

Abstract
Pseudouridine is found in almost all cellular ribonucleic acids (RNAs). Of the multiple characteristics attributed to pseudouridine, making messenger RNAs (mRNAs) highly translatable and non-immunogenic is one such feature that directly implicates this modification in protein synthesis. We report the existence of pseudouridine in the anticodon of Escherichia coli tyrosine transfer RNAs (tRNAs) at position 35. Pseudouridine was verified by multiple detection methods, which include pseudouridine-specific chemical derivatization and gas phase dissociation of RNA during liquid chromatography tandem mass spectrometry (LC-MS/MS). Analysis of total tRNA isolated from E. coli pseudouridine synthase knock-out mutants identified RluF as the enzyme responsible for this modification. Furthermore, the absence of this modification compromises the translational ability of a luciferase reporter gene coding sequence when it is preceded by multiple tyrosine codons. This effect has implications for the translation of mRNAs that are rich in tyrosine codons in bacterial expression systems.
Keywords: chemical modification, mass spectrometry (MS), RNA modification, transfer RNA (tRNA), translation, CMCT, RluF, acrylonitrile, derivatization, pseudouridine
Introduction
Of 150-plus chemical modifications (1, 2) found in cellular RNA, 5-ribosyluridine or pseudouridine (Ψ)3 (3) is the most abundant but still poorly understood (4) modification. Pseudouridine, which has been found in the functionally important regions of RNAs (5), is known to modulate codon-anticodon interactions between mRNA and tRNA (6) and assist assembly of the ribosome (7) and spliceosome (8). The presence of pseudouridine in mRNA makes the message non-immunogenic and highly translatable, thus offering therapeutic opportunities in medical applications (9).
Although the exact mechanism of isomerization is still not clear (4), the incorporation of C5 of the uracil base into the glycosidic bond is catalyzed by either standalone proteins (referred to as Ψ synthases) (10, 11) or RNA-protein complexes (referred to as H/ACA box ribonucleoproteins) (12). Following site-specific recognition, the nitrogen-carbon glycosidic bond is cleaved, and uracil is rotated and then reattached to the sugar, forming a carbon-carbon glycosidic bond on the polyribonucleotide (Fig. 1) by Michael addition-like or glycal mechanisms (13).
FIGURE 1.

Pseudouridine in RNA. Uridine is isomerized into pseudouridine by pseudouridine synthases through base cleavage from ribose sugar and reattachment to generate C–C glycosidic bond.
The Ψ synthases have been initially classified based on the Escherichia coli enzymes RluA, RsuA, TruA, TruB, and TruD (10, 11). A sixth family with members from archaea and eukaryotes but not from bacteria was added after discovery of Pus10 (14). The enzyme active site carries a catalytically essential aspartate, which is the only absolutely conserved residue in all Ψ synthases (15, 16). Substrate recognition by the Ψ synthase is usually in the context of the sequence or structure of the target site in RNA. Site specificity also varies depending on the Ψ synthase. For example, RsuA isomerizes U516 of 16S rRNA (17) exclusively, whereas the universally conserved Ψ55 of the TΨC loop in all elongator tRNAs is catalyzed by TruB (18). RluA catalyzes modification of two distinct RNAs, 23S rRNA (position 746) and some tRNAs (position 32) (19). Members of the RluA and RsuA families share three conserved sequence motifs (Motifs I, II, and III) apart from domains similar to ribosomal protein S4 (20).
Enzymatic activity of pseudouridine synthase is measured by a 3H release assay involving in vitro transcripts (21). Pseudouridine in cellular RNA is assayed quantitatively by enzymatic hydrolysis and thin-layer or liquid chromatography where differences in migration distance or elution time, respectively, distinguish pseudouridine from uridine. Additional specificity in detection is available by coupling liquid chromatography (LC) with electrospray ionization mass spectrometry (MS) (22).
A major drawback of nucleoside hydrolysates is the loss of sequence information. A widely used site-specific detection method involves alkali-stable, Ψ-specific irreversible derivatization using the reagent N-cyclohexyl-N′-β-(4-methylmorpholinium)ethyl carbodiimide p-tosylate (CMCT) followed by extension of a radiolabeled primer with reverse transcriptase, separation, and detection on a denaturing polyacrylamide gel (23). Recently, CMCT-based derivatization has been adapted to high throughput next generation sequencing to detect dynamic pseudouridylations in mRNAs (24, 25). Mass spectrometry-based RNA modification mapping in combination with chemical derivatization is another site-specific strategy (26) wherein a base-specific ribonuclease generates a mixture of oligoribonucleotides whose pseudouridines are chemically derivatized with either CMCT (27–29) or acrylonitrile (30, 31) for detection.
A direct tandem mass spectrometry (MS/MS) approach based on the unique fragmentation pathways of the stable C–C glycosidic bond to generate and detect Ψ-specific fragment/product ions has been developed to confirm the presence of pseudouridine in oligoribonucleotides (32). This selected reaction monitoring (SRM) approach, compatible with MS-based RNA modification mapping, has been used to quantify the levels of pseudouridine in RNA (33). Other quantification methods involve radiolabeling of the phosphate backbone, RNase H-mediated cleavage, and thin layer chromatography of hydrolyzed nucleosides for phosphorimaging (34), a ligation-based method that facilitates the recognition of the modification site and quantification on denaturing PAGE (35), relative quantification from base hydrolysis of oligonucleotides (36), and stable isotope labeling of RNA (37).
The original E. coli tRNATyr sequences described in the literature are reported to contain two pseudouridines at position 39 and 55 (38). Prior reports discuss the possibility of an additional pseudouridine (31) or off-target derivatization (28), but until now, those possibilities remained uninvestigated. Here we have identified the additional pseudouridine derivatization site as residing at position 35 in the anticodon. We then determined that pseudouridine 35 is catalyzed by RluF, which was previously identified as only responsible for modifying 23S rRNA (39). Although deletion of RluF has no growth phenotype, gene expression is negatively affected in a synthetic construct containing multiple tyrosine codons.
Results
LC-MS/MS Analysis of RNase T1 Digest of E. coli tRNATyr
The Tyr I and Tyr II tRNAs from E. coli share an identical sequence except at two nucleotide positions in the variable region (UC versus CA) (Table 1). LC-MS analysis of tRNATyr following treatment with RNase T1 and bacterial alkaline phosphatase (BAP) revealed a single response (40) for the triply charged oligonucleotide anion ACU[Q]UA[ms2i6A]A[Ψ]CUG (m/z 1338.9, where U is the putative site of pseudouridine), which is a signature ion for E. coli tRNATyr (41) (supplemental Fig. S1). Collision-induced dissociation (CID) MS/MS of this mass selected precursor ion yielded all the expected sequence-informative product ion series, viz. cn (sharing the 5′-end) and yn (sharing the 3′-end) (supplemental Table S1), from which the original sequence could easily be reconstructed. This digestion product contains the anticodon, [Q]UA, responsible for decoding tyrosine codons, UAY (Y = U or C), in the mRNA. This underivatized RNA digest did not exhibit any interfering MS signal for m/z values corresponding to additional carbodiimide or cyanoethylation units, indicating the potential for interference-free pseudouridine detection of a derivatized digestion product.
TABLE 1.
Sequences of E. coli tRNATyr I and II that differ at two nucleotide positions (shown in italics)
Nucleotides in bold are part of RNase T1 signature digestion products (see text).
| Sequence | |
|---|---|
| E. coli tRNATyr I | GGUGGGG[s4U]UCCCGAGC[Gm]GCCAAAGGGAGCAGACU[Q]UA[ms2i6A]A[Ψ]CUGCCGUCAUCGACUUCGAAGG [m5U][Ψ]CGAAUCCUUCCCCCACCACCA |
| E. coli tRNATyr II | GGUGGGG[s4U]UCCCGAGC[Gm]GCCAAAGGGAGCAGACU[Q]UA[ms2i6A]A[Ψ]CUGCCGUCACAGACUUCGAAGG [m5U][Ψ]CGAAUCCUUCCCCCACCACCA |
CMCT-treated RNA
LC-MS analysis of CMCT-derivatized T1 and BAP digest of E. coli tRNATyr I or II yielded very little signal for the underivatized oligonucleotide anion but produced a high abundance signal for m/z values that correspond to the digestion product with one and two carbodiimide units (addition of 251 and 502 Da), respectively (Fig. 2, A and B), consistent with previous observations (28). CID MS/MS of these m/z values (Figs. 3 and 4) revealed differences with regard to the location of the second carbodiimide. When m/z values of product ions resulting from the dissociated precursor ion that have the common 3′-end (yn ion series where n refers to nucleotide position) were scored, an additional mass corresponding to the carbodiimide was detected in the MS/MS data from y4 (m/z 1199.8 → m/z 1450.8) through y11, which is consistent with the presence of the known pseudouridine modification at position 39 (38). The cn series yielded no product ions with carbodiimide for uridine at the third position, but carbodiimide-containing product ions were observed from c5 (m/z 1717.1 → m/z 1967.5) through c11 (Fig. 3 and supplemental Table S1). This result led to speculation that there could be pseudouridine at U35 in this tRNA. Furthermore, this observation also indicated that this precursor ion population is a mixture of oligonucleotides with carbodiimide at position 35 or 39 of the tRNA sequence (Fig. 3).
FIGURE 2.

LC-MS analysis of the carbodiimide-tagged RNase T1 digestion product the from anticodon of E. coli tRNATyr(QUA). A, total ion chromatogram representing the elution pattern of all the oligonucleotide digestion products detected as anions. B, XIC for m/z 1338.9 corresponding to the underivatized triply charged oligonucleotide anion (ACU[Q]UA[ms2i6A]A[Ψ]CUG). Note the lower signal intensity for the unreactive product. C, XIC for m/z 1422.7 that corresponds to the digestion product with one carbodiimide unit. D, XIC for m/z 1506.3 that corresponds to the digestion product with two carbodiimide units. E, mass spectrum of the XIC peak (41.2–43.6 min) observed in B where the triply charged oligonucleotide anion with carbodiimide unit is predominantly present. F, mass spectrum of the XIC peak (49–50.3 min) observed in C where the triply charged oligonucleotide anion with two carbodiimide units is predominantly present. Rel, relative; Abs Abund, absolute abundance.
FIGURE 3.

CID-based sequencing of the single carbodiimide-tagged RNase T1 digestion product the from anticodon of E. coli tRNATyr(QUA). Shown is the CID MS/MS spectrum of ACU[Q]UA[ms2i6A]AΨCUG with one carbodiimide (m/z 1422.8) tagged at either one of two different locations. The appearance of unique product ions with the additional carbodiimide mass from c5 (m/z 1967.5) and y4 (m/z 1451.4) defines the region for pseudouridines in the oligonucleotide. Each addition of carbodiimide (+251) is denoted with * for the y product ion series and ♦ for the c product ion series. The secondary structure of E. coli tRNATyr along with the observed pseudouridine modification at position 35 and other previously reported modifications is illustrated.
FIGURE 4.

CID-based sequencing of the double carbodiimide-tagged RNase T1 digestion product from the anticodon of E. coli tRNATyr(QUA). Shown is the CID MS/MS spectrum of ACU[Q]UA[ms2i6A]AΨCUG with two carbodiimide units (+502 Da, m/z 1506.6). The appearance of unique product ions with two carbodiimide adducts, c9 and y8, defines the location of two pseudouridines in the oligonucleotide. Each addition of carbodiimide (+251) is denoted with * for the y product ion series and ♦ for the c product ion series.
Sequencing of ACU[Q]UA[ms2i6A]A[Ψ]CUG with two carbodiimide units also revealed that the carbodiimide-tagged product ions started with y4 (m/z 1450.8) and c5 (m/z 1967.5) (Fig. 4). However, the mass of detected product ions shifted to two carbodiimide units from c9 (m/z 1687.7 → m/z 1813.3) onward and y8 (m/z 1429.0 → m/z 1554.6) onward (supplemental Table S1. These data are in contrast with a previous finding (28) where RNase A and snake venom phosphodiesterase-digested tRNATyr was analyzed by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) after CMCT derivatization. Those earlier data also showed that two carbodiimide units were added to the anticodon region of tRNATyr. However, the exonuclease digestion of the RNase A product (A[ms2i6A]Ap) suggested carbodiimide tagging of ms2i6A at position 37. A key difference between the current study and that in Ref. 28 is the use of CID MS/MS in the present study to precisely define the location of carbodiimide tagging. Although both the present findings and the prior publication identified two sites of carbodiimide tagging in the anticodon loop of tRNATyr, only in this present work is the exact location of an additional pseudouridine revealed.
Given the disparity between the MALDI-MS data from the earlier report and the LC-MS/MS data obtained here, tRNATyr was digested with RNase U2 to yield the digestion product CU[Q]UA>p (m/z 857.7, where >p represents a 2′,3′-cyclic phosphate) (supplemental Fig. S2) that is specific to the anticodon region (42). CMCT-based derivatization and subsequent examination of product ions obtained by CID of m/z 983.1 resulted in a 251-Da mass shift of c4 and y2, which is consistent with RNase T1 derivatization data that the second site for carbodiimide tagging is pseudouridine at position 35.
Acrylonitrile-treated RNA
To confirm the CMCT derivatization results, an alternative derivatization strategy was explored based on the specificity of acrylonitrile for pseudouridine (30, 31). LC-MS analysis of the acrylonitrile-treated T1 digest revealed the presence of triply charged oligonucleotide anions at m/z 1356.8 and 1374.3 that correspond to one and two cyanoethylations (addition of 53 and 106 Da), respectively (supplemental Fig. S3 and supplemental Table S1). CID MS/MS of m/z 1356.8 showed the presence of product ions that correspond to ACU[Q]UA[ms2i6A]A[Ψ]CUG with a mass shift of ∼53 Da from y4 to y11 and c5 to c11. The profile of these product ions in the tandem mass spectrum followed a pattern identical to that observed during carbodiimide derivatization. However, the MS/MS spectrum was more complex due to fragment ions corresponding to side chain fragmentation of queuosine and nucleobase loss for cytosine, adenine, uracil, and ms2i6A. Similarly, the MS/MS spectrum of m/z 1374.3 exhibited a mass shift of ∼53 Da from y4 to y7 and c5 to c8, which thereafter shifted by another 53 Da through y11 and c11 (supplemental Table S1). Thus, two independent derivatization strategies yielded data that point to the presence of pseudouridine at position 35 of tRNATyr.
SRM-based Pseudouridine Detection
Pseudouridine at position 35 of tRNATyr was further validated by subjecting the RNase U2 digest to LC-MS/MS-based SRM analysis. This experiment revealed the presence of the pseudouridine-specific SRM transition (m/z 207 → m/z 164) that corresponded with the extracted ion chromatogram of intact CU[Q]UA>p (supplemental Fig. S4) indicating pseudouridine in this oligomer.
Nucleoside Analysis
The CU[Q]UA>p digestion product was purified after post-column split and digested to nucleosides. LC-MS/MS analysis of the total nucleoside digest confirmed the presence of pseudouridine as it could be distinguished from uridine by its retention time (supplemental Fig. S5). The modified queuosine expected in this RNase U2 digestion product was also detected, providing additional confidence that pseudouridine is associated with the anticodon of tRNATyr.
Analysis of Pseudouridine Synthase Disruption Mutants
After confirming the presence of pseudouridine at position 35 of E. coli tRNATyr, we embarked on identifying the pseudouridine synthase(s) responsible for this modification by analyzing gene knock-out strains for each E. coli pseudouridine synthase. Total tRNA was isolated, digested with RNase T1, and analyzed for the doubly cyanoethylated signature digestion product of tRNATyr by LC-MS/MS. A comparison of the computed peak areas for the singly and doubly cyanoethylated ions of ACU[Q]UA[ms2i6A]A[Ψ]CUG from all of the pseudouridine synthase mutants should indicate the putative enzyme responsible for this modification.
Fig. 5A depicts the peak areas of m/z 1356.8 and 1374.3 from the acrylonitrile-treated total tRNA digest. A signal at m/z 1356.8 is present in all treated samples, although the ion abundance varied among different mutants. In contrast, the peak area corresponding to m/z 1374.3 showed a dramatic decrease for ΔtruA and ΔrluF. To ensure the derivatization conditions were optimal across all samples, the cyanoethylation status of a tRNAPhe signature digestion product (41), AA[ms2i6A]AΨCCCCG, was also monitored. As this oligonucleotide has one pseudouridine (and no other uridines), it is expected to exhibit a mass shift of ∼53 Da upon cyanoethylation (m/z 1619.2 → m/z 1645.8). Fig. 5B depicts the presence of m/z 1645.8 in all tRNA samples except ΔtruA. This result is consistent with the fact that TruA is known to be responsible for pseudouridine modification at position 39 (43), and the absence of this enzyme is expected to result in the loss of modification at this position.
FIGURE 5.
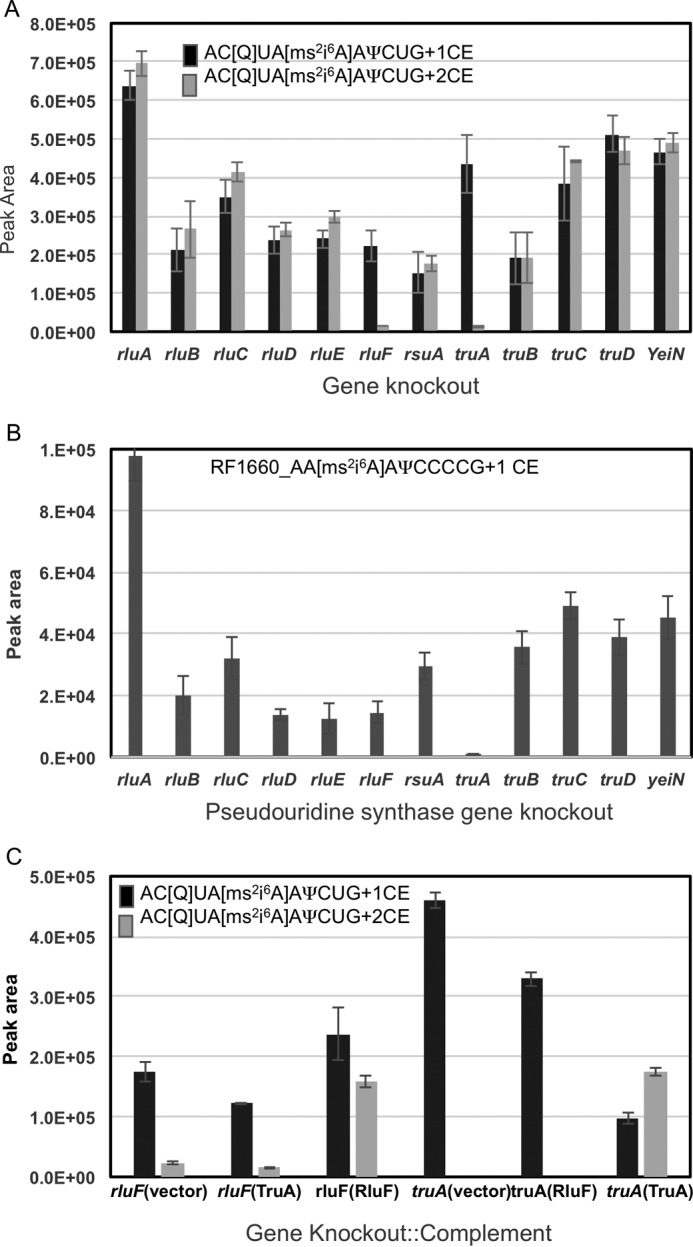
Identification of synthase gene responsible for pseudouridine modification at position 35 of E. coli tRNATyr(QUA). Four micrograms of tRNA isolated from each of the pseudouridine synthase knock-out mutant cell lines (three to five biological replicates) were digested with RNase T1 and BAP before treating with acrylonitrile. The derivatized RNA was subjected to LC-MS. A, peak areas corresponding to one and two cyanoethylation groups on AC[Q]UA[ms2i6A]A[Ψ]CUG of tRNATyr were plotted against each synthase gene. The truA and rluF gene knock-out tRNA samples reveal a significant decrease in cyanoethylation. B, peak areas corresponding to one cyanoethylation group for AA[ms2i6A]A[Ψ]CCCCG from tRNAPhe were plotted against each synthase gene. Only truA exhibited a dramatic decrease in peak area, whereas the mutation in the other genes including rluF strain had no adverse impact on the peak area. C, complementation effects of TruA or RluF gene under truA or rluF genetic background on pseudouridine modification at position 35 of tRNATyr. Note that the complementation ability of the wild type genes is possible only under the respective knock-out mutation background, i.e. rluF::RluF and truA::TruA, but not in the cases of rluF::TruA and truA::RluF. Error bars represent S.D. of replicate measurements.
To distinguish whether TruA or RluF could complement the loss of modification at position 35, the coding regions of TruA and RluF were amplified from an E. coli K12 strain, and the confirmed sequence was cloned into the pET22b vector for subsequent transformation of the ΔrluF or ΔtruA strains. Fig. 5C depicts the cyanoethylation status of the signature digestion product of tRNATyr in complemented lines. The data clearly show that the ΔrluF strain can only be complemented with the RluF coding sequence as the peak area of m/z 1374.3 for this complementation is higher by many orders of magnitude compared with the complementation with TruA. Similarly, the peak area for m/z 1374.3 increased only after complementation of the ΔtruA strain with TruA.
In Vitro Modification of tDNATyr
The in vitro transcribed tDNATyr gene was incubated with purified RluF or TruA, digested with RNase T1 and BAP, and then derivatized by cyanoethylation. Due to the lack of queuosine at position 34 of the in vitro transcript, RNase T1 digestion will generate the oligonucleotide UAAAUCUG (m/z 1245.1) where the 5′-uridine corresponds to position 35 of the native tRNA. LC-MS analysis of both samples yielded the underivatized and cyanoethylated digestion products that correspond to the anticodon region of the tDNA transcript. MS/MS analysis (Fig. 6) revealed differences with respect to the position of cyanoethylation on the nucleotide sequence. RluF-treated RNA exhibited cyanoethylation starting with c1 and continuing through c7 with no mass shift for any y-type product ions (Fig. 6A). In contrast, the TruA-treated RNA exhibited cyanoethylation from c5 through c7 only and from y4 to y7 (Fig. 6B).
FIGURE 6.

CID-based sequencing of the acrylonitrile-tagged RNase T1 digestion product the from anticodon of E. coli tDNATyr transcript. A, MS/MS spectrum of the doubly charged oligonucleotide ion originating from tDNATyr transcript treated with recombinant RluF and acrylonitrile (m/z 1272.4). The cn product ion series is consistent with cyanoethylation (CE), whereas no such addition was significantly observed for the yn product ion series. B, MS/MS spectrum of the doubly charged oligonucleotide ion originating from tDNATyr transcript treated with recombinant TruA and acrylonitrile (m/z 1272.4). Cyanoethylation was observed at y4 and c5 through y7 and c7. Each addition of cyanoethylation (+53) is denoted with * for the y product ion series and ♦ for the c product ion series.
A comparison of the peak areas corresponding to m/z 1245.2 and 1272.2 indicated that of 50 pmol of RNA incubated with 5 pmol of protein about 35% was cyanoethylated upon treatment with RluF and acrylonitrile. In contrast, about 48% of 1500 pmol of RNA (incubated with 0.2 pmol of protein) was cyanoethylated upon treatment with TruA and acrylonitrile. The observed levels of in vitro modification match with the previously reported tritium release assays by other groups (44, 45). When TruA or RluF was omitted but the transcript was treated with acrylonitrile, less than 5% of the RNA was cyanoethylated at random positions (data not shown). The RluF-treated RNA transcript was also analyzed using the pseudouridine-specific SRM assay. SRM signals from the RluF-treated RNA align with the peak corresponding to UAAAUCUG (supplemental Fig. S6).
RluF Modifies 23S rRNA
RluF is known to isomerize uridine to pseudouridine at position 2604 of 23S rRNA. To confirm this behavior remains when detecting pseudouridine at position 35 of tRNATyr, the pseudouridylation status of 23S rRNA was compared between K12 and ΔrluF strains by digesting with RNase T1 and BAP followed by acrylonitrile treatment. Fig. 7, A–C, depicts the presence of the RNase T1 digestion product ΨΨCG with two cyanoethylation groups (m/z 1199.2 + 106 = m/z 1305.2) from K12 23S rRNA. However, 23S rRNA purified from the ΔrluF strain exhibited only one cyanoethylation (m/z 1199.2 + 53 = m/z 1252.1), and MS/MS analysis revealed the mass shift from c2 onward, and only y3 exhibited the 53-Da mass shift (Fig. 7, D–F). These data indicate that 23S rRNA was not modified at position 2604 in the ΔrluF strain.
FIGURE 7.

LC-MS analysis of acrylonitrile-derivatized RNase T1/BAP digests of E. coli 23S rRNA. A, XIC for m/z 1305.2 corresponding to [Ψ][Ψ]CG with two cyanoethylations. B, mass spectrum corresponding to XIC peak at 23.4 min showing the presence of singly charged oligonucleotide [Ψ][Ψ]CG anion with two cyanoethylations. C, MS/MS spectrum of m/z 1305.2. D, XIC for m/z 1252.6 corresponding to [U][Ψ]CG with one cyanoethylation. E, mass spectrum corresponding to XIC peak at 23.1 min showing the presence of singly charged oligonucleotide [U][Ψ]CG anion with one cyanoethylation. F, MS/MS spectrum of m/z 1252.6. Each addition of cyanoethylation (+53) is denoted with * for the w and y product ion series and ♦ for the c product ion series. Rel Abund, relative abundance.
Luciferase Reporter Assay
The above experimental data make a strong case for pseudouridine modification in the anticodon at position 35 of tRNATyr. To understand the role of this modification in decoding Tyr codons, a recombinant reporter gene was constructed with multiple Tyr codons (TAY)10 (to magnify any subtle effects it may have) preceding the open reading frame of the firefly luciferase coding sequence. Luciferase expression was tested under the ΔrluF background in combination with Renilla (Ren) luciferase as an internal control (construct pETDuet “Ren Luc and (TAY)10 FF Luc”). A complementation construct, “RluF and (TAY)10 FF Luc,” tested the ability of the RluF protein to rescue luciferase activity under the ΔrluF background. A third construct containing “Ren Luc and (AAY)10 FF Luc” was also tested to probe the influence of an identical number of multiple codons preceding the reporter gene. As depicted in Fig. 8, transformation of ΔrluF cells with pETDuet Renilla reniformis luciferase (Ren Luc) and (TAY)10 firefly (Photinus pyralis) luciferase (FF Luc) and subsequent induction by isopropyl β-d-1-thiogalactopyranoside (IPTG) at mid-log phase showed very little expression of luciferase as determined by bioluminescence. However, the luminescence from Renilla luciferase was unaffected in these cells. Conversely, when RluF was co-expressed with tyrosine codon- rich FF luciferase ((TAY)10 FF Luc), luciferase could easily be detected, indicating that the induced RluF protein complements the ΔrluF knock-out. The wild type firefly luciferase (without the preceding multiple tyrosine codons) expression under a ΔrluF background is more or less similar to the RluF complement, indicating that ΔrluF is inefficient in translating only those mRNAs that are rich in Tyr codons. Furthermore, luminescence levels were unaffected when recombinant (TAY)10 FF Luc was expressed in BL21 cells (data not shown), indicating that addition of multiple tyrosine codons has no impact on general expression and activity of FF Luc in E. coli under wild type conditions. Thus, these experimental data suggest that the pseudouridine modification at position 35 of tRNATyr has a very subtle impact on mRNA translation under normal conditions, but the impact can be significant if the mRNA is rich in Tyr codons.
FIGURE 8.
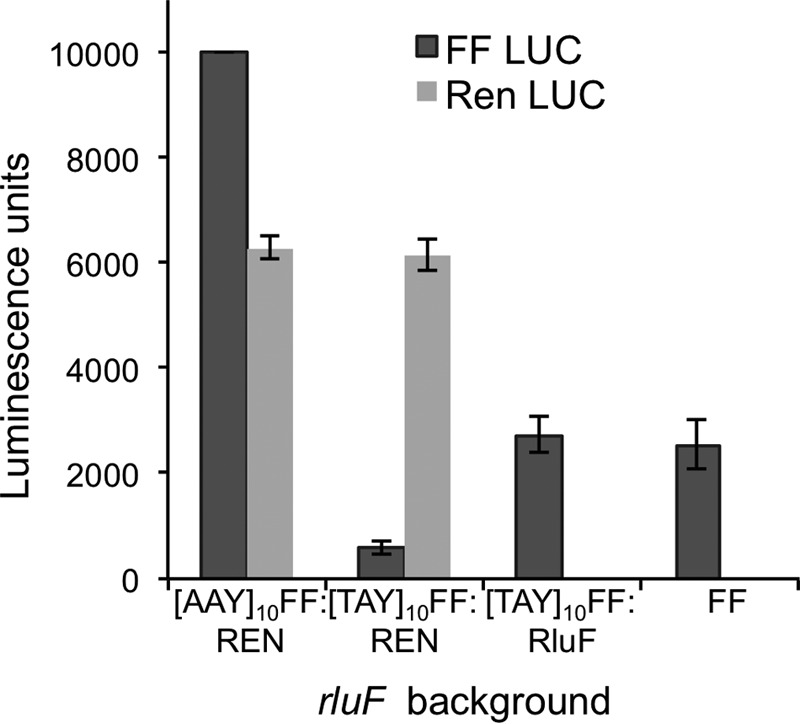
Luciferase assay measuring decreased gene expression under rluF gene knock-out background when the coding sequence is preceded by multiple tyrosine codons. A recombinant firefly luciferase gene was constructed with a set of 10 tyrosine codons ((TAY)10-FF) or 10 asparagine codons ((AAY)10-FF) preceding the firefly luciferase open reading frame. Expression of the luciferase gene in combination with either Renilla luciferase or RluF gene in the rluF gene knock-out background was conducted using a pETDuet vector. The type of recombinant construct used in the luciferase assay is denoted on the x axis. At least four to five biological replicates were tested for each construct. Error bars represent S.D. of replicate measurements.
Discussion
The original E. coli tRNATyr sequences described in the literature are reported to contain two pseudouridines at positions 39 and 55 (38). Prior reports discussed the possibility of an additional pseudouridine (31) or off-target derivatization (28), but until now, those possibilities remained uninvestigated. To clarify this open question, we conducted an exhaustive examination of E. coli tRNATyr using multiple pseudouridine detection strategies. Two different derivatization strategies along with SRM-based direct detection confirmed that the originally annotated uridine at position 35 of tRNATyr is actually modified to pseudouridine. We found no difference in Ψ35 modification levels between the two isoacceptors of tRNATyr (Tyr I and Tyr II), indicating that both tRNAs serve as a substrate for the tRNA:Ψ35 synthase.
E. coli is known to contain 11 pseudouridine synthase genes, and any one of them could be responsible for pseudouridine at position 35 of tRNATyr. Four E. coli genes are known to be tRNA-specific, six are rRNA-specific, and only one (rluA) is known to exhibit dual specificity, catalyzing pseudouridine formation for 23S rRNA (position 746) and tRNAPhe (position 32) (46). We reasoned that screening each of the pseudouridine synthase knock-out strains for the double derivatization status of the tRNATyr signature digestion product (41), ACU[Q]UA[ms2i6A]A[Ψ]CUG, could identify the putative modifying enzyme. Indeed, this strategy identified significantly reduced levels of double cyanoethylated signature digestion products from the ΔtruA and ΔrluF strains.
TruA is known to exhibit broad specificity, catalyzing pseudouridine formation in the anticodon loop and stem of multiple tRNAs (43). Absence of this enzyme would affect the pseudouridylation status of tRNAs beyond tRNATyr. To differentiate TruA effects, the pseudouridylation status of the E. coli tRNAPhe signature digestion product, AA[ms2i6A]A[Ψ]CCCCG, was also examined for each pseudouridine synthase knock-out. Thus, by using mass spectrometry to examine two tRNA-specific signature digestion products, it was found that RluF is the enzyme responsible for pseudouridine modification at position 35 of tRNATyr. Complementation of ΔtruA and ΔrluF strains with their respective wild type sequences confirmed the studies of the knock-outs. Moreover, the site-specific pseudouridine modification of an in vitro tRNATyr transcript by RluF was consistent with all other pseudouridine synthase data. These studies, therefore, have determined that RluF is a second dual specificity pseudouridine synthase responsible for catalyzing the uridine isomerization in both 23S rRNA (position 2604) and tRNATyr (position 35).
As RluF is now found to be a dual specificity synthase, it is interesting to examine the mechanistic differences for pseudouridine synthases. Although RluB modifies U2605 of 23S rRNA through stabilization of the A2602 bulge (47), RluF modifies U2604 through a frameshift in RNA base pairing. This protein binds to an RNA stem-loop to rearrange the base pair in such a way that it moves the A2602 bulge into the stem, thereby translating the target U2604 by flipping it into the active site. Thus, if a mutation facilitates A2602 refolding into the stem, enzymatic activity increases substantially (44).
The pseudouridines at positions 2604 and 2605 of E. coli 23S rRNA are both located in domain V, which is part of the peptidyltransferase center involved in peptide bond formation. It is possible that these modifications could aid peptidyltransferase activity through enhanced base pairing available with pseudouridine rather than uridine, although the precise function of these pseudouridines is still undetermined.
TruA, a functional dimer, modifies uridines at position 38, 39, and/or 40 of tRNAs with highly divergent sequences and structures (43) where the protein utilizes the intrinsic flexibility of the anticodon stem-loop for substrate promiscuity. In contrast, TruB modifies a single uridine at position 55 of the T-loop. This enzyme directly docks to the preformed T-loop through shape complementarity and extensive interactions with nucleotides near the target site. Furthermore, the substrate is accessed by flipping out the uridine and disrupting the tertiary structure involving interaction between the T- and D-loops of the tRNA (48). Although the mechanism of action of RluF on the tDNATyr transcript is not clear in the current study, it can be speculated that it may be recognizing the anticodon loop similarly to TruB based on its site-specific isomerization behavior.
The cytoplasmic tRNATyr of several eukaryotes is known to contain pseudouridine at position 35 (49), which is introduced in an intron-dependent manner (for a review, see Grosjean et al. (50)). Studies with Arabidopsis pre-tRNATyr (49) indicated that the intron sequence helps in orienting and exposing the 5′-exonic consensus sequence U33N34U35A36Pu37 (where N stands for any one of four nucleosides and Pu stands for purine) to the tRNA:Ψ35 synthase. The E. coli tRNATyr anticodon sequence (U[Q]UA[ms2i6A]) matches this exonic consensus sequence. In fact, no other tRNA, except tRNATyr, has this consensus sequence in E. coli, indicating that pseudouridine at position 35 could be unique to tRNATyr. However, RluF catalyzes pseudouridine formation in an intron-independent manner as indicated by the in vitro assay involving the tDNATyr transcript. It is possible that RluF binds to 23S rRNA and tRNATyr by recognizing the stem-loop to flip the base into the active site for modification. Further studies will reveal whether the anticodon loop is sufficient for pseudouridine introduction or RluF requires the whole anticodon stem-loop or other parts of the tRNATyr sequence for recognition and docking.
It has been shown that pseudouridine can stabilize RNA duplexes by forming Ψ-A, Ψ-G, Ψ-U, and Ψ-C pairs by hydrogen bonding through the N3 and N1 imino protons depending on the position of Ψ within the RNA duplex and the type of Watson-Crick pair (51). Such interactions seem to play a crucial role in amino acylation fidelity and efficiency in yeast (52) where Ψ interactions with adenosine strengthen the anticodon-codon/stop codon interactions (51, 53, 54). These findings might explain why incorporation of pseudouridine into mRNA makes it highly stable and translatable, offering a number of opportunities for medical applications (9).
Based on the enhanced base-pairing interactions mediated by pseudouridine (as described above), it is tempting to speculate that such interactions might be the reason for the observed luciferase expression differences in our assays. Here, the expression of luciferase is affected only when its coding sequence is preceded by multiple tyrosine codons; otherwise expression is unaffected in the ΔrluF strain. Complementation of ΔrluF with the wild type RluF gene restores the modification, thereby facilitating the luciferase gene expression even if the protein coding sequence is preceded by multiple tyrosine codons. A cursory examination of the E. coli K12 MG1655 proteome indicates that there are 47 genes with three consecutive tyrosine residues. As the ΔrluF strain does not exhibit any growth phenotype, the impact of three consecutive tyrosine residues on gene expression in this strain might be minimal because the codon-anticodon interactions remain sufficient over such a small region of translation. It would be interesting to study the variations of reporter gene expression if the coding sequence is preceded by four to nine tyrosine codons.
A phylogenetic classification of pseudouridine synthases based on their conserved domains reveals four families: TruA-like (cd00497), TruB-like (cd00506), Rsu_Rlu-like (cd02550), and TruD (cd02552) (supplemental Fig. S7A). RluF falls under the subfamily of Rsu_Rlu-like proteins. However, the subfamily hierarchy of the RluF branch (cd02554) contains 11 more homologous proteins that are of purely bacterial origin (supplemental Fig. S7B) where the homology extends beyond the shared common fold and active site cleft core structure (supplemental Fig. S8). One other interesting feature is that although the 23S rRNA from these 11 bacteria contains uridine at position 2604 pseudouridine is reported only in E. coli, indicating the unknown role of these pseudouridine synthases. In such a situation, it is tempting to speculate that these homologous proteins may be responsible for catalyzing pseudouridine modification at position 35 of tRNATyr. Further experiments are needed to clarify the exact role of these proteins in their respective bacteria.
Moreover, these efforts emphasize the advantages of detection methods of increasing sensitivity and accuracy. As noted by the discovery of Ψ35 in tRNATyr from the well studied model organism E. coli, modern approaches, which provide multiple means of detection/validation, may reveal additional information relating to RNA modifications that have until now remained undiscovered.
Experimental Procedures
RNase T1 and U2 Digestion
RNase T1 (Roche Applied Science) was purified on C18 Sep-Pak cartridges (Waters) as described (55). Two micrograms of E. coli tRNATyr I or II (R0383 or R0258, Sigma-Aldrich) or 4 μg of E. coli total tRNA were digested with purified RNase T1 (25 units/μg of RNA) and BAP (0.01 unit/μg of RNA) (Worthington Biochemical Corp.) in 120 mm ammonium acetate, pH 6.5, at 37 °C for 2 h (56, 57). The digest was dried in a SpeedVac (Thermo Fisher Scientific, Inc.) and further purified for CMCT derivatization using C18 ZipTips (Millipore Corp.) according to the manufacturer's instructions or used as is for acrylonitrile treatment. RNase U2 was purified as described (42). Two micrograms of E. coli tRNATyr I or II were digested with 2 μg of purified ribonuclease U2 in 120 mm ammonium acetate, pH 5.1, at 55 °C for 30 min. The dried RNA digest was purified by C18 ZipTips for subsequent CMCT derivatization.
Chemical Derivatization
For CMCT derivatization, the RNA digest was derivatized with 10 μl of CMCT (10 mg/ml) solution made with 50 mm Tris-HCl, pH 8.3, 4 mm EDTA, and 7 m urea (28). This concentration was found to be optimal based on the independent LC-MS/MS analysis of Ψ-containing (ΨGG, AΨCAG, and UAACΨAΨGACG) and Ψ-lacking (UGG, AUCAG, and UAACUAUGACG) oligonucleotides (Dharmacon, GE Healthcare) following derivatization and alkali treatment (10 μl of 50 mm ammonium bicarbonate, pH 10.4, at 75 °C for 90 min). Incubation of Ψ-lacking oligomers at 37 °C for 16 h at this optimal concentration, ZipTip purification, and alkali treatment resulted in no derivatization at uridine or guanosine. However, alkali-stable Ψ-specific derivatization was observed for >99% of Ψ-containing oligomers (addition of 251.4 Da per pseudouridine; data not shown). The acrylonitrile-based derivatization was similar to the previous procedure (31) except that 1 μl of acrylonitrile (Sigma)/2 μg of RNA digest was used. The procedure involved incubation of the RNA digest (2 μg) in 30 μl of 41% ethanol and 1.1 m triethylamine ammonium acetate (Sigma) and 1 μl of acrylonitrile at 70 °C for 2 h.
LC-MS/MS of RNase Digests
All underivatized and derivatized RNase digestion products were reconstituted in mobile phase A (400 mm hexafluoroisopropanol (Sigma), 16.3 mm triethylamine (Sigma), pH 7.0) before analysis. HPLC was conducted using a 1 × 150-mm XBridge C18 column (Waters) using mobile phase A and mobile phase B (200 mm hexafluoroisopropanol, 8.15 mm triethylamine in 50% methanol (Burdick and Jackson), pH 7.0) at a flow rate of 30 μl/min. The chromatographic gradient and mass spectrometric conditions are identical to the conditions described before (56).
Mongo Oligo Mass Calculator was used to predict the m/z values of RNase T1 and U2 digestion products and associated MS/MS fragment ions that include the addition of carbodiimide (+251.4 Da) or cyanoethylation (+53 Da) when appropriate (supplemental Table S1). The Quan browser feature of Xcalibur was utilized to integrate and quantify the extracted ion chromatogram (XIC) peak areas corresponding to m/z values of interest.
SRM Detection of Pseudouridine
Underivatized RNA was analyzed for pseudouridine essentially as described previously (33). Alignment of the XIC of the m/z corresponding to any particular RNase digestion product (from the full scan) with the Ψ-specific SRM peak (m/z 207–164 or m/z 225–165) denotes the presence of pseudouridine in that RNase digestion product (32).
Nucleoside Analysis by LC-MS
The E. coli tRNATyr RNase U2 digestion product, CUQUA>p (m/z 857.7), was collected during reversed phase ion pair HPLC after a postcolumn split, which allowed for LC-MS monitoring to identify the appropriate digestion product fraction. The fractionated oligomer was digested with snake venom phosphodiesterase and Antarctic phosphatase before subjecting it to subsequent LC-MS (56, 58) with detection in positive polarity.
E. coli Strains, Cell Culture, and RNA Isolation
E. coli strains harboring an insertion mutation in a specific pseudouridine synthase gene as well as the gene associated with pseudouridine degradation (YeiN; supplemental Table S2) were procured from the Yale Coli Genetic Stock Center. The tRNA was isolated from mutant cells grown to mid-log phase (A600 ∼0.5–0.6) using TRI Reagent as described (33, 59). The 23S rRNA was gel-purified from total RNA by electrophoresis on a 1% low melting agarose gel followed by phenol-chloroform extraction and ethanol precipitation.
Genotyping of Insertion Mutants
E. coli strains exhibiting a significant decrease in pseudouridine (based on cyanoethylation) levels at position 35 of tRNATyr were genotyped and validated for kanamycin insertion. This was done by amplifying and sequencing the DNA at the junction of insertion using gene-specific and kanamycin resistance gene-specific (aminoglycoside/neomycin phosphotransferase; GenBankTM accession number V00618.1) primers (supplemental Table S3). Polymerase chain reaction (PCR) conditions were similar to those described (60).
Gene Cloning and Protein Purification
The coding regions of pseudouridine synthase genes TruA (gi|556503834) and RluF (gi|556503834) were amplified using primers (supplemental Table S3) that have compatible restriction sites (BamHI and HindIII) to enable subcloning into the IPTG-inducible gene expression cassette in the pET22b vector (EMD-Millipore) as described (61). Recombinant proteins with C-terminal His6 tag fusion were expressed and purified as described elsewhere (61).
Pseudouridine Synthase Activity Assay
The tDNATyr template required for in vitro transcription was initially amplified and cloned from E. coli K12. The transcription template produced by PCR (using the primers Tyr-5′-T7 and Tyr-3′; supplemental Table S3) was in vitro transcribed, and the RNA was purified by an Ampliscribe kit (Epicenter Biotechnologies) and a MEGAclear kit (Ambion), respectively, as described (62).
The pseudouridine synthase activity assay was performed on the in vitro transcript with recombinant pseudouridine synthase as described (44, 45). Briefly, the pseudouridylation assay mixture consisting of purified protein (50 nm for RluF and 2 nm for TruA) and in vitro transcript (0.5 μm for RluF and 1.5 μm for TruA) in the assay buffer (20 mm Tris-HCl, pH 8.0, 0.1 m NH4Cl, 2 mm DTT) was incubated at room temperature for 2 h. Following digestion with RNase T1 (150 units) and BAP (0.02 unit) at 37 °C for 2 h, the resulting oligonucleotides were extracted with phenol-chloroform, dried in a SpeedVac, and dissolved in sterile water for derivatization with acrylonitrile.
Genetic Manipulations for Luciferase Reporter Gene Assays
The coding regions of FF Luc and Ren Luc were amplified from pGL3 and pRL vectors (Promega), respectively, with primers possessing suitable restriction sites (supplemental Table S3) so that the amplified DNA could be cloned into the pETDuet-1 (EMD Biosciences) vector. Four different constructs were made and tested for luciferase activity in E. coli with the RluF enzyme knock-out background. Construct 1 was pETDuet with FF Luc and/or Ren Luc. For Construct 2, pETDuet Ren Luc and (AAY)10 FF Luc, Ren Luc was cloned into the MCS1 (SacI and HindIII), whereas the annealed and digested (5′ NdeI and BamHI 3′) double-stranded oligomer (AAY)10 along with FF Luc (BglII and XhoI) were cloned into the NdeI-XhoI sites of MCS2. For Construct 3, pETDuet Ren Luc and (TAY)10 FF Luc, cloning was similar to the previous construct except that the oligomer (TAY)10 was used instead of (AAY)10. For Construct 4, pETDuet RluF and (TAY)10 FF Luc, RluF was cloned into the BamHI and HindIII sites of MCS1, and the MCS2 contained (TAY)10 FF Luc.
Luciferase assays were carried out using the Dual-Glo® luciferase assay system (Promega) and a Turner TD-20/20 luminometer. The assay involves transforming the ΔrluF strain of E. coli with each of the four constructs and culturing 10 ml of cells to mid-log phase (0.5–0.6 OD) to induce the expression of cloned genes with IPTG for 2 h at 37 °C. Subsequently, the cells were harvested by centrifugation (6,000 rpm for 3 min) and resuspended in lysis buffer (150 μl of 10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1 mg/ml lysozyme). The cells were lysed by exposure to 4 °C for 10 min, −80 °C for 10 min, and a 25 °C water bath for 5 min (63). The resulting lysate was centrifuged (13,000 × g for 12 min) to remove cell debris. Ten microliters of lysate were added to 50 μl of Luciferase Assay Reagent II and mixed thoroughly by repeated pipetting, and the resulting luminescence for FF Luc was recorded. Subsequently, 50 μl of Stop & Glo reagent was mixed to take the second luminescence measurement for Renilla luciferase.
Author Contributions
B. A. designed, executed, and analyzed the experiments. B. A. and P. A. L. formulated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We acknowledge the guidance received from Sitakanta Pattanaik (Kentucky Tobacco Research AND Development Center) in conducting luciferase assays and Christian Hong (University of Cincinnati Medical Center) for allowing the use of the luminometer.
This work was supported by NIGMS, National Institutes of Health Grant R01 GM 058843 (to P. A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1–S3 and Figs. S1–S8.
- Ψ
- pseudouridine
- CMCT
- N-cyclohexyl-N′-β-(4-methylmorpholinium)ethyl carbodiimide p-tosylate
- MS/MS
- tandem mass spectrometry
- CID
- collision-induced dissociation
- SRM
- selected reaction monitoring
- BAP
- bacterial alkaline phosphatase
- Q
- queuosine
- ms2i6A
- 2-methylthio-N6-isopentenyladenosine
- FF
- firefly
- Luc
- luciferase
- Ren
- Renilla
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- XIC
- extracted ion chromatogram.
References
- 1. Machnicka M. A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., Helm M., Bujnicki J. M., and Grosjean H. (2013) MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 41, D262–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Limbach P. A., Crain P. F., and McCloskey J. A. (1994) Summary: the modified nucleosides of RNA. Nucleic Acids Res. 22, 2183–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohn W. E., and Volkin E. (1951) Nucleoside-5′-phosphates from ribonucleic acid. Nature 167, 483–484 [Google Scholar]
- 4. Spenkuch F., Motorin Y., and Helm M. (2014) Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol. 11, 1540–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charette M., and Gray M. W. (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49, 341–351 [DOI] [PubMed] [Google Scholar]
- 6. Davis D. R., Veltri C. A., and Nielsen L. (1998) An RNA model system for investigation of pseudouridine stabilization of the codon-anticodon interaction in tRNALys, tRNAHis and tRNATyr. J. Biomol. Struct. Dyn. 15, 1121–1132 [DOI] [PubMed] [Google Scholar]
- 7. Cunningham P. R., Richard R. B., Weitzmann C. J., Nurse K., and Ofengand J.(1991) The absence of modified nucleotides affects both in vitro assembly and in vivo function of the 30S ribosomal subunit of Escherichia coli. Biochimie 73, 789–796 [DOI] [PubMed] [Google Scholar]
- 8. Yang C., McPheeters D. S., and Yu Y. T. (2005) ψ35 in the branch site recognition region of U2 small nuclear RNA is important for pre-mRNA splicing in Saccharomyces cerevisiae. J. Biol. Chem. 280, 6655–6662 [DOI] [PubMed] [Google Scholar]
- 9. Boros G., Miko E., Muramatsu H., Weissman D., Emri E., Rózsa D., Nagy G., Juhász A., Juhász I., Van Der Horst G., Horkay I., Remenyik É., Karikó K., and Emri G. (2013) Transfection of pseudouridine-modified mRNA encoding CPD-photolyase leads to repair of DNA damage in human keratinocytes: A new approach with future therapeutic potential. J. Photochem. Photobiol. B Biol. 129, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamma T., and Ferré-D'Amaré A. R. (2006) Pseudouridine synthases. Chem. Biol. 13, 1125–1135 [DOI] [PubMed] [Google Scholar]
- 11. Spedaliere C. J., Ginter J. M., Johnston M. V., and Mueller E. G. (2004) The pseudouridine synthases: revisiting a mechanism that seemed settled. J. Am. Chem. Soc. 126, 12758–12759 [DOI] [PubMed] [Google Scholar]
- 12. Hamma T., and Ferré-D'Amaré A. R. (2010) The box H/ACA ribonucleoprotein complex: interplay of RNA and protein structures in post-transcriptional RNA modification. J. Biol. Chem. 285, 805–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McDonald M. K., Miracco E. J., Chen J., Xie Y., and Mueller E. G. (2011) The handling of the mechanistic probe 5-fluorouridine by the pseudouridine synthase TruA and its consistency with the handling of the same probe by the pseudouridine synthases TruB and RluA. Biochemistry 50, 426–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCleverty C. J., Hornsby M., Spraggon G., and Kreusch A. (2007) Crystal structure of human Pus10, a novel pseudouridine synthase. J. Mol. Biol. 373, 1243–1254 [DOI] [PubMed] [Google Scholar]
- 15. Friedt J., Leavens F. M., Mercier E., Wieden H. J., and Kothe U. (2014) An arginine-aspartate network in the active site of bacterial TruB is critical for catalyzing pseudouridine formation. Nucleic Acids Res. 42, 3857–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramamurthy V., Swann S. L., Paulson J. L., Spedaliere C. J., and Mueller E. G. (1999) Critical aspartic acid residues in pseudouridine synthases. J. Biol. Chem. 274, 22225–22230 [DOI] [PubMed] [Google Scholar]
- 17. Conrad J., Niu L., Rudd K., Lane B. G., and Ofengand J. (1999) 16S ribosomal RNA pseudouridine synthase RsuA of Escherichia coli: deletion, mutation of the conserved Asp102 residue, and sequence comparison among all other pseudouridine synthases. RNA 5, 751–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gutgsell N., Englund N., Niu L., Kaya Y., Lane B. G., and Ofengand J. (2000) Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA 6, 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raychaudhuri S., Niu L., Conrad J., Lane B. G., and Ofengand J. (1999) Functional effect of deletion and mutation of the Escherichia coli ribosomal RNA and tRNA pseudouridine synthase RluA. J. Biol. Chem. 274, 18880–18886 [DOI] [PubMed] [Google Scholar]
- 20. Koonin E. V. (1996) Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res. 24, 2411–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kammen H. O., Marvel C. C., Hardy L., and Penhoet E. E. (1988) Purification, structure, and properties of Escherichia coli tRNA pseudouridine synthase I. J. Biol. Chem. 263, 2255–2263 [PubMed] [Google Scholar]
- 22. Pomerantz S. C., and McCloskey J. A. (1990) Analysis of RNA hydrolyzates by liquid chromatography-mass spectrometry. Methods Enzymol. 193, 796–824 [DOI] [PubMed] [Google Scholar]
- 23. Ofengand J., Del Campo M., and Kaya Y. (2001) Mapping pseudouridines in RNA molecules. Methods 25, 365–373 [DOI] [PubMed] [Google Scholar]
- 24. Carlile T. M., Rojas-Duran M. F., and Gilbert W. V. (2015) Pseudo-Seq: genome-wide detection of pseudouridine modifications in RNA. Methods Enzymol. 560, 219–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carlile T. M., Rojas-Duran M. F., Zinshteyn B., Shin H., Bartoli K. M., and Gilbert W. V. (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Durairaj A., and Limbach P. A. (2008) Mass spectrometry of the fifth nucleoside: a review of the identification of pseudouridine in nucleic acids. Anal. Chim. Acta 623, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Durairaj A., and Limbach P. A. (2008) Matrix-assisted laser desorption/ionization mass spectrometry screening for pseudouridine in mixtures of small RNAs by chemical derivatization, RNase digestion and signature products. Rapid Commun. Mass Spectrom. 22, 3727–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Durairaj A., and Limbach P. A. (2008) Improving CMC-derivatization of pseudouridine in RNA for mass spectrometric detection. Anal. Chim. Acta 612, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patteson K. G., Rodicio L. P., and Limbach P. A. (2001) Identification of the mass-silent post-transcriptionally modified nucleoside pseudouridine in RNA by matrix-assisted laser desorption/ionization mass spectrometry. Nucleic Acids Res. 29, E49–E49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guymon R., Pomerantz S. C., Ison J. N., Crain P. F., and McCloskey J. A. (2007) Post-transcriptional modifications in the small subunit ribosomal RNA from Thermotoga maritima, including presence of a novel modified cytidine. RNA 13, 396–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mengel-Jørgensen J., and Kirpekar F. (2002) Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res. 30, e135/131–e135/137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pomerantz S. C., and McCloskey J. A. (2005) Detection of the common RNA nucleoside pseudouridine in mixtures of oligonucleotides by mass spectrometry. Anal. Chem. 77, 4687–4697 [DOI] [PubMed] [Google Scholar]
- 33. Addepalli B., and Limbach P. (2011) Mass spectrometry-based quantification of pseudouridine in RNA. J. Am. Soc. Mass Spectrom. 22, 1363–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao X., and Yu Y.-T. (2004) Detection and quantitation of RNA base modifications. RNA 10, 996–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dai Q., Fong R., Saikia M., Stephenson D., Yu Y. T., Pan T., and Piccirilli J. A. (2007) Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N6-methyladenosine. Nucleic Acids Res. 35, 6322–6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taucher M., Ganisl B., and Breuker K. (2011) Identification, localization, and relative quantitation of pseudouridine in RNA by tandem mass spectrometry of hydrolysis products. Int. J. Mass Spectrom. 304, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Popova A. M., and Williamson J. R. (2014) Quantitative analysis of rRNA modifications using stable isotope labeling and mass spectrometry. J. Am. Chem. Soc. 136, 2058–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goodman H. M., Abelson J. N., Landy A., Zadrazil S., and Smith J. D. (1970) The nucleotide sequences of tyrosine transfer RNAs of Escherichia coli. Eur. J. Biochem. 13, 461–483 [DOI] [PubMed] [Google Scholar]
- 39. Del Campo M., Kaya Y., and Ofengand J. (2001) Identification and site of action of the remaining four putative pseudouridine synthases in Escherichia coli. RNA 7, 1603–1615 [PMC free article] [PubMed] [Google Scholar]
- 40. Krivos K. L., Addepalli B., and Limbach P. A. (2011) Removal of 3′-phosphate group by bacterial alkaline phosphatase improves oligonucleotide sequence coverage of RNase digestion products analyzed by collision-induced dissociation mass spectrometry. Rapid Commun. Mass Spectrom. 25, 3609–3616 [DOI] [PubMed] [Google Scholar]
- 41. Hossain M., and Limbach P. A. (2007) Mass spectrometry-based detection of transfer RNAs by their signature endonuclease digestion products. RNA 13, 295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Houser W. M., Butterer A., Addepalli B., and Limbach P. A. (2015) Combining recombinant ribonuclease U2 and protein phosphatase for RNA modification mapping by liquid chromatography-mass spectrometry. Anal. Biochem. 478, 52–58 [DOI] [PubMed] [Google Scholar]
- 43. Hur S., and Stroud R. M. (2007) How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Mol. Cell 26, 189–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alian A., DeGiovanni A., Griner S. L., Finer-Moore J. S., and Stroud R. M. (2009) Crystal structure of an RluF-RNA complex: a base-pair rearrangement is the key to selectivity of RluF for U2604 of the ribosome. J. Mol. Biol. 388, 785–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang L., Pookanjanatavip M., Gu X., and Santi D. V. (1998) A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry 37, 344–351 [DOI] [PubMed] [Google Scholar]
- 46. Wrzesinski J., Nurse K., Bakin A., Lane B. G., and Ofengand J. (1995) A dual-specificity pseudouridine synthase: an Escherichia coli synthase purified and cloned on the basis of its specificity for ψ746 in 23S RNA is also specific for ψ32 in tRNA(Phe). RNA 1, 437–448 [PMC free article] [PubMed] [Google Scholar]
- 47. Czudnochowski N., Ashley G. W., Santi D. V., Alian A., Finer-Moore J., and Stroud R. M. (2014) The mechanism of pseudouridine synthases from a covalent complex with RNA, and alternate specificity for U2605 versus U2604 between close homologs. Nucleic Acids Res. 42, 2037–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hoang C., and Ferré-D'Amaré A. R. (2001) Cocrystal structure of a tRNA ψ55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell 107, 929–939 [DOI] [PubMed] [Google Scholar]
- 49. Szweykowska-Kulinska Z., and Beier H. (1992) Sequence and structure requirements for the biosynthesis of pseudouridine (psi 35) in plant pre-tRNA(Tyr). EMBO J. 11, 1907–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grosjean H., Szweykowska-Kulinska Z., Motorin Y., Fasiolo F., and Simos G. (1997) Intron-dependent enzymatic formation of modified nucleosides in eukaryotic tRNAs: a review. Biochimie 79, 293–302 [DOI] [PubMed] [Google Scholar]
- 51. Kierzek E., Malgowska M., Lisowiec J., Turner D. H., Gdaniec Z., and Kierzek R. (2014) The contribution of pseudouridine to stabilities and structure of RNAs. Nucleic Acids Res. 42, 3492–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bare L. A., and Uhlenbeck O. C. (1986) Specific substitution into the anticodon loop of yeast tyrosine transfer RNA. Biochemistry 25, 5825–5830 [DOI] [PubMed] [Google Scholar]
- 53. Davis D. R. (1995) Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res. 23, 5020–5026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zerfass K., and Beier H. (1992) Pseudouridine in the anticodon GψA of plant cytoplasmic tRNA(Tyr) is required for UAG and UAA suppression in the TMV-specific context. Nucleic Acids Res. 20, 5911–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meng Z., and Limbach P. A. (2005) Quantitation of ribonucleic acids using 18O labeling and mass spectrometry. Anal. Chem. 77, 1891–1895 [DOI] [PubMed] [Google Scholar]
- 56. Li M. M., Addepalli B., Tu M. J., Chen Q. X., Wang W. P., Limbach P. A., LaSalle J. M., Zeng S., Huang M., and Yu A. M. (2015) Chimeric microRNA-1291 biosynthesized efficiently in Escherichia coli is effective to reduce target gene expression in human carcinoma cells and improve chemosensitivity. Drug Metab. Dispos. 43, 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang W. P., Ho P. Y., Chen Q. X., Addepalli B., Limbach P. A., Li M. M., Wu W. J., Jilek J. L., Qiu J. X., Zhang H. J., Li T., Wun T., White R. D., Lam K. S., and Yu A. M. (2015) Bioengineering novel chimeric microRNA-34a for prodrug cancer therapy: high-yield expression and purification, and structural and functional characterization. J. Pharmacol. Exp. Ther. 354, 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Russell S. P., and Limbach P. A. (2013) Evaluating the reproducibility of quantifying modified nucleosides from ribonucleic acids by LC-UV-MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 923–924, 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee C., Kramer G., Graham D. E., and Appling D. R. (2007) Yeast mitochondrial initiator tRNA is methylated at guanosine 37 by the Trm5-encoded tRNA (guanine-N1-)-methyltransferase. J. Biol. Chem. 282, 27744–27753 [DOI] [PubMed] [Google Scholar]
- 60. Meeks L. R., Addepalli B., and Hunt A. G. (2009) Characterization of genes encoding poly(A) polymerases in plants: evidence for duplication and functional specialization. PLoS One 4, e8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Addepalli B., Lesner N. P., and Limbach P. A. (2015) Detection of RNA nucleoside modifications with the uridine-specific ribonuclease MC1 from Momordica charantia. RNA 21, 1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Addepalli B., and Hunt A. G. (2007) A novel endonuclease activity associated with the Arabidopsis ortholog of the 30-kDa subunit of cleavage and polyadenylation specificity factor. Nucleic Acids Res. 35, 4453–4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kramer E. B., and Farabaugh P. J. (2007) The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.