

Abstract
AIM
To investigate the role of embryonic liver fordin (ELF) in liver fibrosis by regulating hepatic stellate cells (HSCs) glucose glycolysis.
METHODS
The expression of ELF and the glucose glycolysis-related proteins were evaluated in activated HSCs. siRNA was used to silence ELF expression in activated HSCs in vitro and the subsequent changes in PI3K/Akt signaling and glucose glycolysis-related proteins were observed.
RESULTS
The expression of ELF increased remarkably in HSCs of the fibrosis mouse model and HSCs that were cultured for 3 wk in vitro. Glucose glycolysis-related proteins showed an obvious increase in the activated HSCs, such as phosphofructokinase, platelet and glucose transporter 1. ELF-siRNA, which perfectly silenced the expression of ELF in activated HSCs, led to the induction of glucose glycolysis-related proteins and extracellular matrix (ECM) components. Moreover, pAkt, which is an important downstream factor in PI3K/Akt signaling, showed a significant change in response to the ELF silencing. The expression of glucose glycolysis-related proteins and ECM components decreased remarkably when the PI3K/Akt signaling was blocked by Ly294002 in the activated HSCs.
CONCLUSION
ELF is involved in HSC glucose glycolysis by regulating PI3K/Akt signaling.
Keywords: Liver fibrosis, Embryonic liver fordin, PI3K/Akt signaling, Hepatic stellate cells, Glucose glycolysis
Core tip: The metabolism of activated hepatic stellate cells (HSCs) was reprogrammed. Silence of embryonic liver fordin (ELF) led to the inhibition of PI3K/Akt signaling and decrease of glycolysis in activated HSCs. Glucose glycolysis of activated HSCs was regulated by ELF through PI3K/Akt signaling The present study indicated that metabolism of HSCs may be a novel target for the diagnosis and treatment of liver cirrhosis in clinical practice.
INTRODUCTION
Liver fibrosis occurs in most chronic liver diseases and is characterized by the accumulation of extracellular matrix following sustained inflammation[1]. The main causes of liver fibrosis include chronic HBV and HCV infection, parasitic infections, alcohol abuse, and NASH. Liver fibrosis can lead to the dysfunction of hepatocytes and obstruction of the intrahepatic blood flow, which resulting in the subsequent hepatic insufficiency and hypertension of the portal vein[2].
HSCs, which are the main source of ECM in injured livers, play a crucial role in the progress of liver fibrosis. In normal livers, HSCs are located in the Disse space, between the sinusoidal endothelium and hepatocytes. HSCs become activated and acquire contractile, proinflammatory, and fibrogenic properties when subjected to diverse chronic injuries. Researchers have identified many factors that promote the transdifferentiation of quiescent HSCs into activated HSCs[3].
The TGF-β signaling plays a very important role in various cellular physiological processes through the regulation of downstream proteins[4]. Frequent inactivation of the TGF-β pathway components in liver fibrosis demonstrates a powerful promoting role of the TGF-β pathway. The multifunctional effects of TGF-β in cellular actions occur through the binding of its receptors, TGF-β receptor II and receptor I; the activation of intrinsic kinase activity; and the phosphorylation and translocation of mediators, Smads, followed by TGF-β target gene activation[5]. ELF, also known as β2-spectrin, is important for maintaining function of cellular membranes and polarization of epithelial cells. Meanwhile, ELF also plays an important role in TGF-β pathway.
Smad 3 and Smad 4 are the main proteins of Smad family which is crucial for the activation of TGF-β pathway. The interaction of ELF and Smad proteins was able to facilitate the transport of Smad 3-Smad4 complex into nucleus, which lead to the subsequent activation of TGF-β pathway[6-8].
Our previous study demonstrated that ELF is involved in the activation of HSCs. First, Western blot and RT-qPCR evaluation indicated that ELF expression was increased remarkably in fibrotic mouse model. Moreover, the silence of ELF in activated HSCs significantly reduced the ECM components such as collagen and α-SMA. Clarification of the mechanism is needed[9]. In addition, our study showed that the PI3K/Akt signaling is regulated by ELF in the process of hepatocyte proliferation[10]. The PI3K/Akt signaling is essential in the regulation of glucose metabolism in many types of cells[11,12]. Researchers have identified a novel mechanism for reprogramming quiescent HSCs into activated HSCs that depends on the induction of glycolysis, similar to the Warburg state that has been described in cancer cells[13]. We hypothesized that ELF is involved in glucose metabolism of HSCs through PI3K/Akt signaling.
MATERIALS AND METHODS
Ethics statement
All the processes were approved by the Animal Care and Use Committee of Huazhong University of Science and Technology, Wuhan, China.
Mouse model of liver fibrosis
Eight-week-old male mice(C57/Black/6) that purchased from Animal Center of HUST (Wuhan, China) were challenged by classic subcutaneous injection with carbon tetrachloride (CCl4) diluted at a 3:7 (v/v) ratio in 5 mL/kg olive oil twice weekly for 12 wk as described previously.
HSC isolation
Primary hepatic stellate cells used in this study were isolated from C57/Black/6 mice (including control and fibrosis model) by in situ perfusion and centrifugation. The details were described in our previous study[9].
Preparation and transfection of siRNAs
siRNA used to silence ELF expression was synthesized by Applied Biosystems. The concentration of siRNAs (and scrambled siRNAs) was 50 pmol/L. Lipo2000 (Invitrogen) was used to transduce the siRNA into mice hepatic stellate cells on six wells when the confluence was about 30%-50%. RNA extraction was performed 72 h later. According to previous identification, ELF siRNA s74307 was chosen because of its best efficacy.
Western blot
The total protein was extracted from cell and tissue using RIPA buffer with protease inhibitors. Concentrations of proteins were evaluated by BCA Assay Kit. Total proteins (50 μg) were separated on 10% SDS-PAGE. The immunoblotting was performed. The immune complex was visualized by ECL detection
Immunohistochemistry
Liver specimens for histology and immunohistochemistry were fixed in 10% buffered formalin for 48 h, and then sliced into sections. Staining was performed using ABC kit. Sections were incubated at 4 °C with antibody for 12 h. DAB was used to visualize immunocomplexes.
Measurement of lactate
Whole cell lysates of liver sample and HSCs were prepared with pyruvate assay buffer and then filtered through a 10-kilodalton molecular weight spin filter for deproteinization. Levels of lactate were measured using a lactate assay kit or pyruvate assay kit from BioVision according to the manufacturer’s instructions, and normalized to the control group.
Statistical analysis
The results were presented as the mean ± SD. The differences between groups were tested by Student two-tailed t test, and P < 0.05 was considered statistically significant.
RESULTS
ELF expression is upregulated in fibrotic liver and HSCs
To evaluate whether ELF is involved in liver fibrosis, we generated a fibrotic mouse model. The immunohistochemical (IHC) examination found that ELF expression was increased in the fibrotic livers compared with controls (Figure 1A). Moreover, the ELF expression was observed near the bridging fibrotic areas. In addition, ELF mRNA (with RT-qPCR, approximately 2.5 times) and protein expression by Western blot analysis was increased in the CCl4-treated animals (Figure 1B). HSCs are predominantly located in the areas of bridging fibrosis, and we isolated HSCs from the fibrotic and normal livers. The RT-qPCR and Western blot test of the ELF expression found that there was a more significant increase in the HSCs isolated from the fibrotic livers than from the normal livers (Figure 1C). This finding indicated that the upregulated expression of ELF in HSCs might play an important role in liver fibrosis.
Figure 1.
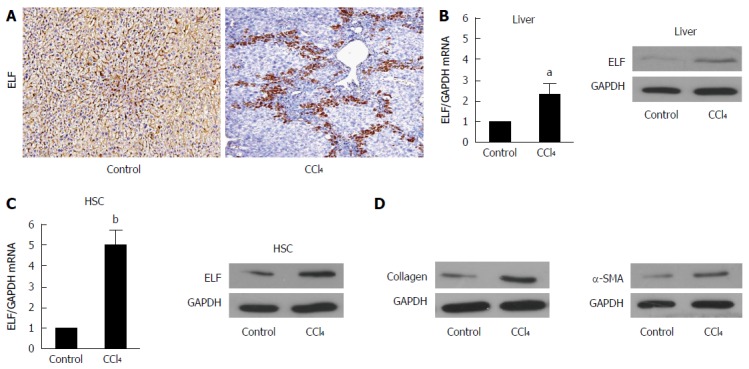
Embryonic liver fordin expression is upregulated in fibrotic livers and hepatic stellate cells. A: The Embryonic liver fordin (ELF) expression in cirrhotic livers was determined by the immunohistochemical analysis. Magnification × 200; B: Real-time RT-PCR and Western blot analysis were used to evaluate the ELF expression in the liver homogenates from the control and CCl4-treated mice. aP < 0.05, the CCl4-treated mice vs the control mice. GAPDH was used as the control; C: Real-time RT-PCR and Western blot analysis were used to evaluate the ELF expression in the primary hepatic stellate cells (HSCs) isolated from the control and CCl -treated mice. bP < 0.01, the CCl4-treated mice vs the control mice; D: The α-SMA and collagen I expression at the protein level were upregulated in the whole liver homogenates from the CCl4-treated mice compared with the controls.
Glycolysis-related genes are upregulated in fibrotic livers
Previous study had demonstrated that glucose metabolism was reprogrammed in fibrotic livers[13]. To confirm whether glycolysis-related genes were changed in fibrotic livers, we selected some key proteins which are involved in glucose glycolysis, such as phosphofructokinase (PFKP), Glut1, PKM2, and MCT4.
PFKP, a gene which encodes the platelet isoform of phosphofructokinase (PFK), plays an important role in glucose glycolysis of various cell types[14,15]. As shown in Figure 2A and 2B, the PFKP expression was increased at both the mRNA and protein levels. Pyruvate kinase isozymes M1/M2 (PKM1/M2) is an enzyme encoded by the PKM2 gene. The function of PKM2 is to catalyze the last step within glycolysis, and leads to the dephosphorylation of phosphoenolpyruvate[16]. Real-time quantitative PCR and Western blot (Figure 2C and D) demonstrated that PKM expression was increased significantly in fibrotic liver at both mRNA and protein levels. Glucose transporter 1 (Glut 1) is a uniporter protein that in humans is encoded by the SLC2A1 gene. Glut 1 is the first glucose transporter which facilitates the transport of glucose from blood into membrane in various kinds of cells[17,18]. We found that Glut1 expression also showed a remarkable increase in fibrotic liver than control mice (Figure 2E and F).
Figure 2.
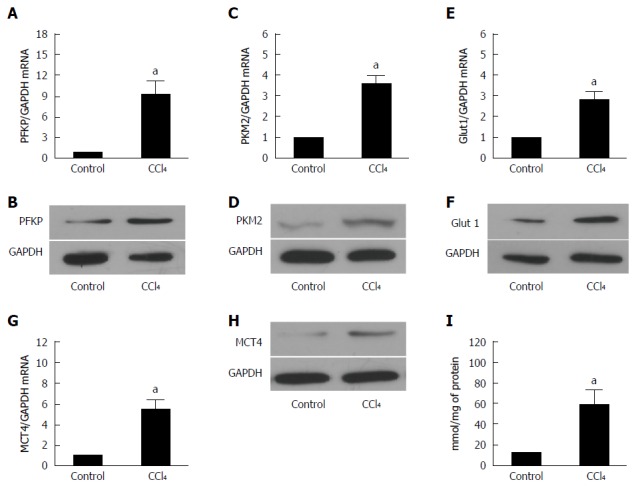
Glycolysis related-genes are upregulated in liver fibrosis. A, C: The real-time RT-PCR analysis evaluated the expression of the hepatic glycolytic enzymes, PFKP and PFKM2 in fibrotic and control mice. The PFKP and PFKM2 expression of the mRNA level was increased compared with that of the control (aP < 0.05 vs control). GAPDH was used as the control; B, D: Western blot analysis indicated that the PFKP and PFKM2 expression of the protein level was upregulated in the fibrotic livers; E: The real-time RT-PCR analysis evaluated the hepatic glucose transporter, Glut 1, expression in the fibrotic livers and the control mice. The expression of Glut 1 mRNA was increased compared with that of the controls (aP < 0.05 vs control); F: Western blot analysis indicated that the expression of the Glut 1 at protein level was upregulated in the fibrotic livers; G: Real-time RT-PCR analysis evaluated the MCT4 hepatic lactate transporter expression in the fibrotic livers and the control mice. The expression of the Glut 1 mRNA was increased compared with that of the controls (aP < 0.05 vs control); H: Western blot analysis indicated that the expression of MCT4 at protein level was upregulated in the fibrotic liver; I: The intracellular lactate was evaluated by a lactate assay kit (aP < 0.05 vs control).
Monocarboxylate transporter 4 (MCT4), the lactate export pump, plays an crucial role in the accumulation of intracellular lactate in various cells[19]. The upregulation of MCT4 in fibrotic livers indicates that lactate accumulation was increased in fibrotic livers compared with the controls (Figure 2G and H). These findings indicated that glycolysis related genes were overexpressed in fibrotic livers. However, the expression of these genes in HSCs remains unknown.
The intracellular lactate was evaluated by a lactate assay kit. As shown in Figure 2I, intracellular lactate level increased significantly in fibrotic liver.
Glycolysis related genes are upregulated significantly in HSCs isolated from fibrotic livers
The liver is composed of many cell types, and further studies should be performed if a certain protein is overexpressed in the liver. Therefore, we first isolated the HSCs from a fibrotic liver by in situ perfusion. Then, the total protein and mRNA were extracted. As demonstrated in Figure 3, the expression of PFKP, GLUT 1, PKM2, and MCT4 increased remarkably (P < 0.01) shown by real time-quantitative PCR (Figure 3A, C, E and G) and Western blot (Figure 3B, D, F and H). Moreover, lactate (Figure 3I) showed a significant increase compared with the controls (P < 0.01). This finding indicates that the glucose metabolism of activated HSCs was reprogrammed. The reprogrammed metabolism might supply energy to HSCs for their proliferation, ECM production, and migration.
Figure 3.
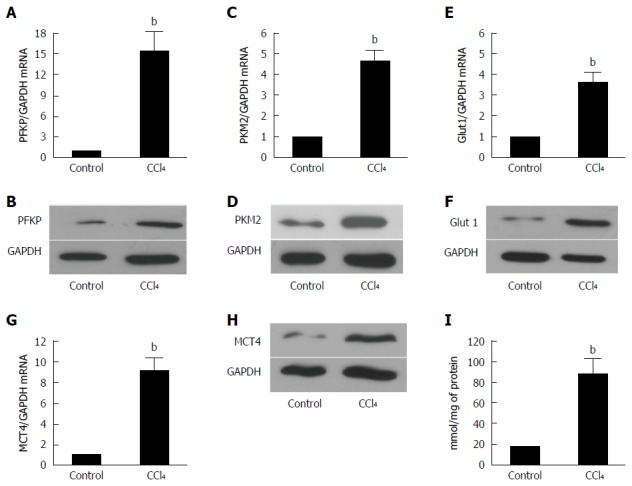
Glycolysis related-genes are upregulated significantly in the HSCs isolated from the fibrotic livers. A, C: The real-time RT-PCR analysis was used to evaluate the expression of the PFKP and PFKM2 hepatic glycolytic enzymes in the fibrotic livers and control mice. The PFKP, pAKT, and PFKM2 expressions of the mRNA level were increased compared with those of the controls (bP < 0.01 vs control). GAPDH was used as the control; B, D: The Western blot analysis indicated that the PFKP and PFKM2 expression at the protein level was upregulated in the fibrotic livers; E: The real-time RT-PCR analysis evaluated the hepatic glucose transporter Glut 1 expression in the fibrotic livers and control mice. The expression of Glut 1 mRNA was increased compare with that of the controls (bP < 0.01 vs control); F: The Western blot analysis indicated that the Glut 1 expression at the protein level was upregulated in the fibrotic livers; G: The real-time RT-PCR analysis evaluated the hepatic lactate transporter MCT4 expression in the fibrotic livers and the control mice. The Glut 1 expression at the mRNA level was increased compared with that of the controls (bP < 0.01 vs control); H: The western blot analysis indicated that the MCT4 expression at the protein level was upregulated in the fibrotic livers; I: The intracellular lactate was evaluated by a lactate assay kit (bP < 0.01 vs control).
Silence of ELF in activated HSCs leads to the inhibition of glycolysis
The efficiency of the knockdown of ELF in activated HSCs by siRNA was tested by Western blot (Figure 4A) and real-time quantitative PCR (Figure 4B). The ELF-siRNA treatment led to a remarkable decrease in the ELF expression. A significant reduction in SMA and collagen 1 expression was observed in the ELF-siRNA treated HSCs (Figure 4H and I). In addition, PFKP, GLUT 1, PKM2, and MCT4 showed a significant decrease in response to the ELF-siRNA (Figure 4C-F). The intracellular lactate also decreased significantly in response to the silence of ELF (Figure 4G). This finding indicates that the silence of ELF leads to the inhibition of glucose glycolysis-related genes. These results indicated that silence of ELF could block HSC activation, reduce ECM production, and inhibit glycolysis.
Figure 4.
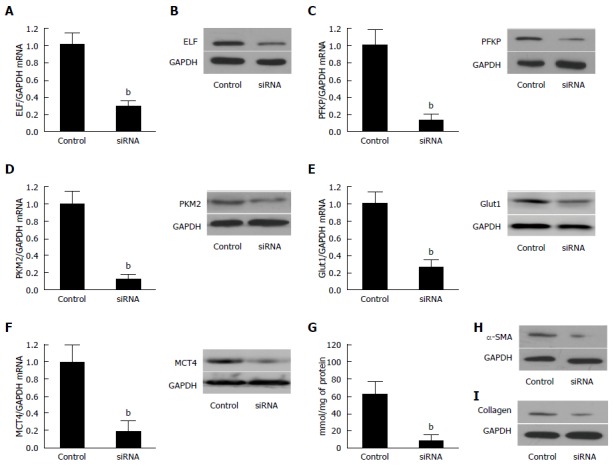
Silencing of Embryonic liver fordin in the activated hepatic stellate cells led to the inhibition of glycolysis. A: The ELF mRNA was reduced in the activated hepatic stellate cells (HSCs) transfected synthetic siRNA against ELF was assessed by real-time RT-PCR. P < 0.01 for the ELF siRNA versus the siRNA controls. GAPDH was used as the control; B: Western blot analysis confirmed that synthetic siRNA inhibited the ELF expression in the activated HSCs; C, D: The hepatic glycolytic enzymes, PFKP and PFKM2, expression of the mRNA and protein level decreased significantly after the ELF siRNA treatment (aP < 0.05 vs control). E, F: The hepatic glycolytic enzymes, Glut 1 and MCT 4, expression of the mRNA at the protein level decreased significantly after the ELF siRNA treatment (aP < 0.05 vs control). G: The intracellular lactate level decreased significantly after the ELF siRNA treatment (aP < 0.05 vs control); H, I: The main components of the extracellular matrix, α-SMA and collagen I, expression showed a remarkable decrease in the activated HSCs, and the HSC transfected synthetic siRNA against ELF.
ELF-PI3K/Akt-glycolysis axis in HSC
Although previous data indicated that ELF might play an important role in HSC activation and glycolysis in the pathogenesis of liver fibrosis, the mechanism needs to be clarified. Since PI3K/Akt signaling is important in glucose glycolysis of many cell types, and based on our previous study of the interaction between ELF and PI3K/Akt pathway, we hypothesized that the cross talk of ELF and PI3K/Akt signaling was involved in the activation of HSCs.
To confirm this finding, we first evaluated Akt, the key protein of PI3K signaling, in ELF-siRNA treated HSCs. As shown in Figure 5A, the expression of pAkt decreased significantly after the silencing of ELF in activated HSCs, however the total AKT was not influenced by ELF-siRNA treatment. Then, we administered activated HSCs with LY294002, a PI3K/Akt signaling inhibitor, and made the following evaluation. The remarkable downregulation of the pAkt expression confirmed that the PI3K/Akt signaling was well blockaded by LY294002 (Figure 5D). According to the Western blot and RT-qPCR testing, we found that inhibition of the PI3K/akt signaling does not affect the ELF expression (Figure 5B and C). Therefore, the key proteins of glycolysis, PFKP and PKM2, showed a significant decrease in the activated HSCs treated with LY294002 at the mRNA (Figure 5E and G) and protein levels (Figure 5F and H). In addition, SMA and collagen, the main components of the extracellular matrix, were effectively repressed by the LY294002 treatment (Figure 5I and J). Based on these data, we first demonstrated that ELF could regulate pAkt and glycolysis. Moreover, the blockade of PI3K/akt led to the repression of glycolysis and ECM production. The ELF was involved in the HSC glycolysis through the regulation of PI3K/Akt signaling.
Figure 5.
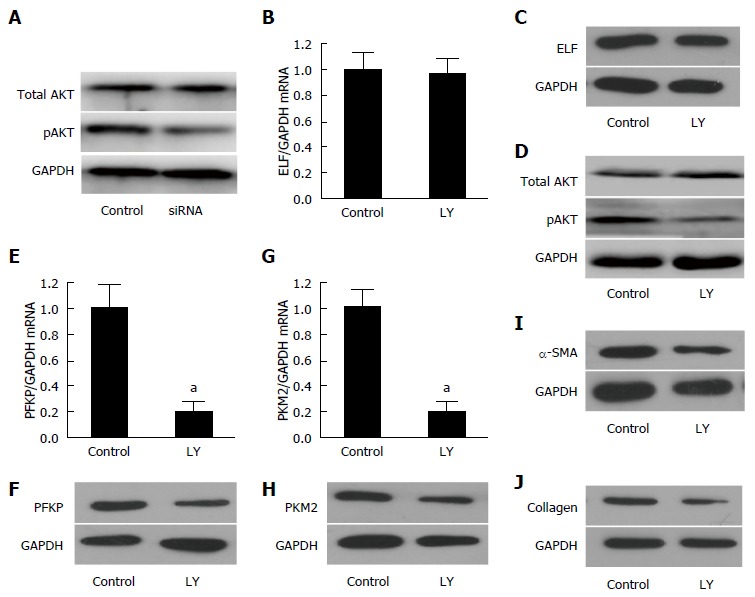
Embryonic liver fordin was involved in the regulation of HSC glycolysis through PI3K/AKT signaling. A: The key protein of PI3K/akt signaling, pAKT, decreased significantly in the embryonic liver fordin (ELF)-siRNA treated hepatic stellate cells (HSCs), but the expression of total AKT was not affected by ELF-siRNA treatment. GAPDH was used as the control; B, C: The Western blot and real-time RT-PCR analysis indicated that the ELF expression was not affected by Ly294002, an inhibitor of the PI3K/akt signaling; D: The pAKT expression decreased obviously after the inhibition of the PI3K/akt signaling in the HSCs; E, F: The hepatic glycolytic enzymes PFKP expression of mRNA and the protein level decreased significantly after the Ly294002 treatment in the activated HSCs (aP < 0.05 vs control); G, H: The hepatic glycolytic enzyme PKM2 expression at mRNA and the protein levels decreased significantly after the Ly294002 treatment in the activated HSCs (aP < 0.05 vs control). I, J: The main components of the extracellular matrix, α-SMA and collagen I expression showed a remarkable decrease in the activated HSCs treated by the PI3K/akt signaling inhibitor, Ly294002.
DISCUSSION
The destruction of the sinusoidal architecture in hepatic fibrosis always leads to hypoxia of the HSCs around the sinusoids. Simultaneously, the HSCs show a strong ability to activate and proliferate, which requires sufficient energy. The mechanism by which the HSCs ensure sufficient energy for activation and proliferation in the anoxic microenvironment is unknown. As shown in recent research, glycolysis was increased significantly in activated HSCs. Cellular metabolism might be a novel aspect in the pathogenesis study of liver fibrosis. To date, most studies of liver fibrosis focused on HSC activation and ECM production. We showed that energy metabolism, particularly glycolysis, represents an innovative role in HSC activation.
In this study, we found a novel role for ELF in the metabolic process of HSC s activation; the PI3K/Akt pathway was shown to regulate the glycolysis of HSCs. We first found that ELF expression was increased in the HSCs of the fibrosis mouse model and the HSCs cultured for 3 wk in vitro. PFKP is the key enzyme in glycolysis and GLU, which showed a significant increase in the activated HSCs compared with the controls. Next, we tested whether increased ELF expression affects glycolysis related-proteins. The silencing of ELF in the activated HSCs leads to decreased expression of glycolysis-related enzymes, such as PFKP, GLUT1, PKM2, and MCT4. Further studies indicated that ELF was involved in HSC glucose metabolism through regulation of pAkt, an important downstream factor in PI3K/Akt signaling.
PI3K signaling, one of the most important pathways that involved various human diseases, provides intense growth and survival signals to many cell types and has profound effects on cellular metabolism[20]. As demonstrated in previous studies, the integration of growth and proliferation signals with alterations to the central metabolism is important for the oncogenic effects of the PI3K pathway[21,22]. Protein kinase B (PKB), also known as Akt, is a serine/threonine-specific protein kinase that plays a key role in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription and cell migration. Akt is the best-studied downstream factor of PI3K signaling, and an important driver of the glycolytic phenotype tumor, and stimulates ATP generation, ensuring that cells have the bioenergetic capacity required to respond to growth signals[23]. Akt was able to phosphorylate key glycolytic enzymes such as hexokinase, phosphofructokinase 2, and Glu 1. This function leads to increased expression and membrane translocation of the glucose transporters[24-26].
Abundant evidence has demonstrated that PI3K/Akt pathway is crucial for the proliferation and metabolism of many kinds of cells. Therefore, many studies have suggested that TGF-β is able to activate PI3K/Akt signaling and leads to the subsequent phosphorylation of the downstream genes without an interaction with Smad proteins[27-29]. TGF-β was shown to downregulate PI3K/Akt signaling activity through Smad proteins[30,31]. Therefore, the PI3K/Akt signaling can also antagonize the Smad-mediated effects in other TGF-β related conditions[32,33]. Our previous studies showed that the activation of PI3K/Akt signaling through insulin stimulation induced the activation of TGF-β/Smad signaling, as indicated by the nuclear translocation of Smad3/4. Moreover, the inhibition of PI3K/Akt signaling by the use of the LY294002 inhibitor led to the blockage of TGF-β/Smad signaling[9]. These findings indicated that the activated PI3k/Akt pathway led to the activation of TGF-β/Smad signaling in the hepatocytes. ELF was shown to play an important role in this process by regulating the localization of Smad3/4 in the nucleus[10]. However, the details underlying the interaction between TGF-β/Smad and PI3K/Akt signaling, particularly in the field of cell energy metabolism, should be investigated in the future.
Multiple intrinsic and extrinsic molecular mechanisms contribute to cellular metabolism and provide support for the three basic needs of dividing cells, as follows: rapid ATP generation to maintain the energy status; increased biosynthesis of macromolecules; and tightened maintenance of appropriate cellular redox status[34,35]. Metabolic changes represent a common feature of all cell types and various diseases including liver fibrosis. This effect is regulated by diverse pathways and factors, such as PI3K signaling, the hypoxia-inducible factor, p53, MYC and AMP-activated protein kinase, liver kinase B1, and the hedgehog pathways[36,37]. Liver fibrosis represents a very complicated process. In recent years, scientists have revealed various pathways that contribute to the pathogenesis of liver fibrosis. Although we showed that PI3K/Akt, which was regulated by ELF, could affect glycolysis in HSCs, other signaling could perform this function. Additional signals that were involved in the regulation of energy metabolism should be investigated in liver fibrosis. Our study could increase the knowledge of the pathogenesis of hepatic fibrosis and might be applied to the diagnosis and treatment of patients.
COMMENTS
Background
The activation of hepatic stellate cells is a key event in pathogenesis of liver fibrosis.
Research frontiers
In recent studies, various factors which were involved in the proliferation, apoptosis, ageing and extracellular matrix secretion were found to be contributive to the activation process of hepatic stellate cells.
Innovations and breakthroughs
This study found that embryonic liver fordin (ELF) was involved in hepatic stellate cells activation by regulating glucose metabolism, which was a novel aspect in the area of liver fibrosis.
Applications
The glucose metabolism of activated hepatic stellate cells provides us a new vision in the diagnosis and therapy of liver fibrosis.
Peer-review
In this study, the authors found that the ELF was involved in the couples of signaling pathways to activate the glucose glycolysis of hepatic stellate cells during the liver fibrosis. The PI3K/Akt signaling and glucose glycolysis-related proteins were evaluated in this study.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Institute of Laboratory Animal Sciences, Huazhong University of Science and Technology, Wuhan, China
Conflict-of-interest statement: The authors declare no conflicts of interest.
Data sharing statement: No additional unpublished data are available.
Peer-review started: June 4, 2016
First decision: July 12, 2016
Article in press: August 19, 2016
P- Reviewer: Duan YN, Shieh KR S- Editor: Qi Y L- Editor: Ma JY E- Editor: Wang CH
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64:830–841. doi: 10.1136/gutjnl-2014-306842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horbelt D, Denkis A, Knaus P. A portrait of Transforming Growth Factor β superfamily signalling: Background matters. Int J Biochem Cell Biol. 2012;44:469–474. doi: 10.1016/j.biocel.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 5.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 6.Macias-Silva M, Li W, Leu JI, Crissey MA, Taub R. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-beta signals during liver regeneration. J Biol Chem. 2002;277:28483–28490. doi: 10.1074/jbc.M202403200. [DOI] [PubMed] [Google Scholar]
- 7.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen LN, Furuya MH, Wolfraim LA, Nguyen AP, Holdren MS, Campbell JS, Knight B, Yeoh GC, Fausto N, Parks WT. Transforming growth factor-beta differentially regulates oval cell and hepatocyte proliferation. Hepatology. 2007;45:31–41. doi: 10.1002/hep.21466. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Liu F, Tu W, Chang Y, Yao J, Wu W, Jiang X, He X, Lin J, Song Y. Embryonic liver fodrin involved in hepatic stellate cell activation and formation of regenerative nodule in liver cirrhosis. J Cell Mol Med. 2012;16:118–128. doi: 10.1111/j.1582-4934.2011.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Song Y, Tu W, He X, Lin J, Liu F. β-2 spectrin is involved in hepatocyte proliferation through the interaction of TGFβ/Smad and PI3K/AKT signalling. Liver Int. 2012;32:1103–1111. doi: 10.1111/j.1478-3231.2012.02812.x. [DOI] [PubMed] [Google Scholar]
- 11.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont R, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–29.e1-11. doi: 10.1053/j.gastro.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 15.Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 16.Wong N, Ojo D, Yan J, Tang D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015;356:184–191. doi: 10.1016/j.canlet.2014.01.031. [DOI] [PubMed] [Google Scholar]
- 17.Rowland AF, Fazakerley DJ, James DE. Mapping insulin/GLUT4 circuitry. Traffic. 2011;12:672–681. doi: 10.1111/j.1600-0854.2011.01178.x. [DOI] [PubMed] [Google Scholar]
- 18.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gogvadze V, Zhivotovsky B, Orrenius S. The Warburg effect and mitochondrial stability in cancer cells. Mol Aspects Med. 2010;31:60–74. doi: 10.1016/j.mam.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 22.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 23.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 24.Cui W, Cai Y, Zhou X. Advances in subunits of PI3K class I in cancer. Pathology. 2014;46:169–176. doi: 10.1097/PAT.0000000000000066. [DOI] [PubMed] [Google Scholar]
- 25.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finlay DK. mTORC1 regulates CD8+ T-cell glucose metabolism and function independently of PI3K and PKB. Biochem Soc Trans. 2013;41:681–686. doi: 10.1042/BST20120359. [DOI] [PubMed] [Google Scholar]
- 27.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 28.Conery AR, Cao Y, Thompson EA, Townsend CM, Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6:366–372. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- 29.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 30.Meulmeester E, Ten Dijke P. The dynamic roles of TGF-β in cancer. J Pathol. 2011;223:205–218. doi: 10.1002/path.2785. [DOI] [PubMed] [Google Scholar]
- 31.Verheyen EM. Opposing effects of Wnt and MAPK on BMP/Smad signal duration. Dev Cell. 2007;13:755–756. doi: 10.1016/j.devcel.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 32.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 33.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 34.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 37.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]