

Abstract
Some members of the class A β-lactamase family are capable of conferring resistance to the last resort antibiotics, carbapenems. A unique structural feature of these clinically important enzymes, collectively referred to as class A carbapenemases, is a disulfide bridge between invariant Cys69 and Cys238 residues. It was proposed that this conserved disulfide bridge is responsible for their carbapenemase activity, but this has not yet been validated. Here we show that disruption of the disulfide bridge in the GES-5 carbapenemase by the C69G substitution results in only minor decreases in the conferred levels of resistance to the carbapenem imipenem and other β-lactams. Kinetic and circular dichroism experiments with C69G-GES-5 demonstrate that this small drop in antibiotic resistance is due to a decline in the enzyme activity caused by a marginal loss of its thermal stability. The atomic resolution crystal structure of C69G-GES-5 shows that two domains of this disulfide bridge-deficient enzyme are held together by an intensive hydrogen-bonding network. As a result, the protein architecture and imipenem binding mode remain unchanged. In contrast, the corresponding hydrogen-bonding networks in NMCA, SFC-1, and SME-1 carbapenemases are less intensive, and as a consequence, disruption of the disulfide bridge in these enzymes destabilizes them, which causes arrest of bacterial growth. Our results demonstrate that the disulfide bridge is essential for stability but does not play a direct role in the carbapenemase activity of the GES family of β-lactamases. This would likely apply to all other class A carbapenemases given the high degree of their structural similarity.
Keywords: antibiotic resistance, crystal structure, disulfide, enzyme kinetics, hydrogen bond, carbapenemase, enzyme stability
Introduction
β-Lactamases are enzymes produced by bacteria for self-protection against various β-lactam antibiotics (1). Based on the conservation of specific motifs in their amino acid sequences, β-lactamases are divided into four molecular classes: A, B, C, and D (2–4). Historically, class A enzymes were the first to be discovered and studied. They were identified in both Gram-positive and Gram-negative bacteria and produced resistance to penicillins and early cephalosporins. To counter the growing problem of resistance, novel β-lactams such as expanded spectrum cephalosporins (including cefotaxime, ceftriaxone, and ceftazidime) and β-lactamase inhibitors (clavulanic acid, sulbactam, and tazobactam) were developed. Extensive use of these drugs for treatment of infectious diseases triggered the selection in Gram-negative bacteria of variants of class A TEM- and SHV-type β-lactamases capable of protecting the microorganisms against these newer antimicrobial agents (5). Fortunately these enzymes failed to evolve to produce resistance to another group of β-lactam antibiotics, carbapenems, which for a long time have been used as the drugs of last resort for the treatment of life-threatening infections caused by bacteria resistant to earlier β-lactams.
However, this situation has changed dramatically following the acquisition by Gram-negative pathogens of novel β-lactamases capable of producing resistance to carbapenems (6). Some of these enzymes, like the KPC and GES types, have disseminated widely in clinical isolates and impose a significant therapeutic challenge, whereas others like NMCA, SFC, and SME are rarer (7–9). Over the years of intensive research, the contribution of some individual amino acid residues to the carbapenemase activity of these enzymes has been demonstrated (10–13). It was also observed that all class A carbapenemases contain a conserved interdomain disulfide bridge between Cys69 and Cys238 (in the standard Ambler numbering for class A enzymes (14)). As this bridge is absent from all previously characterized non-carbapenemases, it was postulated that it is essential for the carbapenemase activity of class A β-lactamases (15). Based on a comparison of the homology models of the NMCA and SME-1 carbapenemases with the structure of the class A β-lactamase, TEM-1, that is devoid of carbapenemase activity, it was proposed that presence of the disulfide bridge changes the geometry of the active site thus resulting in carbapenemase activity (15). To test this hypothesis, random mutagenesis of Cys69 and Cys238 residues in the SME-1 carbapenemase was conducted. However, selection of mutant libraries on plates with ampicillin and imipenem (both good substrates for SME-1) revealed that only clones harboring the wild-type enzyme with the intact disulfide bridge grew in the presence of these antibiotics (16), leaving the question regarding the role of the disulfide bridge in class A carbapenemases unresolved.
Here we substituted conserved cysteine residues in the class A β-lactamases GES-1, GES-2, GES-5, NMCA, SFC-1, and SME-1 and evaluated how the resulting disruption of the disulfide bridge affects activity of these enzymes against the carbapenem imipenem and other β-lactam antibiotics. These experiments, combined with structural studies, allowed us to elucidate the role of the disulfide bridge in the class A carbapenemases.
Results
Disruption of the Disulfide Bridge in the SME-1, NMCA, and SFC Carbapenemases
Previous studies (16) have demonstrated that disruption of the disulfide bridge in SME-1 renders Escherichia coli strains harboring disulfide bridge-deficient variants of the enzyme incapable of growing in the presence of ampicillin or imipenem (both are good substrates for SME-1). To elucidate the reason for the loss of the catalytic activity of these enzymes toward β-lactam substrates, we constructed C69G, C69A, C238G, C238A, and C69G/C238G (Ambler numbering scheme (14)) variants of SME-1 to determine their kinetic parameters and obtain structural information. However, expression of these enzymes in E. coli caused cessation of bacterial growth. Subsequently, we constructed C69G, C69A, C238G, and C238A variants of two other class A carbapenemases, NMCA and SFC-1, and the C69G/C238G variant of SFC-1. Similar to the results with SME-1, no β-lactam-resistant variants could be selected. We also recloned all variant enzymes into the expression vector pET24a+, but no bacterial growth was observed after protein induction, so we were unable to purify even a very small amount of any of them. These results strongly suggest that disruption of the disulfide bridge severely compromises the stability of SME-1, NMCA, and SFC-1 enzymes and show that they are unamenable for studies aimed at the evaluation of the role of the conserved disulfide bond in class A carbapenemases.
Disruption of the Disulfide Bridge in the GES-5 Carbapenemase
We subsequently evaluated the effect of disruption of the disulfide bond on the carbapenemase activity of GES-5, a representative of a large family of class A β-lactamases, the GES-type enzymes, which currently numbers 24 members (17). We substituted the cysteine residue at position 69 of the GES-5 carbapenemase with glycine, alanine, methionine, and leucine. Glycine, the smallest amino acid because of the lack of a side chain at the Cα carbon, occupies the smallest volume and provides maximum flexibility at the Cα carbon. Alanine is the smallest amino acid with a side chain, having more rigidity at the Cα atom and a Cβ atom that mimics the Cβ of a cysteine residue. Methionine is conserved at position 69 of enzymes belonging to the well studied TEM and SHV families of class A β-lactamases, which are very robust against various penicillin substrates but are devoid of carbapenemase activity. Finally, substitution of methionine 69 by leucine in TEM-1 (18) and SHV-1 (19) results in resistance to β-lactamase inhibitors. Plasmids producing variant enzymes were reintroduced into E. coli JM83 for minimal inhibitory concentration (MIC)3 testing. Of all constructed variant enzymes, only GES-5 with a C69G substitution retained the ability to produce resistance to β-lactams, although the MIC values for ampicillin, penicillin G, and cephalothin determined at the physiologically relevant temperature (37 °C) decreased 4-, 4-, and 16-fold, respectively, when compared with those for the wild-type GES-5 (Table 1). MICs of the carbapenem imipenem declined at least 4-fold and reached the background level for the control E. coli JM83 strain that does not produce β-lactamase. The three other substitutions, C69A, C69M, and C69L, compromised the bacterial growth and caused a precipitous decrease in the MIC values of all β-lactams tested, dropping them to the background level. The C69G-GES-5 was the only single amino acid substitution variant that maintained the ability to confer resistance to β-lactam antibiotics. Because the produced MIC levels were below those of the wild-type parental enzyme, we also constructed the C69G/C238G double substituted variant of GES-5 to evaluate whether addition of the substitution at the other cysteine forming the disulfide bridge would restore full activity. Surprisingly, introduction of the plasmid encoding the C69G/C238G enzyme into a recipient E. coli strain resulted in the arrest of bacterial growth.
TABLE 1.
The MICs of different β-lactam antibiotics against E. coli JM83 producing wild-type and variant GES β-lactamases
| Enzyme | MIC |
|||
|---|---|---|---|---|
| Ampicillin | Penicillin G | Cephalothin | Imipenem | |
| μg/ml | ||||
| GES-1 (wt) | 2048 | 1024 | 1024 | 0.25 |
| C69G | 1024 | 1024 | 128 | 0.25 |
| C69A | 4 | 16 | 8 | 0.25 |
| C69L | 4 | 16 | 8 | 0.25 |
| C69M | 4 | 16 | 8 | 0.25 |
| GES-2 (wt) | 2048 | 1024 | 512 | 0.25 |
| C69G | 1024 | 256 | 512 | 0.25 |
| C69A | 4 | 16 | 8 | 0.25 |
| C69L | 4 | 16 | 8 | 0.25 |
| C69M | 4 | 16 | 8 | 0.25 |
| GES-5 (wt) | 2048 (2048)a | 1024 (1024) | 1024 (1024) | 1 (1) |
| C69G | 512 (1024) | 256 (512) | 64 (128) | 0.25 (0.5) |
| C69A | 4 (4) | 16 (16) | 8 (8) | 0.25 (0.25) |
| C69L | 4 (4) | 16 (16) | 8 (8) | 0.25 (0.25) |
| C69M | 4 (4) | 16 (16) | 8 (8) | 0.25 (0.25) |
| Controlb | 4 (4) | 16 (16) | 8 (8) | 0.25 (0.25) |
a MIC values given in parentheses were determined at 30 °C
b Parental E. coli JM83 strain with pHF016 vector (no β-lactamase gene).
Disruption of the Disulfide Bridge in the GES-1 and GES-2 β-Lactamases
Unlike the GES-5 enzyme, which is capable of producing clinically relevant levels of resistance to carbapenems, the GES-2 and, to a larger extent, GES-1 are less efficient carbapenemases (20) such that they are best classified as extended spectrum β-lactamases. The difference in the carbapenemase activity of these enzymes results from changes in the amino acid residue at position 170, which is serine in GES-5, glycine in GES-1, and asparagine in GES-2 (20). To evaluate whether the disruption of the disulfide bond in GES-1 and GES-2 would affect the MIC values of β-lactam antibiotics to the same extent as seen for GES-5, we produced the C69G, C69A, C69M, and C69L variants of these two enzymes. Similar to what was observed with variants of the GES-5 β-lactamase, the disulfide bridge-deficient C69G variants of GES-1 and GES-2 conferred high levels of resistance to the penicillins ampicillin and penicillin G and the cephalosporin cephalothin, whereas the introduction of the C69A, C69M, and C69L substitutions resulted in growth retardation and loss of resistance (Table 1). These data show that the identity of the residue at position 170 of GES-1, GES-2, and GES-5 β-lactamases does not have a significant impact on the produced levels of resistance to β-lactam antibiotics when their disulfide bridge is disrupted by the C69G substitution.
Kinetics and Stability of the Wild-type and the C69G Variant of GES-5
We next evaluated the steady-state kinetic parameters for turnover of β-lactams by the wild-type GES-5 and its C69G variant (Table 2). Although the catalytic efficiency (kcat/Km) of the variant enzyme against ampicillin is very similar to that of the parental enzyme, its turnover rate (kcat) is 2.5-fold lower, and the apparent affinity is 3-fold higher (lower Km value). Against penicillin G, the C69G variant exhibits 2.5-fold lower catalytic activity entirely because of a lower turnover number. Although we were not able to determine the individual kcat and Km values for GES-5 and C69G-GES-5 for cephalothin (because of low affinity of the enzyme against this cephalosporin antibiotic), the catalytic efficiency (kcat/Km) against this substrate was 6-fold lower than that of the wild-type enzyme. A similar (5.4-fold) difference in the catalytic efficiency between the two enzymes was observed for imipenem, with the wild-type GES-5 exhibiting a 31-fold higher turnover number and a 6-fold lower affinity (higher Km value). Contrary to what has been observed for the SME-1 carbapenemase where disruption of the disulfide bridge severely destabilizes the enzyme, such disruption in the GES-5 β-lactamase does not abolish the ability of the C69G variant enzyme to confer resistance to β-lactam antibiotics but renders the enzyme slightly less efficient.
TABLE 2.
Steady-state kinetic parameters for GES5 and GES-5 C69G variant
| Substrate | C69G-GES-5 |
GES-5a |
||||
|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | kcat | Km | kcat/Km | |
| s−1 | μm | m−1 s−1 | s−1 | μm | m−1 s−1 | |
| Ampicillin | 8 ± 1 | 11 ± 3 | (7.3 ± 2.2) × 105 | 27 ± 1 | 32 ± 3 | (8.4 ± 0.9) × 105 |
| Penicillin G | 38 ± 1 | 80 ± 5 | (4.8 ± 0.3) × 105 | 107 ± 2 | 85 ± 5 | (1.3 ± 0.1) × 106 |
| Cephalothin | NAb | NA | (1.2 ± 0.1) × 105 | NA | NA | (7.5 ± 0.1) × 105 |
| Imipenem | 0.014 ± 0.001 | 0.26 ± 0.06 | (5.4 ± 1.3) × 104 | 0.44 ± 0.01 | 1.5 ± 0.3 | (2.9 ± 0.6) × 105 |
a The data for the GES-5 enzyme with imipenem were reported earlier (11).
b NA, not available.
To evaluate the effect of the disulfide bridge disruption on the stability of GES-5, we measured the change in the CD spectra of the wild-type and the C69G enzymes as a function of temperature. Disruption of the disulfide bridge resulted in a decrease of the Tm value of the enzyme from 44.9 to 39.2 °C (Fig. 1A). We subsequently evaluated the rate of ampicillin hydrolysis by the wild-type and the C69G enzymes after their preincubation at various temperatures for 60 min (Fig. 1B). The wild-type GES-5 retained most of its activity at temperatures up to 45 °C and retained some residual activity at 55 °C. The C69G enzyme started losing its activity following preincubation at 35 °C and was completely inactive at 45 °C. To test whether some loss of activity of the C69G-GES-5 enzyme at the physiologically relevant temperature (37 °C) may be partly responsible for the slight decrease in MICs of various β-lactam antibiotics, we re-evaluated the MICs produced by the C69G variant of GES-5 at a lower temperature (30 °C), at which the enzyme is expected to be more stable. Under these conditions, the MICs of all antibiotics consistently increased 2-fold when compared with the correspondent values obtained at 37 °C (Table 1). As a result, when determined at 30 °C, the MIC values of the C69G variant for penicillin G, ampicillin, and imipenem were only 2-fold below those produced by parental wild-type GES-5. These data demonstrate that although the disulfide bridge is important for the overall stability of the GES-5 β-lactamase, it does not contribute significantly to resistance to carbapenems or other β-lactams.
FIGURE 1.
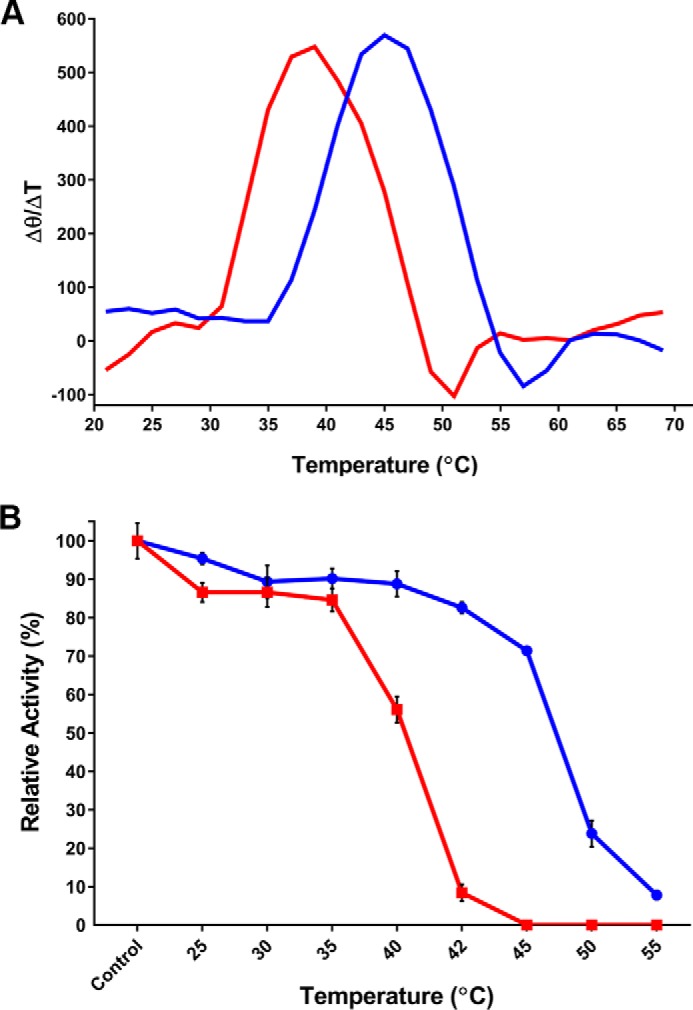
Thermal stability of GES-5 and the C69G variant. A, the first derivative of the thermal-induced unfolding CD curve of wild type (blue) and C69G (red) GES-5. The change in the CD signal as a function of temperature was monitored at 222 nm. The maxima correspond to the melting temperatures (Tm). B, kinetic thermal stability of wild type (blue circles) and C69G (red squares) GES-5. The activity against ampicillin was measured after incubating the enzymes at various temperatures for 60 min. The activity of control reactions that were run with non-incubated enzymes was defined as 100%.
Structure of the C69G-GES-5 Variant
To analyze the structural effects that disulfide bond disruption might have on the GES-5 enzyme, the crystal structure of C69G-GES-5 was determined at atomic resolution (0.96 Å) (Fig. 2A). The crystals of the variant, although grown under different conditions than those previously reported for wild-type GES-5 (12), had the same P21 space group and unit cell dimensions as the reported structure (PDB entry 4GNU) (Table 3), with two independent molecules in the asymmetric unit. Superposition of the apo C69G-GES-5 and wild-type GES-5 structures (rmsd = 0.14 Å for 265 matching Cα atoms) shows that they are essentially identical. The protein molecule has been previously described as comprising two structural domains (21), the first (domain 1, residues 20–59 and 212–285) composed of a five-stranded β-sheet flanked on one face by the N- and C-terminal helices, with the second predominantly α-helical domain (domain 2, residues 60–211) packed tightly against the opposite face of the domain 1 β-sheet (Fig. 2B). The active site is located at one end of the interface between the two structural domains, and residues from both sides of the interface are involved in substrate binding and catalysis (Fig. 2B). Although the disulfide bond is also located at the edge of the structural domain interface, ∼3.5–4 Å from the oxyanion hole that binds the free carbonyl oxygen atom of the acylated substrate, there is no evidence of any direct interaction between the disulfide and the bound substrate intermediate in wild-type GES-5. In the wild-type enzyme, the disulfide bond bridges the two structural domains, linking the N terminus of helix α2 (Cys69) with the loop between strands β3 and β4 (Cys238) (Fig. 3A). Superposition of wild-type and C69G-GES-5 variant structures based upon these two domains gave rmsd values of 0.14 and 0.10 Å for domains 1 and 2, respectively. The close similarity of the rmsd values for the two separate domains compared with the overall structure indicates that there has been negligible relative movement of the domains following the loss of the disulfide bond. The Cα atoms of Gly69 and Cys238 are within 0.2 Å of their positions in the wild-type structure (Fig. 3B). Moreover, the Cα-Cα distance is shorter in the variant than in the wild-type enzyme by ∼0.2 Å. Calculation of the substrate binding site volume using ICM PocketFinder (22) gives estimates of 235 and 230 Å2 for wild-type GES-5 and C69G-GES-5, respectively, which suggests that there is no difference in the size of the active site cleft following removal of the disulfide bond.
FIGURE 2.
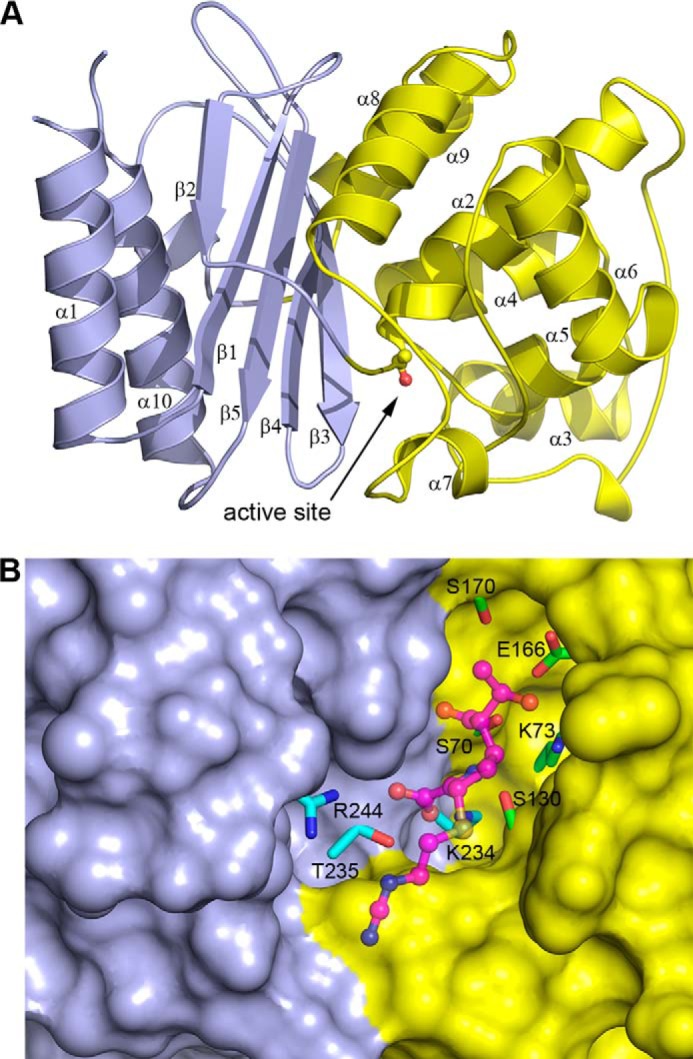
The structure of the C69G-GES-5 variant enzyme. A, ribbon representation of C69G-GES-5 showing the two structural domains colored light blue (domain 1) and yellow (domain 2) and indicating the secondary structure nomenclature. The location of the active site is indicated, with the side chain of the catalytic serine (Ser70) shown in ball and stick. B, surface representation of C69G-GES-5, with the two structural domains colored light blue (domain 1) and yellow (domain 2). The catalytically important residues in the active site are shown, colored cyan for domain 1 and green for domain 2. The bound imipenem substrate in the C69G-GES-5 complex is shown in magenta ball and stick.
TABLE 3.
Data collection and refinement statistics
| apo C69G-GES-5 | Imipenem C69G-GES-5 | |
|---|---|---|
| Data collection | ||
| Unit cell | ||
| a, b, c (Å) | 42.71, 80.94, 71.05 | 42.92, 81.48, 71.72 |
| β (°) | 101.4 | 102.0 |
| Resolution limits (Å) | 37.2–0.96 | 37.3–1.38 |
| Reflections, total/unique | 963,199/80,826 | 328,477/96,496 |
| Rmerge (%) | 5.2 (50.2)a | 4.2 (24.9) |
| Completeness (%) | 97.6 (73.5) | 97.4 (90.8) |
| Mean I/σI | 13.6 (2.2) | 17.1 (4.3) |
| CC½ | 99.7 (71.2) | 99.8 (92.5) |
| Wilson B-factor (Å2) | 6.3 | 12.2 |
| Redundancy | 3.5 (2.6) | 3.4 (2.9) |
| Refinement statistics | ||
| Rwork/Rfree (%) | 11.79/13.39 | 13.96/17.10 |
| Total atoms (protein/solvent) | 4278/950 | 4075/391 |
| B factors | ||
| Protein chain (Å2)b | 8.7 (A), 10.4 (B) | 13.9 (A), 15.9 (B) |
| Solvent (Å2) | 27.3 | 25.2 |
| Substrate (Å2) | 30.0 (A), 30.4 (B) | |
| rmsd values from ideality | ||
| Bonds (Å) | 0.008 | 0.006 |
| Angles (°) | 1.40 | 0.95 |
| Ramachandran plot | ||
| Favored and allowed (%) | 100.0 | 100.0 |
| Number disallowed | 0 | 0 |
a The values in parentheses refer to the highest resolution shell: 0.98–0.96 Å for the apo structure and 1.42–1.38 Å for the imipenem complex.
b The two values given are for the two independent molecules in the asymmetric unit.
FIGURE 3.
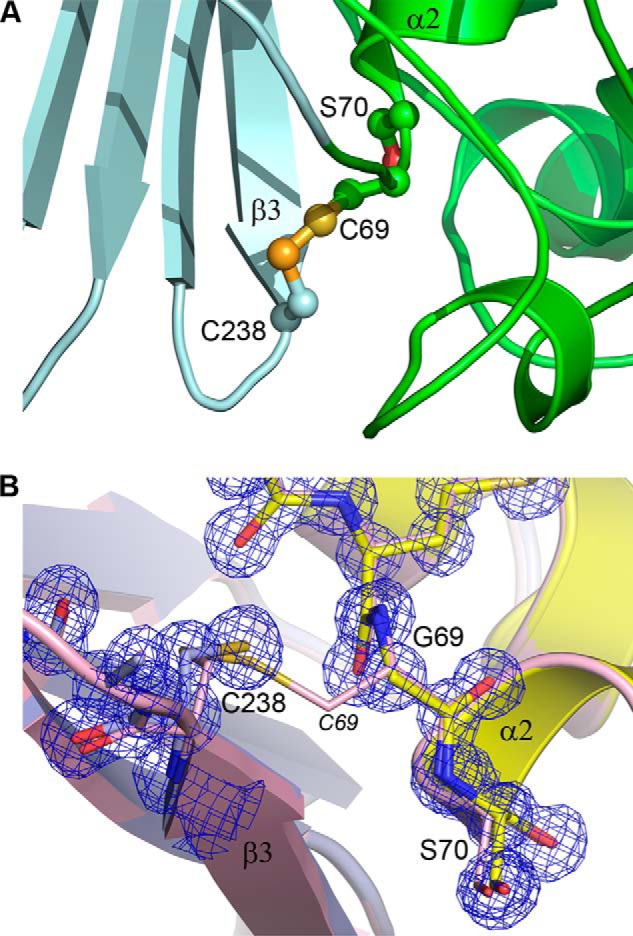
The disulfide bond in GES-5. A, the Cys69–Cys238 disulfide bond linking the N terminus of helix α2 and the C terminus of strand β3 in wild-type GES-5 (12). The two structural domains are light cyan (domain 1) and green (domain 2). B, close-up view of the site of substitution in C69G-GES-5, showing domain 1 on the left in light blue and domain 2 on the right in yellow. The final refined 2Fo − Fc electron density is shown as a blue mesh (contoured at 1.5 σ). The wild-type GES-5 structure is shown as light pink ribbons and sticks superimposed on the C69G-GES-5 structure based upon domain 1 only. The intact C69–Cys238 disulfide in the wild-type structure is shown with the Cys69 labeled in italics.
The C69G-GES-5-Imipenem Complex
Inspection of the active sites of the two independent molecules in the asymmetric unit of the C69G variant of GES-5 following soaking in crystallization buffer containing imipenem, showed strong residual Fo − Fc density near the catalytic Ser70 (Ambler numbering (14)) in both molecules in the asymmetric unit, an indication that acylation had occurred (Fig. 4, A and B). The structure was partially refined, and imipenem was added into both active sites. The occupancies of the substrate were refined using PHENIX.REFINE (23), giving values of 0.72 and 0.76 for the two independent molecules in the asymmetric unit. Final omit Fo − Fc density for the imipenem in molecule A is shown in Fig. 4C. The initial 2Fo − Fc and Fo − Fc electron density prior to the addition of imipenem to the structure showed density for the acyl bond, the free C7 carbonyl group, the 6α-hydroxyethyl side chain, the pyrolline ring, and the sulfur atom attached to the C2 atom (Fig. 5). Although the density for the position of the sulfur atom most strongly suggested the presence of the S stereoisomer of the Δ1 tautomer (Figs. 4 and 5), additional weaker peaks in the two molecules present in the asymmetric unit also indicated the possible presence of both the Δ2 tautomer, with the sulfur atom coplanar with the pyrolline ring (Fig. 4C), and the R stereoisomer of the Δ1 tautomer. Because of difficulties in modeling the three isomeric forms simultaneously, the intermediate was refined as the S stereoisomer, which is the major conformation. The degrees of occupancy of the acyl intermediates are close to those reported in the wild-type enzyme (0.63 and 0.67 for the two independent molecules in the asymmetric unit) (12). For the wild-type GES-5, the equilibration of the enzyme-imipenem system at approximately two-thirds occupancy was indicative of the acylation rate being faster than the rate of deacylation (12). Our structural information with the C69G GES-5 variant is in strong agreement with this proposition.
FIGURE 4.

Imipenem binding in C69G-GES-5. A, initial residual Fo − Fc difference electron density in the active site of C69G-GES-5 soaked in imipenem for molecule A. B, initial residual Fo − Fc difference electron density in the active site of C69G-GES-5 soaked in imipenem for molecule B. In both panels, the density (colored light brown and contoured at 2.5 σ), was calculated following structure solution by molecular replacement but prior to refinement. The location of the final refined imipenem is shown as thin magenta balls and sticks, built as the S stereoisomer of the Δ1 tautomer. The hydrogen bonding interactions with the C7 carbonyl of imipenem in the oxyanion hole are not shown for clarity. The two structural domains are colored light blue (domain 1) and yellow (domain 2). C, stereo view of the omit Fo − Fc difference electron density for the Ser70 side chain and the imipenem in the active site of C69G-GES-5 molecule A. The density (colored blue and contoured at 2.8 σ) was calculated following structure refinement. The final refined imipenem is shown as thin magenta balls and sticks. The imipenem bound to wild-type GES-5 (12) is shown as thin cyan sticks, present in that structure as the Δ2 tautomer.
FIGURE 5.

Chemical structure of imipenem. A, the intact antibiotic with standard atom numbering. B, the hydrolyzed form of imipenem showing the three possible isomers. The S and R stereoisomers of the Δ1 tautomer can interconvert via the Δ2 tautomer.
The pyrroline ring of the imipenem molecule is in an orientation similar to that observed in the wild-type GES-5-imipenem structure (12). The carbonyl oxygen projects into the oxyanion hole adjacent to the catalytic Ser70 and accepts two hydrogen bonds from the amide nitrogen atoms of Ser70 and Thr237. Although the oxyanion hole is adjacent to the disulfide pocket, there is no direct interaction between the carbonyl oxygen and the free Cys238 residue. On one side of the pyrroline ring, the carboxylate moiety of imipenem makes an electrostatic interaction with the guanidinium group of Arg244 and additional hydrogen bonds with the side chains of Thr235 and Thr237 (Fig. 4, A and B). The N4 atom of the pyrroline ring makes a hydrogen bond to the side chain of the universally conserved Ser130. On the opposite side of the ring, the 6α-hydroxyethyl group is observed in two orientations in the two independent molecules in the asymmetric unit: one that is similar to that observed in the wild-type-imipenem complex and another that is rotated by ∼45° (Fig. 6). In both configurations, there is a hydrogen bonding interaction with the side chain of Asn132, but in the latter configuration the methyl group has moved away from the Glu166 side chain sufficiently to allow for the binding of a water molecule in a location between the glutamate side chain and the acylated Ser70 (Fig. 6). The amino tail of imipenem projects into the external milieu and makes no additional interactions with the protein. Moreover, the electron density for the tail is not observed (Fig. 4), which implies a high degree of conformational flexibility in this part of the substrate and may be due to the concurrent presence of the intermediate in all three possible isomeric states. Overall, our data show that acylation of the disulfide bond-deficient variant of GES-5 by the carbapenem substrate imipenem does not result in any changes in the architecture of the active site of the enzyme when compared with that of the acyl-enzyme complex of the wild-type GES-5 with imipenem.
FIGURE 6.

Conformation differences in the bound imipenem. The acylated imipenem substrate in the two independent C69G-GES-5 molecules present in the asymmetric unit is shown. In molecule B, the rotation of the 6α-hydroxyethyl group (6α-HE) provides sufficient space to allow a water molecule to bind between Glu166 and catalytic Ser70. In molecule A, the carbon atom of the 6α-HE blocks this water-binding site.
The Domain Interface in the Class A Carbapenemases
To gain insights into why the disruption of the disulfide bond of GES-5 results in an active enzyme, whereas in SME-1, NMCA, and SFC-1 enzyme activity is completely compromised, we undertook an analysis of the domain interface formation in the wild-type GES-5 and C69G-GES-5, along with three other class A carbapenemases, SME-1, NMCA, and SFC-1. The results are summarized in Table 4. In the wild-type GES-5 structure, a total of 2900 Å2 (21%) of surface area is buried upon formation of the interface between the two domains. Surfaces in contact are very complimentary in their topology (Fig. 7), and a total of 23 hydrogen bonds serve to hold the two sides of the enzyme together, along with 32 additional interactions through 10 bridging water molecules. Following replacement of Cys69 with glycine, there is essentially no difference in the surface area buried or the interdomain interactions (Table 4). Comparison of the same two domains (domain 1, residues 24–65 and 217–291; domain 2, residues 66–216; standard Ambler numbering (14)) of the wild-type structures of SME-1 (PDB code 1DY6) (24), NMCA (PDB code 1BUE) (25), and SFC-1 (PDB code 4EQI) (10) gives approximately the same buried surface area at the interface (2850–2900 Å2). Importantly, however, there are less hydrogen bonding interactions between the two structural domains in SME-1, NMCA, and SFC-1, both protein-protein and water-mediated, and the subsequent decrease in interdomain hydrogen bonding energy may be sufficient to destabilize the interface following the loss of the disulfide bond. Because the active site is at the domain interface, any movement of one part of the molecule relative to the other may alter the structure of the active site, and this could lead to a decrease in or a complete abolition of enzyme activity.
TABLE 4.
Domain-domain interactions in class A carbapenemases
| Enzyme | Domain interface surface area | Interdomain hydrogen bonds |
||
|---|---|---|---|---|
| Protein-protein | Water-mediateda | Total | ||
| Å2 | ||||
| GES-5 C69G | 3050 | 23 | 32 (11) | 55 |
| GES-5 | 2900 | 25 | 27 (10) | 52 |
| SME-1 | 2900 | 18 | 23 (8) | 41 |
| NMCA | 2850 | 17 | 23 (8) | 40 |
| SFC-1 | 2950 | 16 | 29 (12) | 45 |
a The total number of water molecules that interact with both domains is given in parentheses.
FIGURE 7.
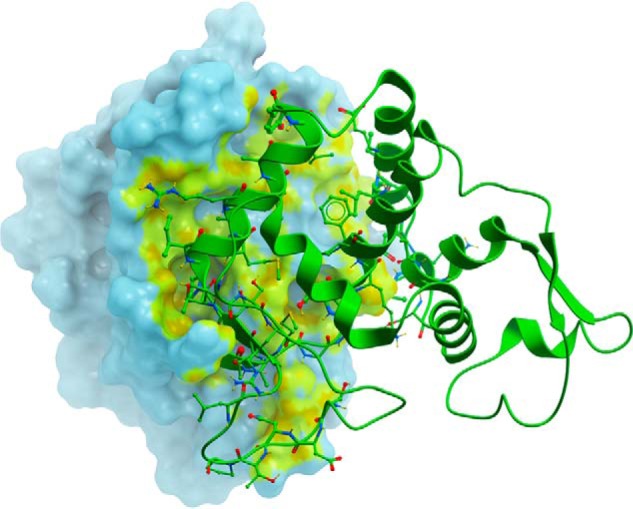
The domain interface in GES-5. The molecular surface representation of domain 1 (light cyan) of wild-type GES-5 shows the region of contact (highlighted yellow-green) with the domain 2 (green ribbons). The residues from domain 2 that are involved in interdomain contacts are shown as green balls and sticks.
To further analyze the effects of disruption of the disulfide bond in GES-5, and to ascertain whether any other amino acid residue could be accommodated in the disulfide pocket, the Cys69 in in the x-ray structure of GES-5 was substituted in silico to 18 common amino acids (excluding cysteine, because that would reform the disulfide bond, and proline, because the main chain conformation of residue 69 is not compatible with a proline in this position). Protein stability following substitution was measured as a difference in the free energy of binding between each simulated variant and the GES-5 enzyme (defined as ΔΔGbinding = ΔGvariant − ΔGGES-5), calculated using ICM-Pro (supplemental Table S1). The substitution of cysteine by all but three amino acid residues (glycine, alanine, and serine) resulted in a net loss in free energy (indicated by a positive ΔΔGbinding value), suggesting that anything other than these residues at position 69 were unfavored. This included leucine and methionine, two of the three of the substitutions generated in this study. Analysis of the simulated C69L and C69M variants shows that their side chains make severe steric clashes with parts of the protein structure surrounding the disulfide pocket.
Because methionine at position 69 is native to the non-carbapenemases TEM-1 and SHV-1, we superimposed these two structures (PDB codes 1BTL and 1SHV, respectively) onto the GES-5 structure to investigate how this residue is accommodated in the non-carbapenemases. Both TEM-1 and SHV-1 have a glycine at position 238, and in these enzymes the loop carrying this residue has moved away from the N terminus of helix α2 by ∼1.0 Å, subsequently opening the disulfide pocket. In silico mutation of Cys238 to glycine in GES-5, followed by calculation of the free energy of binding for the two domains gave positive ΔΔGbinding values for both molecules in the asymmetric unit, which suggests that the disulfide pocket in GES-5 is too small to allow for the Met69/Gly236 pairing and that the β3-β4 loop movement observed in TEM-1 and SHV-1 is necessary to accommodate the methionine.
Substitution by glycine, alanine, and serine resulted in a net gain in free energy (indicated by negative ΔΔGbinding values of −1.82, −2.00, and −2.27, respectively, averaged over both molecules in the asymmetric unit), suggesting that these residues would be favored in this position. Our experimental data confirm that the C69G substitution indeed is well tolerated. Although we did not construct the serine variant at this position, our experimental data show that even the smaller C69A substitution severely compromises bacterial growth, which could result from a severe destabilization of the enzyme. Our high resolution structural data show that disruption of the disulfide bond in GES-5 by the C69G substitution brings the Cα-Cα atoms of residues 69 and 238 between 0.1 and 0.2 Å closer to each other. To investigate whether this would result in a steric hindrance between Cys238 and Ala69, we measured the ΔΔGbinding values for various in silico variants of C69G GES-5. Substitution of Gly69 to any of the 17 amino acid residues (excluding glycine, cysteine, and proline) resulted in a net loss in free energy (supplemental Table S1). The ΔΔGbinding values for the Gly69 variant were consistently higher than those for the wild-type GES-5. As a result, the ΔΔGbinding values for Ala69 and Ser69 became positive, suggesting that anything other than a glycine at position 69 was unfavored. These data support our experimental results that show that glycine at position 69 is indeed well tolerated.
Discussion
A previous study, based on comparison of the structure of the TEM-1 β-lactamase and the homology models of the NMCA and SME-1 carbapenemases, proposed that the disulfide bridge in NMCA and SME-1 plays a role in their carbapenemase activity by altering the structure of strand β3 (relative to TEM-1). This consequently affects the topology of the oxyanion hole, thus allowing for a better accommodation of the carbonyl of carbapenem substrates, which is expected to increase efficiency of deacylation (15). Contrary to this assertion, our analysis of the x-ray crystal structures of the SME-1 and GES-5 carbapenemases shows that strand β3 in these enzymes is in exactly the same location and conformation as the corresponding strand in the TEM-1 β-lactamase. Moreover, the oxyanion hole in TEM-1 is essentially identical in size to that in GES-5 and SME-1, and the disposition of the amide nitrogen atoms that serve as hydrogen bond donors to the carbonyl oxygen of the bound substrate is also identical in all three enzymes. Thus, it is highly unlikely that the flipping out of the carbonyl from the oxyanion hole observed in the TEM-1-imipenem structure (26) is a consequence of the absence of a disulfide bond.
To elucidate the role of the disulfide bridge in class A carbapenemases, we disrupted this bridge in the GES-1, GES-2, GES-5, SME-1, NMCA, and SFC-1 enzymes and evaluated the impact of these disruptions on the conferred levels of resistance, enzyme stability, kinetics, and structures. The single C69G, C69A, C238A, and C238A variants of SME-1, NMCA, and SFC-1 and the double C69G/C238G SME-1 and SFC-1 variants arrested bacterial growth when expressed in E. coli, an indication that stability is severely compromised. These results explain why extensive mutagenesis of cysteines 69 and 238 in SME-1 failed to generate any viable variants capable of producing resistance to the β-lactam antibiotics ampicillin or imipenem (16). In stark contrast to what was observed with the disulfide bridge-deficient variant of SME-1, NMCA, and SFC-1, our studies demonstrate that disruption of the disulfide bridge in GES-5 (a representative of the GES family of class A carbapenemases), by substitution of Cys69 with glycine, yields an active enzyme. Similarly, the C69G substitutions in two other GES-type enzymes, GES-1 and GES-2, had a relatively insignificant impact on levels of resistance to β-lactam antibiotics. Circular dichroism experiments demonstrated that disruption of the disulfide bridge results in the decrease of the Tm value of the enzyme by 5.7 °C, and kinetic studies showed that the C69G substitution in GES-5 diminishes the stability of the enzyme at temperatures above 35 °C. Despite some loss of activity at higher temperatures, the variant enzyme is still capable of producing high levels of resistance to some β-lactam substrates at 37 °C, and at lower temperature (30 °C) the levels of resistance become similar to those conferred by wild-type GES-5. These data show that the disulfide bond contributes to the stability of GES-type enzymes but not their ability to produce resistance to β-lactam antibiotics.
These conclusions are strongly supported by our x-ray crystallographic studies. Structures of the C69G variant of GES-5 and its binary complex with imipenem show that disruption of the disulfide bridge does not disturb the overall structure of the enzyme and has no effect on the binding mode of the carbapenem substrate. Our structural analyses strongly suggest that significantly weaker hydrogen bonding interactions in SME-1, NMCA, and SFC-1 (relative to GES-5) likely cause protein destabilization following loss of the disulfide bond. In GES-5 a wider network of hydrogen bonding interactions could serve to counteract any destabilization that might occur upon loss of the disulfide bridge, albeit only at lower temperatures, because we do observe a decline of activity in the C69G variant when the temperature is raised above 35 °C (Fig. 1B).
It should be noted that of the four variants of wild-type GES enzymes studied, only replacement of Cys69 with a glycine gives rise to active enzymes (Table 1). In silico substitutions of residue 69 in the structure of the wild-type GES-5 show that two of these residues, glycine and alanine, are favored in this position, whereas leucine and methionine are not favored. However, analysis of the structure of the disulfide-deficient C69G-GES-5 shows that two domains of this variant come closer to each other when compared with the wild-type GES-5, which could make alanine at position 69 also unfavored. Indeed, when in silico substitutions of residue 69 were performed using the C69G-GES-5 structure, not only leucine and methionine but also alanine at this position invariably lead to steric clashes that are expected to destabilize the three-dimensional structure of the resultant variant proteins. As a result, substitution of Cys69 in GES-1, -2, and -5 by all three unfavored residues leads to very poor bacterial growth and a precipitous drop in MICs. Moreover, we were unable to express and isolate any of the C69A, C69L, or C69M GES variant enzymes, which indicates that these substitutions significantly compromise enzyme stability.
In conclusion, results of our studies demonstrate that the disulfide bond in the GES family of class A carbapenemases does not play a direct role in their activity against various β-lactam antibiotics, including carbapenems, but rather is important for maintaining the stability of the interdomain interface and, ultimately, the structural integrity of their active sites. We also have shown that disruption of the disulfide bridge in other families of class A carbapenemases severely destabilizes these enzymes, which results in the retardation or arrest of bacterial growth. Close structural similarity between the GES-type and other class A carbapenemases also suggests that the disulfide bridge is not essential for the carbapenemase activity of these enzymes.
Experimental Procedures
Cloning, MIC Testing, Expression, and Purification
The genes for the C69G, C69A, C69M, and C69L variants of GES-1, GES-2, and GES-5 enzymes and C69G, C69A, C238G, and C238A variants of the SME-1, NMCA, and SFC-1 carbapenemases along with the C69G/C238G double mutant variants of GES-5, SME-1, and SFC-1 were custom synthesized (GenScript) with unique NdeI and HindIII restriction sites at the 3′ and 5′ ends, respectively, and cloned into the vector pHF016. Plasmids harboring these genes were subsequently used to electroporate E. coli JM83, and the resulting transformants were used for MIC testing as previously described (11).
For enzyme expression the genes were cloned between the NdeI and HindIII sites of the pET24a (+) vector and transformed into E. coli BL21 (DE3)-competent cells. First, pilot experiments were performed to test for optimal temperature for expression of the enzymes. The cells were grown overnight at 37 °C in 5 ml of LB medium supplemented with 50 μg/ml of kanamycin. The cells were diluted 100-fold into 5 ml of fresh LB broth supplemented with 50 μg/ml of kanamycin and incubated at 37 °C until A600 reached 0.8. The cells were induced with 1 mm isopropyl β-d-thiogalactopyranoside at 12, 23, and 37 °C overnight. The induced cells were harvested by centrifugation at 4 °C at 4500 × g for 15 min, resuspended in 1 ml of 20 mm Tris, pH 7.5, and sonicated. Both the total suspension and the supernatant (obtained after pelleting debris at 16,000 × g for 15 min) were checked for the ability to turn over chromogenic cephalosporin nitrocefin as follows. First, we added 30 μl of the undiluted sample of the total suspension or the supernatant into a microtiter plate and next added 5 μl of 1 mm nitrocefin. Change of the color from yellow to red indicated presence of active enzyme. If the change was too fast to monitor by eye, various dilutions of enzyme-containing samples were made to evaluate which sample contained more active enzyme. Of all the constructed variants only C69G variants of GES-1, GES-2, and GES-5 enzymes were active. All samples also were analyzed by SDS-PAGE to evaluate the amount of produced enzyme.
For large scale isolation of the C69G GES-5, we grew 250 ml of bacterial cells as described above. After induction with isopropyl β-d-thiogalactopyranoside, the cells were incubated overnight at 23 °C. The induced cells were harvested by centrifugation at 4 °C at 4500 × g for 15 min, resuspended in 30 ml of 20 mm Tris, pH 7.5, and sonicated. Following sonication, cell debris was pelleted at 20,000 × g, and the supernatant was applied on the pre-equilibrated Macro-Prep DEAE support anion exchange chromatography column. The column was washed with 300 ml of 20 mm Tris buffer, pH 7.5. The protein was eluted with a linear gradient of NaCl (0–1 m NaCl) between 100 and 200 mm. The eluted fractions were evaluated for the activity against nitrocefin as described above and subsequently analyzed by SDS-PAGE. The most pure fractions were pooled together and dialyzed against 20 mm MES buffer, pH 6.8. The proteins were loaded onto Macro-Prep High S Support cation exchange column. The column was washed with 300 ml of 20 mm MES buffer, pH 6.8, and the protein was eluted with a linear gradient of NaCl (0–1 m) between 400 and 500 mm. The purity of the proteins was analyzed by SDS-PAGE gel. The proteins were dialyzed against 20 mm HEPES buffer, pH 7.6, and concentrated to 8–10 mg/ml.
Enzyme Kinetics
Steady-state kinetic parameters were measured in 50 mm NaPi (pH 7.0), 100 mm NaCl, and varying β-lactam substrates concentrations. The reactions were started by the addition of the enzyme. Changes of absorbance were followed on a Cary UV-visible spectrophotometer at the following wavelengths: ampicillin (λ = 240 nm and Δϵ = −538 m−1 cm−1), penicillin G (λ = −240 nm and Δϵ = − 500 m−1 cm−1), cephalothin (λ = 262 nm and Δϵ = −7960 m−1 cm−1), and imipenem (λ = 297 nm and Δϵ = −10930 m−1 cm−1). The linear phases of progress curves were analyzed, and the steady-state kinetic parameters were calculated as previously described (11). For cephalothin we were not able to measure individual kcat and Km values because of low affinity (high Km value) of the enzyme against this antibiotic. Instead, to determine the kcat/Km value, we monitored the progress curve of full substrate hydrolysis using a substrate concentration well below the Km value. The data were fit to the first order equation At = A∞ + (A0 − A∞)e−kt (27) where At is the absorbance at time t, A0 is the initial absorbance, and A∞ is the final absorbance. In this equation the observed first order rate constant k = kcat/Km[E], where the [E] is the concentration of enzyme in the reaction.
The thermal dependence of enzyme activity was assayed by incubating wild type and C69G-GES-5 (2.5 μm) at various temperatures for 60 min. Next, reactions containing 200 μm ampicillin in 50 mm NaPi (pH 7.0), 100 mm NaCl were initiated by the addition of enzyme (25–100 nm), and the change in absorbance caused by hydrolysis of ampicillin was monitored at 235 nm for 1–2 min. To determine the relative remaining activity, observed rates (kobs = v/[E]) were compared with those obtained with no preincubation of the enzyme.
CD Spectroscopy
CD spectra were measured using a J-815 circular dichroism spectropolarimeter and the Temperature Interval Scan Measurement function of Spectra Manager II (Jasco International Co., Ltd., Tokyo, Japan). Scans were performed with wild type and C69G-GES-5 (5 μm) in 10 mm NaPi (pH 7.0), 20 mm NaCl using a 2-mm-path length quartz cuvette (Starna). Spectra were recorded in triplicate in the range of 190–250 nm using a 100-nm/min scan rate with a 0.5-nm wavelength pitch and 1-nm bandwidth. The thermal dependence of the spectra was accessed by collecting scans every 2 °C from 20 to 70 °C with a 2 °C/min temperature gradient and a 60-s equilibration time. All spectra were background corrected by subtracting the buffer baseline and converted to mean residue ellipticity [θ] by using the relationship [θ] = M/(p × C × n), where M is ellipticity measured in millidegrees, p is the path length in millimeters, C is the molar concentration of protein, and n is the number of amino acid residues (28). The melting temperature (Tm) of the unfolding curve at 222 nm was determined by performing nonlinear regression using Prism 7 (GraphPad Software, Inc.) as described by Greenfield (29).
Crystallization, Data Collection, and Refinement
Crystals of the C69G-GES-5 variant were obtained under at least seven conditions from the PEG/Ion 2 screen (Hampton Research). The crystals from all conditions belonged to space group P21 with similar cell dimensions. The crystals from several conditions were screened for diffraction quality, with those grown from 0.2 m d,l-malic acid, pH 7.0, 20% PEG3350, showing diffraction better than 1.0 Å resolution. Data from a single apo C69G-GES-5 crystal were collected on Beamline BL14–1 at the Stanford Synchrotron Radiation Lightsource (SSRL) using a MX-325 CCD detector (Rayonix). The diffraction data, comprising 360 images (0.5° rotation and 10 s of exposure/image), were indexed using XDS (30) and scaled with AIMLESS (31) from the CCP4 package (32). The data statistics are summarized in Table 3.
The C69G-GES-5 structure was solved by molecular replacement using MOLREP (33) (as implemented in the CCP4 program package), with the refined structure of wild-type GES-5 (PDB code 4GNU) as the starting model, with the Cys69 residue truncated to a glycine. The structure was initially refined using REFMAC5 (34) in the CCP4 package. The structure was rebuilt, and water molecules were added with COOT (35). All atomic displacement parameters were refined anisotropically, along with alternate side chain conformations and occupancies, with PHENIX.REFINE (23). The refinement data are summarized in Table 3.
The C69G-GES-5 imipenem complex was formed by soaking preformed crystals for various times ranging from 1 to 5 min in crystallization buffer augmented with imipenem at either 25 or 50 mm prior to flash cooling in liquid nitrogen. The crystals were then screened for diffraction quality on SSRL Beamline BL7-1 using the Stanford Automated Mounter (36, 37) and the high throughput screening tools in the Blu-Ice software (38, 39). A crystal soaked for 2 min in 50 mm imipenem was chosen for subsequent diffraction data collection on SSRL Beamline BL12-2 using a PILATUS 6M pixel array detector running in shutterless mode. The data comprised 900 images (0.2° rotation and 0.2 s of exposure/image), and was processed with XDS (30) and AIMLESS (31). The structure was solved using the refined apo C69G-GES-5 structure as the starting model for molecular replacement with MOLREP (33) and refined to 1.38 Å resolution with PHENIX.REFINE (23). All protein atoms were refined anisotropically. The data collection and structure refinement statistics are given in Table 3.
Computational Methods
All figures except Figs. 1, 5, and 7 were prepared with PyMOL (40). Fig. 1 was prepared with Prism 7 (GraphPad), Fig. 5 was generated using Chem3D 15.0.0.106 (PerkinElmer), and Fig. 7 was made with ICM-Pro 3.8–4 (Molsoft) (41). The in silico substitutions of Cys69 in GES-5 and Gly69 in C69G-GES-5 was performed using the MolMechanics feature of ICM-Pro.
Accession Numbers
The atomic coordinates and the structure factors for apo C69G-GES-5 and imipenem-C69G-GES-5 were deposited to the Protein Data Bank (42) with PDB codes 5F82 and 5F83, respectively.
Author Contributions
C. A. S. collected diffraction data, solved and analyzed the structure, and wrote the paper. Z. N. conducted kinetic and crystallization experiments. M. T. performed microbiological and crystallization experiments. H. F. and N. K. S. conducted mutagenesis and kinetics experiments. S. B. V. conceived the idea for the project, designed the experiments, and wrote the paper.
Supplementary Material
This work was supported by National Institutes of Health Grant R01AI089726 (to S. B. V.). The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5F82 and 5F83) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Table S1.
- MIC
- minimal inhibitory concentration
- rmsd
- root mean square deviation
- PDB
- Protein Data Bank
- SSRL
- Stanford Synchrotron Radiation Lightsource.
References
- 1. Bush K. (2013) Proliferation and significance of clinically relevant β-lactamases. Ann. N.Y. Acad. Sci. 1277, 84–90 [DOI] [PubMed] [Google Scholar]
- 2. Ambler R. P. (1980) The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 289, 321–331 [DOI] [PubMed] [Google Scholar]
- 3. Jaurin B., and Grundström T. (1981) AmpC cephalosporinase of Escherichia coli K-12 has a different evolutionary origin from that of β-lactamases of the penicillinase type. Proc. Natl. Acad. Sci. U.S.A. 78, 4897–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ouellette M., Bissonnette L., and Roy P. H. (1987) Precise insertion of antibiotic resistance determinants into Tn21-like transposons: Nucleotide sequence of the OXA-1 β-lactamase gene. Proc. Natl. Acad. Sci. U.S.A. 84, 7378–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paterson D. L., and Bonomo R. A. (2005) Extended-spectrum β-lactamases: A clinical update. Clin. Microbiol. Rev. 18, 657–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walther-Rasmussen J., and Høiby N. (2007) Class A carbapenemases. J. Antimicrob. Chemother. 60, 470–482 [DOI] [PubMed] [Google Scholar]
- 7. Nordmann P., Dortet L., and Poirel L. (2012) Carbapenem resistance in Enterobacteriaceae: here is the storm! Trends Mol. Med. 18, 263–272 [DOI] [PubMed] [Google Scholar]
- 8. Nordmann P., Naas T., and Poirel L. (2011) Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 17, 1791–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel G., and Bonomo R. A. (2013) “Stormy waters ahead”: global emergence of carbapenemases. Front. Microbiol. 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fonseca F., Chudyk E. I., van der Kamp M. W., Correia A., Mulholland A. J., and Spencer J. (2012) The basis for carbapenem hydrolysis by class A β-lactamases: A combined investigation using crystallography and simulations. J. Am. Chem. Soc. 134, 18275–18285 [DOI] [PubMed] [Google Scholar]
- 11. Frase H., Shi Q., Testero S. A., Mobashery S., and Vakulenko S. B. (2009) Mechanistic basis for the emergence of catalytic competence against carbapenem antibiotics by the GES family of β-lactamases. J. Biol. Chem. 284, 29509–29513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith C. A., Frase H., Toth M., Kumarasiri M., Wiafe K., Munoz J., Mobashery S., and Vakulenko S. B. (2012) Structural basis for progression toward the carbapenemase activity in the GES family of β-lactamases. J. Am. Chem. Soc. 134, 19512–19515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stewart N. K., Smith C. A., Frase H., Black D. J., and Vakulenko S. B. (2015) Kinetic and structural requirements for carbapenemase activity in GES-type β-lactamases. Biochemistry 54, 588–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ambler R. P., Coulson A. F.., Frère J. M., Ghuysen J. M., Joris B., Forsman M., Levesque R. C., Tiraby G., and Waley S. G. (1991) A standard numbering scheme for the class-A β-lactamases. Biochem. J. 276, 269–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raquet X., Lamotte-Brasseur J., Bouillenne F., and Frère J. M. (1997) A disulfide bridge near the active site of carbapenem-hydrolyzing class A β-lactamases might explain their unusual substrate profile. Proteins 27, 47–58 [DOI] [PubMed] [Google Scholar]
- 16. Majiduddin F. K., and Palzkill T. (2003) Amino acid sequence requirements at residues 69 and 238 for the SME-1 β-lactamase to confer resistance to β-lactam antibiotics. Antimicrob. Agents Chemother. 47, 1062–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tondi D., Cross S., Venturelli A., Costi M. P., Cruciani G., and Spyrakis F. (2016) Decoding the structural basis for carbapenem hydrolysis by class A β-lactamases: fishing for a pharmacophore. Curr. Drug Targets 17, 983–1005 [DOI] [PubMed] [Google Scholar]
- 18. Delaire M., Labia R., Samama J. P., and Masson J. M. (1992) Site-directed mutagenesis at the active site of Escherichia coli TEM-1 β-lactamase: suicide inhibitor-resistant mutants reveal the role of arginine 244 and methionine 69 in catalysis. J. Biol. Chem. 267, 20600–20606 [PubMed] [Google Scholar]
- 19. Helfand M. S., Hujer A. M., Sönnichsen F. D., and Bonomo R. A. (2002) Unexpected advanced generation cephalosporinase activity of the M69F variant of SHV β-lactamase. J. Biol. Chem. 277, 47719–47723 [DOI] [PubMed] [Google Scholar]
- 20. Smith C. A., Caccamo M., Kantardjieff K. A., and Vakulenko S. (2007) Structure of GES-1 at atomic resolution: insights into the evolution of carbapenamase activity in the class A extended-spectrum β-lactamases. Acta Crystallogr. D Biol. Crystallogr. 63, 982–992 [DOI] [PubMed] [Google Scholar]
- 21. Jelsch C., Lenfant F., Masson J. M., and Samama J. P. (1992) β-Lactamase TEM1 of E. coli: crystal structure determination at 2.5 A resolution. FEBS Lett. 299, 135–142 [DOI] [PubMed] [Google Scholar]
- 22. An J., Totrov M., and Abagyan R. (2005) Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell. Proteomics 4, 752–761 [DOI] [PubMed] [Google Scholar]
- 23. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sougakoff W., L'Hermite G., Pernot L., Naas T., Guillet V., Nordmann P., Jarlier V., and Delettré J. (2002) Structure of the imipenem-hydrolyzing class A β-lactamase SME-1 from Serratia marcescens. Acta Crystallogr. D Biol. Crystallogr. 58, 267–274 [DOI] [PubMed] [Google Scholar]
- 25. Swarén P., Maveyraud L., Raquet X., Cabantous S., Duez C., Pédelacq J. D., Mariotte-Boyer S., Mourey L., Labia R., Nicolas-Chanoine M. H., Nordmann P., Frère J. M., and Samama J. P. (1998) X-ray analysis of the NMC-A β-lactamase at 1.64-Å resolution, a class A carbapenemase with broad substrate specificity. J. Biol. Chem. 273, 26714–26721 [DOI] [PubMed] [Google Scholar]
- 26. Maveyraud L., Mourey L., Kotra L. P., Pedelacq J. D., Guillet V., Mobashery S., and Samama J. P. (1998) Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class A β-lactamases from antibiotic-resistant bacteria. J. Am. Chem. Soc. 120, 9748–9752 [Google Scholar]
- 27. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd ed., Wiley-VCH, New York [Google Scholar]
- 28. Greenfield N. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greenfield N. (2006) Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 1, 2527–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 34. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 35. Emsley P., and Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 36. Cohen A. E., Ellis P. J., Miller M. D., Deacon A. M., and Phizackerley R. P. (2002) An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J. Appl. Crystallogr. 35, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Russi S., Song J., McPhillips S. E., and Cohen A. E. (2016) The Stanford Automated Mounter: pushing the limits of sample exchange at the SSRL macromolecular crystallography beamlines. J. Appl. Crystallogr. 49, 622–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith C. A., and Cohen A. E. (2008) The Stanford Automated Mounter: enabling high-throughput protein crystal screening at SSRL. JALA (Charlottesville, VA) 13, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soltis S. M., Cohen A. E., Deacon A., Eriksson T., González A., McPhillips S., Chui H., Dunten P., Hollenbeck M., Mathews I., Miller M., Moorhead P., Phizackerley R. P., Smith C., Song J., et al. (2008) New paradigm for macromolecular crystallography experiments at SSRL: automated crystal screening and remote data collection. Acta Crystallogr. D Biol. Crystallogr. 64, 1210–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeLano W. L. (2002) The PyMOL Molecular Graphics System, San Carlos, CA [Google Scholar]
- 41. Abagyan R., and Totrov M. (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002 [DOI] [PubMed] [Google Scholar]
- 42. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.