

Abstract
Fibroproliferative disorders such as idiopathic pulmonary fibrosis and systemic sclerosis have no effective therapies and result in significant morbidity and mortality due to progressive organ fibrosis. We examined the effect of peptides derived from endostatin on existing fibrosis and fibrosis triggered by two potent mediators, transforming growth factor–β (TGF-β) and bleomycin, in human and mouse tissues in vitro, ex vivo, and in vivo. We identified one peptide, E4, with potent antifibrotic activity. E4 prevented TGF-β–induced dermal fibrosis in vivo in a mouse model, ex vivo in human skin, and in bleomycin-induced dermal and pulmonary fibrosis in vivo, demonstrating that E4 exerts potent antifibrotic effects. In addition, E4 significantly reduced existing fibrosis in these preclinical models. E4 amelioration of fibrosis was accompanied by reduced cell apoptosis and lower levels of lysyl oxidase, an enzyme that cross-links collagen, and Egr-1 (early growth response gene–1), a transcription factor that mediates the effects of several fibrotic triggers. Our findings identify E4 as a peptide with potent antifibrotic activity and a possible therapeutic agent for organ fibrosis.
INTRODUCTION
Excessive deposition of extracellular matrix (ECM) components such as fibronectin (FN) and collagen (Col) by fibroblasts is defined as fibrosis. Organ fibrosis is a final common pathway for many diseases and leads to end-stage organ failure as a result of the loss of normal architecture and loss of function, with consequent high morbidity and mortality. Examples of these diseases are pulmonary fibrosis, subepithelial fibrosis in asthma, systemic sclerosis (SSc), morphea, liver cirrhosis, glomerulosclerosis, and endomyocardial fibrosis. Unfortunately, effective therapy for organ fibrosis is still unavailable (1–3). Uncontrollable wound-healing responses, including acute and chronic inflammation, angiogenesis, activation of resident cells, and ECM remodeling, are thought to be involved in the pathogenesis of fibrosis (3, 4). Transforming growth factor–β (TGF-β) is the prototypical fibrosis-causing cytokine, increasing in fibrotic organs and contributing to the development of fibrosis by stimulating the synthesis of ECM molecules, activating the differentiation of fibroblasts to α smooth muscle actin (α-SMA)–expressing myofibroblasts, and down-regulating matrix metalloproteinases (MMPs) (5, 6). However, a clinical trial of a monoclonal antibody to TGF-β in patients with early SSc failed to show any efficacy (6).
Endostatin is a 20-kD internal fragment of the C terminus of collagen XVIII with potent antiangiogenic activity that was originally identified in the supernatant of a cultured murine hemangioendothelioma cell line (7). Endostatin inhibits endothelial cell proliferation and tube formation in vitro and tumor growth in vivo (7, 8). Recombinant endostatin (rE) has been proposed as a broad-spectrum anticancer therapy because of its endogenous inhibitory effect on angiogenesis, its lack of toxicity, and the fact that it does not seem to promote acquired drug resistance (9, 10). The N-terminal domain of endostatin has been reported to be the functional domain responsible for inhibiting angiogenesis (11). Although the exact molecular mechanism of endostatin’s effects remains unclear, integrins, glypicans, flk-1, and nucleolin have all been named as endostatin receptors (12–15).
Recent studies have shown that endostatin is 2 to 20 times higher in serum and/or bronchoalveolar lavage fluid (BALF) from patients with idiopathic pulmonary fibrosis (IPF) and SSc-associated pulmonary fibrosis than from normal subjects (16–18). Because cytokines and signaling molecules implicated in angiogenesis development have also been identified in purpose, we evaluated the effect of endostatin and endostatin-derived peptides on the generation of fibrosis in vitro using primary human fibroblasts, ex vivo using human skin, and in vivo in TGF-β– and bleomycin-induced dermal and pulmonary fibrosis.
RESULTS
Human rE inhibits FN and Col1 production induced by TGF-β
To evaluate whether endostatin modulates production of ECM components in fibroblasts, we first examined FN and Col1α1 (type I collagen αI chain) protein levels in normal human lung fibroblasts. rE markedly reduced FN and Col1α1 protein levels in fibroblasts pretreated with TGF-β (fig. S1A). Because the skin is affected by fibrosis in SSc, we evaluated the efficacy of endostatin as a potential therapeutic agent for fibrosis using our ex vivo human skin model (19). In untreated human skin, rE did not significantly alter dermal thickness (fig. S1B). Because TGF-β is a well-known profibrotic factor that triggers fibrosis (20), we injected human recombinant TGF-β intradermally ex vivo and assessed the level of resulting fibrosis. TGF-β significantly increased dermal thickness in a dose-dependent manner 1 week after injection (Fig. 1, A and B). We next evaluated whether rE, at the highest concentration that showed no effect on untreated skin, could inhibit fibrosis in TGF-β–treated human skin. rE, administered together with the TGF-β, significantly reduced dermal thickness in a dose-dependent manner when measured after 1 week (Fig. 1, C and D).
Fig. 1.
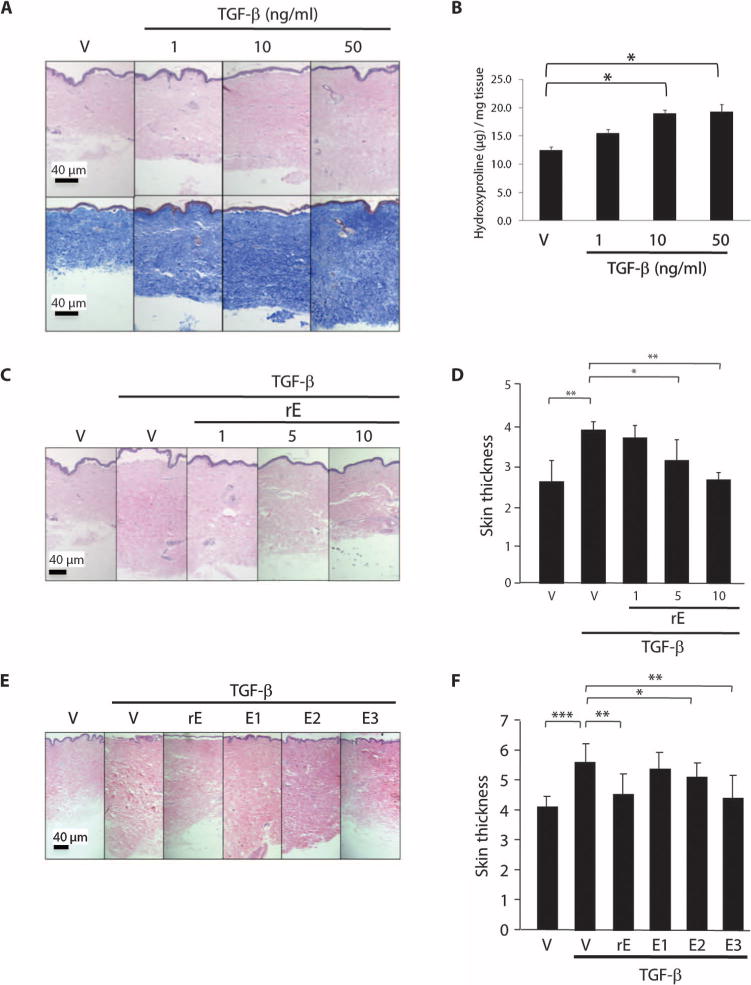
Human rE and its peptides ameliorate TGF-β–induced fibrosis ex vivo. (A) Representative images of hematoxylin and eosin (H&E) (upper)– and Masson trichrome (lower)–stained sections of TGF-β–treated human skin. Recombinant TGF-β or PBS (vehicle) was injected intradermally into human skin ex-plants at a concentration of 1, 10, and 50 ng/ml. Skin was harvested 1 week after injection. V, vehicle control. Scale bar, 40 μm. (B) Graphical summary of hydroxyproline content of human skin tissues shown in (A). *P < 0.05, ANOVA. (C) Representative H&E images of rE-treated human skin. rE was injected with TGF-β ex vivo into human skin in organ culture at a concentration of 1, 5, and 10 μg/ml. Scale bar, 40 μm. (D) Graphical presentation of dermal thickness data shown in (C), representing four independent experiments in triplicate using skin explants from four different donors. Mann-Whitney U test was used for statistical analysis. *P < 0.001; **P < 0.0001. (E) Representative H&E images of human skin injected with vehicle, TGF-β alone (10 ng/ml), or rE, E1, E2, or E3 (10 μg/ml) in combination with TGF-β (10 ng/ml). Scale bar, 40 μm. (F) Graphical presentation of dermal thickness shown in (E). Data represent two independent experiments with skin from two donors, and each experiment was done in triplicate. *P < 0.05; **P < 0.001; ***P < 0.0001, Mann-Whitney U test.
Endostatin peptides reduce TGF-β–induced fibrosis in human skin explants
To define the functional domain of endostatin that mediates its inhibitory effect, we tested three different peptides corresponding to different regions of human endostatin. Similar to rE, a single injection of endostatin peptides did not cause a significant reduction in baseline dermal thickness (fig. S1C). Nevertheless, a fragment from the C terminus of human endostatin (E3) significantly abolished TGF-β–induced fibrosis when compared to TGF-β treatment alone (P = 0.001; Fig. 1, E and F).
To stabilize the E3 peptide and enhance its antifibrotic activity, we generated a homologous peptide identical to E3 except for the addition of an amide modification at the C terminus (E4). This modification protects peptides from proteolytic degradation and thus stabilized E3 to promote retention of its biological activity (21). We first measured FN and Col1α1 in primary human fibroblasts treated with endostatin peptides after prestimulation with TGF-β. E4 decreased FN and Col1α1 induction in TGF-β–pretreated fibroblasts significantly more than did E3 in four independent experiments (fig. S2, A to C). The decrease in ECM production was not due to a decrease in proliferation (fig. S2, D and E), an increase in fibroblast apoptosis (fig. S2, F to H), or modulation of Smad 2/3 (fig. S2I) or mitogen-activated protein kinase (MAPK) signaling cascade (fig. S2J) activation. Furthermore, compared to E3, E4 had enhanced capacity to ameliorate TGF-β–induced fibrosis in human skin ex vivo (Fig. 2, A and B). We next examined the dermal thickness of human skin explants injected with different concentrations of E4 in combination with TGF-β. Concentrations of 5 to 20 μg/ml ameliorated TGF-β–induced skin fibrosis, indicating that the C-terminal domain of endostatin can suppress fibrosis in a dose-dependent manner (Fig. 2, C and D).
Fig. 2.
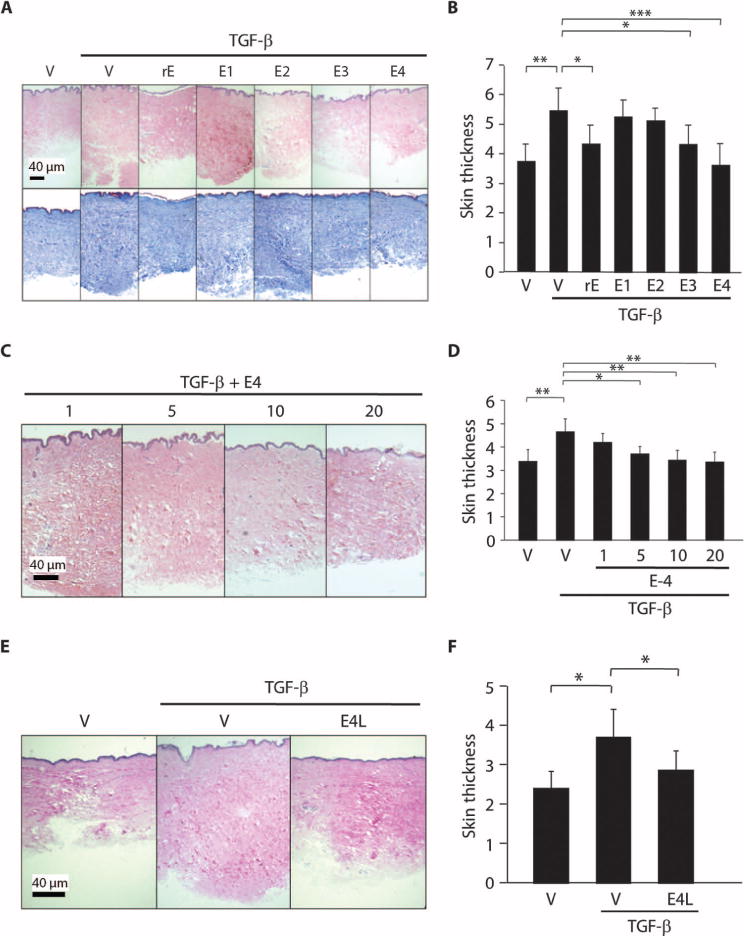
E4 peptide ameliorates ongoing TGF-β–induced fibrosis ex vivo. (A) Representative H&E (upper) and Masson trichrome (lower) images of human skin injected with vehicle, TGF-β alone (10 ng/ml), or rE, E1, E2, E3, or E4 (10 μg/ml) in combination with TGF-β (10 ng/ml). Scale bar, 40 μm. (B) Graphical presentation of dermal thickness in (A). Data represent two independent experiments using human skin explants from two donors, and each experiment was done in triplicate. *P < 0.01; **P < 0.001; ***P < 0.0001, Mann-Whitney U test. (C) Representative H&E images of human skin injected with E4 at a concentration of 1, 5, 10, and 20 μg/ml in the presence of TGF-β (10 ng/ml). Scale bar, 40 μm. (D) Graphical analysis of dermal thickness data in (C). Experiments were conducted in duplicate, and dermal thickness was measured in six fields from each section. *P < 0.02; **P < 0.01, Mann-Whitney U test. (E) Effect of endostatin peptides on existing fibrosis triggered by TGF-β in human skin. Representative H&E images of human skin injected with vehicle (DMSO) or E4 (10 μg/ml) 2 days after administration of TGF-β (10 ng/ml) (E4L). Scale bar, 40 μm. (F) Graphical presentation of dermal thickness data in (E). Data represent two independent experiments using human skin explants from two donors, and each experiment was done in duplicate. *P < 0.01, Mann-Whitney U test.
Endostatin peptides reverse existing fibrosis in human skin
To assess whether endostatin peptides could reverse fibrosis that was already present, we injected E4 into human skin 2 days after a single TGF-β administration, when dermal thickness was already evident (fig. S3, A and B; P = 0.045, TGF-β versus vehicle). Similar to human skin co-treated with E4 and TGF-β, delayed E4 administration also significantly ameliorated TGF-β–induced dermal fibrosis (Fig. 2, E and F). Our findings indicate that the C-terminal peptide of endostatin can prevent the development and progression of fibrosis and can also reverse TGF-β–induced fibrosis in human skin.
Endostatin peptides reduce TGF-β–induced fibrosis in vivo in mouse skin
The antifibrotic effect of endostatin peptides was further assessed to test whether they were still effective in vivo. One week after administration of rE or endostatin peptides, mice appeared healthy and showed no signs of distress. TGF-β significantly increased dermal thickness in mouse skin (P = 0.00001; Fig. 3, A and B), and peptides E3 and E4 prevented the dermal fibrosis induced by TGF-β (P = 0.00007 and 0.00003, respectively), with E4 demonstrating a larger antifibrotic effect (Fig. 3, A and B). These results confirm those obtained in our human skin model and emphasize the importance of the C-terminal domain of endostatin in preventing TGF-β–induced fibrosis in vivo.
Fig. 3.
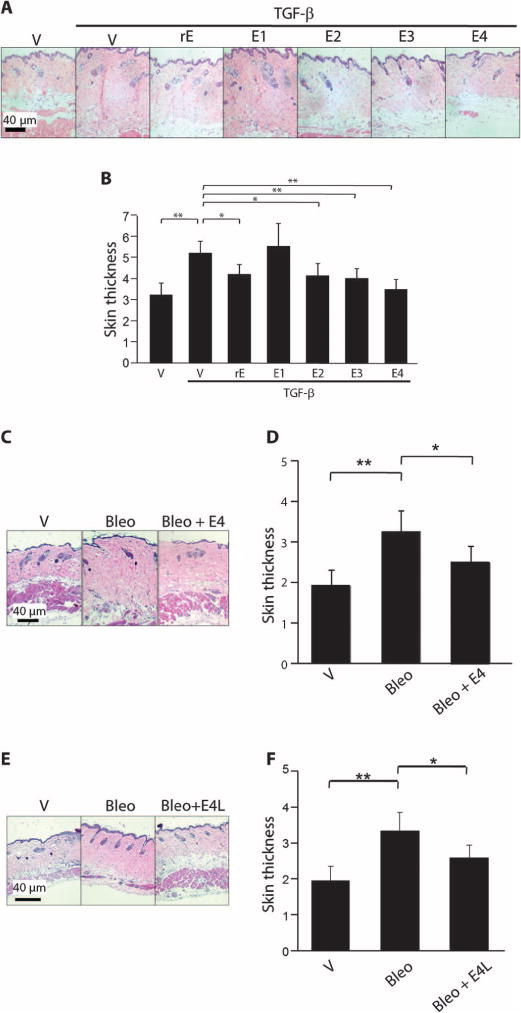
E4 ameliorates fibrosis induced by two different triggers in vivo in mouse skin and reverses ongoing fibrosis. (A) Representative H&E images of mouse skin treated with TGF-β and endostatin peptides. Mice were injected subcutaneously with vehicle, TGF-β alone (10 ng/ml), or rE, E1, E2, E3, and E4 (10 μg/ml) in combination with TGF-β (10 ng/ml). Skin was harvested 1 week after injection. Sections were stained with H&E. Scale bar, 40 μm. (B) Graphical summary of dermal thickness in (A). Data represent four independent experiments, each done in duplicate. *P < 0.001; **P < 0.0001, Mann-Whitney U test. (C) Representative H&E images of mouse skin treated with bleomycin and E4. Mice were injected subcutaneously with 1× PBS as vehicle (V) or bleomycin (Bleo) (20 mg per mice) daily. E4 (10 μg/ml) was mixed with bleomycin on the first day, and daily bleomycin administration was continued without subsequent injections of E4 (Bleo + E4). Skin was harvested after 10 days. Sections were stained with H&E. Scale bar, 40 μm. (D) Graphical summary of dermal thickness data in (C). n = 3. *P < 0.001; **P < 0.0001, Mann-Whitney U test. (E) Representative H&E images of mouse skin injected with E4 3 days after bleomycin (E4L). Mice were injected intradermally with bleomycin and the peptide was administered 3 days later with bleomycin. Skin was harvested on day 10. Sections were stained with H&E. Magnification, ×100. Scale bar, 40 μm. (F) Graphical summary of dermal thickness in (E). *P < 0.001, **P < 0.0001, Mann-Whitney U test.
E4 ameliorates skin fibrosis induced by bleomycin in vivo
To confirm the antifibrotic effect of E4 peptide on fibrosis in a separate model with a different fibrotic trigger, we used bleomycin, a chemotherapeutic agent with known profibrotic effects. A single injection of E4 (Bleo + E4) significantly reduced dermal thickness compared to bleomycin alone (P = 0.0006) (Fig. 3, C and D). In a separate experiment, E4 was administered daily 3 days after initiation of the bleomycin injections. This delayed administration of E4 significantly decreased bleomycin-induced dermal thickness (P = 0.0007) (Fig. 3, E and F).
E4 ameliorates pulmonary fibrosis induced by bleomycin in vivo
To determine whether the antifibrotic effects of E4 were organ-specific, we administered bleomycin intratracheally to induce pulmonary fibrosis. E4 was administered either concomitantly with bleomycin or 3 days afterwards. We assessed lung histology and measured collagen content with the Sircol assay 10 and 21 days after treatment with bleomycin. Compared to mice treated with bleomycin only, mice treated simultaneously with E4 and bleomycin or first with bleomycin followed 3 days later by E4 had reduced pulmonary fibrosis, as assessed histologically on hematoxylin and eosin (H&E) sections (Fig. 4A and fig. S4A), or by Masson trichrome stain (Fig. 4B and fig. S4B). In addition, soluble collagen content of lung tissues was significantly decreased in E4-treated mice (Fig. 4C). Similar results were obtained whether E4 was administered as a single injection 5 or 8 days after bleomycin (fig. S5, A to C). Thus, the antifibrotic effects of E4, whether for preventing or reversing fibrosis, were confirmed not only in TGF-β–induced fibrosis but also in bleomycin-induced dermal and pulmonary fibrosis. On the basis of histology and collagen content, administration of E4 3 days after bleomycin had a more pronounced therapeutic effect than did concomitant administration of the peptide at both time points, suggesting that E4 may be a viable therapeutic option for fibrosis because it can reverse ongoing fibrosis in both skin and lung.
Fig. 4.
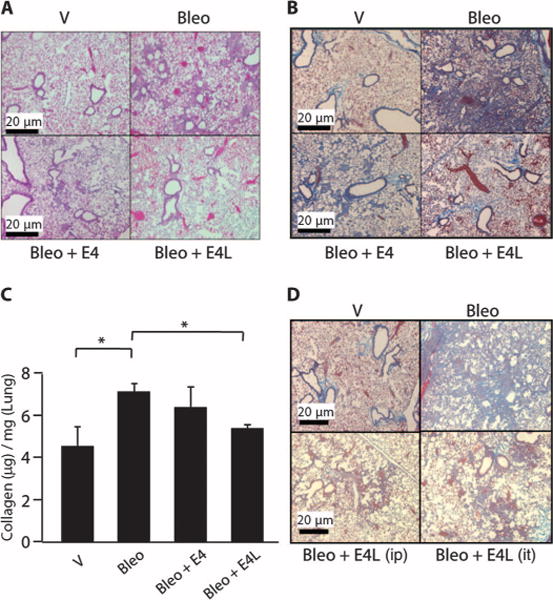
E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo whether administered concomitantly or after a fibrotic trigger. (A and B) Representative H&E images of mouse lung tissues treated with E4 after bleomycin. Bleomycin (60 μg) was administered intratracheally in combination with DMSO as a vehicle (Bleo) or E4 (Bleo + E4; 10 μg/ml). E4 (10 μg/ml) was also administered intratracheally 3 days after bleomycin (Bleo + E4L). Lungs were harvested 10 days after treatment. Images of H&E (A)– and Masson trichrome (B)–stained sections. n = 3. Scale bars, 20 μm. (C) Quantification of acid-soluble collagen obtained from mouse lungs treated as in (B). Collagen values are presented as micrograms per milligram of lung from three independent experiments. *P < 0.05, unpaired t test. (D) Representative Masson trichrome images of mouse lung. Mice were treated as in (A) and (B), except that E4 was administered 3 days after bleomycin (E4L) either intraperitoneally (ip) or intratracheally (it), and mice were sacrificed 21 days later. Scale bars, 20 μm.
To determine whether the mode of E4 administration affects the antifibrotic effects of E4, we gave the mice bleomycin intratracheally followed by administration of E4 intraperitoneally or intratracheally. Mice were euthanized after 21 days. E4 exerted similar antifibrotic effects irrespective of the mode of administration (Fig. 4D and fig. S6).
E4 reduces bleomycin-induced apoptosis in vivo
Endostatin inhibits wound repair and reduces cell viability and proliferation in vitro in distal lung epithelial cells and primary type II cells (22). We thus examined the effects of E4 on apoptosis in vivo. E4 resulted in a significant decrease in bleomycin-induced cell apoptosis in mouse lungs (fig. S7, A and B), suggesting that it may partly ameliorate fibrosis by preventing cell death.
E4 reduces bleomycin-induced lysyl oxidase expression in vitro and in vivo
Lysyl oxidase (LOX) is a copper enzyme that cross-links collagen, thus contributing to fibrosis by rendering collagen and ECM components more resistant to proteolytic degradation. LOX is induced in vivo by bleomycin (23) and other fibrotic factors. We examined levels of secreted LOX in normal primary adult human fibroblasts stimulated with TGF-β and then treated with E4. E4 resulted in a reduction of LOX levels (Fig. 5, A and B). Similar findings were obtained in vivo in lung tissues of mice treated with bleomycin and E4, where E4 resulted in a reduction of LOX levels whether administered simultaneously with bleomycin or 3 days afterward (Fig. 5C).
Fig. 5.
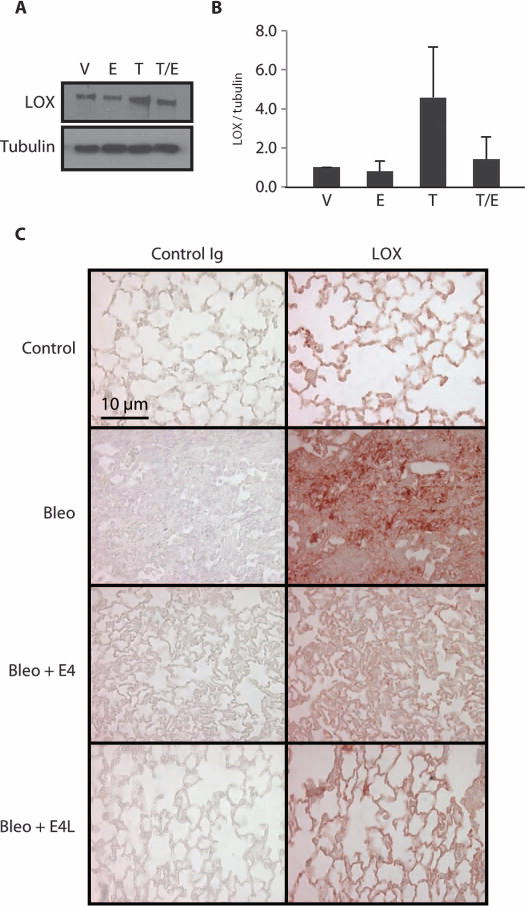
E4 reduces levels of LOX. (A) Immunoblotting of LOX in medium conditioned by primary human lung fibroblasts treated with vehicle (V), E4 (E), TGF-β (T), or TGF-β followed by E4 (T/E). Tubulin was used as an internal standard. (B) Graphical summary of data in (A) depicted as change relative to the internal standard. n = 4. (C) Detection of LOX in mouse lungs. Lung tissues of mice treated as described for Fig. 4 were stained for LOX by immunohistochemistry. Mice were sacrificed 10 and 21 days after treatment. Representative images of lung tissues from mice treated for 10 days are shown. Scale bar, 10 μm. Ig, immunoglobulin.
E4 reduces Egr-1 expression in vitro and in vivo
Early growth response gene–1 (Egr-1) is a central transcription factor that mediates the response of multiple triggers of fibrosis including TGF-β and bleomycin (24). Egr-1 levels induced by TGF-β were reduced by E4 in primary human lung fibroblasts (Fig. 6, A and B). E4 also reduced mRNA and protein levels of Egr-1 in vivo in bleomycin-treated mouse lungs (Fig. 6, C and D). These results demonstrate that E4 acts on a central regulator of fibrosis, which explains its ability to counteract the effects of more than one fibrotic trigger.
Fig. 6.
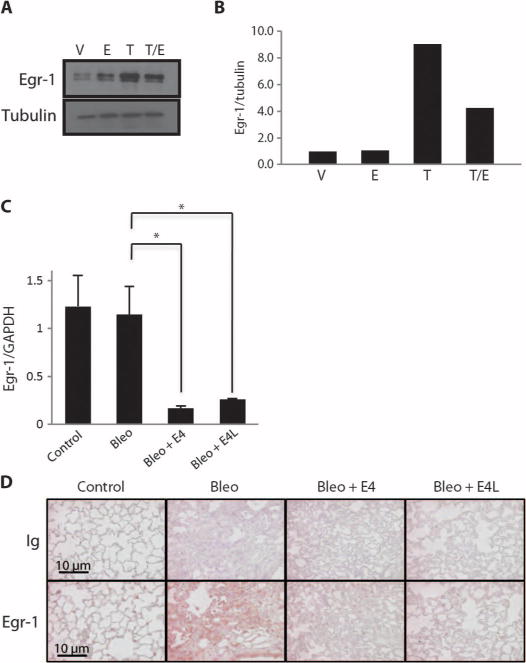
E4 reduces Egr-1 production. (A) Immunoblotting of Egr-1 in primary fibroblast lysates. Primary fibroblasts were treated as in Fig. 5A. Egr-1 was detected in cellular lysates by immunoblotting. Tubulin was used as an internal standard. (B) Graphical summary of data in (A) depicted as change relative to the internal standard. n = 2. (C) Measurement of Egr-1 mRNA. Mice were treated as in Fig. 4. Total RNA was extracted from lung tissues 10 days after treatment and used for the measurement of Egr-1 mRNA by real-time PCR. Data are shown as a ratio of Egr-1 to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels. *P < 0.001, ANOVA. (D) Immunohistochemical detection of Egr-1 in lung tissues from mice treated as in Fig. 5. Scale bars, 10 μm.
The C-terminal peptide of endostatin has modest antiangiogenic activity
The antiangiogenic effect of endostatin is attributed to its N-terminal domain (11). To evaluate the antiangiogenic activity of the C-terminal region of endostatin, we examined the effect of E4 on in vitro tubule formation. The ability of E4 to suppress angiogenesis was minimal (fig. S8, A and B). This conclusion is supported by the presence of histologically normal blood vessels in mouse and human skin, as well as in mouse lung treated with E4 (fig. S8C). These findings suggest that the region of endostatin corresponding to E4 does not significantly contribute to its antiangiogenic activity.
DISCUSSION
We have demonstrated that E3 and its amidated form E4, peptides corresponding to the C-terminal region of endostatin, prevent and reverse TGF-β– and bleomycin-induced dermal and pulmonary fibrosis. E4 suppressed TGF-β–induced ECM production in primary lung fibroblasts in vitro. Ex vivo and in vivo analyses revealed that E4 impedes the increase in dermal thickness triggered by TGF-β and bleomycin. Together, our findings suggest that the C-terminal domain of endostatin is responsible for its antifibrotic activity.
Elevated levels of serum and BALF endostatin are seen in fibrotic disorders such as IPF and SSc (16–18). Endostatin was increased in IPF patients with severe respiratory dysfunction and in SSc patients with pulmonary fibrosis and severe skin fibrosis compared to patients without those clinical manifestations (16–18). In addition, the amount of collagen XVIII was increased in cultured dermal fibroblasts of SSc patients (25) and in whole-lung extracts of patients with IPF (26). In this regard, because endostatin is a proteolytic product of collagen XVIII cleaved by several proteases including MMPs and cathepsin L (27, 28) and because MMPs are also up-regulated in SSc and IPF (17, 29), it is plausible that cleaved endostatin concentrations are elevated in those patients. On the basis of our findings, we propose that increased endostatin in fibrotic tissues may constitute a negative feedback regulatory loop, which, although unsuccessful, is directed at halting the progression of fibrosis. MMPs, also increased in SSc and IPF (17, 29), do not reverse fibrosis despite their matrix-degrading activity. Alternatively, the presence of a yet unidentified endostatin inhibitory molecule that blocks its antifibrotic effects might explain the inefficacy of endogenous endostatin to block fibrosis.
Our observations are partially supported by previous studies. Bloch et al. reported reduced connective tissue but normal vessel density in rE-treated mouse skin in a wound-healing model (30). Furthermore, a peptide from the N-terminal region of endostatin prevented the progression of peritoneal sclerosis in a mouse model (31). In contrast, we found that the C-terminal region, but not the N terminus, of endostatin was responsible for its antifibrotic effects. The peptide corresponding to the N-terminal domain of endostatin contributed to the fibrotic phenotype.
The antiangiogenic activity of endostatin has been the focus of numerous investigations directed at the development of antitumor therapy (7,10). Studies defining the specific amino acid sequence responsible for endostatin’s antiangiogenic capacity (17, 32, 33) have shown that the entire angio-suppressive activity of endostatin is located in a 27–amino acid peptide in the N-terminal domain (17). Our findings suggest that the functional domain of endostatin that mediates its antifibrotic activity is distinct from that responsible for its antiangiogenic capacity. Indeed, E4 had little or no antiangiogenic activity in our preclinical models compared to endostatin. The long-term effects of E4 on angiogenesis have not yet been explored, however.
The N-terminal peptide of endostatin has been reported to ameliorate peritoneal sclerosis by reducing expression of TGF-β and α-SMA (31). Neutralization of endostatin exaggerated tissue remodeling and interstitial fibrosis with increased tissue collagen and MMP-2 and MMP-9 activities in a rat myocardial infarction model (34). These reports suggest that endostatin might interact with some profibrotic factors in fibroproliferative circumstances. In our experiments, E3 and E4 reduced dermal thickness of mouse and human skin treated with TGF-β and also inhibited bleomycin-induced skin and lung fibrosis. This suggests that the antifibrotic effect of the endostatin C-terminal peptide may be due, in part, to its blockade of TGF-β activity.
The use of endostatin as a therapeutic strategy is attractive because of its lack of toxicity and the fact that it is an endogenous protein. Numerous studies have focused on endostatin for cancer therapy with regression of tumor growth with no drug resistance (35). In 2005, Endostar, a human recombinant endostatin purified from Escherichia coli, was approved for the treatment of non–small cell lung cancer in China (36). Despite its effectiveness, the treatment had several disadvantages, including a requirement for high doses and the protein’s short half-life, poor stability, and easy inactivation (10, 35, 37). A small synthetic peptide derived from endostatin may overcome these obstacles and render this therapeutic approach more appealing. In addition, a functional small peptide may have more pronounced beneficial effects because the intact recombinant protein contains both angio-suppressive and angio-stimulatory domains that could counteract each other’s effects (38). Indeed, our results show that E4 significantly inhibited fibrosis compared to rE and even E3 in vitro, in vivo, and ex vivo. In addition, E4 had minimal antiangiogenic activity compared to rE, confirming that the antiangiogenic activity of endostatin resides in its N-terminal domain. The only difference between E3 and E4 was the presence of an amide bond in the C terminus of E4. This amide renders the peptide more resistant to C-terminal degradation by carboxypeptidases or other degrading molecules (21).
Our findings show that E4 reduces bleomycin-induced apoptosis. Epithelial cell death combined with aberrant reepithelialization and wound healing is believed to contribute to the pathogenesis of lung fibrosis in IPF (39). On the basis of this theory, a reduction in epithelial cell apoptosis would block the onset of pulmonary fibrosis, perhaps explaining how E4 could prevent and reverse fibrosis. It is unlikely that E4 blocks early signaling events downstream of TGF-β such as Smad 2/3 or MAPK activation because these signaling cascades are activated within minutes, whereas E4 is effective even if administered days after the fibrotic trigger, long after the activation of these signaling cascades has subsided. Additional possible mechanisms for the antifibrotic activity of E4 are a reduction in Egr-1, a central transcription factor that mediates the effects of multiple fibrogenic triggers, and a reduction in LOX levels. A decrease in Egr-1 attenuates the effects of TGF-β and bleomycin, thus counteracting the fibrotic trigger. A reduction in LOX reduces cross-linking of collagen, rendering the ECM more fragile and thus susceptible to proteolytic degradation. E4 modulation of Egr-1 and LOX, both of which are induced by multiple fibrotic triggers, may underlie its ability to counteract the fibrogenic effects of both TGF-β and bleomycin in more than one organ. Thus, E4 may reduce even established fibrosis through a multi-pronged effect: decreasing levels of an essential fibrosis-mediating transcription factor, reducing matrix cross-linking by means of LOX inhibition, and preventing epithelial cell apoptosis.
There are currently no effective therapies for organ fibrosis. We demonstrate that the C-terminal domain of endostatin, corresponding to amino acid sequences 133 to 180, is a promising therapeutic agent for fibrotic disorders such as IPF, SSc, and morphea. Because E4 prevented and reversed both dermal and pulmonary fibrosis, it may also be useful in other organs such as subepithelial fibrosis in asthma and liver cirrhosis.
MATERIALS AND METHODS
Reagents and antibodies
The full-length human rE was purchased from Sigma-Aldrich. Recombinant human TGF-β was from R&D Systems Inc. Bleomycin was from Enzo Life Sciences Inc. Anti-human FN, anti-human Col1α1, anti–Egr-1, and anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibodies were from Santa Cruz Biotechnology. Anti-LOX antibody was from Novus Biologicals LLC. Anti-tubulin antibody was from Abcam Inc.
Synthesis of human endostatin peptides
Peptides were synthesized in the Peptide Synthesis Facility, University of Pittsburgh Genomics and Proteomics Core Laboratories. Sequences of the peptides are as follows: E1, amino acids 1 to 45 (H-1HSHRDFQPVLHLVALNSPLSGGMRGIRGADFQCFQQARAVGLAGT45-OH); E2, amino acids 71 to 115 (H-71IVNLKDELLFPSWEALFSGSEGPLKPGARIFSFDGKDVLRHPTWP115-OH); E3, amino acids 133 to 180 (H-133SYCETWRTEAPSATGQASSLLGGRLLGQSAASCHHAYIVLCIENSFMT180-OH); E4, which differs from E3 by the presence of a C-terminal amide, amino acids 133 to 180 (H-133SYCETWRTEAPSATGQASSLLGGRLLGQSAASCHHAYIVLCIENSFMT180-CONH2). The purity of all peptides was >98%. All peptides were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 5 μg/ml and diluted in 1× phosphate-buffered saline (PBS) to 1 to 20 μg/ml.
Primary fibroblast culture
Human primary fibroblasts from normal organ donors were cultured as previously described (40) under a protocol approved by the University of Pittsburgh Institutional Review Board. All the cells were used between passages 3 and 6.
Western blot analysis
Cellular lysates were obtained from cultured fibroblasts as described (40). Briefly, 2.0 × 105 primary fibroblasts were cultured in 35-mm wells in 0.5% fetal bovine serum–containing medium supplemented with human recombinant TGF-β (10 ng/ml) or PBS as vehicle control for 24 hours, after which human rE (5 μg/ml), endostatin peptides (E1 to E4), or DMSO (vehicle) was added for 48 hours. Cellular lysates were analyzed by Western blot. The intensity of individual bands with expected molecular sizes was semiquantitatively analyzed with the ImageJ software (National Institutes of Health) and normalized to individual GAPDH or tubulin intensity.
Ex vivo human skin assays
Human abdominal skin was obtained from cosmetic plastic surgery. All tissues were obtained according to the guidelines of the University of Pittsburgh and under a protocol approved by the Institutional Review Board of the University of Pittsburgh, as described (19). The following were injected intradermally in a total volume of 100 μl of 1× PBS: rE alone (1 to 10 μg/ml), endostatin peptides alone (10 μg/ml), rE or endostatin peptides (1 to 20 μg/ml) in combination with TGF-β (10 ng/ml), or TGF-β alone (10 ng/ml). Where indicated, human skin samples were preinjected with TGF-β for 48 hours followed by administration of peptides in the same injection site as TGF-β. Skin samples were cultured in an air-liquid interface epidermal side up. After 1 week, skin tissue corresponding to an area with 8-mm diameter centered around the injection site was harvested with disposable 8-mm Acu-Punch (Acuderm Inc.). Skin tissue was frozen or fixed in 10% formalin and embedded in paraffin.
In vivo experiments
TGF-β treatment
Six- to 8-week-old C57BL/6J male mice (The Jackson Laboratory) were used for all the in vivo experiments. Mouse experiments were done following guidelines and a protocol approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Human rE (10 μg/ml) or endostatin peptides (10 μg/ml) in combination with TGF-β (10 ng/ml), or TGF-β alone, was injected subcutaneously on the back of mice in a total volume of 100 μl of 1× PBS. Mice were injected in two different skin sites and sacrificed 1 week after injection. Skin surrounding the injection site was harvested.
Bleomycin treatment
Dermal or pulmonary fibrosis was induced in male 6- to 8-week-old C57BL/6J mice by local injection or intratracheal administration of bleomycin, respectively. To induce skin fibrosis, we subcutaneously injected 20 μg of bleomycin into the upper back of mice daily for 10 consecutive days in a total volume of 100 μl. E4 (10 μg/ml) was administered in the same total volume, with bleomycin on day 1 only, or from day 3 to day 10 of the bleomycin treatment. Mice were euthanized by CO2 asphyxiation, and skin surrounding the injection site was harvested. For inducing pulmonary fibrosis, 60 μg of bleomycin in a total volume of 50 μl was administered intratracheally, and lungs were harvested after 10 and 21 days. E4 (10 μg/ml) was administered simultaneously with or 3 to 8 days after bleomycin.
Measurement of skin dermal thickness
Six-micrometer sections of paraffin-embedded human and mouse skin tissues were stained with H&E. Images were taken on a Nikon Eclipse 800 microscope. For each section, the thickness of the dermis was measured from the epidermal-dermal junction to the dermal-fat junction with the ImageJ software. Measurements from six random fields were averaged.
Measurement of collagen levels
Sircol assay
Acid-soluble collagen levels were measured with the Sircol collagen assay kit (Biocolor) following the manufacturer’s recommendations.
Hydroxyproline assay
The hydroxyproline content of skin tissues was measured as described (41).
Immunohistochemistry
Immunohistochemistry of 6-μm sections of paraffin-embedded mouse lung tissues was performed as described (40). Images were taken on an Olympus Provis III microscope (Olympus America Inc.) with identical settings.
Quantification of apoptosis in vivo using TUNEL
Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) was performed with the ApopTag Peroxidase In Situ Apoptosis Detection kit (Millipore) following the manufacturer’s protocol. The number of positive cells in 10 noncoincident microscopic fields of each section was counted at a magnification of ×400, and the mean of the numbers of TUNEL-positive cells was calculated.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from frozen lung tissues of mice with QIAzol Lysis Reagent and purified with RNeasy Kit (Qiagen). Reverse transcription was performed with SuperScript II (Invitrogen) followed by quantitative polymerase chain reaction (PCR) amplification with the TaqMan method (ABI Prism 7300; Applied Biosystems). Premixed PCR primers and TaqMan probes for mouse Egr-1 and GAPDH were obtained from Applied Biosystems. Gene expression levels were normalized to GAPDH and compared with the 2−ΔΔCt method.
Statistical analysis
All continuous variables were expressed as means ± SD. Comparisons between two groups were tested for statistical significance with the paired t test, unpaired t test, or Mann-Whitney U test, as appropriate. Comparison among three or more groups was performed with analysis of variance (ANOVA) followed by Bonferroni correction.
Supplementary Material
Fig. S1. Recombinant human endostatin ameliorates TGF-β–induced fibrosis in vitro and ex vivo.
Fig. S2. E4 peptide ameliorates ongoing TGF-β–induced fibrosis in vitro.
Fig. S3. E4 peptide ameliorates ongoing TGF-β–induced fibrosis ex vivo.
Fig. S4. E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo.
Fig. S5. E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo whether administered concomitantly or following a fibrotic trigger.
Fig. S6. E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo whether administered intraperitoneally or intratracheally.
Fig. S7. E4 reduces bleomycin-induced apoptosis in vivo.
Fig. S8. The C-terminal peptide of endostatin has modest antiangiogenic activity.
Acknowledgments
We thank M.-H. Fan for assistance with the hydroxyproline assay.
Funding: Supported by NIH grant AR050840. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author contributions: C.A.F.-B. and Y.Y. designed the research, performed the experiments, and wrote and edited the manuscript; T.T., R.A.C., and K.L.V. performed the experiments and edited the manuscript; and A.T.L. performed the experiments and contributed to manuscript preparation and editing.
Competing interests: Y.Y. and C.A.F.-B. hold a provisional patent (WO 2011/050311 A1) on the use of E4 for the treatment of fibrosis, filed through the University of Pittsburgh. The other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/4/136/136ra71/DC1 Materials and Methods
REFERENCES AND NOTES
- 1.Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199–203. doi: 10.1164/ajrccm.157.1.9704130. [DOI] [PubMed] [Google Scholar]
- 2.Varga J, Abraham D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalluri R, Sukhatme VP. Fibrosis and angiogenesis. Curr Opin Nephrol Hypertens. 2000;9:413–418. doi: 10.1097/00041552-200007000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Branton MH, Kopp JB. TGF-β and fibrosis. Microbes Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 6.Varga J, Pasche B. Transforming growth factor b as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. 2009;5:200–206. doi: 10.1038/nrrheum.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 8.Dhanabal M, Volk R, Ramchandran R, Simons M, Sukhatme VP. Cloning, expression, and in vitro activity of human endostatin. Biochem Biophys Res Commun. 1999;258:345–352. doi: 10.1006/bbrc.1999.0595. [DOI] [PubMed] [Google Scholar]
- 9.Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 10.Folkman J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp Cell Res. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Tjin Tham Sjin RM, Satchi-Fainaro R, Birsner AE, Ramanujam VM, Folkman J, Javaherian K. A 27-amino-acid synthetic peptide corresponding to the NH2-terminal zinc-binding domain of endostatin is responsible for its antitumor activity. Cancer Res. 2005;65:3656–3663. doi: 10.1158/0008-5472.CAN-04-1833. [DOI] [PubMed] [Google Scholar]
- 12.Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by avb3 and a5b1 integrins. Proc Natl Acad Sci USA. 2003;100:4766–4771. doi: 10.1073/pnas.0730882100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 13.Karumanchi SA, Jha V, Ramchandran R, Karihaloo A, Tsiokas L, Chan B, Dhanabal M, Hanai JI, Venkataraman G, Shriver Z, Keiser N, Kalluri R, Zeng H, Mukhopadhyay D, Chen RL, Lander AD, Hagihara K, Yamaguchi Y, Sasisekharan R, Cantley L, Sukhatme VP. Cell surface glypicans are low-affinity endostatin receptors. Mol Cell. 2001;7:811–822. doi: 10.1016/s1097-2765(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 14.Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, Gho YS, Kwon YG. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. 2002;277:27872–27879. doi: 10.1074/jbc.M202771200. [DOI] [PubMed] [Google Scholar]
- 15.Shi H, Huang Y, Zhou H, Song X, Yuan S, Fu Y, Luo Y. Nucleolin is a receptor that mediates antiangiogenic and antitumor activity of endostatin. Blood. 2007;110:2899–2906. doi: 10.1182/blood-2007-01-064428. [DOI] [PubMed] [Google Scholar]
- 16.Sumi M, Satoh H, Kagohashi K, Ishikawa H, Sekizawa K. Increased serum levels of endostatin in patients with idiopathic pulmonary fibrosis. J Clin Lab Anal. 2005;19:146–149. doi: 10.1002/jcla.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richter AG, McKeown S, Rathinam S, Harper L, Rajesh P, McAuley DF, Heljasvaara R, Thickett DR. Soluble endostatin is a novel inhibitor of epithelial repair in idiopathic pulmonary fibrosis. Thorax. 2009;64:156–161. doi: 10.1136/thx.2008.102814. [DOI] [PubMed] [Google Scholar]
- 18.Hebbar M, Peyrat JP, Hornez L, Hatron PY, Hachulla E, Devulder B. Increased concentrations of the circulating angiogenesis inhibitor endostatin in patients with systemic sclerosis. Arthritis Rheum. 2000;43:889–893. doi: 10.1002/1529-0131(200004)43:4<889::AID-ANR21>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 19.Yasuoka H, Yamaguchi Y, Larregina AT, Feghali-Bostwick CA. Human skin culture as an ex vivo model for assessing the fibrotic effects of insulin-like growth factor binding proteins. Open Rheumatol J. 2008;2:17–22. doi: 10.2174/1874312900802010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pohlers D, Brenmoehl J, Löffler I, Müller CK, Leipner C, Schultze-Mosgau S, Stallmach A, Kinne RW, Wolf G. TGF-β and fibrosis in different organs—Molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Bode JW. Emerging methods in amide- and peptide-bond formation. Curr Opin Drug Discov Devel. 2006;9:765–775. [PubMed] [Google Scholar]
- 22.Richter AG, McKeown S, Rathinam S, Harper L, Rajesh P, McAuley DF, Heljasvaara R, Thickett DR. Soluble endostatin is a novel inhibitor of epithelial repair in idiopathic pulmonary fibrosis. Thorax. 2009;64:156–161. doi: 10.1136/thx.2008.102814. [DOI] [PubMed] [Google Scholar]
- 23.Counts DF, Evans JN, Dipetrillo TA, Sterling KM, Jr, Kelley J. Collagen lysyl oxidase activity in the lung increases during bleomycin-induced lung fibrosis. J Pharmacol Exp Ther. 1981;219:675–678. [PubMed] [Google Scholar]
- 24.Bhattacharyya S, Wu M, Fang F, Tourtellotte W, Feghali-Bostwick C, Varga J. Early growth response transcription factors: Key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol. 2011;30:235–242. doi: 10.1016/j.matbio.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan FK, Hildebrand BA, Lester MS, Stivers DN, Pounds S, Zhou X, Wallis DD, Milewicz DM, Reveille JD, Mayes MD, Jin L, Arnett FC., Jr Classification analysis of the transcriptosome of nonlesional cultured dermal fibroblasts from systemic sclerosis patients with early disease. Arthritis Rheum. 2005;52:865–876. doi: 10.1002/art.20871. [DOI] [PubMed] [Google Scholar]
- 26.Yang IV, Burch LH, Steele MP, Savov JD, Hollingsworth JW, McElvania-Tekippe E, Berman KG, Speer MC, Sporn TA, Brown KK, Schwarz MI, Schwartz DA. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med. 2007;175:45–54. doi: 10.1164/rccm.200601-062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J. The generation of endostatin is mediated by elastase. Cancer Res. 1999;59:6052–6056. [PubMed] [Google Scholar]
- 28.Felbor U, Dreier L, Bryant RA, Ploegh HL, Olsen BR, Mothes W. Secreted cathepsin L generates endostatin from collagen XVIII. EMBO J. 2000;19:1187–1194. doi: 10.1093/emboj/19.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toubi E, Kessel A, Grushko G, Sabo E, Rozenbaum M, Rosner I. The association of serum matrix metalloproteinases and their tissue inhibitor levels with scleroderma disease severity. Clin Exp Rheumatol. 2002;20:221–224. [PubMed] [Google Scholar]
- 30.Bloch W, Huggel K, Sasaki T, Grose R, Bugnon P, Addicks K, Timpl R, Werner S. The angiogenesis inhibitor endostatin impairs blood vessel maturation during wound healing. FASEB J. 2000;14:2373–2376. doi: 10.1096/fj.00-0490fje. [DOI] [PubMed] [Google Scholar]
- 31.Tanabe K, Maeshima Y, Ichinose K, Kitayama H, Takazawa Y, Hirokoshi K, Kinomura M, Sugiyama H, Makino H. Endostatin peptide, an inhibitor of angiogenesis, prevents the progression of peritoneal sclerosis in a mouse experimental model. Kidney Int. 2007;71:227–238. doi: 10.1038/sj.ki.5002040. [DOI] [PubMed] [Google Scholar]
- 32.Cattaneo MG, Pola S, Francescato P, Chillemi F, Vicentini LM. Human endostatin-derived synthetic peptides possess potent antiangiogenic properties in vitro and in vivo. Exp Cell Res. 2003;283:230–236. doi: 10.1016/s0014-4827(02)00057-5. [DOI] [PubMed] [Google Scholar]
- 33.Xu HL, Tan HN, Wang FS, Tang W. Research advances of endostatin and its short internal fragments. Curr Protein Pept Sci. 2008;9:275–283. doi: 10.2174/138920308784534050. [DOI] [PubMed] [Google Scholar]
- 34.Isobe K, Kuba K, Maejima Y, Suzuki J, Kubota S, Isobe M. Inhibition of endostatin/collagen XVIII deteriorates left ventricular remodeling and heart failure in rat myocardial infarction model. Circ J. 2010;74:109–119. doi: 10.1253/circj.cj-09-0486. [DOI] [PubMed] [Google Scholar]
- 35.Hu B, Zhu HW, Zhu LP, Li C, Rong ZG, Xu JM, Wu ZW, Wang JJ, Xu GX. Bioactivity, pharmacokinetics, and immunogenicity assays in preclinical and clinical trials for recombinant human endostatin. Acta Pharmacol Sin. 2008;29:1357–1369. doi: 10.1111/j.1745-7254.2008.00865.x. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Wang J, Liu Y, Song X, Zhang Y, Li K, Zhu Y, Zhou Q, You L, Yao C. Results of phase III trial of rh-endostatin (YH-16) in advanced non-small cell lung cancer (NSCLC) patients. J Clin Oncol. 2005;23:7138. [Google Scholar]
- 37.Crystal RG. The body as a manufacturer of endostatin. Nat Biotechnol. 1999;17:336–337. doi: 10.1038/7890. [DOI] [PubMed] [Google Scholar]
- 38.Morbidelli L, Donnini S, Chillemi F, Giachetti A, Ziche M. Angiosuppressive and angio-stimulatory effects exerted by synthetic partial sequences of endostatin. Clin Cancer Res. 2003;9:5358–5369. [PubMed] [Google Scholar]
- 39.Selman M, King TE, Pardo A, American Thoracic Society; European Respiratory Society; American College of Chest Physicians Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 40.Pilewski JM, Liu L, Henry AC, Knauer AV, Feghali-Bostwick CA. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am J Pathol. 2005;166:399–407. doi: 10.1016/S0002-9440(10)62263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Recombinant human endostatin ameliorates TGF-β–induced fibrosis in vitro and ex vivo.
Fig. S2. E4 peptide ameliorates ongoing TGF-β–induced fibrosis in vitro.
Fig. S3. E4 peptide ameliorates ongoing TGF-β–induced fibrosis ex vivo.
Fig. S4. E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo.
Fig. S5. E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo whether administered concomitantly or following a fibrotic trigger.
Fig. S6. E4 ameliorates bleomycin-induced pulmonary fibrosis in vivo whether administered intraperitoneally or intratracheally.
Fig. S7. E4 reduces bleomycin-induced apoptosis in vivo.
Fig. S8. The C-terminal peptide of endostatin has modest antiangiogenic activity.