

Abstract
Iron is an essential element for life, but its soluble form is scarce in the environment and is rarer in the human body. Mtb (Mycobacterium tuberculosis) produces two aryl-capped siderophores, mycobactin (MBT) and carboxymycobactin (cMBT), to chelate intracellular iron. The adenylating enzyme MbtA catalyzes the first step of mycobactin biosynthesis in two half-reactions: activation of the salicylic acid as an acyl-adenylate and ligation onto the acyl carrier protein (ACP) domain of MbtB to form covalently salicylated MbtB-ACP. We report the first apo-MbtA structure from Mycobacterium smegmatis at 2.3 Å. We demonstrate here that MbtA activity can be reversibly, post-translationally regulated by acetylation. Indeed the mycobacterial Pat (protein lysine acetyltransferase), Rv0998, specifically acetylates MbtA on lysine 546, in a cAMP-dependent manner, leading to enzyme inhibition. MbtA acetylation can be reversed by the NAD+-dependent DAc (deacetyltransferase), Rv1151c. Deletion of Pat and DAc genes in Mtb revealed distinct phenotypes for strains lacking one or the other gene at low pH and limiting iron conditions. This study establishes a direct connection between the reversible acetylation system Pat/DAc and the ability of Mtb to adapt in limited iron conditions, which is critical for mycobacterial infection.
Keywords: acetylation, enzyme structure, iron, Mycobacterium tuberculosis, post-translational modification (PTM), MbtA, mycobactin
Introduction
Iron is the most abundant element in the earth's core and represent 5% of the earth's crust (1). However, because ferric iron, Fe(III), is poorly soluble in water at neutral pH, its biological availability is scarce (10−18 m in water) (2). Iron is a vital nutrient for most prokaryotes and eukaryotes with a notable exception among well studied pathogenic organisms being the Lyme disease pathogen, Borrelia burgdorferi, which uses manganese (3). Iron is required for DNA replication, oxygen transport, energy generation, enzymatic redox reactions, and oxidative stress protection. Because of the scarcity of free iron, essentially all living species have evolved ways to acquire and store iron and regulate iron levels. In humans, the majority of iron is found in the porphyrin ring of heme proteins, in iron/sulfur-containing enzymes or sequestered in the ferritin protein. To prevent pathogen colonization, humans deplete free iron availability to a concentration of 10−24 m in serum using the transferrin protein (4), and macrophages decrease the expression of ferritin and the transferrin receptor to keep intracellular macrophage iron levels low (5). To circumvent host protection mechanisms, the intracellular pathogen Mtb (Mycobacterium tuberculosis) has fine-tuned an effective iron-chelating arsenal during its 70,000-year-old cohabitation with humans (6). Mtb acquires iron in two distinct ways: a high affinity heme acquisition system (7, 8) and a siderophore-based system (9–11). Mtb synthesizes two related aryl-capped polyketide-polypeptide siderophores, called mycobactin (MBT)2 and carboxymycobactin (cMBT) (Fig. 1A) (12). These siderophores differ only in the composition of their aliphatic tails, which determine the localization of the siderophore: long chain fatty acylated MBT is membrane-associated, whereas cMBT contains short chain dicarboxy fatty acids and is more water-soluble and is secreted by the pathogen (13, 14). Two MBT gene clusters, Mbt-1 and Mbt-2 (Fig. 1B), are composed of a mixture of nonribosomal peptide synthetase and polyketide synthase enzymes (15). Three specific enzymes, MbtA, MbtM (also known as FadD33), and MbtK, are essential for mature mycobactin biosynthesis (16). MbtA catalyzes the first step of MBT core synthesis by activating salicylic acid as an acyladenylate before subsequent ligation to the pantothenyl group of the acyl carrier protein domain of MbtB (Fig. 1C) (12). FadD33 activates the long chain fatty acid before its transfer onto the central N-hydroxy lysine residue of the MBT core (16). MbtK is a GCN5-related N-acetyl-transferase (GNAT) N-acetyltransferase that transfers the long chain fatty acid onto the mycobactin core (16).
FIGURE 1.

Salicyl-capped mycobactin siderophore structure, genetic loci and first biosynthesis reaction. A, lipophilic MBT and hydrophilic cMBT share a common core structure but differ in the length of the alkyl substitution (R group). Atoms in blue are involved in the hexadentate ferric iron coordination. B, mbt-1 gene cluster produces mycobactin core, whereas mbt-2 gene cluster assembles and loads acyl fatty acid onto mycobactin lysine core. The black boxes indicate the presence of IdeR binding sequences, which cause genes repression upon iron binding on IdeR. C, adenylation-ligation reaction catalyzed by MbtA. ACP, acyl carrier protein domain of MbtB.
Iron-dependent transcriptional regulators, IdeR and HupB, control the transcription level of genes involved in iron metabolism including Mbt-1 and Mbt-2 to avoid excessive iron uptake, storage, and toxic overload in the bacterial cell (17, 18). Another level of MBT regulation was recently characterized as a post-translational acetylation of FadD33 by the Pat (protein lysine acetyltransferase) in Msmeg (Mycobacterium smegmatis) (19). Pat acetylates and inactivates FadD33 in a cAMP-dependent manner, which is then reversed by a NAD+-dependent Sirtuin-like deacetylase, DAc1, to restore FadD33 activity. Pat has been structurally characterized and consists of an N-terminal cAMP-binding domain fused to a C-terminal GNAT domain (20, 21). The N-terminal domain allows Pat to be activated by the second messenger, cAMP, produced by the adenylate cyclase (AC) enzymes. The Mtb genome encodes a total of 16 ACs that are sensitive to diverse environmental stimuli, including pH, hypoxia, fatty acids, and carbon dioxide level (22). This complex response system allows mycobacteria to rapidly adapt to external changes by relaying this information, via a specific cAMP pathway, to downstream effectors.
In this study, we demonstrate that the first committed step in mycobactin biosynthesis, catalyzed by MbtA, can also be reversibly post-translationally acetylated by Pat with the loss of its enzymatic activity and reactivated by DAc through deacetylation. Deletion of Pat and DAc genes in Mtb highlights the importance of those specific two genes for normal MBT production during iron starvation.
Results
Cloning, Expression, and Purification of MbtA
To avoid solubility problems, Mtb and Msmeg N-terminal His6-tagged MbtA were expressed at low temperature. Heterologous expression in Escherichia coli gave good quantities of soluble MbtA. After purification by nickel affinity chromatography an apparent molecular mass of ∼59 kDa was observed by SDS-PAGE, which is consistent with the predicted 58,983 and 59,280 Da molecular masses for Msmeg and Mtb, respectively. Mtb and Msmeg MbtA share 69.2% sequence identity and 85.0% sequence similarity.
Cloning, Expression, and Purification of MbtB
The Mtb ACP domain of MbtB module was expressed in E. coli but was mostly insoluble. Using denaturing conditions, small quantities of soluble apo-MbtB-ACP were obtained after nickel affinity chromatography. As assessed by SDS-PAGE, apo-MbtB-ACP displayed an apparent molecular mass of ∼11 kDa, consistent with the molecular mass of 11,670 Da calculated from the amino acid sequence. For the subsequent activity assay with MbtA, apo-MbtB-ACP is required to be phosphopantetheinylated (holo-MbtB-ACP form). Sfp, a phosphopantetheinyl transferase from Bacillus subtilis was used to covalently transfer the 4′-phosphopantetheinyl group from coenzyme A onto apo-MbtB-ACP as described under “Experimental Procedures.” The conversion of apo-MbtB-ACP to holo-MbtB-ACP was confirmed by the addition of a 340-Da moiety, corresponding to the phosphopantetheine arm by Fourier transform mass spectral analysis (data not shown).
Specific Acetylation of MbtA by Protein Lysine Acetyltransferase
Recently, Pat has been identified as a regulator of one of the later step of mycobactin biosynthesis via post-translational modification of fatty acyl-AMP ligase FadD33 (19). MbtA and FadD33 both catalyze an adenylation reaction followed by thioesterification of the substrate onto an ACP. FadD33 is acetylated by Pat on Lys511 and amino acid sequence comparison of MbtA and FadD33 highlights a conserved lysine residue, Lys546, in MbtA (Fig. 2A). Moreover, like FadD33-K511, MbtA-K546 is also flanked by a preceding glycine, as well as two nearby downstream basic residues, which are believed to lower the steric hindrance between Pat and MbtA and reduce the pKa value of the NH2 group of Lys546, respectively (Fig. 2A). Based on these observations, we probed MbtA as a potential substrate for Pat. For this experiment, Msmeg Pat (MSMEG_5458) was used instead of Mtb Pat (Rv0998) because protein refolding issues occurred after protein purification therefore affecting Mtb Pat activity. Western blotting analysis using anti-acetyllysine antibody shows that MbtA is acetylated by Pat in the presence of acetyl-CoA and cAMP (Fig. 2B, lane 2). Negative controls without Pat or acetyl-CoA show no MbtA acetylation (Fig. 2B, lanes 3 and 4). Only in the negative control without cAMP, a light MbtA acetylation is observed (Fig. 2B, lane 5), likely because of copurification of Pat with some bound cAMP as noted previously (23). Msmeg Pat also acetylates Mtb MbtA in the same manner (data not shown). To determine the site(s) of acetylation, we created a single amino acid change in MbtA, changing Lys546 to an alanine. Lys546 appears to be the main acetylation site because the K546A mutant completely loses the ability to be acetylated by Pat by Western blotting (Fig. 2B, lane 1). To confirm this unique acetylation site, in vitro acetylated MbtA was analyzed by mass spectrometry. Good sequence coverage (85%) allowed the identification of Lys546 as the single acetylation site (Fig. 2C) and confirms that Pat acetylates MbtA on Lys546.
FIGURE 2.

Pat acetylates specifically MbtA on Lys546. A, partial alignments of MbtA proteins from M. smegmatis (MSMEG_4516) and M. tuberculosis (Rv2384) versus M. smegmatis FadD33 (MSMEG_2132). Acetylated lysines are indicated in bold type. Conserved residues and basic residues flanking acetylated sites are underlined. B, wild type MbtA or MbtA K546A mutant was incubated with multiple reaction components as indicated in the table. The samples were analyzed by Western blotting (bottom panel) with acetyl-lysine antibody (α-AcK), and total protein content was determined by Ponceau red (top panel). C, MbtA unique acetylation site was identified by MS/MS as Lys546. Shown in an MS/MS spectrum charged tryptic peptide from Ms MbtA (TTAVGKAcIDKK) bearing an acetylated lysine. The acetylated lysine is indicated as KAc.
Effect of Acetylation on MbtA Activity
MbtA enzymatic activity was followed by monitoring the formation of AMP in the presence of 2,3-dihydroxybenzoate and acceptor holo-MbtB-ACP. To test acetylation effects, the activity of Msmeg MbtA was monitored overtime in the presence of Msmeg Pat, acetyl-CoA and cAMP. After 5 h of incubation, Msmeg MbtA activity was reduced by 80%, whereas MbtA activity without either acetyl-CoA or Pat remained unchanged (Fig. 3A). The gradual loss of activity is directly mediated by MbtA acetylation in a Pat- and acetyl-CoA-dependent manner. Using the same Mtb MbtA and Msmeg Pat ratio as used previously for Msmeg MbtA, 12 h of incubation were necessary to achieve complete Mtb MbtA inactivation (data not shown). For all subsequent experiments MbtAs were incubated overnight with Msmeg Pat to ensure full inactivation. The Mtb genome encodes one known sirtuin-like deacetylase DAc (Rv1151c) in contrast to the Msmeg genome, which contains two sirtuin-like deacetylases named DAc1 (Msmeg_5175) and DAc2 (Msmeg_4620). DAc and DAc1 are very similar and were previously shown to deacetylate acetylated forms of acetyl-CoA synthetase (ACS) and FadD33 (19, 23). Western blotting analysis of acetylated Mtb and Msmeg MbtA after overnight incubation with DAc deacetylase demonstrates that both MbtAs are deacetylated by DAc (Fig. 4, A, lane 2, and B, lane 3). To check whether deacetylation restores MbtA activity, acetylated MbtA was incubated with DAc and NAD+. Over time, inactive acetylated MbtA regained its activity when incubated with NAD+ and DAc. However, omission of either NAD+ or deacetylase prevented reactivation (Fig. 3B). These results suggest that MbtA catalytic activity is regulated by Pat and DAc via the reversible acetylation and deacetylation of MbtA at position Lys546.
FIGURE 3.

Acetylation inhibits and deacetylation enhances the MbtA enzyme activity. A, time-dependent inactivation of Msmeg MbtA by acetylation. Msmeg MbtA activity was monitored at different time intervals with the additional components: ●, cAMP, Msmeg Pat and acetyl-CoA; ▴, cAMP and acetyl-CoA; ▵, cAMP and Msmeg Pat. B, time-dependent reactivation of acetylated Mtb MbtA by deacetylation. After acetylation by Msmeg Pat, Mtb MbtA was then incubated with the following components: ●, NAD+ and Mtb DAc; ▴, Mtb DAc; ○, NAD+.
FIGURE 4.
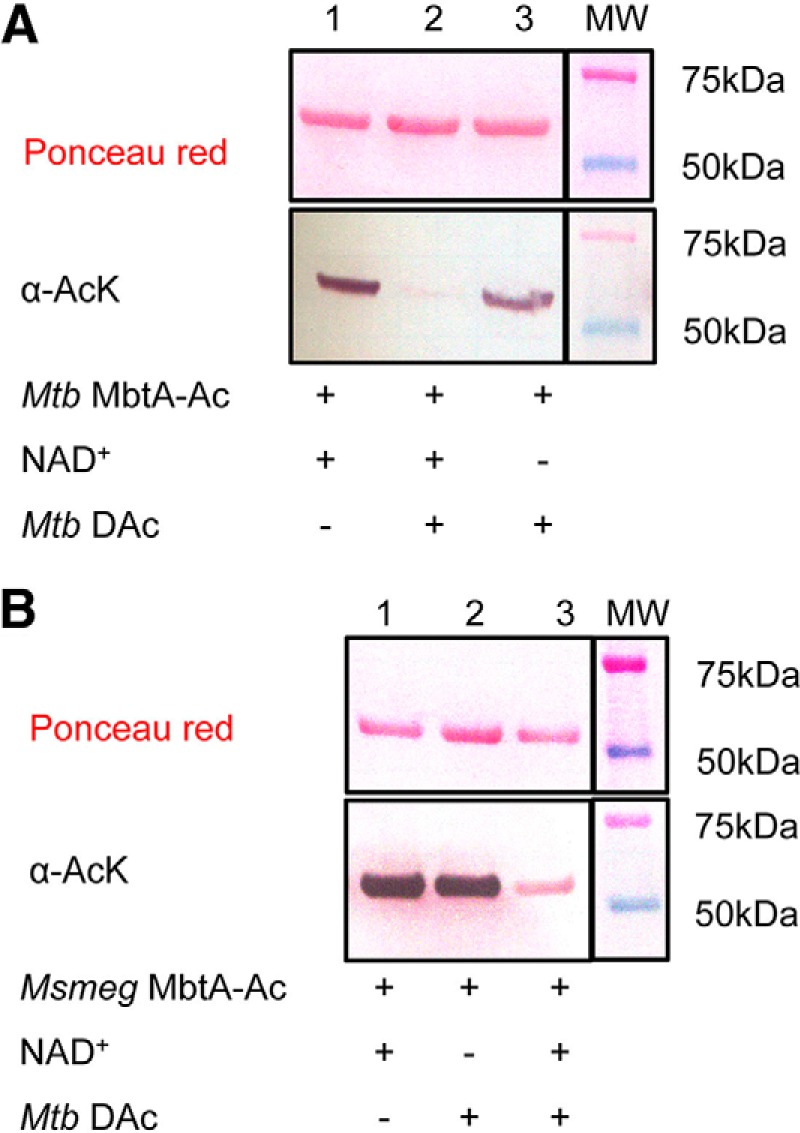
Mtb DAc (Rv1151c) enzyme deacetylates both MbtA homologs. A, acetylated Mtb MbtA were analyzed by Western blotting (bottom panel) with acetyl-lysine antibody (α-AcK), and total protein content was determined by Ponceau red (top panel). B, acetylated Msmeg MbtA were analyzed by Western blotting (bottom panel) with acetyl-lysine antibody (α-AcK), and total protein content was determined by Ponceau red (top panel).
Crystal Structure of Msmeg MbtA apo
Msmeg MbtA was crystallized in an apo form, and the structure was solved to 2.3 Å (Table 1). The Msmeg MbtA structure was solved by molecular replacement using the DhbE structure (Protein Data Bank code 1MDB), which is the adenylation domain in the bacillibactin siderophore biosynthesis (24). Molecular replacement led to a partial solution for MbtA with a defined N-terminal domain and two C-terminal domain fragments (residues 450–462 and 478–503). To complete the Msmeg MbtA structure, the C-terminal domain of DhbE was aligned with the MbtA C-terminal fragments. Through multiple cycles of refinements and manual corrections, a Msmeg MbtA model was generated containing one chain in the asymmetric unit with residues 18–151, 157–313, and 321–556 observed and with an Rwork value of 18.9% and an Rfree value of 24.5%. The overall structure of Msmeg MbtA exhibits the typical fold observed for the ANL (acyl/aryl, non-ribosomal peptide synthetases, and luciferase) superfamily of adenylating enzymes (25). The structure includes two main domains (Fig. 5A): a large N-terminal domain (1–457) and a smaller C-terminal domain (458–558). The N-terminal domain is composed of three subdomains: two β-sheet subdomains (a and b) and a β-barrel subdomain (c). Subdomains a and b form a five-layered αβαβα sandwich. Subdomain a includes six β-strands and five α-helices, whereas the second β-sheet subdomain contains eight β-strands and seven α-helices. The β-barrel subdomain c abuts subdomains a and b and leads to the compact C-terminal domain through a short hinge (including residue Lys457). The compact C-terminal domain consists of three α-helices and five β-strands and harbors Lys546, the lysine acetylated by Pat.
TABLE 1.
Data collection and refinement statistics
| MsMbtA apo | |
|---|---|
| Data collection | |
| Protein Data Bank code | 5KEI |
| Space group | P212121 |
| Unit cell dimensions | |
| a, b, c (Å) | 56.4; 85.8; 104.7 |
| α, β, γ (°) | 90.0; 90.0; 90.0 |
| Resolution range (Å) | 50.0–2.3 |
| Wavelength (Å) | 0.97931 |
| Rmerge (%) (highest shell) | 12.8 (79.3) |
| CC1/2 (highest shell) | 99.3 (65.5) |
| I/σI (highest shell) | 8.7 (1.6) |
| Completeness (%) (highest shell) | 99.6 (98.8) |
| Multiplicity (highest shell) | 3.9 (3.8) |
| Total reflections (unique) | 163,230 (41,974) |
| Refinement statistics | |
| Rwork/Rfree | 18.9/24.5 |
| Number of non-hydrogen atoms | |
| Protein | 3943 |
| Water | 102 |
| Average B factors (Å2) | 40.3 |
| Protein | 40.3 |
| Water | 35.6 |
| Wilson B factor (Å2) | 35.8 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.169 |
| Ramachandran plot | |
| Favored (%) | 95.0 |
| Outliers (%) | 0.6 |
FIGURE 5.
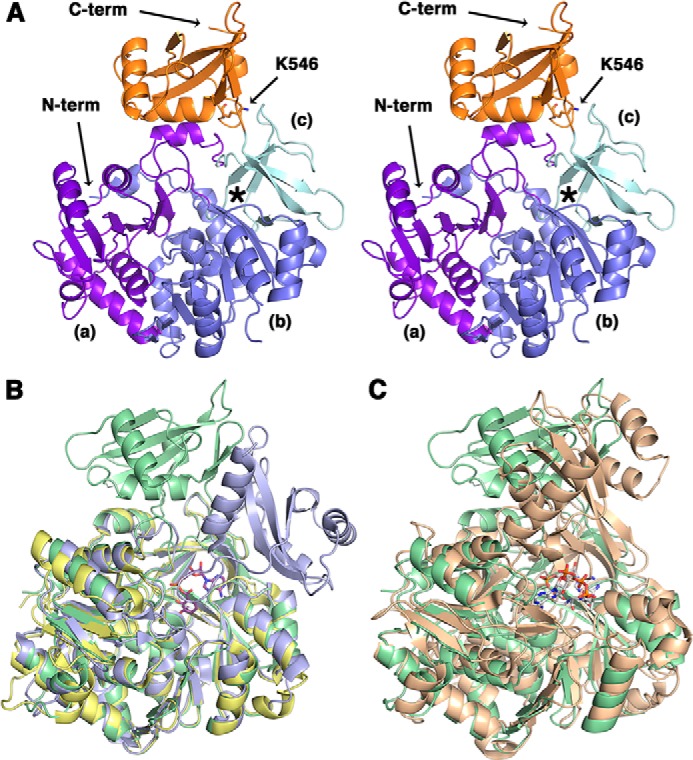
Crystal structure of apo Msmeg MbtA. A, stereo view of the overall structure of apo Msmeg MbtA. The three subdomains parts of the N-terminal domain (N-term) are colored in purple (a), blue (b), and cyan (c), respectively, whereas the C-terminal domain (C-term), or lid, is represented in orange. The location of the active site is highlighted by an asterisk. The residue Lys546, demonstrated in this manuscript to be acetylated, is rendered as sticks colored by CPK, with carbon atoms in orange. B, superimposition of the apo Msmeg MbtA (green), B. subtilis DhbE in complex with DHB adenylated (blue; Protein Data Bank code 1MDB) and A. baumannii BasE N-terminal domain (yellow; Protein Data Bank code 3O83) structures. The DHB-adenylate product is displayed as sticks colored by CPK, with carbon atoms in magenta. C, superimposition of the apo Msmeg MbtA (green) and S. enterica acetyl-CoA synthetase in complex with CoA and AMP (orange; Protein Data Bank 2P2F) structures. The CoA and AMP ligands are rendered as sticks colored by CPK, with carbon atoms in white.
The Msmeg MbtA structure is very similar to the B. subtilis DhbE (44% sequence identity) and the Acinetobacter baumannii BasE N-terminal domain (39% sequence identity) structures. Superimposition of MbtA with DhbE and BasE N-terminal domain shows low root mean square displacement values for the Cα atom positions of 1.0 and 0.7 Å, respectively (Fig. 5B) (24, 26). The highly conserved P-loop motif (Ser212–Lys221) is implicated in the ATP phosphate moiety binding and adopts a slightly different conformation compared with the BasE N-terminal domain or the DhbE structures caused by the lack of substrate in the MbtA active site (24, 26). Salmonella enterica ACS and MbtA both catalyzed adenylation reactions, and structural superimposition indicates a fairly high structure similarity (4.9 Å root mean square displacement for the Cα atom positions) (Fig. 5C) (27). Moreover, Pat acetylates ACS-K609 and MbtA-K546, which are both localized on the same C-terminal domain loop. The main difference between the Msmeg MbtA structure and the other structures (DhbE and ACS) lies in the conformation adopted by the C-terminal domain: the latter is rotated by 91° and 102° compared with the DhbE and the ACS C-terminal domains, respectively. The C-terminal domain forms a “lid,” which can close above the β-barrel subdomain and plays a significant role in the transfer of the adenylated product to the acyl-carrier protein (24, 28). This domain has been shown to adopt different orientations depending on which half-reaction is catalyzed, either the adenylation or the thioesterification reactions (25, 29).
Generation of Precise Null Deletion Mutants in Genes Encoding Pat (Rv0098) and DAc (Rv1151c) in Mtb
Prior to this study, the potential for regulation of mycobacterial siderophore production via post-translational acetylation of enzymes required for mycobactin biosynthesis has been demonstrated biochemically. Here, we also examine the in vivo impact of genetically removing Pat or Dac (Fig. 6, A and C), under conditions encountered in the intraphagosomal environment. Toward this end, using a BLS2-safe M. tuberculosis H37Rv derivative, which is a double auxotroph for leucine and pantothenate, we engineered ΔPat and ΔDAc Mtb mutant strains (Fig. 6, B and D). The genes were deleted by the specialized transduction methodology described under “Experimental Procedures.” To verify the deletions, PCR amplification using three diagnostic independent primer pairs shows expected amplicon sizes for ΔPat + pMV261 and ΔDAc + pMV261 (Fig. 6, E and F).
FIGURE 6.

Construction and verification of Mtb Pat (Rv0998) and Mtb DAc (Rv1151c) deletion mutants. A and C, Rv0998 and Rv1151c adjacent gene organizations are shown. B and D, the ORF removal in ΔRv0998 and ΔRv1151c deletion strains are replaced by sacB-hygR cassette. E and F, left and right flanking PCR were used to confirm deletion of Rv0998 and Rv1151c with specific set of primers.
ΔPat and ΔDAc Share Distinct Phenotypes at Low pH and Limited Iron Conditions
Pat exhibits a unique structural feature with an N-terminal cAMP-binding domain fused to a C-terminal GNAT domain. Our current study and past studies (19, 23) have shown that Pat acetylation activity is dependent on cAMP generated by multiple ACs. To investigate the effect of Pat and DAc deletions in ΔPat and ΔDAc, we tested these strains under different physiological conditions such as low pH, at which specific ACs induce cAMP production (22). As a reference point, we first determined growth curves for the five strains in Sauton medium at pH 7. We observed no significant growth difference between the Mtb WT + pMV261 (expression vector), Mtb ΔPat + pMV261, Mtb ΔDAc + pMV261, Mtb ΔPat + pMV261-Pat, and Mtb ΔDAc + pMV261-DAc (Fig. 7A). All strains carried the pMV261 expression vector to facilitate a comparative analysis. In contrast, when these same strains were grown in Sauton medium at pH 6, Mtb ΔPat + pMV261 showed a growth advantage compared with the parent and complemented strains, whereas Mtb ΔDAc + pMV261 displayed a slight growth defect (Fig. 7B). These results indicate that deletion of Pat leads to an acceleration of growth and suggests that lack of acetylation of Pat substrates at pH 6 is beneficial for Mtb. Because Pat is able to potentially inhibit mycobactin biosynthetic enzymes in vivo, we next cultured the five strains in Sauton medium with limiting iron (100-fold less than standard Sauton) to test the same hypothesis that inhibition of siderophore production under these conditions would inhibit growth in vivo. In iron-limited Sauton medium at pH 7, we observed no significant difference in the growth of the strains, but a much slower growth rate was noted as compared with iron-sufficient medium (Fig. 7C). If we now compared the growth pattern in Sauton pH 6 with limited iron, we observed a distinct growth advantage for Mtb ΔPat + pMV261 compared with the parent and complemented stains, whereas Mtb ΔDAc + pMV261 displayed a major growth defect (Fig. 7D). These results support the idea that in vivo, at lower pH and under elevated cAMP levels, deletion of Mtb Pat likely prevents the acetylation and inhibition of MbtA and FadD33, thus allowing for mycobactin production, whereas deletion of Mtb DAc likely prevents the deacetylation of these enzymes and inhibits mycobactin production.
FIGURE 7.

Iron level and pH effects on Mtb Pat and Mtb DAc deletion mutant phenotypes. Four growth conditions are presented: Sauton pH 7 medium (A), Sauton pH 6 medium (B), limited iron Sauton pH 7 medium (C), and limited iron Sauton pH 6 medium for five constructs (D). Black, Mtb wild type + pMV261; solid red, Mtb ΔPat + pMV261; dotted red, Mtb ΔDAc + pMV261; solid green, Mtb ΔPat + pMV261-Pat; dotted green, Mtb ΔDAc + pMV261-DAc. Error bars represent standard deviations from the mean of results from biological duplicates. Mtb wild type corresponds to mc26206.
Discussion
The acquisition of iron from the environment, its incorporation into iron-containing proteins and enzymes, and the regulation of intracellular iron levels are essential for the growth and virulence of Mtb. Although several methods for iron acquisition exist in the organism, the major mechanism is the production of the iron siderophore, mycobactin. This secondary metabolite has a number of unusual features. A hydroxyphenyloxazoline moiety, made from salicylate and serine, forms a N-cap to the adjacent ϵN-hydroxylysine residue that is additionally acylated on the ϵ-amino group. The remainder of the molecule contains a hydroxybutyryl group and a terminal ϵN-hydroxylysine residue that is cyclized into a seven-membered hydroxamate ring. The hexadentate coordination of ferric iron occurs from the six oxygen and nitrogen atoms in the molecule shown in blue in Fig. 1A. Acylation of the central hydroxylysine residue occurs with saturated and Δ2-unsaturated long chain fatty acids to generate mycobactin or with short chain dicarboxylic acids to generate carboxymycobactins. It is thought that the carboxymycobactins are secreted and acquire host iron and then deliver this iron to the membrane-associated mycobactins (10).
It has been recognized for some time that bacteria tightly regulate the production of siderophores such as mycobactin. In Mtb, a major regulator of mycobactin production is the iron-binding transcriptional regulator IdeR (Rv2711). This 230-residue protein is essential for mycobacterial growth and is a member of the diphtheria toxin repressor family and exists as a homodimer in solution (30). It binds to a highly conserved 19-base pair inverted repeat that is found adjacent to genes whose transcription is repressed at high iron concentrations. In both the Mbt-1 and Mbt-2 gene clusters, multiple IdeR binding sites are observed adjacent to the transcriptional start sites for the genes in the two clusters. Because iron is acquired by the mycobactins and the intracellular concentration increases, the iron-bound IdeR binds to these regions, effectively preventing transcription of these genes (31). This is a general mechanism of regulation in many bacteria. There is a recent report that the HupB protein can serve as a positive regulator of mycobactin synthesis under conditions of low intracellular iron by binding to a region 5′ to that of the IdeR binding site and recruiting RNA polymerase to the transcription start site (32).
In the current study, we have analyzed the potential for non-transcriptional regulation of mycobactin biosynthesis. We have previously reported the reversible, post-translational monoacetylation of both the Mtb ACS and the mbtM-encoded acyl-ACP synthetase (FadD33) (19, 23). These enzymes are mechanistically related and use ATP to form an intermediate acetyl or fatty acyl adenylate, which is then nucleophilically attacked by the thiol of CoASH or the phosphopantetheine arm of an ACP domain. Knowing the primary sequence around the lysine residue that is acetylated in both these enzymes, we identified a highly similar sequence in the Mtb and Msmeg MbtA enzymes. It was previously shown that MbtA-K546 lysine equivalents in FadD33 and ACS are required for catalysis in the first adenylation half-reaction (19, 23). The mechanism of MbtA is similar to that of both ACS and FadD33 in that a carboxyl group of the substrate is initially adenylated for activation, followed by the attack of a thiol group on the mixed carboxylic-phosphoric anhydride, although MbtA uniquely uses an aryl carboxylate, salicylate. We now show that MbtA is also reversibly acetylated and that this leads to enzyme activity loss.
MbtA has become a well established druggable target in the last decade. The production of mycobactin is essential, and the tight binding of hydrolytically stable, isosteric analogs of the salicyladenylate has been demonstrated (33). The intermediate analog, 5′-O[N-(salicyl)sulfamoyl]adenosine was shown independently by three groups to inhibit MbtA with a Ki value of ∼6 nm (33–35). This compound exhibits antimycobacterial activity in vitro with a MIC value of 0.4 mm and exhibited in vivo activity in an acute mouse model. Because MbtA is the first and committed step in mycobactin synthesis, it is perhaps not surprising that its inhibition would be so deleterious.
To support our biochemical analysis, we also performed genetic analysis to investigate the role of acetylation in the Mtb physiological contest. We generated two strains in which either the Pat or DAc genes were knocked out and strains in which the knockouts were complemented. To mimic the environment inside the macrophage phagosome, strains were grown at low pH and limiting iron condition. At pH 7 (Sauton pH 7 and limited iron pH 7), all strains exhibit a similar growth phenotype, but at relevant physiological pH (pH 6 and under limiting iron conditions), the ΔPat strain grows significantly faster than the WT, and the ΔDAc grows considerably slower. Both complemented strains grow approximately as well as WT. This suggests that when acetylation of MbtA or FadD33 is prevented in the ΔPat strain, the resulting active enzymes are capable of generating the required levels of mycobactin. On the other hand, in the ΔDAc strain, MbtA and FadD33 are likely to be functioning at less than full activity, even less activity than the WT strain, resulting in insufficient mycobactin levels. Thus mutant phenotypes suggest that acetylation may influence Mtb survival in the macrophage.
It is clear that the transcriptional regulation of the mycobactin biosynthetic gene clusters is the major mechanism by which mycobactin biosynthesis is regulated. However, once the proteins are made, they remain constitutively active, potentially enabling the production of excess mycobactin and allowing for the accumulation of intracellular iron in excess of its ability to be incorporated into iron-dependent proteins. This would lead to free iron in the cell, and in the reducing, but aerobic, intracellular environment of the cell, the potential for the generation of reactive oxygen species through the action of Fenton-type chemistry. In support of this, a recent report showed that Mtb is particularly sensitive to ascorbic acid, vitamin C, in comparison with other Gram-negative and positive species (36). The ability to rapidly inactivate MbtA could be used to break the ATP-consuming production of mycobactin very quickly when the organism senses that sufficient iron is present. This secondary level of regulation could be used to fine-tune iron levels, allowing just enough as required and preventing the overaccumulation of iron to toxic levels.
Experimental Procedures
Cloning, Expression, and Purification of MbtA
MbtA (MSMEG_4516 and Rv2384) was amplified from Msmeg mc2155 and Mtb H37Rv genomic DNA using the primer pairs Msmeg_MbtA_F (5′-GGAATTCCATATGACTCTGACCACGCCCCAC-3′) and Msmeg_MbtA_R (5′-CCCAAGCTTCCCGCCGAGCTGACGCACGA-3′) or Mtb_MbtA_F (5′-GGAATTCCATATGCCACCGAAGGCGGCAGAT-3′) andMtb_MbtA_R (5′-CCCAAGCTTATGGCAGCGCTGGGTCGTCAC-3′) containing NdeI and HindIII sites, respectively. The PCR amplicon was ligated into the pET-28a (+) vector and then transformed into E. coli DH5α competent cells to create pET-28a (+)::MbtA N-terminally His6 tag plasmid. A sequence-verified construct was transformed in E. coli T7 express lysy/Iq for protein expression. A 4-ml preculture was used to inoculate 1 liter of Luria-Bertani medium supplemented with 50 μg/ml kanamycin. The culture was grown to midlog phase (A600 ∼ 0.6) at 37 °C, then induced by the addition of 0.4 mm isopropyl 1-thio-β-d-galactopyranoside. After 18 h of additional incubation (20 °C), the cells were harvested by centrifugation (6,000 × g, 20 min) and stored at −80 °C. The cell pellet was thawed and resuspended in lysis buffer (25 mm Tris, pH 8.0, containing 150 mm NaCl, 10 mm imidazole) supplemented with DNase I (0.1 μg/ml), lysozyme (2 mg/ml), and EDTA-free protease inhibitors mixture (Roche). Resuspended cells were disrupted by sonication, and cellular debris was removed from the lysate by centrifugation at 16,000 rpm for 50 min. MbtA was purified by nickel affinity chromatography. After an extensive wash with 25 mm Tris, pH 8.0, containing 150 mm NaCl and 10 mm imidazole buffer, the bound protein was eluted with a linear imidazole gradient (30–250 mm). Pure fractions, as determined by SDS-PAGE, were pooled together for buffer exchange into 25 mm Tris, pH 8.0, containing 150 mm sodium chloride and 10% glycerol. For crystallization purpose, the N-terminal polyhistidine tag of Msmeg MbtA was cleaved using thrombin protease. The cleavage reaction was dialyzed overnight against 50 mm Tris (pH 8.0), 150 mm sodium chloride, 1 mm dithiothreitol, and 1 mm calcium chloride. The enzyme was further purified using size exclusion chromatography (HiLoadTM 26/60 SuperdexTM 75 prep grade) in the buffer described above without calcium chloride and then concentrated to 12 mg/ml using a 30-kDa Amicon ultra centrifugal filter.
Crystallization
The apo enzyme was screened against the MCSG suite (Microlytic, 384 conditions). A Crystal Gryphon robot (Art Robbins) was used to dispense both the reservoir solutions and the sitting drops (1:1 ratio). Crystal trays (Intelli-Plates 96, Art Robbins) were then sealed and incubated at 20 °C. Preliminary crystals were obtained in the condition 1D2 containing 0.2 m sodium chloride, 0.1 m bis-tris (pH 6.5), and 25% (w/v) PEG 3350. The crystallization condition containing 0.1 m bis-tris (pH 6.5), 0.4 m sodium chloride, and 17.5% (w/v) PEG 3350 improved the quality of the crystal. The latter was cryoprotected by the addition of 30% (w/v) PEG and flash-cooled in liquid nitrogen.
Data Collection and Structure Determination
Diffraction data were collected at the Lilly Research Laboratories Collaborative Access Team Beamline at the Advance Photon Source Argonne National Laboratory at 0.97931 Å wavelength radiation. Diffraction was observed to 2.3 Å. The data were indexed, integrated, and finally scaled using the XDS package (37). Matthews' coefficient analysis indicated the presence of one molecule in the asymmetric unit (43% solvent). The structure of DhbE from B. subtilis (Protein Data Bank code 1MDF) was used to carry out molecular replacement with PHASER-MR (24, 38). The solution obtained by molecular replacement was used as a template for the Autobuild tool to build 78% of the model (39). The rest of the model was manually built using COOT and iterative cycles of refinements (rigid body refinement, simulated annealing, positional, and B-factor refinements) (40, 41). Three residues were observed in the disallowed region of the Ramachandran plot (Val197, Val544, and Arg554).
Cloning, Expression, and Purification of MbtB-ACP
The acyl carrier protein domain of MbtB enzyme, MbtB-ACP (Rv2383c) was amplified from Mtb genomic DNA using the primer pairs Mtb_MbtB-ACP_F (5′-GGAGGGCATATGGTGCATGCTACGGCG-3′) and Mtb_MbtB-ACP_R (5′-CCCAAGCTTTGCGGCAACTGCCGTGGG-3′) containing NdeI and HindIII sites, respectively. The PCR amplicon was ligated into the pET-28a (+) vector and then transformed into E. coli DH5α competent cells to create pET-28a (+)::MbtB-ACP N-terminally His6 tag plasmid. A sequence-verified construct was transformed in E. coli T7 express lysy/Iq for protein expression. MbtB-ACP domain was expressed as per MbtA, and cells were stored at −80 °C. The cell pellet was thawed and resuspended in lysis buffer (25 mm Tris, pH 8.0) supplemented with DNase I (0.1 μg/ml) and EDTA-free protease inhibitors mixture (Roche). Resuspended cells were disrupted by homogenization using the EmulsiFlex-C3 (Avestin) at 10,000 p.s.i., and cellular debris was removed from the lysate by centrifugation at 16,000 rpm for 50 min. MbtB-ACP was purified by nickel affinity chromatography. After extensive wash with 4 m urea, 25 mm Tris, pH 8.0, 10 mm imidazole buffer, the bound protein was eluted with a linear imidazole gradient (30–250 mm) with 4 m urea. Pure fractions, as determined by SDS-PAGE were pooled together for buffer exchange first in 1 m urea, 50 mm Tris, pH 8.0, 150 mm NaCl and then in 50 mm Tris, pH 8.0, 150 mm NaCl to allow refolding. MbtB-ACP was concentrated using an Amicon pressure concentrator with a 3-kDa filter to 100 μm.
Cloning, Expression, and Purification of Sfp
Sfp was amplified from B. subtilis genomic DNA using the primer pairs Bsub_Sfp_F (5′-GGTATTGAGGGTCGCATGAAGATTTACGGAATT-3′) and Bsub_Sfp_R (5′-AGAGGAGAGTTAGAGCCTTATAAAAGCGCTTCGTA-3′). The PCR amplicon was ligated into the pET-30 Xa/LIC (+) vector and then transformed into E. coli DH5α competent cells to create pET-30 Xa (+)::Sfp N-terminally His6 tag plasmid. A sequence-verified construct was transformed in E. coli T7 express lysy/Iq for protein expression. A 4-ml preculture was used to inoculate 1 liter of Luria-Bertani medium supplemented with 50 μg/ml kanamycin. The culture was grown to midlog phase (A600 = ∼0.6) at 37 °C and then induced by the addition of 0.1 mm isopropyl 1-thio-β-d-galactopyranoside. After 18 h of additional incubation (30 °C), the cells were harvested by centrifugation (6,000 × g, 20 min) and stored at −80 °C. The cell pellet was thawed and resuspended in lysis buffer (50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl, 10 mm imidazole) supplemented with DNase I (0.1 μg/ml), lysozyme (2 mg/ml), and EDTA-free protease inhibitors mixture (Roche). Resuspended cells were disrupted by sonication, and cellular debris was removed from the lysate by centrifugation at 16,000 rpm for 50 min. Sfp was purified by nickel affinity chromatography. After an extensive wash with 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 10 mm imidazole buffer, the bound protein was eluted with a linear imidazole gradient (30–250 mm). Pure fractions, as determined by SDS-PAGE, were pooled together for buffer exchange into 50 mm sodium phosphate, pH 8.0, containing 150 mm NaCl, 5 mm DTT, 10 mm MgCl2, and 10% glycerol. Sfp was concentrated using an Amicon pressure concentrator with a 10-kDa filter to 250 μm.
Phosphopantetheinylation of MbtB-ACP by Sfp
To convert the apo form of MbtB-ACP to the phosphopantetheinylated form, holo-MbtB-ACP, the phosphopantetheinyl transferase Sfp was used. 100 μm Apo-MbtB-ACP was incubated with 0.2 μm Sfp in 50 mm Tris, pH 7.8, with 150 mm NaCl, 1 mm CoASH, and 2 mm DTT. The reaction was allowed to proceed at 25 °C for 18 h before buffer exchange into 50 mm Tris, pH 8.0, 150 mm NaCl. Traces of Sfp were removed during MbtB-ACP concentration using two successive Amicon filtrations with 30- and 3-kDa cut-off filters, respectively.
Measurement of Enzymatic Activity
The enzymatic activity of MbtA was determined spectrophotometrically by coupling the formation of AMP to the reactions of myokinase, pyruvate kinase, and lactate dehydrogenase as described previously (42). The reactions were performed in 100 mm Hepes, pH 7.5, 10 mm MgCl2, 250 mm NaCl, 1 mm phosphoenolpyruvate, 0.15 mm NADH, 300 μm holo-MbtB-ACP, 18 units of myokinase, 18 units of pyruvate kinase, and 18 units of lactate dehydrogenase in a final volume of 100 μl. Typically 0.5 μm of Msmeg or Mtb MbtA was used and incubated for 5 min at 25 °C with the reaction mix prior to reaction initiation by substrate addition with 2,3-dihydroxybenzoate, a salicylic acid analog. The reaction was monitored at 340 nm (ϵ340 = 6220 m−1 cm−1) using a Shimadzu spectrophotometer (UV-2450).
Site-directed Mutagenesis and Purification of MbtA-K546A
The K546A mutation was introduced into the pET-28a (+)::MbtA N-terminally His6 tag plasmid using the QuikChange mutagenesis kit (Stratagene) with the following primers: Mtb_MbtA_K542A_F (5′-CAACGCCGATCGGGGCGATCGACAAACGAG-3′), Mtb_MbtA_K542A_R (5′-CTCGTTTGTCGATCGCCCCGATCGGCGTTG-3′), Msmeg_MbtA_K546A_F(5′-CACGGCCGTCGGCGCGATCGACAAGAAG-3′), and Msmeg_MbtA_K546A_R (5′-CTTCTTGTCGATCGCGCCGACGGCCGTG-3′). The mutation was confirmed by DNA sequencing. The expression and purification of MbtA mutant was the same as described for the wild type.
In Vitro Acetylation Assay
10 μm MbtA or the mutant protein was incubated with 1 mm cAMP, 100 μm AcCoA, and 1 μm Msmeg Pat at 37 °C for 5 h. Samples were then analyzed by SDS-PAGE. Proteins were transferred to a nitrocellulose membrane. Western blotting was performed using an anti-acetyllysine antibody (Cell Signaling Technology Inc. and ImmuneChem Pharmaceuticals Inc., dilution of 1:2000) and goat anti-rabbit IgG AP conjugate (Bio-Rad, dilution of 1:2000). Development was carried out according to the manufacturer's instructions.
Time-dependent Inactivation and Reactivation of MbtA
20 μm MbtA was incubated with 1 mm cAMP and 100 μm AcCoA with 5 μm Msmeg Pat in 50 mm HEPES, pH 7.5, 100 mm NaCl at 37 °C. Aliquots of the reaction mixture were withdrawn every hour. The residual activity of MbtA was measured as described above. Control experiments were performed when Msmeg Pat or AcCoA were not included. MbtA acetylation was also assessed by Western blotting. For reactivation time course, the acetylated MbtA was then purified by nickel-nitrilotriacetic acid chromatography and used in the deacetylation assay. A typical reaction mixture contained 10 μm acetylated MbtA, 2 mm NAD+, and 5 μm Rv1151c (deacetylase). Aliquots of the reaction mixture were withdrawn every hour, and MbtA activity was measured as described above. Control experiments were performed without adding NAD+ or the deacetylase. MbtA deacetylation was also assessed by Western blotting.
Mass Spectrometric Analysis of Acetylated MbtA
The mass spectrometric analysis was performed at the Laboratory for Macromolecular Analysis and Proteomics of Albert Einstein College of Medicine. The same protocol as described in Refs. 19 and 43 was implemented for identification of MbtA acetylation sites.
Media and Bacterial Cultures
Our growth studies utilized a BSL2-safe derivative strain of Mtb H37Rv called mc26206 (H37Rv ΔpanCD ΔleuCD), which is routinely used by the Mtb community (44). mc26206 is a strain auxotrophic for both pantothenate and leucine (pan− leu−) and was maintained in Middlebrook 7H9 broth (Difco) supplemented with 10% ADC (5% BSA, 2% dextrose, 5% catalase) 0.025% tyloxapol, 50 μg/ml of pantothenate, and 50 μg/ml of leucine. Antibiotics (hygromycin at 50 μg/ml and kanamycin at 25 μg/ml) were included as appropriate for strains harboring antibiotic resistance cassettes.
For growth in iron-limited medium, mc26206 pan− leu− strains were grown in deferreted Sauton. Sauton contains 0.2% citric acid, 0.05% K2HPO4, 0.4% l-asparagine, 0.2% dextrose, 6% glycerol, 0.025% tyloxapol, 50 μg/ml of pantothenate, and 50 μg/ml of leucine. To remove metal contamination, Sauton medium was treated with Chelex-100 (Bio-Rad) according to the manufacturer's instructions. Chelex was removed by filtration, and the medium was supplemented with 1 mg/liter ZnSO4, 0.5 g/liter MgSO4, and 16.2 mg/liter ferric chloride. Iron limited Sauton medium was supplemented with ZnSO4, MgSO4 as described above, and 0.162 mg/liter of ferric chloride. When required, kanamycin (25 μg/ml) and hygromycin (50 μg/ml) were added to the medium. Sauton, 7H9, and Sauton iron-limited medium pHs were adjusted to 7.0 or 6.0 and 100 mm MES was added to maintain a constant pH during growth curve.
Rv0998 and Rv1151c Mutant Constructions in Mtb
The gene knockouts of Pat acetyltransferase (Rv0998) and Sirtuin-like deacetylase DAc (Rv1151c) were created by specialized transduction methodology in mc26206 (44, 45). Constructs for allelic exchange were generated by amplifying the upstream and downstream flanking regions of each of the two genes using the primer pairs listed in Table 2. The upstream and downstream flanking regions were cloned into suicidal delivery vector pYUB1471 to create an allelic exchange vectors harboring a selectable/counterselectable cassette (γδ(sacB-Hyg)γδ) between the two flanking regions. The allelic exchange constructs were incorporated into shuttle mycobacteriophage vector phAE159; the phasmid constructs used in this work were obtained as part of a collaboration with Genomics Institute of the Novartis Research Foundation to generate a set of gene deletion constructs for Mtb (46). Phasmid DNA was electroporated into Msmeg to obtain plaques at the permissive temperature of 30 °C. Specialized transducing phages were picked and amplified at 30 °C to generate high titer mycobacteriophage. mc26206 was transduced with high titer phages at the non-permissive temperature of 37 °C to delete genes of interest by specialized transduction (44, 45). The transductants were plated on selective medium: Middlebrook 7H10 medium (Difco) containing 10% oleate-albumin-dextrose-catalase enrichment (0.5 g of oleic acid, 50 g of albumin, 20 g of dextrose, 0.04 g of catalase, 8.5 g of sodium chloride in 1 liter of water), 0.5% glycerol, 50 μg/ml leucine, 24 μg/ml pantothenate, and 50 μg/ml hygromycin. Genomic DNA prepared from transductants was screened by a three-primer PCR to confirm gene deletion, using primers in Table 3.
TABLE 2.
PCR primers for cloning flanks of the indicated loci to generate allelic exchange substrates
| Locus | Flank | Primer name | Primer sequence |
|---|---|---|---|
| Rv1151c | Left | DRv1151c LL | TTTTTTTTCCATAAATTGGGCCGCGGACCTGGTAAATAA |
| Left | DRv1151c LR | TTTTTTTTCCATTTCTTGGTACAAGAATTGCAGGCTGTGGATGTCTCACTGAGGTCTCTTCTTGTCATCGCGGAACGTC | |
| Right | DRv1151c RR | TTTTTTTTCCATCTTTTGGCCAAGATCCATGCCCTGACC | |
| Right | DRv1151c RL | TTTTTTTTCCATAGATTGGCTGGTCTAGTAGTGTATAGCCGAGTGTCTGGTCTCGTAGGACGATCAGCATCCGCGAGT | |
| Rv0998 | Left | DRv0998 LL | TTTTTTTTCCATAAATTGGACGAAGGCATTCCGGTCAAA |
| Left | DRv0998 LR | TTTTTTTTCCATTTCTTGGAATCGTATGACACGCGCTTGGATGTCTCACTGAGGTCTCTCCGGACATCCCTGAAAGACG | |
| Right | DRv0998 RR | TTTTTTTTCCATCTTTTGGAGCTGCGTCCCGGAAAAAGT | |
| Right | DRv0998 RL | TTTTTTTTCCATAGATTGGTTAATGATTGTGGCTGACGCCGAGTGTCTGGTCTCGTAGGTCCGGGTGAGCTGAGCTTG |
TABLE 3.
PCR primers used to screen for deletions at the indicated loci
AES, allelic exchange substrate.
| Locus | Flank | Primer name | Binding site | Primer sequence |
|---|---|---|---|---|
| Rv1151c | Left | 1151c L Flank F = P7 | Upstream of L flank, in wild-type and mutant | CAT GCC GTT CAG CAT GTC |
| sacBout_LR = P10 | Within AES construct, mutant only | GAT GTC TCA CTG AGG TCT CT | ||
| 1151c L Flank R = P8 | Within deleted region, wild-type only | GGT GAT GAC GCT GAC CTC | ||
| Right | 1151c R Flank F = P5 | Within deleted region, wild-type only | CGT CAT CAC CCA GAA TGT C | |
| hygout_RR = P9 | Within AES construct, mutant only | CGA GTG TCT GGT CTC GTA G | ||
| 1151c R Flank R = P6 | Downstream of R flank, in wild-type and mutant | GGC ACT GTC GGA TTA CAA G | ||
| Rv0998 | Left | 0998 L Flank F = P1 | Upstream of L flank, in wild-type and mutant | CGT TGT GTC TAC TGC TCG AC |
| sacBout_LR = P9 | Within AES construct, mutant only | GAT GTC TCA CTG AGG TCT CT | ||
| 0998 L Flank R = P2 | Within deleted region, wild-type only | GAT GAT CGC AAC ACC ATC | ||
| Right | 0998 R Flank F = P3 | Within deleted region, wild-type only | CGG TTC ATG TCG GCT CGT GTT C | |
| hygout_RR = P10 | Within AES construct, mutant only | CGA GTG TCT GGT CTC GTA G | ||
| 0998 R Flank R = P4 | Downstream of R flank, in wild-type and mutant | TGT GCG GTA CAT CGA CCA CCT C |
For complementation, the Rv0998 and Rv1151c genes were amplified by PCR from H37Rv genomic DNA using the following primers Rv00998_F (TTTTTTTTGGATCCATTGGACGGGATAGCCGAATTG), Rv0998_R (TTTTTTTTGTTAACTCAGCCGACGGCCTCGATCAC), Rv1151c_F (TTTTTTTTGGATCCAATGCGAGTGGCGGTGCTCAG), and Rv1151c_R (TTTTTTTTGTTAACCTATTTCAGCAGGGCGGGCAG) containing BamHI and HpaI, respectively. PCR products were digested with BamHI and HpaI and cloned into the mycobacterial episomal pMV261 vector using the BamHI and HpaI restriction sites (47). After sequence verification, complementation plasmids were electroporated into the relevant deletion mutant strains, with transformants selected on 7H10 medium as noted above, also containing 25 μg/ml kanamycin. To generate control strains for comparison in growth studies, the pMV261 empty vector was similarly electroporated into mc26206 parental, mc26206ΔPat, and mc26206ΔDAc.
Growth Assays
Mycobacterial strains were inoculated in liquid media, 7H9 pH 7, and then transferred into fresh 7H9, Sauton, or low iron Sauton media at either pH 7 or pH 6. After one culture passage in the final medium to allow strain adaptation, growth curves were started out at an A600 = 0.05, and optical densities were measured at regular intervals for 20–40 days.
Author Contributions
O. V., H. X., J. M. T., J. S. B., and W. R. J. planned and designed the research. O. V. purified and L. F. crystallized MbtA protein and determined its x-ray structure. O. V. and H. X. performed the in vitro enzyme activity. J. M. T. designed mutant constructs, and mutants were obtained by H. X. and O. V. O. V., H. X., and A. A. M. analyzed the mutant phenotypes. O. V., L. F., and J. S. B. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. Myrasol Callaway (Albert Einstein College of Medicine) for assistance in mass spectrometry. We thank Dr. Steven Almo and his laboratory for use of the crystallography equipment. We also thank Laura Cole and Annie Dai for technical assistance.
This work was supported by National Institutes of Health Grants AI60899 (to J. S. B.) and AI26170, AI098925, and P01AI63537 (to W. R. J.). This research used resources of the Advanced Photon Source, a U.S. Department of Energy Office of Science User Facility operated for the Department of Energy Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team Beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MBT
- mycobactin
- cMBT
- carboxymycobactin
- ACP
- acyl carrier protein
- GNAT
- GCN5-related N-acetyl-transferase
- AC
- adenylate cyclase
- ACS
- acetyl-CoA synthetase
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. McDonough W. F., and Sun S. S. (1995) Composition of the earth. Chem. Geol. 120, 223–253 [Google Scholar]
- 2. Raymond K. N., and Carrano C. J. (1979) Coordination chemistry and microbial iron transport. Acc. Chem. Res. 12, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Posey J. E., and Gherardini F. C. (2000) Lack of a role for iron in the Lyme disease pathogen. Science 288, 1651–1653 [DOI] [PubMed] [Google Scholar]
- 4. Raymond K. N., Dertz E. A., and Kim S. S. (2003) Enterobactin: an archetype for microbial iron transport. Proc. Natl. Acad. Sci. U.S.A. 100, 3584–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Byrd T. F., and Horwitz M. A. (1993) Regulation of transferrin receptor expression and ferritin content in human mononuclear phagocytes. Coordinate upregulation by iron transferrin and downregulation by interferon gamma. J. Clin. Invest. 91, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Comas I., Coscolla M., Luo T., Borrell S., Holt K. E., Kato-Maeda M., Parkhill J., Malla B., Berg S., Thwaites G., Yeboah-Manu D., Bothamley G., Mei J., Wei L., Bentley S., et al. (2013) Out-of-Africa migration and neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones C. M., and Niederweis M. (2011) Mycobacterium tuberculosis can utilize heme as an iron source. J. Bacteriol. 193, 1767–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tullius M. V., Harmston C. A., Owens C. P., Chim N., Morse R. P., McMath L. M., Iniguez A., Kimmey J. M., Sawaya M. R., Whitelegge J. P., Horwitz M. A., and Goulding C. W. (2011) Discovery and characterization of a unique mycobacterial heme acquisition system. Proc. Natl. Acad. Sci. U.S.A. 108, 5051–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Voss J. J., Rutter K., Schroeder B. G., Su H., Zhu Y., and Barry C. E. 3rd (2000) The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 97, 1252–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gobin J., and Horwitz M. A. (1996) Exochelins of Mycobacterium tuberculosis remove iron from human iron-binding proteins and donate iron to mycobactins in the M. tuberculosis cell wall. J. Exp. Med. 183, 1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ryndak M. B., Wang S., Smith I., and Rodriguez G. M. (2010) The Mycobacterium tuberculosis high-affinity iron importer, IrtA, contains an FAD-binding domain. J. Bacteriol. 192, 861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quadri L. E., Sello J., Keating T. A., Weinreb P. H., and Walsh C. T. (1998) Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 5, 631–645 [DOI] [PubMed] [Google Scholar]
- 13. Luo M., Fadeev E. A., and Groves J. T. (2005) Mycobactin-mediated iron acquisition within macrophages. Nat. Chem. Biol. 1, 149–153 [DOI] [PubMed] [Google Scholar]
- 14. Wells R. M., Jones C. M., Xi Z., Speer A., Danilchanka O., Doornbos K. S., Sun P., Wu F., Tian C., and Niederweis M. (2013) Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 9, e1003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chavadi S. S., Stirrett K. L., Edupuganti U. R., Vergnolle O., Sadhanandan G., Marchiano E., Martin C., Qiu W. G., Soll C. E., and Quadri L. E. (2011) Mutational and phylogenetic analyses of the mycobacterial mbt gene cluster. J. Bacteriol. 193, 5905–5913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krithika R., Marathe U., Saxena P., Ansari M. Z., Mohanty D., and Gokhale R. S. (2006) A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2069–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodriguez G. M. (2006) Control of iron metabolism in Mycobacterium tuberculosis. Trends Microbiol. 14, 320–327 [DOI] [PubMed] [Google Scholar]
- 18. Pandey R., and Rodriguez G. M. (2014) IdeR is required for iron homeostasis and virulence in Mycobacterium tuberculosis. Mol. Microbiol. 91, 98–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vergnolle O., Xu H., and Blanchard J. S. (2013) Mechanism and regulation of mycobactin fatty acyl-AMP ligase FadD33. J. Biol. Chem. 288, 28116–28125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee H. J., Lang P. T., Fortune S. M., Sassetti C. M., and Alber T. (2012) Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat. Struct. Mol. Biol. 19, 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Favrot L., Blanchard J. S., and Vergnolle O. (2016) Bacterial GCN5-related N-acetyltransferases: from resistance to regulation. Biochemistry 55, 989–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDonough K. A., and Rodriguez A. (2012) The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 10, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu H., Hegde S. S., and Blanchard J. S. (2011) Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 50, 5883–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. May J. J., Kessler N., Marahiel M. A., and Stubbs M. T. (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 99, 12120–12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gulick A. M. (2009) Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drake E. J., Duckworth B. P., Neres J., Aldrich C. C., and Gulick A. M. (2010) Biochemical and structural characterization of bisubstrate inhibitors of BasE, the self-standing nonribosomal peptide synthetase adenylate-forming enzyme of acinetobactin synthesis. Biochemistry 49, 9292–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gulick A. M., Starai V. J., Horswill A. R., Homick K. M., and Escalante-Semerena J. C. (2003) The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 [DOI] [PubMed] [Google Scholar]
- 28. Conti E., Stachelhaus T., Marahiel M. A., and Brick P. (1997) Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reger A. S., Carney J. M., and Gulick A. M. (2007) Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46, 6536–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmitt M. P., Predich M., Doukhan L., Smith I., and Holmes R. K. (1995) Characterization of an iron-dependent regulatory protein (IdeR) of Mycobacterium tuberculosis as a functional homolog of the diphtheria toxin repressor (DtxR) from Corynebacterium diphtheriae. Infect Immun. 63, 4284–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez G. M., Voskuil M. I., Gold B., Schoolnik G. K., and Smith I. (2002) ideR, an essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 70, 3371–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandey S. D., Choudhury M., Yousuf S., Wheeler P. R., Gordon S. V., Ranjan A., and Sritharan M. (2014) Iron-regulated protein HupB of Mycobacterium tuberculosis positively regulates siderophore biosynthesis and is essential for growth in macrophages. J. Bacteriol. 196, 1853–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ferreras J. A., Ryu J. S., Di Lello F., Tan D. S., and Quadri L. E. (2005) Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 1, 29–32 [DOI] [PubMed] [Google Scholar]
- 34. Nelson K. M., Viswanathan K., Dawadi S., Duckworth B. P., Boshoff H. I., Barry C. E. 3rd, and Aldrich C. C. (2015) Synthesis and pharmacokinetic evaluation of siderophore biosynthesis inhibitors for Mycobacterium tuberculosis. J. Med. Chem. 58, 5459–5475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lun S., Guo H., Adamson J., Cisar J. S., Davis T. D., Chavadi S. S., Warren J. D., Quadri L. E., Tan D. S., and Bishai W. R. (2013) Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother. 57, 5138–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vilchèze C., Hartman T., Weinrick B., and Jacobs W. R. Jr. (2013) Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat. Commun. 4, 1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pfleiderer G., Kreiling A., and Wieland T. (1960) [On pantothenic acid synthetase from E. coli: II. quantitative enzymatic microdetermination of pantoyl acid, β-alanine and pantothenic acid]. Biochem. Z. 333, 308–310 [PubMed] [Google Scholar]
- 43. Zheng R., and Blanchard J. S. (2001) Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry 40, 12904–12912 [DOI] [PubMed] [Google Scholar]
- 44. Jain P., Hsu T., Arai M., Biermann K., Thaler D. S., Nguyen A., González P. A., Tufariello J. M., Kriakov J., Chen B., Larsen M. H., and Jacobs W. R. Jr. (2014) Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. MBio 5, e01245–e01214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bardarov S., Bardarov S. Jr., Pavelka M. S. Jr., Sambandamurthy V., Larsen M., Tufariello J., Chan J., Hatfull G., and Jacobs W. R. Jr. (2002) Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017 [DOI] [PubMed] [Google Scholar]
- 46. Tufariello J. M., Malek A. A., Vilchèze C., Cole L. E., Ratner H. K., González P. A., Jain P., Hatfull G. F., Larsen M. H., and Jacobs W. R. Jr. (2014) Enhanced specialized transduction using recombineering in Mycobacterium tuberculosis. MBio 5, e01179–01114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stover C. K., de la Cruz V. F., Fuerst T. R., Burlein J. E., Benson L. A., Bennett L. T., Bansal G. P., Young J. F., Lee M. H., and Hatfull G. F. (1991) New use of BCG for recombinant vaccines. Nature 351, 456–460 [DOI] [PubMed] [Google Scholar]