

Abstract
Primary hepatic lymphoma (PHL) is a rare malignancy. We aimed to assess the clinical profile, outcome and prognostic factors in PHL through the Rare Cancer Network (RCN). A retrospective analysis of 41 patients was performed. Median age was 62 years (range, 23-86 years) with a male-to-female ratio of 1.9:1.0. Abdominal pain or discomfort was the most common presenting symptom. Regarding B-symptoms, 19.5% of patients had fever, 17.1% weight loss, and 9.8% night sweats. The most common radiological presentation was multiple lesions. Liver function tests were elevated in 56.1% of patients. The most common histopathological diagnosis was diffuse large B-cell lymphoma (65.9%). Most of the patients received Chop-like (cyclophosphamide, doxorubicin, vincristine, and prednisone) regimens; 4 patients received radiotherapy (dose range, 30.6-40.0 Gy). Median survival was 163 months, and 5- and 10-year overall survival rates were 77 and 59%, respectively. The 5- and 10-year disease-free and lymphoma-specific survival rates were 69, 56, 87 and 70%, respectively. Multivariate analysis revealed that fever, weight loss, and normal hemoglobin level were the independent factors influencing the outcome. In this retrospective multicenter RCN study, patients with PHL had a relatively better prognosis than that reported elsewhere. Multicenter prospective studies are still warranted to establish treatment guidelines, outcome, and prognostic factors.
Key words: Non-Hodgkin’s lymphoma, primary hepatic lymphoma, chemotherapy
Introduction
Primary hepatic lymphoma (PHL) is a rare malignancy, although secondary liver involvement in patients with advanced stages of lymphoma is common. PHL represents 0.016% of all non-Hodgkin’s lymphomas and 0.4% of all extranodal lymphomas.1 Because of the low number of reported cases, the disease is poorly understood and few clinical studies have been conducted to help elucidate the pathogenesis, variability of the clinical presentation, natural course of the disease, optimal therapy, response to therapy, and survival.2,3
Regarding the exact definition of PHL, no consensus has been achieved. Some reports have been based on cases in which staging investigations failed to show extrahepatic disease, whereas others have accepted cases as being primary if the disease was confined to the liver at presentation.4 The criteria for diagnosis defined by Lei are: i) symptoms caused mainly by liver involvement at presentation; ii) absence of distant lymphadenopathy, palpable clinically at presentation or detected during staging radiologic studies; and iii) absence of leukemic blood involvement in the peripheral blood smear.5
The pathogenesis is not clear but several etiologic factors for PHL have been proposed such as an increased incidence of PHL in patients with hepatitis B and C virus infection and exposure to chemicals.6-8 Most of the patients diagnosed with PHL are middle-aged men, majority of cases are diffuse large B-cell type, and presenting complaints consists of abdominal pain, fatigue, and constitutional symptoms, hepatomegaly being frequent.9
Although there is little consistency in the literature regarding the treatment, options include surgery, chemotherapy, radiation therapy or combination of these modalities. The reported median survival rates vary widely in the literature, however, it is believed that the prognosis for patients with PHL is dismal, with early disease recurrence at extrahepatic sites and short survival.5,9
The purpose of this study was to assess the clinical profile, outcome and prognostic factors in PHL in an attempt to gather additional information on this rare entity through the Rare Cancer Network (RCN).
Materials and Methods
A retrospective analysis of patients with PHL diagnosed between 1977 and 2014 was performed in the framework of RCN. Seven institutions within 5 countries of the RCN group contributed cases of PHL, and 41 patients met the criteria for PHL suggested by Lei.5 Patients with splenic and bone marrow involvement were excluded. This study was approved by Mayo Foundation Institutional Review Board and at each participating center independently. The medical records of all patients were reviewed to identify patients and tumor characteristics, treatment details, and follow-up information. All original pathology reports were reviewed using the World Health Organization (WHO) classification of lymphoma. The cut-off date for the survival study was December 2014. The histologic slides were not centrally reviewed.
Survival curves were computed using the Kaplan-Meier method. The events were death irrespective of cause for overall survival (OS) and any relapse for disease-free survival (DFS), and time was measured from the date of diagnosis to the event. The lymphoma-specific survival (LSS) was calculated to the date of death related to lymphoma from the date of diagnosis. Log-rank and Wilcoxon tests were used to determine prognostic factors and assess differences between groups. Cox multiple regression analysis was done for prognostic factors having a P-value ≤0.25 in univariate analyses. A P-value<0.05 was considered to be significant. The statistical analyses were performed using JMP v11 (SAS Corp., Cary, NC, USA).
Results
Patients’ clinicopathological and laboratory characteristics are summarized in Table 1. Median age was 62 years (range, 23-86 years) with a male-to-female ratio of 1.9:1.0 (27/14). Median follow-up time was 116 months (range, 1-225 months). Abdominal pain or discomfort was the most common presenting symptom, and occurred in 22 patients (53.7%). The other symptoms were fatigue (22.0%), jaundice (14.6%), anorexia (14.6%), nausea and/or vomiting (17.1%). Regarding the B-symptoms, 19.5% of patients had fever, 17.1% had weight loss, and 9.8% had night sweats. Hepatomegaly was found in 17.1% of patients, and splenomegaly was less common (9.8%). Thirteen patients (31.7%) had a history of preexisting liver disease, 6 patients had chronic hepatitis (14.6%), 4 patients with cirrhosis (9.8%), one patient had polycystic liver disease (2.4%), and 4 patients were diagnosed after liver transplantation (9.8%). The imaging modalities used evolved over time and location, most patients (63.4%) had computed tomography (CT) or ultrasonography (56.1%), and only 6 patients had positron emission tomography-CT (14.6%). The most common radiological presentation was multiple lesions (53.7%) followed by solitary lesion (36.6%), and diffuse infiltration was present in 7.3% of patients. Liver function tests, including lactate dehydrogenase, alkaline phosphatase and bilirubin, were elevated in 56.1% of patients who were tested. Hemoglobin levels in complete blood count were lower than normal in 11 patients (26.8%) who were tested. Two patients were Hepatitis B Ag positive, and 2 patients were HIV positive among patients who were tested. Bone-marrow aspiration results were recorded, and were normocellular if available (26 patients). The most common histopathological diagnosis was diffuse large B-cell lymphoma, which was observed in 27 patients (65.9%) according to the WHO classification system. Four tumors were marginalzone B-cell lymphoma, 3 were follicular lymphoma, 2 were precursor-cell lymphoblastic lymphoma, 2 were mature T-cell lymphoma, 1 was malignant lymphoma small lymphocytic, 1 was small lymphocytic type T-cell lymphoma, and 1 was Burkitt lymphoma. Combination chemotherapy was administered to 30 patients (73.2%). Chemotherapy regimens were different according to the year of diagnosis and the center. Most of the patients received CHOPlike (cyclophosphamide, doxorubicin, vincristine, and prednisone) regimens (Table 2). Four patients had also surgery, and only 4 patients were treated with radiotherapy or chemo-radiotherapy. Total radiotherapy doses ranged from 30.6-40.0 Gy in 1.8-2.0 Gy/fraction.
Table 1.
Patients’ clinicopathological and laboratory characteristics.
| Characteristics | No. (%) |
|---|---|
| Median age (at the time of diagnosis) | 62 (range, 23-86 years) |
| Gender (male-to-female ratio) | 1.9:1.0 |
| Male | 27 (65.9) |
| Female | 14 (34.1) |
| Presenting symptoms | |
| Abdominal pain or discomfort | 22 (53.7) |
| Fatigue | 9 (22.0) |
| Jaundice | 6 (14.6) |
| Anorexia | 6 (14.6) |
| Nausea and/or vomiting | 7 (17.1) |
| B symptoms | |
| Fever | 8 (19.5) |
| Weight loss | 7 (17.1) |
| Night sweats | 4 (9.8) |
| Hepatomegaly | 7 (17.1) |
| Splenomegaly | 4 (9.8) |
| Pre-existing liver disease | |
| Chronic hepatitis | 6 (14.6) |
| Cirrhosis | 4 (9.8) |
| Liver transplantation | 4 (9.8) |
| Polycystic liver disease | 1 (2.4) |
| Radiological presentation | |
| Multiple lesions | 22 (53.7) |
| Solitary lesion | 15 (36.6) |
| Diffuse infiltration | 3 (7.3) |
| Histopathological diagnosis | |
| Diffuse large B-cell lymphoma | 27 (65.9) |
| Marginal zone B-cell lymphoma | 4 (9.8) |
| Follicular lymphoma grade 1 | 3 (7.3) |
| Precursor cell lymphoblastic lymphoma | 2 (4.9) |
| Mature T-cell lymphoma | 2 (4.9) |
| Small lymphocytic type T-cell lymphoma | 1 (2.4) |
| Malignant lymphoma small lymphocytic | 1 (2.4) |
| Burkitt lymphoma | 1 (2.4) |
| Treatment | |
| Combination chemotherapy | 30 (73.2) |
| Radiotherapy/chemo-radiotherapy | 4 (9.8) |
| Surgery | 4 (9.8) |
Table 2.
Chemotherapy regimens.
| Regimen | No. (%) |
|---|---|
| CHOP-like | |
| CHOP | 8 (19.5) |
| R-CHOP | 8 (19.5) |
| CHOEP | 1 (2.4) |
| CHEP | 1 (2.4) |
| COP | 1 (2.4) |
| CHOP-Bleo | 1 (2.4) |
| Others | 10 (24.4) |
CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; R, rituximab; E, etoposide; Bleo, bleomycin.
The median survival was 163 months, and 5- and 10-year OS rates were 77% [95% confidence interval (CI): 64-90%] and 59% (95% CI: 42-76%), respectively (Figure 1). The 5- and 10-year DFS and LSS rates were 69% (95% CI: 55-83%) and 56% (95% CI: 40-72%), and 87% (95% CI: 76-98%) and 70% (95% CI: 53-87%), respectively. In univariate analyses (log rank test), patients with fever had a better outcome (P=0.03). There was a positive trend (P=0.07) for better OS in younger patients (≤62 years). Gender, abdominal pain or discomfort, weight loss, night sweats, preexisting liver disease, multiple or diffuse lesions, elevated liver enzymes, low hemoglobin levels, hepatosplenomegaly, histopathology, and type of treatment did not show any influence on the outcome, most probably due to the small sample size (Table 3). In multivariate analysis (Cox model), the presence of fever [relative risk (RR)=2.16; P=0.005], absence of weight loss (RR=1.22; P=0.048), and normal hemoglobin level (RR=1.07; P=0.047) were the independent factors influencing the outcome (Table 4).
Figure 1.
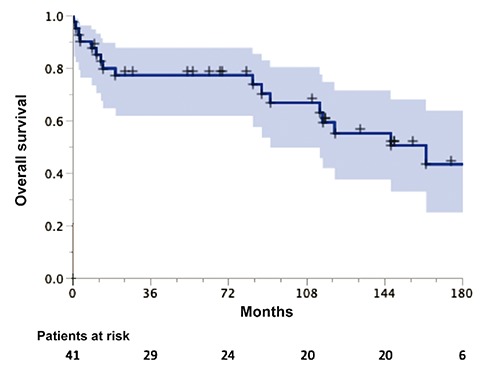
Overall survival for 41 patients with primary hepatic lymphoma.
Table 3.
Univariate analyses in 41 patients with primary hepatic lymphoma.
| Variable | No. | 5-year OS, % | 95% CI | 10-year OS,% | 95% CI | P value* |
|---|---|---|---|---|---|---|
| All patients | 41 | 77 | 64-90 | 59 | 42-76 | |
| Age | ||||||
| >62 | 21 | 70 | 50-90 | 49 | 25-73 | 0.07 |
| <62 | 20 | 84 | 68-100 | 69 | 46-92 | |
| Gender | ||||||
| Male | 27 | 77 | 61-93 | 60 | 39-81 | 0.90 |
| Female | 14 | 79 | 57-100 | 57 | 27-87 | |
| Surgery | ||||||
| Yes | 4 | 100 | - | 100 | - | 0.12 |
| No | 37 | 75 | 61-89 | 56 | 38-74 | |
| Hemoglobin level | ||||||
| Normal | 30 | 86 | 73-99 | 65 | 45-85 | 0.13 |
| Lower than normal | 11 | 55 | 26-85 | 44 | 14-74 | |
| Liver function tests | ||||||
| Normal | 18 | 78 | 59-97 | 69 | 46-92 | 0.86 |
| Elevated | 23 | 77 | 59-95 | 52 | 29-75 | |
| Radiological presentation | ||||||
| Solitary | 15 | 71 | 47-95 | 71 | 47-95 | 0.52 |
| Multiple or diffuse | 26 | 80 | 64-96 | 54 | 32-76 | |
| Pre-existing liver disease | ||||||
| Yes | 13 | 76 | 52-99 | 63 | 33-93 | 0.86 |
| No | 28 | 78 | 62-94 | 58 | 38-79 | |
| Splenomegaly | ||||||
| Yes | 4 | 38 | 0-94 | 38 | 0-94 | 0.37 |
| No | 37 | 81 | 68-94 | 61 | 43-79 | |
| Hepatomegaly | ||||||
| Yes | 7 | 71 | 38-100 | 54 | 15-93 | 0.78 |
| No | 34 | 78 | 64-92 | 60 | 41-79 | |
| Night sweats | ||||||
| Yes | 4 | 100 | - | 75 | 33-100 | 0.24 |
| No | 37 | 75 | 61-89 | 57 | 39-75 | |
| Weight loss | ||||||
| Yes | 7 | 57 | 20-94 | 57 | 20-94 | 0.14 |
| No | 34 | 81 | 68-94 | 59 | 40-78 | |
| Fever | ||||||
| Yes | 8 | 100 | - | 86 | 60-100 | 0.03 |
| No | 33 | 72 | 56-88 | 53 | 33-73 | |
| B-symptoms (night sweats, weight loss, fever) | ||||||
| Yes | 13 | 77 | 54-99 | 68 | 42-94 | 0.67 |
| No | 28 | 78 | 62-94 | 54 | 32-76 | |
| Nausea/vomiting | ||||||
| Yes | 7 | 86 | 60-100 | 57 | 20-94 | 0.55 |
| No | 34 | 75 | 60-90 | 61 | 42-80 | |
| Anorexia | ||||||
| Yes | 6 | 67 | 29-100 | 67 | 29-100 | 0.80 |
| No | 35 | 79 | 65-93 | 57 | 38-76 | |
| Jaundice | ||||||
| Yes | 6 | 83 | 53-100 | 83 | 53-100 | 0.88 |
| No | 35 | 76 | 62-90 | 54 | 35-73 | |
| Fatigue | ||||||
| Yes | 9 | 78 | 51-100 | 47 | 10-84 | 0.19 |
| No | 32 | 77 | 62-92 | 63 | 44-82 | |
| Abdominal pain or discomfort | ||||||
| Yes | 22 | 72 | 53-91 | 49 | 26-72 | 0.25 |
| No | 19 | 83 | 66-100 | 74 | 51-97 | |
| Treatment modality | ||||||
| Chemotherapy alone | 26 | 77 | 61-93 | 61 | 40-82 | 0.59 |
| Other | 15 | 79 | 58-100 | 56 | 26-86 | |
| Histopathology | ||||||
| Diffuse large B-cell | 27 | 81 | 66-96 | 65 | 45-85 | 0.69 |
| Others | 14 | 71 | 47-95 | 45 | 12-78 | |
*Log rank test. OS, overall survival; CI, confidence interval.
Table 4.
Multivariate analysis in 41 patients with primary hepatic lymphoma (Cox model).
| Variable | RR | SE | P-value |
|---|---|---|---|
| Fever (yes better) | 2.16 | 1.04 | 0.005 |
| Weight loss (no better) | 1.22 | 0.57 | 0.048 |
| Hemoglobin level (normal better) | 1.07 | 0.52 | 0.047 |
RR, relative risk; SE, standard error.
Discussion
To our knowledge, this is one of the largest PHL series reported in the literature. PHL was first described by Ata and colleagues in 1965.10 The published articles to date are mostly case reports, and the largest series reported in the English literature are those of El-Sharkawi and colleagues11 with 22 patients and Page and colleagues9 with 24 patients. Also there are review articles collecting and summarizing the data about pathogenesis, clinical and pathological features, and the results of treatment of PHL patients.2,12,13 Therefore, clinico-pathological features of patients remain unclear, and we proposed this study in an attempt to gather additional information on this rare entity through the RCN.
The age of presentation varies but PHL commonly presents in the 5th decade with a male predominance according to the published literature. In our study, median age was 62 years, which was slightly older than the literature with a male predominance. Clinical presentation is generally nonspecific. The most common presenting symptom of PHL patients is abdominal pain and the other symptoms include fatigue, weight loss, jaundice, anorexia, nausea, malaise, night sweats, and vomiting.2 Abdominal pain or discomfort was the most common presenting symptom in our patients, and occurred in more than 50% of patients. The B symptoms of fever, weight loss, and night sweats were less common. Hepatomegaly is common, and on examination can be found in at least 50% of patients;2,5,13 however, only 17.1% of our patients had hepatomegaly. Although enlargement of spleen by lymphomatous involvement indicates a disease process, congestive splenomegaly may occur because of hepatic dysfunction and portal hypertension in PHL patients,2 and splenomegaly was less common in our series (9.8% of patients).
The etiologic factors and the pathogenesis are not clear, and there are several possible factors that have been proposed. A number of reports have suggested that PHL could evolve in patients with chronic hepatitis, cirrhosis, systemic lupus erythematosus, and in transplant patients who were being treated with immunosuppressive drugs.14-25 Thirteen patients had a history of preexisting liver disease in our series (31.7%); 6 patients had chronic hepatitis (14.6%), 4 patients cirrhosis (9.8%), and 1 patient polycystic liver disease (2.4%); 4 patients were diagnosed after liver transplantation (9.8%). Lymphoproliferative disorders after transplantation have been developed in transplant recipients.26 The possible factor is continuous B-cell proliferation, which is normally inhibited by T-lymphocytes.5,13 Acquired immune deficiency syndrome has also been suggested to play a role in the pathogenesis of PHL.27 Two patients were Hepatitis B Ag positive and 2 patients were HIV positive among patients who were tested in our study.
PHL may radiologically present in 3 possible ways in patients: a solitary lesion, multiple lesions, or diffuse infiltration of the liver. The most common presentation in the literature is a solitary lesion, which occurs in 55-60% of patients.2 In contrast to the previous reports, the majority of our patients had multiple lesions (53.7%). The complete blood count was usually within normal range when lymphoma was confined to the liver; and liver function tests, including lactate dehydrogenase, alkaline phosphatase and bilirubin, were usually elevated.13 Liver function tests were elevated in 56.1% of our patients who were tested. Hemoglobin levels were lower than normal limits in eleven patients.
PHL is a pathologic diagnosis. Most of the PHL cases (46-68%) reported in the literature are diffuse large B-cell lymphoma, and other histologies have been described in less than 5% of cases including diffuse mixed large and small cell, lymphoblastic, diffuse immunoblastic, diffuse histiocytic, mantle cell, and small non-cleaved or Burkitt lymphoma.2,5,9
Immunohistochemistry, gene rearrangement, karyotyping, and flow cytometry help in further characterization of the tumor.2 The most common histopathological diagnosis was diffuse large B-cell lymphoma, which was observed in 27 patients (65.9%) in our series. T-cell PHL is less common and the described subtypes include peripheral T-cell lymphoma, anaplastic T-cell lymphoma, and hepatosplenic T-cell lymphoma.28 Three of our patients had T-cell lymphoma including two mature T-cell and small lymphocytic type T-cell lymphomas. There were also other subtypes including marginal zone B-cell lymphoma in 4 patients, follicular lymphoma grade 1 in 3 patients, precursor cell lymphoblastic lymphoma in 2 patients, malignant lymphoma small lymphocytic type in 1 patient, and Burkitt lymphoma in 1 patient. The tumor may have a nodular or diffuse growth pattern microscopically in which lymphoma cells expand into the liver parenchyma.29
The optimal therapy is still unclear and treatment options include surgery, chemotherapy, radiation therapy, or combination of these therapies.2 Surgical resection might be performed for patients with localized disease with good results.30 Past indications for surgical treatment have included localized disease, which can be completely resected, or for debulking prior to chemotherapy.13 However, there may be a bias towards better results in patients treated surgically since they had a better performance status, preserved liver function, low volume, and no comorbidities.2 Chemotherapy is the recommended treatment option for extranodal diffuse large B-cell lymphoma in general, making it a choice also for the treatment of PHL when it is diagnosed preoperatively.31 Chemotherapy can be used either as an adjunct to surgery or as a single modality. In a review by Avlonitis and colleagues, a median survival of 20.7 months, ranging from 10-123.6 months has been described in 14 patients treated surgically followed by adjuvant chemotherapy.13 Adjuvant radiation therapy, or a combination of adjuvant chemotherapy and radiation, have been utilized in some studies.32 Early and aggressive anthracycline-based combination chemotherapy may result in prolonged remissions in properly selected PHL patients.2 The addition of rituximab, a chimeric monoclonal antibody against the CD20 B-cell antigen, to the CHOP regimen increases the complete-response rate, and prolongs event-free and overall survival in elderly patients with diffuse large-B-cell lymphoma, without a clinically significant increase in toxicity.33 Combination chemotherapy was administered to 30 patients (73.2%) in our study, although chemotherapy regimens differed according to the date of diagnosis and the center. The majority of patients received CHOPlike regimens.
Most of the PHL patients had poor prognostic features that include advanced age, constitutional symptoms, elevated liver enzymes, and comorbid diseases.2 The median survival varies considerably in the literature, and reported survival ranges from 3-123.6 months.13 In our study, the median survival was 163 months, which was higher than previously reported rates. In a study by Page and colleagues, reviewing the experience of MD Anderson Cancer Center, 24 PHL patients were treated with chemotherapy and the overall 5-year survival was 83%.9 The 5-year survival rate in our study was 77%.
The absence of fever and presence of weight loss and low hemoglobin levels were found to be the worse independent prognostic factors influencing the outcome. In univariate analyses, it is interesting to note that among the B-symptoms, fever (P=0.03) and night sweats (P=0.24) seemed to have a better influence but weight loss (P=0.14) a worse influence on the outcome. In general, fever is a result of immune response by the body to a foreign invader. The fever response is executed by integrated physiological and neuronal circuitry, and confers a survival benefit during infection.34 A major goal of cancer immunology is to stimulate the generation of long-lasting, tumor antigen-specific immune responses that recognize and destroy tumor cells, and accumulating evidence indicates that survival benefits are accorded to individuals who achieve an increase in body temperature (i.e., fever) following infection.35
Conclusions
In this retrospective multicenter RCN study, patients with PHL had a relatively better prognosis than those reported elsewhere. It is important to recognize this rare disease entity, as it responds to therapy, and has a better prognosis than hepatocellular carcinoma or systemic lymphomas secondarily involving the liver. Multicenter prospective studies are still warranted to establish treatment guidelines, outcome, and prognostic factors.
References
- 1.Loddenkemper C, Longerich T. Lymphoma of the liver. Cavalli F, Stein H, Zucca E, eds. Extranodal Lymphomas, Pathology and Management. Abingdon, UK: CRC Press; 2008. pp 277-88. [Google Scholar]
- 2.Noronha V, Shafi NQ, Obando JA, Kummar S. Primary non-Hodgkin’s lymphoma of the liver. Crit Rev Oncol Hematol 2005;53:199-207. [DOI] [PubMed] [Google Scholar]
- 3.Emile JF, Azoulay D, Gornet JM, et al. Primary non-Hodgkin’s lymphomas of the liver with nodular and diffuse infiltration patterns have different prognoses. Ann Oncol 2001;12:1005-10. [DOI] [PubMed] [Google Scholar]
- 4.Lei KI, Chow JH, Johnson PJ. Aggressive primary hepatic lymphoma in Chinese patients. Presentation, pathologic features, and outcome. Cancer 1995;76:1336-43. [DOI] [PubMed] [Google Scholar]
- 5.Lei KI. Primary non-Hodgkins lymphoma of the liver. Leuk Lymphoma 1998;29:293-9. [DOI] [PubMed] [Google Scholar]
- 6.Chim CS, Choy C, Ooi GC, Liang R. Primary hepatic lymphoma. Leuk Lymphoma 2001;40:667-70. [DOI] [PubMed] [Google Scholar]
- 7.Bronowicki JP, Bineau C, Feugier P, et al. Primary lymphoma of the liver: clinical-pathological features and relationship with HCV infection in French patients. Hepatology 2003;37:781-7. [DOI] [PubMed] [Google Scholar]
- 8.DeMent SH, Mann RB, Staal SP, et al. Primary lymphomas of the liver: report of six cases and review of the literature. Am J Clin Pathol 1987;88:255-63. [DOI] [PubMed] [Google Scholar]
- 9.Page RD, Romaguera JE, Osborne B, et al. Primary hepatic lymphoma: favorable outcome after combination chemotherapy. Cancer 2001;92:2023-9. [DOI] [PubMed] [Google Scholar]
- 10.Ata AA, Kamel IA. Primary reticulum cell sarcoma of the liver. A case report. J Egypt Med Assoc 1965;48:514-21. [PubMed] [Google Scholar]
- 11.El-Sharkawi D, Ramsay A, Cwynarski K, et al. Clinico-pathologic characteristics of patients with hepatic lymphoma diagnosed using image-guided liver biopsy techniques. Leuk Lymphoma 2011;52:2130-4. [DOI] [PubMed] [Google Scholar]
- 12.Aozasa K, Mishima K, Ohsawa M. Primary malignant lymphoma of the liver. Leuk Lymphoma 1993;10:353-7. [DOI] [PubMed] [Google Scholar]
- 13.Avlonitis VS, Linos D. Primary hepatic lymphoma: a review. Eur J Surg 1999;165:725-9. [DOI] [PubMed] [Google Scholar]
- 14.Lisker-Melman M, Pittaluga S, Pluda JM, et al. Primary lymphoma of the liver in a patient with acquired immune deficiency syndrome and chronic hepatitis B. Am J Gastroenterol 1989;84:1445-8. [PubMed] [Google Scholar]
- 15.Mohan C, Alurkar SS, Sharma OP, Advani SH. Primary liver lymphoma: a diagnostic dilemma. AJR Am J Roentgenol 1991;157:413. [DOI] [PubMed] [Google Scholar]
- 16.Mohamed M, Fernando R. Diagnostic and therapeutic quandaries in a patient with primary hepatic lymphoma and concurrent hepatitis C infection. Indian J Hematol Blood Transfus 2014;30:394-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekiguchi Y, Yoshikawa H, Shimada A, et al. Primary hepatic circumscribed Burkitt’s lymphoma that developed after acute hepatitis B: report of a case with a review of the literature. J Clin Exp Hematop 2013;53:167-73. [DOI] [PubMed] [Google Scholar]
- 18.Somaglino C, Pramaggiore P, Polastri R. Primary hepatic lymphoma in a patient with chronic hepatitis B and C infection: diagnostic pitfalls and therapeutic challenge. Updates Surg 2014;66:89-90. [DOI] [PubMed] [Google Scholar]
- 19.Kikuma K, Watanabe J, Oshiro Y, et al. Etiological factors in primary hepatic B-cell lymphoma. Virchows Arch 2012;460:379-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama S, Yokote T, Kobayashi K, et al. Primary hepatic MALT lymphoma associated with primary biliary cirrhosis. Leuk Res 2010;34:e17-20. [DOI] [PubMed] [Google Scholar]
- 21.Sato S, Masuda T, Oikawa H, et al. Primary hepatic lymphoma associated with primary biliary cirrhosis. Am J Gastroenterol 1999;94:1669-73. [DOI] [PubMed] [Google Scholar]
- 22.Chowdhary VR, Crowson CS, Poterucha JJ, Moder KG. Liver involvement in systemic lupus erythematosus: case review of 40 patients. J Rheumatol 2008;35:2159-64. [DOI] [PubMed] [Google Scholar]
- 23.Sutton E, Malatjalian D, Hayne OA, Hanly JG. Liver lymphoma in systemic lupus erythematosus. J Rheumatol 1989;16:1584-8. [PubMed] [Google Scholar]
- 24.Honda H, Franken EA, Jr, Barloon TJ, Smith JL. Hepatic lymphoma in cyclosporinetreated transplant recipients: sonographic and CT findings. AJR Am J Roentgenol 1989;152:501-3. [DOI] [PubMed] [Google Scholar]
- 25.Palazzo JP, Lundquist K, Mitchell D, et al. Rapid development of lymphoma following liver transplantation in a recipient with hepatitis B and primary hemochromatosis. Am J Gastroenterol 1993;88:102-4. [PubMed] [Google Scholar]
- 26.Rostaing L, Suc B, Fourtanier G, et al. Liver B cell lymphoma after liver transplantation. Transplant Proc 1995;27:1781-2. [PubMed] [Google Scholar]
- 27.Caccamo D, Pervez NK, Marchevsky A. Primary lymphoma of the liver in the acquired immunodeficiency syndrome. Arch Pathol Lab Med 1986;110:553-5. [PubMed] [Google Scholar]
- 28.Stancu M, Jones D, Vega F, Medeiros LJ. Peripheral T-cell lymphoma arising in the liver. Am J Clin Pathol 2002;118:574-81. [DOI] [PubMed] [Google Scholar]
- 29.Steller EJ, van Leeuwen MS, van Hillegersberg R, et al. Primary lymphoma of the liver - A complex diagnosis. World J Radiol 2012;4:53-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scoazec JY, Degott C, Brousse N, et al. Non-Hodgkin’s lymphoma presenting as a primary tumor of the liver: presentation, diagnosis and outcome in eight patients. Hepatology 1991;13:870-5. [PubMed] [Google Scholar]
- 31.Sehn LH, Donaldson J, Chhanabhai M, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol 2005;23:5027-33. [DOI] [PubMed] [Google Scholar]
- 32.Chowla A, Malhi-Chowla N, Chidambaram A, Surick B. Primary hepatic lymphoma in hepatitis C: case report and review of the literature. Am Surg 1999;65:881-3. [PubMed] [Google Scholar]
- 33.Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235-42. [DOI] [PubMed] [Google Scholar]
- 34.Evans SS, Repasky EA, Fisher DT. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol 2015;15:335-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Repasky EA, Evans SS, Dewhirst MW. Temperature matters! And why it should matter to tumor immunologists. Cancer Immunol Res 2013;1:210-6. [DOI] [PMC free article] [PubMed] [Google Scholar]