

Abstract
Nuclear factor E2-related factor 2 (Nrf2) is considered a promising target against diabetic complications such as cardiovascular diseases and diabetic nephropathy. Herein, we investigated the effects of a potential Nrf2 modulator, salvianolic acid A (SAA), which is a natural polyphenol, on diabetes-associated macrovascular and renal injuries in streptozotocin-induced diabetic mice. Given that lowering glucose is the first objective of diabetic patients, we also examined the effects of SAA combined with metformin (MET) on both complications. Our results showed that SAA significantly increased the macrovascular relaxation response to acetylcholine and sodium nitroprusside in diabetic mice. Interestingly, treatment with SAA alone only provided minor protection against renal injury, as reflected by minor improvements in impaired renal function and structure, despite significantly reduced oxidative stress observed in the diabetic kidney. We demonstrated that decreased oxidative stress and NF-κB p65 expression were associated with SAA-induced expression of Nrf2-responsive antioxidant enzymes heme oxygenase-1 (HO-1), NAD(P)H dehydrogenase (quinone) 1 (NQO-1), and glutathione peroxidase-1 (GPx-1) in vivo or in vitro, which suggested that SAA was a potential Nrf2 modulator. More significantly, compared with treatment with either SAA or MET alone, we found that their combination provided further protection against the macrovascular and renal injury, which was at least partly due to therapeutic activation of both MET-mediated AMP-activated protein kinase and SAA-mediated Nrf2/antioxidant-response element pathways. These findings suggested that polyphenol Nrf2 modulators, especially combined with drugs activating AMP-activated protein kinase, including hypoglycemic drugs, are worthy of further investigation to combat diabetic complications.
Keywords: diabetes, diabetic nephropathy, metformin, Nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), oxidative stress, diabetic macrovascular injury, salvianolic acid A
Introduction
To date, diabetes-associated complications, especially cardiovascular and chronic renal diseases, remain a tremendous threat to a diabetic patient's survival and quality of life despite the use of currently available therapies. It is known that oxidative stress and chronic systemic inflammation are common pathogenic mechanisms of cardiovascular and chronic renal diseases (1, 2). With oxidative stress and chronic systemic inflammation inseparably interconnected, inhibiting oxidative stress was theoretically an effective strategy to delay diabetes-related macrovascular and renal diseases.
The prevention of oxidative stress and inflammation can be achieved through activating the nuclear factor E2-related factor 2 (Nrf2)/antioxidant-response element (ARE)2 signaling pathway, which is known to play a pivotal role in the up-regulation of cellular antioxidant enzymes such as heme oxygenase-1 (HO-1), glutathione peroxidase-1 (GPx-1), and NAD(P)H dehydrogenase (quinone) 1 (NQO-1) (3, 4). Currently, a number of small molecule modulators of Nrf2 have been identified (5), and some of them, such as triterpenoid derivatives bardoxolone methyl and its analogs, have been shown to improve renal or vascular injuries in diabetic rodent models in phase 2 clinical trials (6–9). Recently, however, bardoxolone methyl therapy, after the phase 3 clinical trial, did not reduce the risk of end-stage renal disease or death from cardiovascular causes, but instead it increased the rate of cardiovascular events (10), which thus presented a challenge of Nrf2 modulators, especially triterpenoid derivatives, in the therapy of diabetic renal and cardiovascular complications. Accordingly, the investigation of the effects of therapeutic activation of the Nrf2/ARE pathway by other Nrf2 modulators on both complications was necessary.
Salvianolic acid A (SAA), a polyphenol derivative extracted from the root of Salvia miltiorrhiza, is known to show a variety of pharmacological activities, including antioxidant (11–13), anti-inflammatory (14, 15), and antiplatelet properties (16, 17). Specifically, SAA's antioxidant mechanism has attracted significant attention. A recent study showed that SAA alleviated H2O2-mediated oxidative stress via activation of Nrf2/HO-1 signaling (18), suggesting that SAA might be a potential Nrf2 modulator.
Currently, the effects of several Nrf2 modulators on diabetic complications were studied in diabetic animal models without consideration of lowering glucose (6, 19, 20), which is the first choice for diabetic patients. Thus it might be more significant for clinical reference to observe efficacy of tested drugs when combined with a hypoglycemic drug in diabetic animal models. Metformin (MET), an oral hypoglycemic agent, is widely used in patients with type 2 diabetes. Interestingly, MET has recently been shown to attenuate diabetic nephropathy and atherosclerosis in streptozotocin (STZ)-induced diabetic mice (21, 22). Additionally, MET treatment lowered blood glucose levels in STZ-induced diabetic animals (23, 24). In view of this, we investigated the effects of SAA alone or combined with MET on diabetic macrovascular complication and diabetic nephropathy in STZ-induced diabetic mice.
Our results indicated that SAA significantly improved macrovascular injury, but only made minor improvements in diabetic renal injury. Furthermore, we found that SAA combined with MET further improved both complications. In addition to SAA action, further improvements from their combination might be the consequence of MET-mediated lowering glucose or AMP-activated protein kinase (AMPK) activation by which MET has shown protection against chronic renal and cardiovascular diseases (25–27). In addition, we provided in vivo and in vitro evidence that SAA was a natural polyphenol Nrf2 modulator, which confirmed the previous result in cell-based experiments (18).
Results
General Parameters of Diabetic Mice
Compared with non-diabetic mice, all diabetic mice showed significantly decreased body weights with significantly increased food and water intake regardless of treatment. SAA and MET had no effect on food and water intake and body weights; however, their combination mildly reduced food and water intake of diabetic mice although it did not reach statistical significance (Table 1). Serum triglyceride (TG) and total cholesterol (CHO) levels in vehicle-treated diabetic mice were significantly increased. Treatment with SAA alone or in combination with MET almost completely recovered serum TG and total CHO to normal levels. Although MET therapy also showed a trend toward mildly decreasing serum TG and CHO in diabetic mice, this decrease fell just short of statistical significance. Malonaldehyde (MDA), a marker of lipid oxidation, was markedly increased in diabetic mice. SAA and its combination therapy significantly reduced the MDA level, whereas MET had a minor effect on MDA in diabetic mice (see Table 1).
TABLE 1.
Effects of SAA, MET, or their combination on general parameters of STZ-induced diabetic mice at conclusion of a 21-week study
The following abbreviations are used: ND, nondiabetic mice; D, diabetic mice; D + MET, diabetic mice treated with metformin; D + SAA, diabetic mice treated with SAA; D + MET + SAA, diabetic mice treated with SAA combined with metformin. TG, triglyceride; CHO, cholesterol; MDA, malonaldehyde. Data are mean ± S.D. n = 6–12 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus ND; #, p < 0.05; ##, p < 0.01 versus D; γ, p < 0.01 versus D + MET.
| ND (n = 12) | D (n = 6) | D +MET (n = 7) | D + SAA (n = 7) | D + MET + SAA (n = 11) | |
|---|---|---|---|---|---|
| Blood glucose, mm | 4.7 ± 0.60 | 22.4 ± 4.2*** | 15.4 ± 2.1***,## | 20.8 ± 2.8*** | 14.3 ± 4.6***,### |
| Food intake, g/24 h | 6.1 ± 0.7 | 14.8 ± 1.5*** | 12.5 ± 2.5** | 14.2 ± 1.6** | 11.6 ± 3.3* |
| Water intake, g/24 h | 7.8 ± 1.1 | 53.6 ± 4.8*** | 54.2 ± 9.5*** | 59.0 ± 11.2*** | 51.5 ± 12.3*** |
| TG, mm | 1.0 ± 0.2 | 1.9 ± 0.3*** | 1.6 ± 0.3** | 1.1 ± 0.3###, γ | 1.2 ± 0.2##, γ |
| Total CHO, mm | 2.4 ± 0.6 | 3.4 ± 0.3** | 2.7 ± 0.4 | 2.1 ± 0.3### | 2.3 ± 0.6## |
| MDA, nm | 3.7 ± 0.7 | 9.1 ± 2.0*** | 7.4 ± 2.1** | 4.9 ± 1.8## | 5.2 ± 1.3## |
Macrovascular Function in Diabetic Mice
In diabetic ICR mice, aorta relaxation to the endothelium-dependent acetylcholine (ACh) was greatly decreased, up to ∼30% relaxation compared with non-diabetic mice. Impaired endothelial function was partially recovered after treatment with SAA, MET, or a combination, and the combination treatment showed the biggest extent of recovery with the curve closest to that of non-diabetic mice, then followed by SAA treatment. Additionally, relaxation to the endothelium-independent SNP was partially impaired in diabetic mice, and therapy with all drugs provided a nearly complete recovery of relaxation (Fig. 1A). Furthermore, the artery structure showed that the local vascular wall of the diabetic mice became thin, with numerous lipid deposition compared with non-diabetic mice (Fig. 1B). Treatment with SAA or combined with MET improved impaired vascular structure.
FIGURE 1.

Impaired vascular function and structure were improved by SAA, MET, or a combination. Isometric tension studies were performed in diabetic or non-diabetic ICR mice and aorta relaxation in response to endothelium-dependent ACh and endothelium-independent SNP. Data were representative of 6–10 mice per group (A). H&E-stained aortas in mice were examined at ×400 (B). Data were expressed as mean ± S.E. *, p < 0.05; **, p < 0.01 versus ND; #, p < 0.05 versus D; γ, p < 0.05 at the same dose points of ACh and SNP. ND, non-diabetic mice; D, diabetic mice; D+MET, diabetic mice treated with metformin; D+SAA, diabetic mice treated with SAA; D+MET+SAA, diabetic mice treated with SAA combined with metformin; ACh, acetylcholine; SNP, sodium nitroprusside.
VCAM-1 and HO-1 Expression in High Glucose-treated Human Umbilical Vein Endothelial Cells
Pro-inflammatory cytokines also play an important role in diabetic vascular damage in addition to oxidative stress. In human umbilical vein endothelial cells (HUVECs) treated with high glucose (25 mm), we examined the effects of SAA, MET, and their combination on vascular cell adhesion protein 1 (VCAM-1), a pro-inflammatory cytokine that functions in leukocyte-endothelial cell signal transduction. The result indicated that the expression of VCAM-1 was significantly increased in high glucose (HG)-treated HUVECs compared with normal glucose (NG)-treated cells. SAA treatment reduced VCAM-1 expression in HG-treated HUVECs (Fig. 2, A and B). Given that up-regulation of Nrf2-mediated HO-1 was known to inhibit VCAM-1 expression in vascular disease (28, 29), HO-1 up-regulation caused by SAA (Fig. 2, A and C) was a possible mechanism underlying the decreased VCAM-1 expression. In addition, MET also mildly reduced VCAM-1 expression, but it had no effect on HO-1 expression (see Fig. 2, A–C). Combined treatment further reduced VCAM-1 expression.
FIGURE 2.

Expression of VCAM-1 was decreased and HO-1 was increased by SAA and SAA combined with MET. HUVECs were treated with NG, Man, HG, HG + MET, HG + SAA, or HG + MET + SAA for 48 h. Man was used as an osmotic control. The expression of HO-1 and VCAM-1 in cell lysates was analyzed by Western blotting (A), and the quantitation of HO-1 and VCAM-1 is shown in B and C, respectively. Data were expressed as mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus NG; #, p < 0.05; ##, p < 0.01 versus HG, γ, p < 0.05. NG, 5.5 mm normal glucose; HG, 25 mm high glucose; Man, 19.5 mm mannitol; MET, 2 mm metformin; SAA, 5 μm salvianolic acid A.
Kidney Function and Structure in Diabetic Mice
Compared with non-diabetic mice, proteinuria was strikingly increased in diabetic mice. Proteinuria levels in diabetic mice were reduced in all therapeutic groups, with SAA in combination with MET indicating the greatest reduction. SAA treatment revealed a trend toward a decrease in diabetic mice despite no significance (Fig. 3A). Similarly, left and right kidney indices revealed a trend toward a significant increase in diabetic mice. This trend was mitigated after treatment with MET or SAA in combination with MET. However, SAA treatment had no significant effect on this trend (Fig. 3, B and C). Similar results in glomerular size were observed (Fig. 3E).
FIGURE 3.

Impaired kidney function and structure in diabetic mice were improved by SAA in combination with MET. Proteinuria, expressed as urinary protein excreted relative to urinary creatinine, was decreased after SAA treatment in combination with MET (A). SAA in combination with MET significantly reduced kidney index, which was indicative as kidney weight (mg)/body weight (g) (B and C). PAS-stained glomeruli are shown (D). SAA in combination with MET significantly reduced glomerular size, glomerular PAS area, and GSI in STZ-induced ICR mice, respectively (E–G). PAS-stained tissue was examined at ×400. Data were expressed as mean ± S.E. n = 6–10 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus ND; #, p < 0.05; ##, p < 0.01 versus D; γ, p < 0.05. ND, non-diabetic mice; D, diabetic mice; D+MET, diabetic mice treated with metformin; D+SAA, diabetic mice treated with SAA; D+MET+SAA, diabetic mice treated with SAA combined with metformin; GSI, glomerulosclerosis index.
In glomeruli, the percentage of the periodic acid-Schiff (PAS)-stained area, which was indicative of mesangial matrix expansion, was increased compared with non-diabetic mice (Fig. 3, D and F). In comparison with vehicle-treated diabetic mice, the PAS-stained area was reduced in all diabetic mice that received SAA or MET therapy either alone or in combination; however, only the combination therapy reached statistical significance (Fig. 3F). Similarly, the glomerulosclerosis index (GSI) also revealed a trend toward an increase in diabetic mice (Fig. 3G). This trend was also alleviated after receiving SAA or MET treatment either alone or in combination, and the combination therapy resulted in the greatest reduction in diabetic mice.
Renal Fibrosis in Diabetic Mice
In line with increased GSI in the diabetic kidney, collagen deposition as shown in Masson's trichrome staining (Fig. 4A, top panel) was significantly increased compared with a normal kidney. This was also reflected by increased collagen I and α-SMA expression, which was evaluated by immunohistochemistry (Fig. 4, A and B). Treatment with SAA or MET alone slightly decreased collagen deposition, collagen I, and α-SMA; however, SAA treatment in combination with MET significantly decreased these three indicators, which explained improved renal function and structure in diabetic mice treated with SAA in combination with MET.
FIGURE 4.

Diabetes-associated renal fibrosis was attenuated by SAA treatment in combination with MET. Fixed kidney tissues were stained with Masson's trichrome, and fibrosis areas were stained blue (A, top panel, ×200). Renal fibrosis-associated collagen I and α-SMA expression were examined using immunohistochemistry, and representative images (×200) are shown (A). The quantitative analysis (B) showed that the blue area in Masson's trichrome and expression of collagen I and α-SMA were significantly reduced by SAA in combination with MET. Data were mean ± S.E. n = 5 to 6 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus ND; #, p < 0.05; ##, p < 0.01 versus D. ND, non-diabetic mice; D, diabetic mice; D+MET, diabetic mice treated with metformin; D+SAA, diabetic mice treated with SAA; D+MET+SAA, diabetic mice treated with SAA combined with metformin.
Oxidative Stress and Antioxidant Enzyme Expression in the Kidney
Nitrotyrosine (NT), a marker of reactive nitrogen species-induced nitrative stress, was significantly increased in both the renal tubules and glomeruli in STZ-induced diabetes compared with non-diabetic mice (Fig. 5, A–C). Similarly, the fluorescence of dihydroethidine (DHE), a cell-permeable probe that is often used to monitor superoxide (O2˙̄) production, increased ∼2-fold in diabetic mice (Fig. 5, A and D). Treatment with SAA or SAA in combination with MET significantly reduced both the NT level and DHE fluorescence. Although MET therapy also revealed a decreased trend in the NT-stained area, it did not reach a significant difference compared with vehicle-treated diabetic control. Furthermore, MET therapy had no effect on DHE fluorescence in the diabetic kidney (see Fig. 5, A and D). These data suggested that SAA, and not MET, significantly attenuated oxidative stress in STZ-induced diabetic mice with SAA treatment or a combination.
FIGURE 5.

Attenuation of oxidative stress by SAA or SAA in combination with MET was linked to the up-regulation of antioxidant enzymes. Oxidative stress in the kidney cortex was examined by NT immune staining in paraformaldehyde-fixed kidney and DHE fluorescent dye in kidney cryosections (A). The quantitative results in NT and DHE staining were assessed by Image-Pro Plus 6.0 (B–D). The protein level for NOX-4 was examined by Western blotting with its quantitation shown (E). Similarly, protein levels of several antioxidant enzymes, HO-1, NQO-1, and GPx-1 are shown (F–H). Data were mean ± S.E. n = 6–10 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus ND; #, p < 0.05; ##, p < 0.01 versus D. γ, p < 0.05. ND, non-diabetic mice; D, diabetic mice; D+MET, diabetic mice treated with metformin; D+SAA, diabetic mice treated with SAA; D+MET+SAA, diabetic mice treated with SAA combined with metformin.
The protein level of NADPH oxidase 4 (NOX-4), which is an important source of reactive oxygen species (ROS) formation in renal pathogenic conditions (30), was also examined in our study. The results revealed that the kidney's NOX-4 expression was significantly increased in diabetic conditions compared with non-diabetic mice. NOX-4 expression was significantly decreased after treatment with SAA alone or in combination with MET. Nevertheless, NOX-4 expression was not obviously changed in diabetic mice with MET treatment alone (Fig. 5E).
The expressions of Nrf2-mediated anti-oxidative enzymes, including HO-1, NQO-1, and GPx-1, were significantly increased in the diabetic kidney when treated with SAA alone or in combination with MET. However, MET therapy alone had almost no effect on these enzymes (Fig. 5, F–H). Altogether, these findings suggested that SAA-induced reduction of oxidative stress was linked to the up-regulation of anti-oxidative enzymes.
Renal Inflammation in Diabetic Mice and Survival of Diabetic Mice
Inflammatory cell infiltration in the diabetic kidney was visualized using hematoxylin and eosin (H&E) stain (Fig. 6A, left panel), with significantly increased inflammatory cell infiltration observed in diabetic mice compared with normal mice (Fig. 6B). Consistent with this, Ly6G, a granulocyte marker, was also markedly increased in the diabetic kidney (Fig. 6, A, right panel, and C). Similarly, NF-κB p65 and monocyte chemoattractant protein-1 (MCP-1) levels were elevated in the diabetic kidney (Fig. 6, D and E). Treatment with SAA or MET alone mildly reduced inflammatory cell infiltration, Ly6G-positive cells, NF-κB p65, and MCP-1 levels, but they fell out of statistical significance. However, combined therapy with SAA and MET significantly reduced these indicators (see Fig. 6, D and E). These results suggested that SAA treatment in combination with MET reduced diabetic renal tubulointerstitial inflammation, possibly in a synergistic manner.
FIGURE 6.

Effects of SAA, MET, or a combination on renal tubulointerstitial inflammation and survival of STZ-induced diabetic mice. The renal pathological changes (at ×200) were assessed by H&E (HE) staining (A, left panel). The score of renal tubulointerstitial inflammation is presented (B). Ly6G, a marker of granulocytes, was examined using immunohistochemistry (A, right panel), and the quantitation is shown in C. The MCP-1 level in the kidney was measured by ELISA (D), and NF-κB protein level was also examined by Western blotting (E). One hundred and forty days after STZ treatment, animal survival was analyzed using the Kaplan-Meier survival analysis (F). Data were mean ± S.E. n = 6–10 per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus ND; #, p < 0.05 versus D; ##, p < 0.01 versus D. ND, non-diabetic mice; D, diabetic mice; D+MET, diabetic mice treated with metformin; D+SAA, diabetic mice treated with SAA; D+MET+SAA, diabetic mice treated with SAA combined with metformin.
One hundred and forty days after STZ treatment, the survival of the diabetic mouse that received no therapy significantly decreased to less than 40%. SAA and MET therapy delayed the death of the diabetic mouse but did not show a significant advantage of survival compared with vehicle-treated groups. However, SAA and MET in combination significantly increased the survival of the diabetic mouse (Fig. 6F).
Combination of SAA and MET Inhibited HG-induced Harmful Effects in Human Kidney-2 Cells by MET-mediated AMPK Activation and SAA-induced Antioxidant Enzyme
To support the previous in vivo results, HK-2 cells in HG conditions were treated with SAA, MET, or a combination of both. In addition to lowering glucose action, MET is known to protect cells from HG-induced damage through several mechanisms (26, 31–33), most of which were mainly due to the activation of AMPK. Thus, we first examined the effect of MET on p-AMPKα. Our findings showed that MET significantly increased p-AMPKα expression in diabetic kidneys (Fig. 7A). Similarly, p-AMPKα expression was markedly increased in HG-exposed HK-2 cells treated with MET (Fig. 7B). In contrast, SAA induced antioxidant enzyme HO-1 up-regulation in HK-2 cells (Fig. 7C). Not surprisingly, we observed significantly increased p-AMPKα and HO-1 expression in HK-2 cells treated with SAA in combination with MET (see Fig. 7, B and C).
FIGURE 7.

Combination of SAA and MET inhibited HG-induced harmful effects in HK-2 cells through MET-mediated AMPK activation and SAA-induced HO-1 up-regulation. Expression of AMPK and p-AMPK in the diabetic kidney was analyzed by Western blotting, showing that MET activated AMPK (A) (n = 6 mice per group). HK-2 cells were treated with NG, Man, HG, HG + MET, HG + SAA, HG + MET + SAA for the indicated times. After a 12-h incubation, cell lysates were used to detect AMPK and p-AMPK expression by Western blotting, and the quantitation showed metformin-activated AMPK (B). After a 24-h incubation, HO-1 expression was analyzed by Western blotting, which showed that SAA induced HO-1 up-regulation (C). After a 48-h incubation, the α-SMA and p65 expression were analyzed by Western blotting, with their expression showing a trend toward further decreases under the SAA treatment in combination with MET (D). Similarly, HK-2 cells were treated with the above agents, or a combination for 48 h, and then evaluated by fluorescent microscopy after DHE and DCFH-DA treatments for 30 min (E, ×200). Results were mean ± S.E. (B–E, n = 3 independent experiments). *, p < 0.05; **, p < 0.01; ***, p < 0.001. NG, 5.5 mm normal glucose; HG, 25 mm high glucose; Man, 19.5 mm mannitol; MET, 2 mm metformin; SAA, 5 μm salvianolic acid A.
Consistent with the reported findings that activation of AMPK or the Nrf2/ARE pathway reduced HG-induced harmful effects in several types of cells (34–36), our results showed that treatment with SAA, MET, and their combination reduced increased expression of NF-κB p65 and α-SMA in HG-exposed HK-2 cells, with the biggest decrease observed in SAA treatment in combination with MET (Fig. 7D). Similarly, HG-induced ROS in HK-2 cells, which was assessed by DHE and 2′,7′-dichlorofluorescin diacetate (DCFH-DA), was significantly reduced by SAA treatment alone and in combination with MET (Fig. 7E). MET mildly reduced increased ROS, although there was a lack of statistical significance (Fig. 7E). These findings suggested that SAA treatment in combination with MET was helpful in further improving HG-induced injury, which partially supported our in vivo results.
SAA-induced Down-regulation of NF-κB p65 Expression and ROS Level Depended on Nrf2 in HG-exposed HK-2 Cells
We explored nuclear translocation of Nrf2 in HK-2 cells to determine whether SAA-induced HO-1 up-regulation was associated with Nrf2 activation. As shown in Fig. 8A, SAA promoted nuclear accumulation of Nrf2 from cytoplasm, basically in a dose-dependent manner. This result was further confirmed in the Nrf2 DNA binding assay, which showed that ARE-driven luciferase activity response to Nrf2 binding was increased by SAA treatment (Fig. 8B). These findings suggested that SAA does activate the Nrf2/ARE pathway, subsequently leading to HO-1 up-regulation.
FIGURE 8.

SAA reduced NF-κB expression and ROS levels in HG-exposed HK-2 cells through Nrf2-mediated HO-1 up-regulation. HK-2 cells were treated with 1, 5, and 20 μm SAA for 4 h. The total cytoplasmic and nuclear Nrf2 proteins were analyzed by Western blotting (A). Nrf2 DNA binding activity was analyzed by firefly luciferase activity following normalization against Renilla luciferase activity and presented as fold Nrf2 DNA binding activity relative to its basal levels in HK-2 cells (B, n = 3). HK-2 cells were transfected with Nrf2 siRNA or SCR siRNA and then were treated with or without 5 μm SAA in the presence of NG or HG for 48 h. Cell lysates were used to detect expression of Nrf2, HO-1, and p65 (C, n = 3) and DHE and DCFH-DA fluorescence intensities were evaluated by fluorescent microscopy (D, ×200, n = 3). NG, 5.5 mm normal glucose; HG, 25 mm high glucose; SCR siRNA, negative scrambled siRNA. Results were mean ± S.E. *, p < 0.05; **, p < 0.01.
Next, to determine whether Nrf2 was required for SAA protection from HG-induced harmful effects, Nrf2 small interfering RNA (siRNA) was used to silence Nrf2 expression in HK-2 cells. As shown in Fig. 8C, Nrf2 siRNA reduced Nrf2 expression by ∼70% in the SAA-treated cells, which resulted in down-regulation of HO-1 expression. This result suggested that SAA-induced activation of the Nrf2/ARE pathway was prevented by Nrf2 siRNA. Accordingly, SAA-induced inhibition of NF-κB p65 was partly reversed in HG-exposed HK-2 cells treated with Nrf2 siRNA. HG-induced ROS formation was also restored by Nrf2 siRNA in HG-exposed HK-2 cells with SAA treatment (Fig. 8D). These findings suggested that SAA protection from HG-induced injury depended on Nrf2.
Discussion
SAA in this study activated Nrf2-mediated HO-1 expression in HK-2 and HUVEC cells, as judged by assays of ARE luciferase activities, Nrf2 nuclear translocation, and Nrf2 siRNA. This was in agreement with a previous report showing that SAA prevented H2O2-induced oxidative injury by up-regulating Nrf2-mediated HO-1 expression (18). Furthermore, our findings in the diabetic mice showed that SAA induced the expression of antioxidant enzymes HO-1, GPx-1, and NQO-1. These findings suggested that SAA, a natural polyphenol derivative, was a potential Nrf2 modulator. In fact, many other natural polyphenols, such as resveratrol and epigallocatechin gallate, have also proven to be potential Nrf2 modulators (37). The common mechanism underlying induction of Nrf2 nuclear translocation among these polyphenols might be their ability to interact with cysteine residues present in Keap1 (37, 38).
In our previous study, SAA improved diabetes-associated vascular damage in diabetic rats (39). Similarly, this study again showed that SAA significantly alleviated the impairment of vascular relaxation response to ACh and SNP in diabetic mice, which was possibly associated with significantly decreased serum MDA, TG, and total CHO. Furthermore, SAA inhibited VCAM-1 expression in HG-treated HUVECs possibly by the mechanism underlying the up-regulation of HO-1. These findings supported the improvement of SAA in diabetic macrovascular injury. Additionally, a recent study showed that resveratrol, another polyphenol derivative with the ability to activate the Nrf2/ARE pathway (40), also exerted beneficial effects on diabetic vascular damage (41). Taken together, these findings supported the concept that polyphenol Nrf2 modulators are promising candidates for preventing or delaying diabetic macrovascular injury.
However, SAA treatment just mildly improved renal function and structure in diabetic mice, which was somewhat discordant with the significant improvements in macrovascular dysfunction. This was reflected by minor improvement in the rate of urinary protein to creatinine, kidney index, glomerular size, and the extent of the mesangial matrix expansion, glomerulosclerosis, and renal fibrosis. Interestingly, oxidative stress in the diabetic kidney was significantly reduced after SAA treatment, with restoration to an almost normal level as observed in non-diabetic mice, as judged by significantly decreased NT level and DHE fluorescence intensity in the kidney via induction of several antioxidant enzyme expressions.
Recently, two studies (20, 42) thought that harmful or no effect of the Nrf2 inducer dh404 on diabetic renal damage resulted from off-target effects of high dose dh404, which led to increased NF-κB and MCP-1 expression, although oxidative stress significantly decreased (20). However, this was not an appropriate explanation in our study because 3 mg/kg SAA did not increase NF-κB and MCP-1 expression, but instead reduced their expression despite a lack of statistical significance. For minor improvements observed in this study, we thought that a more possible explanation was complex pathogenic mechanisms of diabetic nephropathy. In addition to oxidative damage, high blood glucose itself and the other factors that resulted, such as hypertension, metabolism disorders, and energy dysfunction (43, 44), were also important contributors to diabetic nephropathy, which was likely associated with the multiple cell types and the various physiological roles in the kidney (44). In fact, in several published studies, diabetic animals receiving Nrf2 modulators showed obvious renal injury despite significant improvements observed in comparison with vehicle-treated diabetic mice (6, 20). Taken together, these findings suggested that the efficacy of Nrf2-mediated antioxidant therapy alone was limited in preventing or delaying diabetic renal injury.
Therefore, Nrf2 modulator therapy combined with different mechanism-based drugs might be a better strategy to prevent diabetic renal disease. Given that high blood glucose is the source of the onset and progression of diabetic complications and glycemic control is always the first choice for diabetic patients, we thus examined the effects of SAA combined with MET on macrovascular and renal injury in diabetic mice. It should be pointed out that although MET was used in the treatment of patients with type 2 diabetes, it lowered blood glucose in animals with STZ-induced type 1 diabetes (23), which was involved in decreased hepatic glucose production, increased peripheral glucose disposal, and reduced intestinal glucose absorption. In line with this, MET lowered HbA1c and decreased insulin dosage in teens with type 1 diabetes (45). Thus, it was reasonable to use MET treatment in our study.
We found that MET treatment alone, as a reference in this study, only mildly improved the renal injury in the diabetic mouse, which still showed severe injury. Similar results were observed in the vascular relaxation response to ACh. The results were reasonable because the blood glucose remained high, about 15 mm, although MET significantly reduced the level in the diabetic mouse.
As expected, SAA combined with MET improved the renal injury and was superior to that of SAA or MET alone, which was reflected in the extent of improvements in renal function, mesangial matrix expansion, glomerulosclerosis, and interstitial fibrosis observed in the combined therapy group. Glomerulosclerosis, which is caused by accumulation of extracellular matrix, is an important factor contributing to progression of diabetic nephropathy to end-stage kidney disease (58). It is known that hyperglycemia results in increased advanced glycation end product, oxidative stress generation, and expression of TGF-β1 and α-SMA, which subsequently leads to enhanced extracellular matrix production via a number of signaling pathways (58–60). In this study, treatment with SAA and MET combination significantly reduced mesangial matrix expansion, GSI, and collagen I in STZ-induced diabetic mice, which was thus linked to decreased oxidative stress and α-SMA expression. Consistent with this, we found that treatment with the combination of SAA and MET markedly reduced oxidative stress α-SMA expression in HG-exposed HK-2 cells. These findings suggested that the improved glomerulosclerosis was at least partly associated with reduced oxidative stress and α-SMA expression.
Similarly, the curve of the vascular relaxation response to ACh in diabetic mice treated with this combination was the closest to that of normal mice. Furthermore, after 18 weeks of therapy, this combination significantly increased the survival rate of the diabetic mouse compared with the vehicle-treated group, whereas SAA or MET alone had no significant effects on the survival rate.
On the one hand, we thought that the efficacy of SAA combined with MET was the consequence of reduced oxidative stress and inflammation resulting from SAA treatment as discussed previously. On the other hand, MET's glucose-lowering action was a likely contributor to the further improvement of the combination-treated group's complications, given the fact that glycemic control decreased the risk of complications in patients with diabetes (46, 47). It should be noted that the improvement that resulted from MET might also be linked to non-glucose-lowering mechanisms, most of which are known to be associated with AMPK activation (48, 49). AMPK, as a major cellular energy sensor, plays a positive role in diabetes-associated complications (50, 51). Recently, AMPK activation has been shown to reduce HG-induced α-SMA expression by inhibiting nuclear translocation of Smad4 (60). Similarly, we showed that MET activated AMPK in diabetic mice and HG-induced HK-2 cells, which resulted in the reduction of α-SMA and NF-κB p65 expression. This partly explained the improvement of STZ-induced diabetic renal injury. Importantly, MET-mediated activation of AMPK and SAA-mediated activation of the Nrf2/ARE pathway further improved the diabetic renal kidney. Obviously, the different mechanisms between SAA and MET (Fig. 9) are likely to be the key reasons for further improvements in both complications. Our study thus supports the notion that therapeutic activation of both the AMPK and the Nrf2/ARE pathway might be a good strategy to improve diabetic nephropathy.
FIGURE 9.
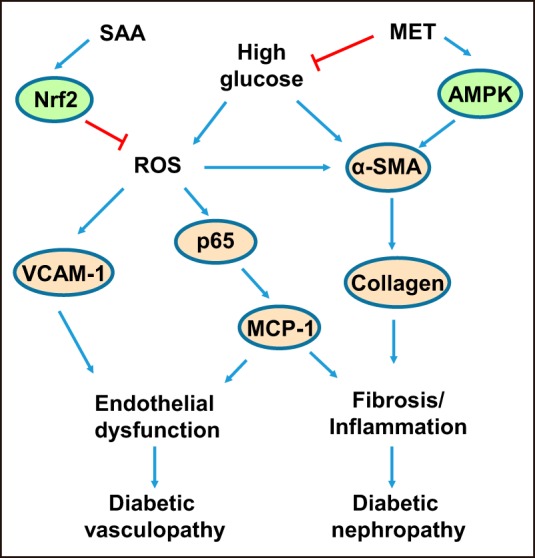
Schematic diagram showing that treatment with the combination of SAA and MET alleviates diabetes-associated vasculopathy and nephropathy. SAA, salvianolic acid A; MET, metformin; Nrf2, nuclear factor E2-related factor 2; AMPK, AMP-activated protein kinase; ROS, reactive oxygen species; α-SMA, α-smooth muscle actin; p65, nuclear factor NF-κB p65 subunit; VCAM-1, vascular cell adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1.
In conclusion, our findings indicated that SAA was a natural polyphenol Nrf2 modulator that induced Nrf2-mediated expression of antioxidant enzymes in vitro and in vivo. SAA showed marked protection against STZ-induced diabetic macrovascular injury. However, SAA showed only minor improvements in the renal injury, which we believe resulted from the other pathogenic mechanisms of diabetic nephropathy in addition to oxidative stress. Along with this notion (i.e. that combination therapy based on different mechanisms might be a better strategy to further prevent the disease), we showed that SAA combined with MET further improved both complications. Collectively, we believe that therapeutic induction of Nrf2 is effective in preventing diabetes-associated macrovascular complications, but to achieve further improvements, Nrf2 modulators combined with drugs that activate AMPK might also need to be considered, especially in diabetic nephropathy.
Experimental Procedures
Drugs and Chemicals
SAA (HPLC, 98%) was provided as a lyophilized powder by the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China). MET hydrochloride was purchased from Sino-American Shanghai Squibb Pharmaceuticals Ltd. (Shanghai, China). STZ, phenylephrine, SNP, and ACh were obtained from Sigma. All other chemicals were of analytical grade.
Animals
Five-week-old male ICR mice were obtained from Vital River Laboratory Animal Center (Beijing, China). The animals were housed under a 12-h light/dark cycle at a temperature of 22 ± 3 °C and humidity of 55 ± 5% with free access to food and water for 7 days before the experiment. All procedures using animals were approved by the animal care and use committee of the Institute of Materia Medica, Chinese Academy of Medical Sciences.
Preparation of Diabetic Animals
Diabetes was induced by intraperitoneal injections of STZ at 100 mg/kg/day on 2 consecutive days (20). Control mice received an equal volume of vehicle (citrate buffer, 0.01 mol/liter, pH 4.5). Diabetes in mice was confirmed 21 days after STZ injection by fasting blood glucose measurement with an ACCU-CHEK® active glucometer (Roche Applied Science, Hoffmann, Germany).
Diabetic mice were randomly subdivided to be gavaged with either saline, 3 mg/kg SAA, 200 mg/kg MET, or 3 mg/kg SAA + 200 mg/kg MET for 18 weeks. Age-matched normal mice received an equal volume of saline. Body weights, food and water intakes, and blood glucose were determined weekly during the entire study. Mice were killed at 18 weeks after drug treatment. Collected plasma and urine were stored at −80 °C. Plasma TG and total CHO were determined with a commercially available enzyme kit (BioSino Bio-technology & Science Inc., Beijing, China). MDA in serum was determined according to the previously described method (52) using a thiobarbituric acid assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Isometric Tension Studies
Endothelial dysfunction was assessed as described previously (39, 53, 54). Briefly, the thoracic aortas isolated from the mice were cut into 2-mm-long circular segments and mounted between two stainless steel hooks in the bath chambers, which contained 8 ml of K-H solution (118 NaCl, 4.7 KCl, 1.5 CaCl2, 25 NaHCO3, 1.1 MgSO4, 1.2 KH2PO4, and 5.6 glucose in mm) at 37 °C and gassed with 95% O2 and 5% CO2 for the measurement of isometric tension with a force displacement transducer connected to a BIOPAC polygraph (MP100A, BIOPAC Systems, Inc., Goleta, CA). After a 45-min equilibration with a resting tension of 1.2 G, rings were stimulated twice by high KCl (60 mm) solution and then equilibrated for 15 min in K-H solution. Subsequently, the aortic rings were contracted by phenylephrine (10−6 m) and then relaxed by ACh (10−8 to 10−5 m) and the NO donor SNP (10−10 to 10−7 m). Additionally, the left thoracic aortas were cut into several sections fixed into 4% paraformaldehyde for H&E staining. Sections were imaged under a Nikon Eclipse Ti-U fluorescent microscope and assessed by a pathologist from Beijing Bsea Biotechnology Co., Ltd.
Kidney Function and Histological Assessment
The urinary protein-to-creatinine ratio was measured using a commercial kit (BioSino Bio-technology & Science Inc., Beijing, China). Kidney paraffin sections were stained with PAS and Masson's trichrome. PAS-stained sections were used to assess glomerular injury using Image-Pro Plus version 6.0, which was expressed as a percentage of PAS-stained area per glomerulus. The glomerulosclerosis index was graded on a scale of 0 to 4 according to the PAS-stained area in each of the assessed glomeruli; a score of 0 was assigned to normal glomeruli, 1 for up to 25% involvement, 2 for up to 50%, 3 for up to 75%, and 4 for greater than 75% sclerosis. The glomerulosclerosis index was calculated as GSI = ((1 × N1) + (2 × N2) + (3 × N3) + (4 × N4))/(N0 + N1 + N2 + N3 + N4), where Nx represented the number of glomeruli with each given score (55).
Renal Inflammation Assessment
Renal interstitial inflammation was assessed by H&E staining and imaged under the Nikon Eclipse Ti-U fluorescent microscope. The degree of inflammation was assessed by a pathologist and was graded on a scale of 0 to 3; 0 was assigned to normal renal interstitium, 1 for a few scattered infiltration of inflammatory cells in renal interstitium, 2 for mild diffuse infiltration of inflammatory cells in renal interstitium, and 3 for marked diffuse infiltration of inflammatory cells in renal interstitium. The level of MCP-1 in the kidney was determined by ELISA kit (eBioscience, San Diego).
Reactive Oxygen Species Determination in the Kidney
ROS levels in kidney cryosections were determined by 10 μm DHE (Sigma) fluorescence microtopography as in the method described previously (56). Sections were imaged under a Nikon Eclipse Ti-U fluorescent microscope with a high resolution digital camera. Quantitative analysis was performed in a blinded manner with the Image-Pro Plus version 6.0.
Immunohistochemical Analysis
Kidney tissues were fixed in neutral buffered formalin and embedded in paraffin. The deparaffinized and rehydrated sections were boiled in sodium citrate buffer, pH 6. The sections were stained with the primary antibody against nitrotyrosine (Santa Cruz Biotechnology), Ly6G, α-SMA, and collagen I (Abcam, Cambridge, UK). The peroxidase-conjugated secondary antibodies were then applied and developed with 3,3′-diaminobenzidine for immunoperoxidase staining. Sections were imaged under a Nikon Eclipse Ti-U fluorescent microscope with a high resolution digital camera. Quantitative analysis was performed in a blinded manner with the Image-Pro Plus version 6.0.
Cell Cultures, Reactive Oxygen Species Detection, Luciferase Assays, Gene Silencing
HUVECs were purchased as cryopreserved aliquot in passage 2 from Lifeline Cell Technologies (Frederick, MD). HUVECs (≤ passage 5) were maintained in VascuLife VEGF cell culture medium with 2% serum. For high glucose experiments, NG media were supplemented with glucose to a final concentration of 25 mm, or mannitol (Man) to a final concentration of 19.5 mm as an osmotic control for high glucose. After HUVECs grown in 6-well plates reached 80% confluence, the cells were maintained in serum-free VascuLife VEGF cell culture medium with 5.5 mm NG or 25 mm HG and treated with or without SAA, MET, or a combination (Sigma). After a 48-h incubation, the cells were lysed in M-PER mammalian protein extraction reagent (Thermo Fisher) for Western blotting analysis.
HK-2 (ATCC, CRL-2190) cells were maintained in Dulbecco's modified Eagle's medium (DMEM), Ham's F-12 media (1:1, Gibco BRL) with 10% FBS. For experiments, HK-2 cells were first cultured through two passages in NG (1 g/liter, 5.5 mm) DMEM (Gibco). For high glucose conditions, treatment was similar with that in HG-exposed HUVECs described previously. For analysis of protein expressions, cells grown in 6-well plates were maintained in serum-free DMEM containing NG or HG and treated with SAA, MET, or a combination for 12, 24, or 48 h.
The intracellular ROS level was determined according to the method described previously (57). Briefly, HK-2 cells grown on glass coverslips were treated with NG, mannitol, HG, HG + MET, HG + SAA, or HG + SAA + MET for 48 h. The cells were then incubated with 10 μm DHE or DCFH-DA for 30 min at 37 °C. ROS formation was analyzed at 488/515 nm for DCFH-DA or 510/580 nm for DHE with a Nikon Eclipse Ti-U fluorescent microscope. The mean fluorescence intensity per cell was calculated using Image-Pro Plus version 6.0 with at least five random fields analyzed.
For Nrf2 nuclear translocation analysis, HK-2 cells were grown in 6-well plates for 4 h in the presence of SAA. The nuclear and cytosolic extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific, Waltham, MA).
For Nrf2 binding activity assay, HK-2 cells grown in a white 96-well plate (Corning Costar, Corning, NY) were co-transfected with pGL4.37(luc2P/ARE/Hygro) containing four copies of the ARE enhancer sequences (5′-TAGCTTGGAAATGACATTGCTAATGGTGACAAAGCAACTTT-3′) (Promega, Mullion, WI) with firefly luciferase and pRL-SV40 vector with Renilla luciferase in a 10:1 mass ratio. After treatment of SAA for 24 h, firefly luminescence and Renilla luminescence were measured using the Dual-Glo luciferase assay system (Promega, Madison, WI).
For the assay of gene silencing, siRNAs specifically targeting human Nrf2 and negative scrambled (SCR) siRNA (Santa Cruz Biotechnology) were used. HK-2 cells grown in 6-well cell plates were transfected with Nrf2 siRNA or SCR siRNA using siRNA transfection medium and reagent (Santa Cruz Biotechnology) according to the manufacturer's instructions. After incubation at 37 °C for 6 h, the transfection solution was aspirated and replaced by DMEM containing NG. After an additional 18-h incubation, cells were maintained in serum-free DMEM with NG, HG, and HG + SAA for 48 h. Cells were then used to assess DCFH-DA fluorescence intensity or collected for Western blotting analysis.
Western Blotting Analysis
The kidney cortex was frozen and homogenized in ice-cold RIPA buffer containing proteases and phosphatase inhibitors. Kidney and cell lysates were subjected to SDS-PAGE and then transferred onto PVDF membrane (Millipore, Billerica, MA). After blocking, immunoblotting was incubated with the following primary antibodies: polyclonal rabbit β-actin (Sigma) as a control for loading and transfer; polyclonal goat NADPH oxidase-4 (NOX-4) (Santa Cruz Biotechnology); polyclonal rabbit HO-1, polyclonal GPx-1, and NQO-1 (Santa Cruz Biotechnology); polyclonal rabbit α-SMA antibody (Abcam); monoclonal rabbit AMPKα antibody, monoclonal rabbit phospho-AMPKα (Thr172) antibody, polyclonal rabbit VCAM-1 antibody, and polyclonal rabbit NF-κB p65 antibody (Cell Signaling Technology, Beverly, MA). Finally, HRP-conjugated secondary antibodies were applied, and the signals were detected using an enhanced chemiluminescence kit (GE Healthcare).
Statistical Analyses
Data were expressed as means ± S.E. or S.D. and analyzed by one-way analysis of variance with Newman-Keuls post hoc testing. Statistical analyses were performed using GraphPad Prism version 6.0 (GraphPad, La Jolla, CA). p < 0.05 was considered statistically significant.
Author Contributions
P. W. designed, performed, analyzed all the experiments, and wrote the paper. Y. Y. performed the animal experiment and revised the paper. L. M. provided the agents and revised the paper. B. H., Y. H., Z. N., and X. P. performed the experiments. L. Z. and J. S. provided advice on the graphics presentation. X. Y. designed the experiments and revised the paper. G. D. conceived ideas, oversaw the entire research, and revised the paper.
Acknowledgments
We gratefully acknowledge the technical services and pathological analysis on vascular and renal histological assessment and immunohistochemistry provided by Beijing Bsea Biotechnology Co., Ltd.
This work was supported by National Science and Technology Major Projects for “Major New Drugs Innovation and Development” Grants 2013ZX09508104 and 2013ZX09402203 (to G. D.), Doctoral Innovation Fund of Peking Union Medical College Grant 10023-1007-1020 (to P. W.), and National Natural Science Foundation of China Grant 81470159 (to X. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
- ARE
- antioxidant-response element
- Nrf2
- nuclear factor E2-related factor 2
- SAA
- salvianolic acid A
- MET
- metformin
- HO-1
- heme oxygenase-1
- GPx-1
- glutathione peroxidase-1
- NQO1
- NAD(P)H dehydrogenase (quinone) 1
- STZ
- streptozotocin
- SNP
- sodium nitroferricyanide
- ACh
- acetylcholine
- TG
- triglyceride
- CHO
- cholesterol
- MDA
- malonaldehyde
- PAS
- periodic acid-Schiff
- MCP-1
- monocyte chemoattractant protein-1
- NT
- nitrotyrosine
- DHE
- dihydroethidine
- DCFH-DA
- dichlorofluorescin diacetate
- VCAM-1
- vascular cell adhesion protein 1
- NOX-4
- NADPH oxidases 4
- AMPK
- adenosine monophosphate-activated protein kinase
- HUVEC
- human umbilical vein endothelial cell
- GSI
- glomerulosclerosis index
- HG
- high glucose
- NG
- normal glucose
- ROS
- reactive oxygen species
- Man
- mannitol
- α-SMA
- α-smooth muscle actin.
References
- 1. Vikram A., Tripathi D. N., Kumar A., and Singh S. (2014) Oxidative stress and inflammation in diabetic complications. Int. J. Endocrinol. 2014, 679754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mittal M., Siddiqui M. R., Tran K., Reddy S. P., and Malik A. B. (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 20, 1126–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaiswal A. K. (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol Med. 36, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 4. Johnson D. A., and Johnson J. A. (2015) Nrf2–a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 88, 253–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kobayashi M., Li L., Iwamoto N., Nakajima-Takagi Y., Kaneko H., Nakayama Y., Eguchi M., Wada Y., Kumagai Y., and Yamamoto M. (2009) The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 29, 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chin M., Lee C. Y., Chuang J. C., Bumeister R., Wigley W. C., Sonis S. T., Ward K. W., and Meyer C. (2013) Bardoxolone methyl analogs RTA 405 and dh404 are well tolerated and exhibit efficacy in rodent models of type 2 diabetes and obesity. Am. J. Physiol. Renal Physiol. 304, F1438–F1446 [DOI] [PubMed] [Google Scholar]
- 7. Ungvari Z., Bagi Z., Feher A., Recchia F. A., Sonntag W. E., Pearson K., de Cabo R., and Csiszar A. (2010) Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 299, H18–H24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pergola P. E., Krauth M., Huff J. W., Ferguson D. A., Ruiz S., Meyer C. J., and Warnock D. G. (2011) Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b-4 CKD. Am. J. Nephrol. 33, 469–476 [DOI] [PubMed] [Google Scholar]
- 9. Pergola P. E., Raskin P., Toto R. D., Meyer C. J., Huff J. W., Grossman E. B., Krauth M., Ruiz S., Audhya P., Christ-Schmidt H., Wittes J., Warnock D. G., and BEAM Study Investigators. (2011) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 365, 327–336 [DOI] [PubMed] [Google Scholar]
- 10. de Zeeuw D., Akizawa T., Audhya P., Bakris G. L., Chin M., Christ-Schmidt H., Goldsberry A., Houser M., Krauth M., Lambers Heerspink H. J., McMurray J. J., Meyer C. J., Parving H. H., Remuzzi G., Toto R. D., et al. (2013) Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 369, 2492–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin T. J., Zhang K. J., and Liu G. T. (1996) Effects of salvianolic acid A on oxygen radicals released by rat neutrophils and on neutrophil function. Biochem. Pharmacol. 51, 1237–1241 [DOI] [PubMed] [Google Scholar]
- 12. Zhang H. A., Gao M., Zhang L., Zhao Y., Shi L. L., Chen B. N., Wang Y. H., Wang S. B., and Du G. H. (2012) Salvianolic acid A protects human SH-SY5Y neuroblastoma cells against H(2)O(2)-induced injury by increasing stress tolerance ability. Biochem. Biophys. Res. Commun. 421, 479–483 [DOI] [PubMed] [Google Scholar]
- 13. Xu H., Li Y., Che X., Tian H., Fan H., and Liu K. (2014) Metabolism of salvianolic acid A and antioxidant activities of its methylated metabolites. Drug Metab. Dispos. 42, 274–281 [DOI] [PubMed] [Google Scholar]
- 14. Huang J., Qin Y., Liu B., Li G. Y., Ouyang L., and Wang J. H. (2013) In silico analysis and experimental validation of molecular mechanisms of salvianolic acid A-inhibited LPS-stimulated inflammation, in RAW264.7 macrophages. Cell Prolif. 46, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang X. C., Chen J. Q., and Li B. (2014) Salvianolic acid A suppresses CCL-20 expression in TNF-α-treated macrophages and ApoE deficient mice. J. Cardiovasc. Pharmacol. 64, 318–325 [DOI] [PubMed] [Google Scholar]
- 16. Fan H. Y., Fu F. H., Yang M. Y., Xu H., Zhang A. H., and Liu K. (2010) Antiplatelet and antithrombotic activities of salvianolic acid A. Thromb. Res. 126, e17–e22 [DOI] [PubMed] [Google Scholar]
- 17. Huang Z. S., Zeng C. L., Zhu L. J., Jiang L., Li N., and Hu H. (2010) Salvianolic acid A inhibits platelet activation and arterial thrombosis via inhibition of phosphoinositide 3-kinase. J. Thromb. Haemost. 8, 1383–1393 [DOI] [PubMed] [Google Scholar]
- 18. Zhang H., Liu Y. Y., Jiang Q., Li K. R., Zhao Y. X., Cao C., and Yao J. (2014) Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic. Biol. Med. 69, 219–228 [DOI] [PubMed] [Google Scholar]
- 19. Zoja C., Corna D., Nava V., Locatelli M., Abbate M., Gaspari F., Carrara F., Sangalli F., Remuzzi G., and Benigni A. (2013) Analogs of bardoxolone methyl worsen diabetic nephropathy in rats with additional adverse effects. Am. J. Physiol. Renal Physiol. 304, F808–F819 [DOI] [PubMed] [Google Scholar]
- 20. Tan S. M., Sharma A., Stefanovic N., Yuen D. Y., Karagiannis T. C., Meyer C., Ward K. W., Cooper M. E., and de Haan J. B. (2014) Derivative of bardoxolone methyl, dh404, in an inverse dose-dependent manner lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 63, 3091–3103 [DOI] [PubMed] [Google Scholar]
- 21. Alhaider A. A., Korashy H. M., Sayed-Ahmed M. M., Mobark M., Kfoury H., and Mansour M. A. (2011) Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chem. Biol. Interact. 192, 233–242 [DOI] [PubMed] [Google Scholar]
- 22. Zhang M., Song P., Guzman M. R., Asfa S., and Zou M.-H. (2009) Metformin attenuates atherosclerosis in streptozotocin-induced diabetic ApoE-deficient mice through AMP-activated protein kinase. Diabetes 58, A190 [Google Scholar]
- 23. Cheng J. T., Huang C. C., Liu I. M., Tzeng T. F., and Chang C. J. (2006) Novel mechanism for plasma glucose-lowering action of metformin in streptozotocin-induced diabetic rats. Diabetes 55, 819–825 [DOI] [PubMed] [Google Scholar]
- 24. Erejuwa O. O., Sulaiman S. A., Wahab M. S., Sirajudeen K. N., Salleh M. S., and Gurtu S. (2011) Glibenclamide or metformin combined with honey improves glycemic control in streptozotocin-induced diabetic rats. Int. J. Biol. Sci. 7, 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vasamsetti S. B., Karnewar S., Kanugula A. K., Thatipalli A. R., Kumar J. M., and Kotamraju S. (2015) Metformin inhibits monocyte-to-macrophage differentiation via AMPK mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 64, 2028–2041 [DOI] [PubMed] [Google Scholar]
- 26. Kim D., Lee J. E., Jung Y. J., Lee A. S., Lee S., Park S. K., Kim S. H., Park B. H., Kim W., and Kang K. P. (2013) Metformin decreases high-fat diet-induced renal injury by regulating the expression of adipokines and the renal AMP-activated protein kinase/acetyl-CoA carboxylase pathway in mice. Int. J. Mol. Med. 32, 1293–1302 [DOI] [PubMed] [Google Scholar]
- 27. Calvert J. W., Gundewar S., Jha S., Greer J. J., Bestermann W. H., Tian R., and Lefer D. J. (2008) Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 57, 696–705 [DOI] [PubMed] [Google Scholar]
- 28. Lin C. C., and Yang W. C. (2009) Prognostic factors influencing the patency of hemodialysis vascular access: literature review and novel therapeutic modality by far infrared therapy. J. Chin. Med. Assoc. 72, 109–116 [DOI] [PubMed] [Google Scholar]
- 29. Kim J. H., Choi Y. K., Lee K. S., Cho D. H., Baek Y. Y., Lee D. K., Ha K. S., Choe J., Won M. H., Jeoung D., Lee H., Kwon Y. G., and Kim Y. M. (2012) Functional dissection of Nrf2-dependent phase II genes in vascular inflammation and endotoxic injury using Keap1 siRNA. Free Radic. Biol. Med. 53, 629–640 [DOI] [PubMed] [Google Scholar]
- 30. Paravicini T. M., and Touyz R. M. (2008) NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 31, S170–S180 [DOI] [PubMed] [Google Scholar]
- 31. Zheng Z., Chen H., Li J., Li T., Zheng B., Zheng Y., Jin H., He Y., Gu Q., and Xu X. (2012) Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 61, 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Detaille D., Guigas B., Chauvin C., Batandier C., Fontaine E., Wiernsperger N., and Leverve X. (2005) Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 54, 2179–2187 [DOI] [PubMed] [Google Scholar]
- 33. Pilmore H. L. (2010) Review: metformin: potential benefits and use in chronic kidney disease. Nephrology, 15, 412–418 [DOI] [PubMed] [Google Scholar]
- 34. Lee H. J., Mariappan M. M., Feliers D., Cavaglieri R. C., Sataranatarajan K., Abboud H. E., Choudhury G. G., and Kasinath B. S. (2012) Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J. Biol. Chem. 287, 4451–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H., Wang F., Zhang L., Cao Y., Liu W., Hao J., Liu Q., and Duan H. (2011) Modulation of Nrf2 expression alters high glucose-induced oxidative stress and antioxidant gene expression in mouse mesangial cells. Cell. Signal. 23, 1625–1632 [DOI] [PubMed] [Google Scholar]
- 36. Csiszar A., Bagi Z., Feher A., Recchia F. A., Sonntag W. E., Ungvari Z., Pearson K., and de Cabo R. (2011) Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. FASEB J. 25, 1093–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar H., Kim I. S., More S. V., Kim B. W., and Choi D. K. (2014) Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 31, 109–139 [DOI] [PubMed] [Google Scholar]
- 38. Suzuki T., and Yamamoto M. (2015) Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 88, 93–100 [DOI] [PubMed] [Google Scholar]
- 39. Wang S. B., Yang X. Y., Tian S., Yang H. G., and Du G. H. (2009) Effect of salvianolic acid A on vascular reactivity of streptozotocin-induced diabetic rats. Life Sci. 85, 499–504 [DOI] [PubMed] [Google Scholar]
- 40. Haskó G., and Pacher P. (2010) Endothelial Nrf2 activation: a new target for resveratrol? Am. J. Physiol. Heart Circ. Physiol. 299, H10–H12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo R., Liu B., Wang K., Zhou S., Li W., and Xu Y. (2014) Resveratrol ameliorates diabetic vascular inflammation and macrophage infiltration in db/db mice by inhibiting the NF-κB pathway. Diab. Vasc. Dis. Res. 11, 92–102 [DOI] [PubMed] [Google Scholar]
- 42. Vaziri N. D., Liu S., Farzaneh S. H., Nazertehrani S., Khazaeli M., and Zhao Y. Y. (2015) Dose-dependent deleterious and salutary actions of the Nrf2 inducer dh404 in chronic kidney disease. Free Radic. Biol. Med. 86, 374–381 [DOI] [PubMed] [Google Scholar]
- 43. Eelen G., de Zeeuw P., Simons M., and Carmeliet P. (2015) Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 116, 1231–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Forbes J. M., and Cooper M. E. (2013) Mechanisms of diabetic complications. Physiol. Rev. 93, 137–188 [DOI] [PubMed] [Google Scholar]
- 45. Hamilton J., Cummings E., Zdravkovic V., Finegood D., and Daneman D. (2003) Metformin as an adjunct therapy in adolescents with type 1 diabetes and insulin resistance: a randomized controlled trial. Diabetes Care 26, 138–143 [DOI] [PubMed] [Google Scholar]
- 46. Stratton I. M., Adler A. I., Neil H. A., Matthews D. R., Manley S. E., Cull C. A., Hadden D., Turner R. C., and Holman R. R. (2000) Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321, 405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Selvin E., Marinopoulos S., Berkenblit G., Rami T., Brancati F. L., Powe N. R., and Golden S. H. (2004) Meta-analysis: glycosylated hemoglobin and cardiovascular disease in diabetes mellitus. Ann. Intern. Med. 141, 421–431 [DOI] [PubMed] [Google Scholar]
- 48. Kinaan M., Ding H., and Triggle C. R. (2015) Metformin: an old drug for the treatment of diabetes but a new drug for the protection of the endothelium. Med. Princ. Pract. 24, 401–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Foretz M., Guigas B., Bertrand L., Pollak M., and Viollet B. (2014) Metformin: from mechanisms of action to therapies. Cell Metab. 20, 953–966 [DOI] [PubMed] [Google Scholar]
- 50. Yao F., Zhang M., and Chen L. (2016) 5′-Monophosphate-activated protein kinase (AMPK) improves autophagic activity in diabetes and diabetic complications. Acta Pharm. Sin. B 6, 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin J. T., Chen H. M., Chiu C. H., and Liang Y. J. (2014) AMP-activated protein kinase activators in diabetic ulcers: from animal studies to Phase II drugs under investigation. Expert Opin. Invest. Drugs 23, 1253–1265 [DOI] [PubMed] [Google Scholar]
- 52. Zhao Q., Shao L., Hu X., Wu G., Du J., Xia J., and Qiu H. (2013) Lipoxin a4 preconditioning and postconditioning protect myocardial ischemia/reperfusion injury in rats. Mediators Inflamm. 2013, 231351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Csiszar A., Labinskyy N., Podlutsky A., Kaminski P. M., Wolin M. S., Zhang C., Mukhopadhyay P., Pacher P., Hu F., de Cabo R., Ballabh P., and Ungvari Z. (2008) Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am. J. Physiol. Heart Circ. Physiol. 294, H2721–H2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taguchi K., Hida M., Matsumoto T., Ikeuchi-Takahashi Y., Onishi H., and Kobayashi T. (2014) Effect of short-term polyphenol treatment on endothelial dysfunction and thromboxane A2 levels in streptozotocin-induced diabetic mice. Biol. Pharm. Bull. 37, 1056–1061 [DOI] [PubMed] [Google Scholar]
- 55. Tan S. M., Stefanovic N., Tan G., Wilkinson-Berka J. L., and de Haan J. B. (2013) Lack of the antioxidant glutathione peroxidase-1 (GPx1) exacerbates retinopathy of prematurity in mice. Invest. Ophthalmol. Vis. Sci. 54, 555–562 [DOI] [PubMed] [Google Scholar]
- 56. Oelze M., Daiber A., Brandes R. P., Hortmann M., Wenzel P., Hink U., Schulz E., Mollnau H., von Sandersleben A., Kleschyov A. L., Mülsch A., Li H., Förstermann U., and Münzel T. (2006) Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 48, 677–684 [DOI] [PubMed] [Google Scholar]
- 57. Shah A., Xia L., Goldberg H., Lee K. W., Quaggin S. E., and Fantus I. G. (2013) Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J. Biol. Chem. 288, 6835–6848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qian Y., Feldman E., Pennathur S., Kretzler M., and Brosius F. C. 3rd (2008) From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 57, 1439–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mishra R., Cool B. L., Laderoute K. R., Foretz M., Viollet B., and Simonson M. S. (2008) AMP-activated protein kinase inhibits transforming growth factor-β-induced Smad3-dependent transcription and myofibroblast transdifferentiation. J. Biol. Chem. 283, 10461–10469 [DOI] [PubMed] [Google Scholar]
- 60. Zhao J., Miyamoto S., You Y. H., and Sharma K. (2015) AMP-activated protein kinase (AMPK) activation inhibits nuclear translocation of Smad4 in mesangial cells and diabetic kidneys. Am. J. Physiol. Renal Physiol. 308, F1167–F1177 [DOI] [PMC free article] [PubMed] [Google Scholar]