain (Life Technologies, Inc.).
NMR Spectroscopy
15N-Labeled WT and variant Sry domains were prepared by growth of a corresponding overexpression strain of E. coli (12) in minimal medium containing [15N]ammonium sulfate as sole nitrogen source. The proteins were dissolved in a nitrogen-purged buffer containing 10 mm potassium phosphate buffer (10% D2O (pH 6.5)) and 50 mm NaCl and placed in a 280-μl Shigemi NMR tube. Homonuclear 2D 1H nuclear Overhauser effect (NOESY), total correlation (TOCSY), and double quantum-filtered correlation spectra were obtained of the free domains and their specific DNA complexes; a 15-bp DNA duplex was employed with sequence 5′-TCGGTGATTGTTCAG-3′ and complement (core target sequence underlined and site of Ile-68-cantilever insertion in boldface). To obtain a spectrum of the unfolded state, the WT 15N-labeled domain was also dissolved in phosphate-buffered saline (PBS) solution containing 5.4 m urea (10% D2O (pH 7.4)). Two-dimensional 1H-15N heteronuclear single-quantum coherence (HSQC) spectra were acquired at 25 °C using a Bruker Avance 700 MHz spectrometer.
Mammalian Plasmids
Plasmids expressing full-length human SRY or variants were constructed by PCR (13). Following the initiator Met, the cloning site encoded a hemagglutinin (HA) tag in triplicate to enable WB and chromatin immunoprecipitation (ChIP; below). Mutations in SRY were introduced using QuikChangeTM (Stratagene). Constructions were verified by DNA sequencing.
Rodent Cell Culture
CH15− and CH34 cells (kindly provided by Dr. P. K. Donahoe, Massachusetts General Hospital, Boston, MA) (14, 46) were cultured in DMEM containing 5% heat-inactivated fetal bovine serum at 37 °C under 5% CO2. For proteasome-inhibitor studies, transfected cells were maintained for 24 h in serum-free conditions and then treated with proteasome inhibitor MG132 for 6 h followed by 18 h of incubation in 5% serum-containing medium.
Human Cell Lines
Hs1.TES cells (derived from second-trimester human fetal testis; ATCC® CRL 7002TM) were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium containing 10% FBS in 5% CO2 atmosphere. HEK 293T cells (derived from the human embryonic kidney (47)) were obtained from ATCC and grown in Dulbecco's modified Eagle's medium with 10% FBS.
Transient Transfections
Transfections were performed using FuGENE 6 as described by the vendor (Hoffmann-La Roche, Nutley, NJ). After 24 h in serum-free medium, cells were recovered using fresh DMEM containing 5% heat-inactivated fetal bovine serum. Transfection efficiencies were determined by ratio of GFP-positive cells to untransfected cells following co-transfection with pCMX-SRY and pCMX-GFP in equal amounts (14). Subcellular localization was visualized by immunostaining for 24 h post-transfection following treatment with 0.01% trypsin (Invitrogen) and plating on 12-mm coverslips. SRY expression was monitored by Western blotting via its triplicate HA tag (above).
Cycloheximide Assay and Western Blotting
24 h following transient transfection, cells were split evenly into 6-well plates and treated with cycloheximide to a final concentration of 20 μg/ml in DMEM for the indicated times; cells were then lysed by radioimmunoprecipitation assay buffer (Hoffmann-La Roche). After protein normalization, cell lysates were subjected to 12% SDS-PAGE and WB using anti-HA antiserum (Sigma) at a dilution ratio of 1:5000 with α-tubulin antiserum providing a loading control. Quantification was performed by Image J software (rsbweb.nih.gov/ij).
Plasmid Dilution Protocol
Dilution of the WT or variant SRY expression plasmid by the parent plasmid (to provide a constant DNA mass per transient transfection) was employed in CH34 cells to modulate the intracellular concentration of SRY as described (13, 14). Dilution ratios ranged from 1 to 50×, where 1× represents the undiluted expression plasmid and 50× represents a 1:50 ratio of the expression plasmid concentration (in μg/ml) to the parent plasmid concentration.
Estimation of Intracellular SRY Concentration
The intracellular concentration of the WT or variant epitope-tagged SRY was estimated following transient transfection with dilution ratio 1–50× as described (13, 14). In brief, α-tubulin was employed in each case as an internal standard. In step 1, we assumed that a typical mammalian cell contains by weight the following amounts of tubulin: 300 × 10−12 g × 3% = 9 × 10−12 g. In step 2, we next estimated the number of tubulin molecules (molecular mass 110 kDa) as (9 × 10−12 g/110 × 103 g/ mol) × 6.02 × 1023 molecules/mol = 5 × 107 molecules. In step 3, we in turn estimated the number of transfected SRY molecules/cell in the 1× transfection using its Western blotting signal strength relative to α-tubulin: 5 × 107/100 (the tubulin-blotting dilution ratio)/2.5 (relative band intensity/tubulin/1× SRY signal)/28.9% (transfection efficiency), which yields an estimate of 7 × 105 molecules (predominantly in the nucleus in the case of WT SRY and F109S SRY). Finally, in step 4, we estimated the number of transfected SRY molecules/cell following 50-fold plasmid dilution using relative signal strength between 1 and 50× HA-SRY Western blottings: 7 × 105 molecules/390, which yields an estimate of near 1900 molecules per cell. This degree of expression is within the middle of the range of cellular abundances expected of a lineage- and stage-specific transcription factor (102–104 molecules per nucleus). This four-step procedure assumed that the affinities of the anti-HA and anti-tubulin antisera were similar as indicated by the vendor.
Anti-ubiquitin Western Blottings
To compare relative extents of ubiquitination of HA-tagged SRY variants in the above cell lines on transient transfection, the cells were grown in the absence or presence of MG132 and lysed with radioimmunoprecipitation assay buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, 0.05% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride; PMSF) containing protease inhibitors (Hoffmann-La Roche). Lysates were precipitated with monoclonal anti-HA-agarose beads (catalogue no. E6779; Sigma) and resolved by SDS-PAGE (4–20% gradient gel; Bio-Rad). Ubiquitinated proteins within the immunoprecipitation were detected by immunoblotting using a monoclonal anti-Ub antibody (catalogue no. MAB1510; Millipore). To monitor the total SRY input control, the anti-HA-agarose beads were also subjected to immunoblotting using 10% SDS-PAGE as probed by a polyclonal anti-HA antiserum (catalogue no. H6908; Sigma). WB using an antiserum against α-tubulin (catalogue no. T6074; Sigma) provided general loading controls. To facilitate quantification of band intensities, successive dilutions of the immunoprecipitated proteins were tested followed by optimization of film exposure times. Four or five biological replicates were obtained for each cell line. Images were analyzed using ImageJ in triplicate (technical replicates) to estimate relative levels of ubiquitination. Statistical analyses employed the Student's t test.
Default Degradation of SRY HMG Boxes by the 20S Core Proteasome
The purified (>95%) rabbit 20S core proteasome was obtained from Sigma (catalogue no. P3988) and used without further purification. In vitro digestion of WT or F109S variant HMG boxes was performed at 37 °C as described by Yamada et al. (102). The domains were in each case made 100 μm in a reaction volume of 100 μl in a buffer consisting of 10 mm Tris-HCl (pH 8.0), 10 mm MgCl2, 1 mm 2-mercaptoethnol, and 0.025% (w/v) SDS. Domains were pre-incubated at 37 °C for 15 min, and ∼2 μg (final concentration is 0.03 μm) of the 20S proteasome was then added to each reaction. At indicated times, 3-μl aliquots of the reaction were quenched at room temperature in Laemmli sample buffer (Bio-Rad). Samples were resolved by SDS-PAGE (4–20% gradient gels; Bio-Rad). Following Coomassie staining, band intensities were measured using ImageJ software. For each domain, three biological replicates were obtained; for each replicate, three technical replicates of the SDS-polyacrylamide gels were obtained and quantified.
Immunocytochemistry
Transfected CH34 and HEW 293T cells were plated evenly on 12-mm coverslips, fixed with 3% para-formaldehyde in PBS (pH 7.4) on ice for 30 min, treated with cold permeability buffer solution (PBS containing 10% goat serum and 1% Triton X-100; Sigma) for 10 min, blocked with 10% goat serum and 0.1% Tween 20 in cold PBS (Sigma), and incubated overnight at 4 °C with FITC-conjugated anti-HA antibody (diluted to 1:400 ratio; Santa Cruz Biotechnology, Santa Cruz, CA). After washing and 4′,6-diamidino-2-phenylindole (DAPI) staining, cells were visualized by fluorescence microscopy. Nuclear localization of F109S SRY was evaluated in relation to WT SRY in CH34 cells by the ratio of cells exhibiting nuclear HA-tagged SRY to the total number of GFP-positive cells. To enable robust statistical analysis, ∼500 SRY-positive cells were counted in each case (exclusive of non-expressing cells with mean transfection efficiency in the range 37–41%) and categorized as predominantly nuclear or pan-cellular in accordance with past studies (14).
TESCO Chromatin Immunoprecipitation
Cells were transfected with plasmids encoding epitope-tagged WT or variant SRY under 1× conditions (undiluted by the parent plasmid) and subjected to ChIP. Recovered cells were cross-linked in wells by formaldehyde, collected, and lysed after quenching the cross-linking reaction. Lysates were sonicated to generate 300–400-bp fragments and immunoprecipitated with anti-HA antiserum (Sigma) containing a protein A slurry (Santa Cruz Biotechnology) for pre-clearing. A nonspecific antiserum (control IgG; Santa Cruz Biotechnology) served as nonspecific control. After de-cross-linking at 65 °C overnight, fragments were treated with proteinase K and RNase (Hoffmann-La Roche), and then extracted by using 1:1 solution of phenol with a 24:1 solution of chloroform and isoamyl alcohol. PCR and qPCR protocols were as described (15). The following pairs of forward (F) and reverse (R) DNA oligonucleotide primers were employed to probe TESCO subsites as defined by Sekido and Lovell-Badge (49). Rat-specific primer sets a–c (pertinent to cell lines CH15 and CH34) are as follows: set a (with conserved SRY-binding sites), F 5′-GGAACTCCAACTACGTAC-3′ and R 5′-CCTGTAGTTGGTAGCTGC-3′; set b (as negative control), F 5′-ATCTCTACAGCTGACTTC-3′ and R 5′-TAGCTGGGCTCATATCG-3′; and set c (with conserved SRY-binding sites), F 5′-CTGAGAGCAATCTGAGC-3′ and R 5′-CACACCGTGCAAATGTA-3′. Human-specific primer sets a–c (pertinent to cell lines HEK 293T and Hs1.TES) are as follows: set a (with conserved SRY-binding sites), F 5′-CTAAACTCCAACTACACACGAA-3′ and R 5′-CCTTGACAGCTGCCACCT-3′; set b (as negative control), F 5′-GTTTTATGCAGCTGATTCT-3′ and R 5′-CTTGTCTGTGTAGCC-3′; and set c (with conserved SRY-binding sites), F 5′-GTGGAATGAATGTGCAC-3′ and R 5′-GGTTAGTCTTTTAATTTTTG-3′.
ChIP Studies of Histone Modifications
To probe histone modifications within rat or human TESCO associated with activated (mono-methylation, di-methylation, or tri-methylation of lysine H3K4 (designated me1, me2, and me3), mono-methylation of lysine H3K9 or H3K27) or repressed chromatin (di-methylation or tri-methylation of lysine H3K9 or H3K27), ChIP was employed with primer set a in the appropriate species (above) following immunoprecipitation with corresponding anti-me1, anti-me2, or anti-me3 IgG (Millipore Corp., Billerica, MA). Signals were quantified by qPCR as above.
Transcriptional Activation Assay
Following transient transfection (above), SRY-mediated transcriptional activation of Sox9 was measured in triplicate by qPCR as described (13). Cellular RNA was extracted using RNeasy as described by the vendor (Qiagen, N.V., Hilden, Germany). Transfection protocols were performed with the following: (i) SRY expression plasmid only, which contains 1 μg of WT or variant SRY-encoded plasmid per million cells (“1×” conditions) and (ii) a mixture of the empty mammalian parent (0.98 μg) and target SRY-encoded plasmid (0.02 μg), with the overall transfected mass of plasmids retained at 1 μg (“50×” conditions). Such dilution provided a control for potential artifacts of TF overexpression (14). The following sets of primers were used: Sox2, 5′-GCCGAGTGGAAACTTTTGTCG-3′ and 5′-CGGGAAGCGTGTACTTATCCT-3′; Sox4, 5′-GCAAGAAAAGAAGCCAAGCT-3′ and 5′-TGACCAAGAGGCAAAATAAAATCAA-3′; Sox9, 5′-AGCACTCCGGGCAATCT-3′ and 5′-CGGCAGGTATTGGTCAAACT-3′; GAPDH, 5′-GACATGCCGCCTGGAGAA-3′ and 5′-GCCCAGGATGCCCTTTAGT-3′; TFIID, 5′-CTGAGGGGGCAATGTCTAAC-3′ and 5′-GGGCAGCTAGTGAGATGAGC-3′.
Author Contributions
J. D. R. and Y. Y. performed the biochemical and biophysical studies; Y.-S. C. performed the cell biological studies; J. D. R., Y.-S. C., Y. Y., and M. A. W. undertook an integrated analysis of the data; J. D. R., Y.-S. C., and M. A. W. wrote the manuscript; N. B. P. coordinated studies and analysis of data for the revised manuscript; and M. A. W. designed and coordinated the research.
Acknowledgments
We thank C. M. Haqq and P. K. Donahoe (Massachusetts General Hospital and Harvard Medical School, Boston) for cell lines CH15 and CH34, M. Karplus and M. Ptashne for encouragement, and B. Baker, P. DeHaseth, E. Haas, H.-Y. Kao, M. Montano, C. O. Pabo, D. Samols, D. Schlesinger, and V. Yee for discussion. M. A. W. is grateful to P. Koopman, V. Lefebvre, R. Lovell-Badge, R. Sekido, D. Wilhelm and members of the international SOX community for advice and encouragement. We thank a reviewer for his or her suggestion to assess the relative extent of cellular ubiquitination of the WT and variant proteins.
This work, a contribution from the Cleveland Center for Membrane and Structural Biology, was supported in part by National Institutes of Health Grant GM080505 (to M. A. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TF
- transcription factor
- DSD
- disorders (or differences) of sex development
- HMG
- high mobility group
- NCS
- nucleocytoplasmic shuttling
- NLS
- nuclear localization signal
- qPCR
- quantitative real time polymerase chain reaction
- TES
- testis-specific enhancer
- TESCO
- testis-specific enhancer of Sox9 core
- Ub
- ubiquitin
- WB
- Western blotting
- F
- forward
- R
- reverse
- PDB
- Protein Data Bank
- HSQC
- heteronuclear single-quantum coherence.
References
- 1. Atkinson M. R., Savageau M. A., Myers J. T., and Ninfa A. J. (2003) Development of genetic circuitry exhibiting toggle switch or oscillatory behavior in Escherichia coli. Cell 113, 597–607 [DOI] [PubMed] [Google Scholar]
- 2. Johnson A. D., Poteete A. R., Lauer G., Sauer R. T., Ackers G. K., and Ptashne M. (1981) λ repressor and cro—components of an efficient molecular switch. Nature 294, 217–223 [DOI] [PubMed] [Google Scholar]
- 3. Tian T., and Burrage K. (2006) Stochastic models for regulatory networks of the genetic toggle switch. Proc. Natl. Acad. Sci. U.S.A. 103, 8372–8377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ptashne M. (2004) A Genetic Switch: Phage λ Revisited, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 5. Meyer B. J., Maurer R., and Ptashne M. (1980) Gene regulation at the right operator (O R) of bacteriophage λ: II. OR1, OR2, and OR3: Their roles in mediating the effects of repressor and cro. J. Mol. Biol. 139, 163–194 [DOI] [PubMed] [Google Scholar]
- 6. Davidson E. H. (2010) Emerging properties of animal gene regulatory networks. Nature 468, 911–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wuensche A. (2004) in Modularity in Development and Evolution (Schlosser G., and Wagner G. P., eds) pp. 600, University of Chicago Press, Chicago [Google Scholar]
- 8. Payne J. L., Moore J. H., and Wagner A. (2014) Robustness, evolvability, and the logic of genetic regulation. Artif. Life 20, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keyes L. N., Cline T. W., and Schedl P. (1992) The primary sex determination signal of Drosophila acts at the level of transcription. Cell 68, 933–943 [DOI] [PubMed] [Google Scholar]
- 10. Graves J. A. (2013) How to evolve new vertebrate sex determining genes. Dev. Dyn. 242, 354–359 [DOI] [PubMed] [Google Scholar]
- 11. Li B., Zhang W., Chan G., Jancso-Radek A., Liu S., and Weiss M. A. (2001) Human sex reversal due to impaired nuclear localization of SRY. A clinical correlation. J. Biol. Chem. 276, 46480–46484 [DOI] [PubMed] [Google Scholar]
- 12. Phillips N. B., Jancso-Radek A., Ittah V., Singh R., Chan G., Haas E., and Weiss M. A. (2006) SRY and human sex determination: The basic tail of the HMG box functions as a kinetic clamp to augment DNA bending. J. Mol. Biol. 358, 172–192 [DOI] [PubMed] [Google Scholar]
- 13. Phillips N. B., Racca J., Chen Y. S., Singh R., Jancso-Radek A., Radek J. T., Wickramasinghe N. P., Haas E., and Weiss M. A. (2011) Mammilian testis-determining factor SRY and the enigma of inherited human sex reversal. J. Biol. Chem. 286, 36787–36807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Y. S., Racca J. D., Phillips N. B., and Weiss M. A. (2013) Inherited human sex reversal due to impaired nucleocytoplasmic trafficking of SRY defines a male transcriptional threshold. Proc. Natl. Acad. Sci. U.S.A. 110, E3567–E3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Racca J. D., Chen Y.-S., Maloy J. D., Wickramasinghe N., Phillips N. B., and Weiss M. A. (2014) Structure-function relationships in human testis-determining factor SRY: an aromatic buttress underlies the specific dna-bending surface of a high mobility group (HMG) box. J. Biol. Chem. 289, 32410–32429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park S. Y., and Jameson J. L. (2005) Minireview: transcriptional regulation of gonadal development and differentiation. Endocrinology 146, 1035–1042 [DOI] [PubMed] [Google Scholar]
- 17. Graves J. A. (2002) Evolution of the testis-determining gene—the rise and fall of SRY. Novartis Found. Symp. 244, 86–97 [PubMed] [Google Scholar]
- 18. Sequeira P., Chen Y-S, and Weiss M. A. (2015) in Evolutionary Biology: Biodiversification from Genotype to Phenotype (Pontarotti P., ed) pp. 141–164, Springer [Google Scholar]
- 19. Sinclair A. H., Berta P., Palmer M. S., Hawkins J. R., Griffiths B. L., Smith M. J., Foster J. W., Frischauf A. M., Lovell-Badge R., and Goodfellow P. N. (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346, 240–244 [DOI] [PubMed] [Google Scholar]
- 20. Koopman P., Gubbay J., Vivian N., Goodfellow P., and Lovell-Badge R. (1991) Male development of chromosomally female mice transgenic for Sry. Nature 351, 117–121 [DOI] [PubMed] [Google Scholar]
- 21. Sekido R. (2010) SRY: a transcriptional activator of mammalian testis determination. Int. J. Biochem. Cell Biol. 42, 417–420 [DOI] [PubMed] [Google Scholar]
- 22. Knower K. C., Kelly S., Ludbrook L. M., Bagheri-Fam S., Sim H., Bernard P., Sekido R., Lovell-Badge R., and Harley V. R. (2011) Failure of SOX9 regulation in 46XY disorders of sex development with SRY, SOX9 and SF1 mutations. PLoS ONE 6, e17751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ner S. S. (1992) HMGs everywhere. Curr. Biol. 2, 208–210 [DOI] [PubMed] [Google Scholar]
- 24. Harley V. R., Jackson D. I., Hextall P. J., Hawkins J. R., Berkovitz G. D., Sockanathan S., Lovell-Badge R., and Goodfellow P. N. (1992) DNA binding activity of recombinant SRY from normal males and XY females. Science 255, 453–456 [DOI] [PubMed] [Google Scholar]
- 25. Werner M. H., Bianchi M. E., Gronenborn A. M., and Clore G. M. (1995) NMR spectroscopic analysis of the DNA conformation induced by the human testis determining factor SRY. Biochemistry 34, 11998–12004 [DOI] [PubMed] [Google Scholar]
- 26. Werner M. H., Huth J. R., Gronenborn A. M., and Clore G. M. (1995) Molecular basis of human 46X,Y sex reversal revealed from the three-dimensional solution structure of the human SRY-DNA complex. Cell 81, 705–714 [DOI] [PubMed] [Google Scholar]
- 27. Murphy E. C., Zhurkin V. B., Louis J. M., Cornilescu G., and Clore G. M. (2001) Structural basis for SRY-dependent 46-X,Y sex reversal: modulation of DNA bending by a naturally occuring point mutation. J. Mol. Biol. 312, 481–499 [DOI] [PubMed] [Google Scholar]
- 28. Jäger R. J., Harley V. R., Pfeiffer R. A., Goodfellow P. N., and Scherer G. (1992) A familial mutation in the testis-determining gene SRY shared by both sexes. Hum. Genet. 90, 350–355 [DOI] [PubMed] [Google Scholar]
- 29. Draghi J. A., Parsons T. L., Wagner G. P., and Plotkin J. B. (2010) Mutational robustness can facilitate adaptation. Nature 463, 353–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arkin A., Ross J., and McAdams H. H. (1998) Stochastic kinetic analysis of developmental pathway bifurcation in phage λ-infected Escherichia coli cells. Genetics 149, 1633–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Correa S. M., Washburn L. L., Kahlon R. S., Musson M. C., Bouma G. J., Eicher E. M., and Albrecht K. H. (2012) Sex reversal in C57BL/6J XY mice caused by increased expression of ovarian genes and insufficient activation of the testis determining pathway. PLoS Genet. 8, e1002569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eicher E. M., and Washburn L. L. (2001) Does one gene determine whether a C57BL/6J-YPOS mouse will develop as a female or as an hermaphrodite? J. Exp. Zool. 290, 322–326 [DOI] [PubMed] [Google Scholar]
- 33. Eicher E. M., Washburn L. L., Schork N. J., Lee B. K., Shown E. P., Xu X., Dredge R. D., Pringle M. J., and Page D. C. (1996) Sex-determining genes on mouse autosomes identified by linkage analysis of C57BL/6J-YPOS sex reversal. Nat. Genet. 14, 206–209 [DOI] [PubMed] [Google Scholar]
- 34. Sekido R., and Lovell-Badge R. (2009) Sex determination and SRY: down to a wink and a nudge? Trends Genet. 25, 19–29 [DOI] [PubMed] [Google Scholar]
- 35. Harley V. R., Layfield S., Mitchell C. L., Forwood J. K., John A. P., Briggs L. J., McDowall S. G., and Jans D. A. (2003) Defective importin β recognition and nuclear import of the sex-determining factor SRY are associated with XY sex-reversing mutations. Proc. Natl. Acad. Sci. U.S.A. 100, 7045–7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li B., Phillips N. B., Jancso-Radek A., Ittah V., Singh R., Jones D. N., Haas E., and Weiss M. A. (2006) SRY-directed DNA bending and human sex reversal: reassessment of a clinical mutation uncovers a global coupling between the HMG box and its tail. J. Mol. Biol. 360, 310–328 [DOI] [PubMed] [Google Scholar]
- 37. Smith J. M., and Koopman P. A. (2004) The ins and outs of transcriptional control: nucleocytoplasmic shuttling in development and disease. Trends Genet. 20, 4–8 [DOI] [PubMed] [Google Scholar]
- 38. Poulat F., Girard F., Chevron M. P., Gozé C., Rebillard X., Calas B., Lamb N., and Berta P. (1995) Nuclear localization of the testis determining gene product SRY. J. Cell Biol. 128, 737–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forwood J. K., Harley V., and Jans D. A. (2001) The C-terminal nuclear localization signal of the sex-determining region Y (SRY) high mobility group domain mediates nuclear import through importin β1. J. Biol. Chem. 276, 46575–46582 [DOI] [PubMed] [Google Scholar]
- 40. Sim H., Argentaro A., and Harley V. R. (2008) Boys, girls and shuttling of SRY and SOX9. Trends Endocrinol. Metab. 19, 213–222 [DOI] [PubMed] [Google Scholar]
- 41. Kirschner M. (1999) Intracellular proteolysis. Trends Cell Biol. 9, M42–M45 [PubMed] [Google Scholar]
- 42. Livneh I., Cohen-Kaplan V., Cohen-Rosenzweig C., Avni N., and Ciechanover A. (2016) The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 26, 869–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dahlmann B. (2016) Mammalian proteasome subtypes: their diversity in structure and function. Arch. Biochem. Biophys. 591, 132–140 [DOI] [PubMed] [Google Scholar]
- 44. Haqq C. M., King C. Y., Ukiyama E., Falsafi S., Haqq T. N., Donahoe P. K., and Weiss M. A. (1994) Molecular basis of mammalian sexual determination: activation of Müllerian inhibiting substance gene expression by SRY. Science 266, 1494–1500 [DOI] [PubMed] [Google Scholar]
- 45. Knower K. C., Kelly S., and Harley V. R. (2003) Turning on the male–SRY, SOX9 and sex determination in mammals. Cytogenet. Genome Res. 101, 185–198 [DOI] [PubMed] [Google Scholar]
- 46. Haqq C. M., and Donahoe P. K. (1998) Regulation of sexual dimorphism in mammals. Physiol. Rev. 78, 1–33 [DOI] [PubMed] [Google Scholar]
- 47. Graham F. L., Smiley J., Russell W. C., and Nairn R. (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59–74 [DOI] [PubMed] [Google Scholar]
- 48. Zhang L., Ding X., Nie S., Li-Ling J., Zhang Y., Zhang H., Chen L., Li L., and Ding M. (2015) Association of hsa-miR-145 overexpression in human testicular cells with male infertility. Mol. Med. Rep. 11, 4365–4372 [DOI] [PubMed] [Google Scholar]
- 49. Sekido R., and Lovell-Badge R. (2008) Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453, 930–934 [DOI] [PubMed] [Google Scholar]
- 50. Ikeguchi M., Kuwajima K., and Sugai S. (1986) Ca2+ alteration in the unfolding behavior of α-lactalbumin. J. Biochem. 99, 1191–1201 [DOI] [PubMed] [Google Scholar]
- 51. Albrecht K. H., Young M., Washburn L. L., and Eicher E. M. (2003) Sry expression level and protein isoform differences play a role in abnormal testis development in C57BL/6J mice carrying certain Sry alleles. Genetics 164, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ivanov V. I., Minchenkova L. E., Schyolkina A. K., and Poletayev A. I. (1973) Different conformations of double-stranded nucleic acid in solution as revealed by circular dichroism. Biopolymers 12, 89–110 [DOI] [PubMed] [Google Scholar]
- 53. King C. Y., and Weiss M. A. (1993) The SRY high-mobility-group box recognizes DNA by partial intercalation in the minor groove: a topological mechanism of sequence specificity. Proc. Natl. Acad. Sci. U.S.A. 90, 11990–11994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hessa T., Sharma A., Mariappan M., Eshleman H. D., Gutierrez E., and Hegde R. S. (2011) Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature 475, 394–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saini K. S., Summerhayes I. C., and Thomas P. (1990) Molecular events regulating messenger RNA stability in eukaryotes. Mol. Cell. Biochem. 96, 15–23 [DOI] [PubMed] [Google Scholar]
- 56. Ennis H., and Lubin M. (1964) Cycloheximide: aspects of inhibition of protein synthesis in mammalian cells. Science 146, 1474–1476 [DOI] [PubMed] [Google Scholar]
- 57. Komander D. (2009) The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953 [DOI] [PubMed] [Google Scholar]
- 58. Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 59. Asher G., Reuven N., and Shaul Y. (2006) 20S proteasomes and protein degradation “by default”. BioEssays 28, 844–849 [DOI] [PubMed] [Google Scholar]
- 60. Goentoro L., Shoval O., Kirschner M. W., and Alon U. (2009) The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol. Cell 36, 894–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rosenfeld J. A., Wang Z., Schones D. E., Zhao K., DeSalle R., and Zhang M. Q. (2009) Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics 10, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weiss M. A., Ukiyama E., and King C. Y. (1997) The SRY cantilever motif discriminates between sequence- and structure-specific DNA recognition: alanine mutagenesis of an HMG box. J. Biomol. Struct. Dyn. 15, 177–184 [DOI] [PubMed] [Google Scholar]
- 63. Hersmus R., de Leeuw B. H., Stoop H., Bernard P., van Doorn H. C., Brüggenwirth H. T., Drop S. L., Oosterhuis J. W., Harley V. R., and Looijenga L. H. (2009) A novel SRY missense mutation affecting nuclear import in a 46,XY female patient with bilateral gonadoblastoma. Eur. J. Hum. Genet. 17, 1642–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kurnit D. M., Layton W. M., and Matthysse S. (1987) Genetics, chance, and morphogenesis. Am. J. Hum. Genet. 41, 979–995 [PMC free article] [PubMed] [Google Scholar]
- 65. Chen Y-S, Racca J. D., Sequeira P. W., and Weiss M. A. (2015) Inherited sex-reversal mutations in SRY define a functional threshold of gonadogenesis: biochemical and evolutionary implications of a rare monogenic syndrome. Rare Disorders Diag. Ther. 1, 1–12 [Google Scholar]
- 66. Laudet V., Stehelin D., and Clevers H. (1993) Ancestry and diversity of the HMG box superfamily. Nucleic Acids Res. 21, 2493–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murphy F. V. 4th, Sweet R. M., and Churchill M. E. (1999) The structure of a chromosomal high mobility group protein-DNA complex reveals sequence-neutral mechanisms important for non-sequence-specific DNA recognition. EMBO J. 18, 6610–6618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jiang T., Hou C. C., She Z. Y., and Yang W. X. (2012) The SOX gene family: function and regulation in testis determination and male fertility maintenance. Mol. Biol. Rep. 316, 359–370 [DOI] [PubMed] [Google Scholar]
- 69. Kapp O. H., Moens L., Vanfleteren J., Trotman C. N., Suzuki T., and Vinogradov S. N. (1995) Alignment of 700 globin sequences: extent of amino acid substitution and its correlation with variation in volume. Protein Sci. 4, 2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Matthews B. W. (1996) Structural and genetic analysis of the folding and function of T4 lysozyme. FASEB J. 10, 35–41 [DOI] [PubMed] [Google Scholar]
- 71. Gianni S., Dogan J., and Jemth P. (2014) Distinguishing induced fit from conformational selection. Biophys. Chem. 189, 33–39 [DOI] [PubMed] [Google Scholar]
- 72. Liu Z., and Huang Y. (2014) Advantages of proteins being disordered. Protein Sci. 23, 539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Meyer J., Südbeck P., Held M., Wagner T., Schmitz M. L., Bricarelli F. D., Eggermont E., Friedrich U., Haas O. A., Kobelt A., Leroy J. G., Van Maldergem L., Michel E., Mitulla B., Pfeiffer R. A., et al. (1997) Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum. Mol. Genet. 6, 91–98 [DOI] [PubMed] [Google Scholar]
- 74. Pingault V., Bodereau V., Baral V., Marcos S., Watanabe Y., Chaoui A., Fouveaut C., Leroy C., Vérier-Mine O., Francannet C., Dupin-Deguine D., Archambeaud F., Kurtz F. J., Young J., Bertherat J., et al. (2013) Loss-of-function mutations in SOX10 cause Kallmann Syndrome with deafness. Am. J. Hum. Genet. 92, 707–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bowles J., Cooper L., Berkman J., and Koopman P. (1999) Sry requires a CAG repeat domain for male sex determination in Mus musculus. Nat. Genet. 22, 405–408 [DOI] [PubMed] [Google Scholar]
- 76. Chen Y. S., Racca J. D., Sequeira P. W., Phillips N. B., and Weiss M. A. (2013) Microsatellite-encoded domain in rodent Sry functions as a genetic capacitor to enable the rapid evolution of biological novelty. Proc. Natl. Acad. Sci. U.S.A. 110, E3061–E3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhao L., Ng E. T., Davidson T.-L., Longmuss E., Urschitz J., Elston M., Moisyadi S., Bowles J., and Koopman P. (2014) Structure-function analysis of mouse Sry reveals dual essential roles of the C-terminal polyglutamine tract in sex determination. Proc. Natl. Acad. Sci. U.S.A. 111, 11768–11773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dubin R. A., and Ostrer H. (1994) Sry is a transcriptional activator. Mol. Endocrinol. 8, 1182–1192 [DOI] [PubMed] [Google Scholar]
- 79. Eicher E. M., and Washburn L. L. (1983) Inherited sex reversal in mice: identification of a new primary sex-determining gene. J. Exp. Zool. 228, 297–304 [DOI] [PubMed] [Google Scholar]
- 80. Ben-Nissan G., and Sharon M. (2014) Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ptashne M. (1984) Repressors. Trends Biochem. Sci. 9, 142–145 [Google Scholar]
- 82. Volker C., and Lupas A. N. (2012) in The Proteasome: Ubiquitin Protein Degradation Pathway (Zwickl P., and Baumeister W., eds) pp. 1–19, Springer Science & Business Media, GmBH, Berlin [Google Scholar]
- 83. Dice J. (1987) Molecular determinants of protein half-lives in eukaryotic cells. FASEB J. 1, 349–357 [DOI] [PubMed] [Google Scholar]
- 84. Peterson C. L., Laniel M.-A. (2004) Histones and histone modifications. Curr. Biol. 14, R546–R551 [DOI] [PubMed] [Google Scholar]
- 85. Kobayashi A., Chang H., Chaboissier M. C., Schedl A., and Behringer R. R. (2005) Sox9 in testis determination. Ann. N.Y. Acad. Sci. 1061, 9–17 [DOI] [PubMed] [Google Scholar]
- 86. Desclozeaux M., Poulat F., de Santa Barbara P., Capony J.-P., Turowski P., Jay P., Méjean C., Moniot B., Boizet B., and Berta P. (1998) Phosphorylation of an N-terminal motif enhances DNA-binding activity of the human SRY protein. J. Biol. Chem. 273, 7988–7995 [DOI] [PubMed] [Google Scholar]
- 87. Gilbert W., and Müller-Hill B. (1966) Isolation of the lac repressor. Proc. Natl. Acad. Sci. U.S.A. 56, 1891–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stent G. S., and Calendar R. (1978) Molecular Genetics: An Introductory Narrative, pp. 204–237, W. H. Freeman & Co., New York [Google Scholar]
- 89. Johnson P. R., Swanson R., Rakhilina L., and Hochstrasser M. (1998) Degradation signal masking by heterodimerization of MATα2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell 94, 217–227 [DOI] [PubMed] [Google Scholar]
- 90. Gehring W. J., Qian Y. Q., Billeter M., Furukubo-Tokunaga K., Schier A. F., Resendez-Perez D., Affolter M., Otting G., and Wüthrich K. (1994) Homeodomain-DNA recognition. Cell 78, 211–223 [DOI] [PubMed] [Google Scholar]
- 91. Paoletti M., Seymour F. A., Alcocer M. J., Kaur N., Calvo A. M., Archer D. B., and Dyer P. S. (2007) Mating type and the genetic basis of self-fertility in the model fungus Aspergillus nidulans. Curr. Biol. 17, 1384–1389 [DOI] [PubMed] [Google Scholar]
- 92. Czaja W., Miller K. Y., Skinner M. K., and Miller B. L. (2014) Structural and functional conservation of fungal MatA and human SRY sex-determining proteins. Nat. Commun. 5, 5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wagner T., Wirth J., Meyer J., Zabel B., Held M., Zimmer J., Pasantes J., Bricarelli F. D., Keutel J., Hustert E., Wolf U., Tommerup N., Schempp W., and Scherer G. (1994) Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79, 1111–1120 [DOI] [PubMed] [Google Scholar]
- 94. Foster J. W., Dominguez-Steglich M. A., Guioli S., Kwok C., Weller P. A., Stevanović M., Weissenbach J., Mansour S., Young I. D., Goodfellow P. N., Brook D. J., and Schafer A. J. (1994) Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372, 525–530 [DOI] [PubMed] [Google Scholar]
- 95. Warr N., and Greenfield A. (2012) The molecular and cellular basis of gonadal sex reversal in mice and humans. Wiley Interdiscip. Rev. Dev. Biol. 1, 559–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee C.-H., and Taketo T. (1994) Normal onset, but prolonged expression, of Sry gene in the B6.YDOM sex-reversed mouse gonad. Dev. Biol. 165, 442–452 [DOI] [PubMed] [Google Scholar]
- 97. Eicher E. M., Wahsburn L. L., Whitney J. B., and Morrow K. E. (1982) Mus poschiavinus Y chromosome in the C57BL/6J murine genome causes sex reversal Science 217, 535–537 [DOI] [PubMed] [Google Scholar]
- 98. Wilhelm D., Washburn L. L., Truong V., Fellous M., Eicher E. M., and Koopman P. (2009) Antagonism of the testis- and ovary-determining pathways during ovotestis development in mice. Mech. Dev. 126, 324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Graves J. A. (2015) In retrospect: Twenty-five years of the sex-determining gene. Nature 528, 343–344 [DOI] [PubMed] [Google Scholar]
- 100. Phillips N. B., Nikolskaya T., Jancso-Radek A., Ittah V., Jiang F., Singh R., Haas E., and Weiss M. A. (2004) Sry-directed sex reversal in transgenic mice is robust to enhanced DNA bending: comparison of human and murine in HMG boxes. Biochemistry 43, 7066–7081 [DOI] [PubMed] [Google Scholar]
- 101. Privalov P. L., Jelesarov I., Read C. M., Dragan A. I., and Crane-Robinson C. (1999) The energetics of HMG box interactions with DNA: thermodynamics of the DNA binding of the HMG box from mouse sox-5. J. Mol. Biol. 294, 997–1013 [DOI] [PubMed] [Google Scholar]
- 102. Yamada S., Hojo K., Yoshimura H., and Ishikawa K. (1995) Reaction of 20S proteasome: shift of SDS-dependent activation profile by divalent cations. J. Biochem. 117, 1162–1169 [DOI] [PubMed] [Google Scholar]
- 103. Lodish H., Berk A., and Zipursky S. (2000) Molecular Cell Biology, 4th Ed., pp. 129–188, Garland Science, New York [Google Scholar]
- 104. Hiller G., and Weber K. (1978) Radioimmunoassay for tubulin: a quantitative comparison of the tubulin content of different established tissue culture cells and tissues. Cell 14, 795–804 [DOI] [PubMed] [Google Scholar]
- 105. Domenice S., Yumie Nishi M., Correia Billerbeck A. E., Latronico A. C., Aparecida Medeiros M., Russell A. J., Vass K., Marino Carvalho F., Costa Frade E. M., Prado Arnhold I. J., and Bilharinho Mendonca B. (1998) A novel missense mutation (S18N) in the 5′ non-HMG box region of the SRY gene in a patient with partial gonadal dysgenesis and his normal male relatives. Hum. Genet. 102, 213–215 [DOI] [PubMed] [Google Scholar]
- 106. Assumpção J. G., Benedetti C. E., Maciel-Guerra A. T., Guerra G. Jr., Baptista M. T., Scolfaro M. R., and de Mello M. P. (2002) Novel mutations affecting SRY DNA-binding activity: the HMB box N65H associated with 46, XY pure gonadal dysgenesis and the familial non-HMG box R301 associated with variable phenotypes. J. Mol. Med. 80, 782–790 [DOI] [PubMed] [Google Scholar]
- 107. Imai A., Takagi A., and Tamaya T. (1999) A novel sex-determining region on Y (SRY) missense mutation identified in a 46,XY female and also in the father. Endocr. J. 46, 735–739 [DOI] [PubMed] [Google Scholar]
- 108. Dörk T., Stuhrmann M., Miller K., and Schmidtke J. (1998) Independent observation of SRY mutation I90M in a patient with complete gonadal dysgenesis. Hum. Mutat. 11, 90–91 [DOI] [PubMed] [Google Scholar]
- 109. Jordan B. K., Jain M., Natarajan S., Frasier S. D., and Vilain E. (2002) Familial mutation in the testis-determining gene SRY shared by an XY female and her normal father. J. Clin. Endocrinol. Metab. 87, 3428–3432 [DOI] [PubMed] [Google Scholar]
- 110. Tajima T., Nakae J., Shinohara N., and Fujieda K. (1994) A novel mutation localized in the 3′ non-HMG box region of the SRY gene in 46,XY gonadal dysgenesis. Hum. Mol. Genet. 3, 1187–1189 [DOI] [PubMed] [Google Scholar]
- 111. Love J. J., Li X., Case D. A., Giese K., Grosschedl R., and Wright P. E. (1995) Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature 376, 791–795 [DOI] [PubMed] [Google Scholar]