

Abstract
Aims
Cardiopoiesis is a conditioning programme that aims to upgrade the cardioregenerative aptitude of patient‐derived stem cells through lineage specification. Cardiopoietic stem cells tested initially for feasibility and safety exhibited signs of clinical benefit in patients with ischaemic heart failure (HF) warranting definitive evaluation. Accordingly, CHART‐1 is designed as a large randomized, sham‐controlled multicentre study aimed to validate cardiopoietic stem cell therapy.
Methods
Patients (n = 240) with chronic HF secondary to ischaemic heart disease, reduced LVEF (<35%), and at high risk for recurrent HF‐related events, despite optimal medical therapy, will be randomized 1:1 to receive 600 × 106 bone marrow‐derived and lineage‐directed autologous cardiopoietic stem cells administered via a retention‐enhanced intramyocardial injection catheter or a sham procedure. The primary efficacy endpoint is a hierarchical composite of mortality, worsening HF, Minnesota Living with Heart Failure Questionnaire score, 6 min walk test, LV end‐systolic volume, and LVEF at 9 months. The secondary efficacy endpoint is the time to cardiovascular death or worsening HF at 12 months. Safety endpoints include mortality, readmissions, aborted sudden deaths, and serious adverse events at 12 and 24 months.
Conclusion
The CHART‐1 clinical trial is powered to examine the therapeutic impact of lineage‐directed stem cells as a strategy to achieve cardiac regeneration in HF populations. On completion, CHART‐1 will offer a definitive evaluation of the efficacy and safety of cardiopoietic stem cells in the treatment of chronic ischaemic HF.
Trial registration: NCT01768702
Keywords: Cardiopoiesis, Heart failure, Ischaemic cardiomyopathy, Regenerative medicine, Stem cell
Introduction
Heart failure (HF) is still a common disorder associated with significant morbidity and mortality despite recent advances in therapy.1 Current approaches do not, however, address the fundamental issues of myocyte loss and microvascular ischaemia that underscore the progression of ischaemic cardiomyopathy, warranting treatment‐innovative approaches,2 which is at the foundation of more recently proposed regenerative therapies aimed to limit remodelling and restore parenchymal integrity.3, 4 One such approach is stem cell therapy that has shown some promise in proof‐of‐concept and humans studies, where signs of functional recovery were observed.5, 6, 7 Yet, these effects were not consistently shown.8
Meta‐analyses of stem cell‐based trials in cardiovascular disease suggest that the procedures are generally safe, yet clinical benefit remains more uncertain.9, 10, 11 Recent data have revealed that only rare patients harbour stem cells with an innate reparative capacity.12 The restorative proficiency of autologous preparations can either be harvested through pre‐selection of such rare patient cohorts or alternatively rescued by pre‐emptive programming within a youthful conditioning environment.13 As pre‐selection limits stem cell therapy to only the rare few, pre‐emptive specification has been pursued as an avenue to generalize therapeutic proficiency in the setting of HF.14
In ‘guided cardiopoiesis’, non‐regenerative patient‐derived stem cells are expanded and directed towards the cardiac lineage using a cardiogenic conditioning medium without administration of genetic material.15 Clinical grade cardiopoietic stem cells (C3BS‐CQR‐1, Celyad S.A., Mont‐Saint‐Guibert, Belgium) represent a bio‐therapeutic in which autologous bone marrow‐derived mesenchymal stem cells (MSCs) have been programmed to ensure therapeutic performance. The safety and feasibility of C3BS‐CQR‐1 therapy was evaluated in 45 patients with HF of ischaemic origin in the C‐CURE (Cardiopoietic stem Cell therapy in heart failURE) trial.16 In this first‐in‐man, open label study, intramyocardial administration of C3BS‐CQR‐1 was associated with some improvements in LVEF, LV end‐systolic volume (LVESV), 6‐min walk test (6MWT) distance, and in a composite clinical score of clinical and functional parameters compared with standard care alone. Cell administration was well tolerated, without signs of increased cardiac or systemic toxicity.
Based on these initial results, the impact of cardiopoietic stem cell preparations on patient‐centred outcomes will be investigated in a prospective randomized sham‐controlled trial evaluation. Accordingly, the Congestive Heart failure cArdiopoietic Regenerative Therapy (CHART‐1) trial has been designed to assess the benefits of C3BS‐CQR‐1 in patients with chronic HF secondary to ischaemic heart disease.
Study design
Key objectives
The primary objective of CHART‐1 (ClinicalTrials.gov Identifier: NCT01768702, EudraCT number 2011‐001117‐13) is to evaluate the efficacy of cardiopoietic stem cells (C3BS‐CQR‐1, Celyad S.A.; Mont‐Saint‐Guibert, Belgium) delivered using an endoventricular injection catheter (C‐Cathez®) in comparison with a sham procedure on a hierarchical outcome comprising measures of mortality, morbidity, and changes in quality of life, 6MWT distance, functional capacity, and LV structure and function at 39 weeks (9 months) post‐procedure.
The secondary objective is to assess safety by comparing the incidence of serious adverse events between study groups at 52 weeks (12 months) and all‐cause mortality at 104 weeks (24 months) post‐procedure.
Eligible patients with chronic HF secondary to ischaemic heart disease who consent to participate will be randomized 1:1 to either intramyocardial injection of cardiopoietic stem cells (Treatment Group) or sham procedure (Control Group) following successful expansion of MSCs from autologous bone marrow (Figure 1). Follow‐up will occur at 4, 13, 26, 39, 52, and 104 weeks post‐procedure. The primary efficacy endpoint is a Finkelstein–Schoenfeld hierarchical composite endpoint comprising all‐cause mortality, worsening HF (WHF) events, and changes in Minnesota Living with Heart Failure Questionnaire (MLHFQ) score, 6MWT distance, LVESV, and LVEF by transthoracic echocardiography (TTE) at 39 weeks.17, 18, 19
Figure 1.

Study flow chart. MSC, mesenchymal stem cells.
Study population
A minimum of 240 patients with chronic HF secondary to ischaemic heart disease fulfilling all inclusion and exclusion criteria (Table 1) will be enrolled at ∼50 centres in Europe and Israel. The study will be conducted in compliance with the requirements of governmental regulatory bodies and ethics committees of each participating centre. Study criteria seek to identify a population of NYHA class III–IV patients with limited functional capacity and low LVEF (<35%), who are treated with optimal medical and revascularization therapies. Patients must have had an episode of worsened HF, requiring either hospital admission or outpatient treatment with i.v. vasoactive medication or diuretic for WHF, in the last 12 months. The absence of recent acute coronary syndrome, non‐ischaemic causes of LV dysfunction, significant mitral or aortic valve disease, peripheral vascular disease, and co‐morbidities or cardiac transplant listing that would impact the ability to complete the trial successfully is required. Screening procedures (Table 2) include a comprehensive medical history, health status assessment, physical examination, laboratory tests, 12‐lead ECG, TTE, 6MWT, and MLHFQ. In addition to decreased systolic function, to qualify for study intervention, the left ventricle must be without aneurysm and with suitable wall thickness characteristics (see ‘Study procedure’ below). Patients who meet eligibility criteria will be scheduled for bone marrow harvest.
Table 1.
Study eligibility
| Key inclusion criteria | Key exclusion criteria |
|---|---|
| Age ≥18 and <80 years | Pregnancy |
| Chronic ischaemic HF, without need for revascularization | Acute coronary syndrome or PCI within 90 days, or CABG within 180 days |
| Systolic dysfunction with LVEF ≤35% | Cardiac resynchronization therapy within 180 days |
| Hospitalization or outpatient with i.v. therapy for HF within the previous 12 months | Moderate to severe valvular disease |
| Be or must have been within the previous 12 months in NYHA class III or IV or in INTERMACS class 4, 5, 6, or 7, and at the time of inclusion, must be in NYHA class II or greater | Prosthetic mitral or aortic valve |
| Total MLHFQ score >30 | LV thrombus |
| 6MWT distance >100 and ≤400 m | LV wall thickness <8 mm visualized in >50% of the left ventricle |
| Stable medical regimen, including ACE inhibitor and/or ARB, beta‐blocker, aldosterone blocker, and diuretic for at least 1 month | Sustained VT or VF within 90 days |
| Willing and able to give written informed consent | BMI <19 or >45 kg/m2 |
| Inability to perform a 6MWT due to physical limitations other than HF including: stroke, peripheral vascular disease, pulmonary disease | |
| Immunosuppressive therapy | |
| Chronic infection or active malignancy | |
| Renal dysfunction with serum creatinine >3.0 mg/dL (>0.265 mmol/L) | |
| Haematocrit <28% | |
| Sero‐positivity for HIV 1 or 2, hepatitis B or C, HTLV 1 or 2 | |
| Prior cell or angiogenic therapy within 60 days | |
| Any illness other than HF which might reduce life expectancy to <2 years | |
| Allergies to dextran or other plasma volume expanders |
BMI, body mass index; CABG, coronary artery bypass graft; HF, heart failure; HIV, human immunodeficiency virus; HTLV, human T‐cell lymphotropic virus; MLHFQ, Minnesota Living with Heart Failure Questionnaire; 6MWT, 6‐min walk test; VF, ventricular fibrillation; VT, ventricular tachycardia.
Table 2.
Study schedule
| Patient procedures | Screening | Bone marrow harvest | Index procedure (C3BS‐CQR‐1 injection or sham) | 4 weeks (±7 days) | 13 weeks (±7 days) | 26 weeks (±14 days) | 39 weeks (+14 days) | 52 weeks (+30 days) | 104 weeks (+30 days) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Pre‐procedure (baseline) | Post‐procedure | |||||||||
| Informed consent | X | |||||||||
| Inclusion/exclusion criteria | X | |||||||||
| Demographics and medical history including cardiac risk factors | X | |||||||||
| Pregnancy testa | X | |||||||||
| Clinical examination | X | X | X | X | X | X | X | X | X | X |
| NYHA class | X | X | X | X | X | |||||
| INTERMACS patient profile | X | X | X | X | X | |||||
| Concomitant medications | X | X | X | X | X | X | X | X | X | X |
| Routine laboratory tests | X | X | X | |||||||
| HIV1, HIV2, HBV, HCV, HTLV‐1b, HTLV‐2b and syphilis | X | |||||||||
| Blood sampling for central laboratory assessment of CK‐MB, troponin T | X | Xc | ||||||||
| Blood sampling for central laboratory assessment of NT‐proBNP | X | X | ||||||||
| 12‐lead ECG | X | X | X | X | X | X | X | X | X | |
| Echocardiography | Xd | X | Xd | Xd | X | X | X | |||
| AICD interrogatione | X | X | X | X | X | X | X | X | ||
| Six‐minute walk test | X | X | X | X | X | |||||
| MLHFQ | X | X | X | X | X | |||||
| Adverse events | X | X | X | X | X | X | X | X | Xf | |
| Clinical events for adjudication | X | X | X | X | X | X | ||||
AICD, automatic implantable cardioverter‐defibrillator; CK, creatine kinase; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HTLV, human T‐cell lymphotropic virus; MLHFQ, Minnesota Living with Heart Failure Questionnaire.
Females of child‐bearing potential only.
If required per local regulations.
To be done at 6 and 24 h post‐procedure, even if discharged within 24 h of the index procedure.
Echocardiography can be assessed locally.
For patients with an existing AICD only.
Serious adverse events and clinical events only (non‐serious events recorded up to 52 weeks).
Cell procurement and processing
A volume of 65–85 mL of bone marrow aspirated from the iliac crest will be transported at 2–8°C to a central Good Manufacturing Practice (GMP) production facility where an initial MSC expansion and subsequent cardiopoietic stem cell formulation will occur (Figure 2). Subjects who undergo successful expansion of bone marrow to 24 × 106 MSCs will be randomized in a 1:1 fashion using a web‐based randomization system (Sealed Envelope™), according to a central randomization scheme stratified for each study centre. Patients from whom aspirates are <65 mL or yield <24 × 106 MSCs will be eligible for a repeat bone marrow harvest. If the second attempt again yields low bone marrow volume or insufficient MSC numbers, the subject will fail the screening process and will not be randomized.
Figure 2.
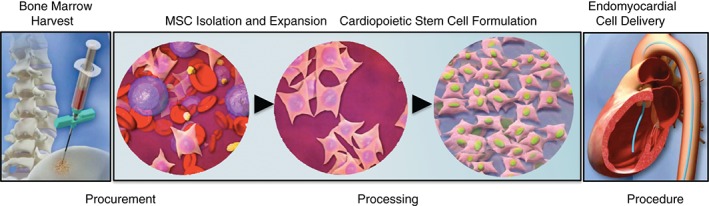
Cardiopoietic stem cell procurement and processing prior to the delivery procedure. Following bone marrow harvest, mesenchymal stem cells (MSCs) are purified and expanded. Following standard operating procedures, cardiopoiesis is imposed for lineage guidance of MSCs to derive cardiopoietic stem cells. Stem cells meeting pre‐defined release criteria are delivered in an autologous fashion to patients with ischaemic cardiomyopathy using a catheter‐based endomyocardial delivery procedure.
The MSCs from subjects randomized to the Treatment Group will be processed to undergo lineage specification, and released as cardiopoietic stem cells (C3BS‐CQR‐1). Following closed system cryopreservation, the C3BS‐CQR‐1 product will be thermally packaged and shipped to clinical sites at −130°C or lower. For each patient, aliquots will be bio‐banked for quality control, as well as for future high‐throughput molecular studies at the Mayo Clinic. If release specifications (purity, identity, homogeneity, and sterility)16 are not met, the cell product will be withheld and the subject allocated to the Process Failure Group (Figure 1).
Study procedure
In order to maintain a double‐blind design, each site will identify two independent investigative teams: (i) a blinded assessment team which will perform all non‐procedural study visits and assessments; and (ii) an unblinded interventional team which will perform the procedure. Through repeated onsite training and review, communications between teams will be limited. All patients will undergo retrograde cardiac catheterization and biplane contrast left ventriculography according to best practices. Unfractionated or low molecular weight heparin will be used during the study to maintain low‐therapeutic anticoagulation levels (activated clotting time at ∼250 s where possible).
The Treatment Group will receive cardiopoietic stem cells by endomyocardial injections (Figure 2). In preparation for this, frozen stem cell product will be reconditioned by unblinded team members on the day of the procedure. Based on echocardiography and contrast left ventriculography, regions of the left ventricle to be targeted for injection will be delineated onto real‐time fluoroscopy images. An 8Fr deflectable tip catheter containing a 28 gauge retractable Nitinol alloy needle, featuring a curved tip with seven side holes (Figure 3) will be used for injections (C‐Cathez®). This catheter has demonstrated cell retention‐enhanced properties following endomyocardial injections.20 Both tip deflection and needle advancement are controlled at the proximal handle (Figure 3). Once prepared and primed with cells, the catheter is inserted via the femoral artery and advanced into the left ventricle in retro‐aortic fashion. Twenty injections, each of 0.5 mL and separated by ∼1 cm will be distributed over non‐apical, non‐basal portions of the left ventricle (segments 7–16 on the standard bulls‐eye echocardiogram display 17 ASE model)21 that have wall thickness >8 mm.
Figure 3.

Catheter‐based delivery system. Endomyocardial delivery catheter designed for enhanced endomyocardial retention of stem cells (C‐Cathez®) features a thumbwheel control for distal catheter tip deflection (upper left panel), a curved needle design with side holes (inset), and a luer lock connector for study injection with adjustable needle advancing mechanism (lower left).
Patients from the Control and Process Failure groups will undergo a sham procedure, without intramyocardial injections (Figure 1). The sham procedure, conducted over 30–60 min, consists of insertion and removal of an introducer sheath into the femoral artery following standard percutaneous intervention procedures, with the patient sedated per local practice. If the patient is minimally sedated, a 30‐min mock injection procedure is performed.
Immediately following the procedure, all subjects will undergo echocardiography to assess for procedural complications, and blood samples will be taken for measurement of cardiac biomarkers. All patients will be observed in the catheterization laboratory for at least 30 min after the procedure and then for a minimum of 12 h in a telemetry unit.
Follow‐up
All follow‐up visits will be performed by the blinded team, scheduled for 4, 13, 26, 39, 52, and 104 weeks following the procedure (Table 2). Clinical assessments include grading of HF signs and symptoms, NYHA class, and INTERMACS classification, echocardiography, MLHFQ score, and 6MWT distance. Blood samples for central laboratory measurement of cardiac markers; creatine kinase (CK)‐MB, troponin‐T, and NT‐proBNP will be taken at week 39. Automatic implantable cardioverter‐defibrillator (AICD) interrogation will be conducted at each follow‐up visit.
Adverse events and the occurrence of clinical events of interest, including death, WHF, sudden death, AICD activation, myocardial infarction (MI), and stroke, will be collected through week 52. Serious adverse events (SAEs) will be collected through week 104.
Statistical methods
The primary efficacy outcome is a hierarchical composite18 constructed from the following endpoints evaluated at 39 weeks following the study procedure in patients treated as randomized:
Mortality = days alive out of 39 weeks.
Number of WHF events: 0, 1, or ≥ 2,
MLHFQ ≥10 point improvement, no meaningful change, ≥10 point deterioration.
6MWT ≥40 m improvement, no meaningful change.
LVESV ≥15 mL improvement, no meaningful change.
LVEF ≥4% absolute improvement, no meaningful change, ≥4% absolute deterioration.
Worsening heart failure is defined as new or worsening HF signs or symptoms requiring additional or increased treatment—including initiation or up‐titration of intravenous therapy, mechanical or surgical intervention, or ultrafiltration, haemofiltration, or dialysis—in either an urgent outpatient setting or hospital admission of at least 24 h duration (or overnight if exact times are not available). WHF will be adjudicated by the Clinical Events Committee (CEC; see Appendix 1).
Each patient is compared with every other patient and classified as having done better (+1), worse (−1), or the same, starting with the first endpoint in the hierarchy. For a given pair, if the patients are tied at the first level in the hierarchy they are compared with respect to the second level, and, if tied, are compared at the third level, and so on. Treatment groups are then compared among treated patients at the two‐sided 5% significance level using a modification of the generalized Wilcoxon test constructed from the sum of the scores for each patient.
Effects were hypothesized based on results reported for effective therapies in similar patient populations (e.g. CRT), results of the C‐CURE trial,16 and clinical judgement regarding expected results given the target patient population. Power with 120 patients per group is ∼87% at the two‐sided 5% significance level, with assumed correlations among the endpoints and with the following assumed treatment group differences (Treatment vs. Control):
Mortality of 7.5% vs. 10%.
Number of WHF events of 83.5% vs. 78% with 0, 11% vs. 16% with 1, and 5.5% vs. 6% with ≥2 events.
Change in MLHFQ total score of −14 vs. –5 points with a common SD of 20 points.
Change in 6MWT distance of 45 m vs. 10 m with an SD of 120 m.
Change in LVESV of −10 mL vs. 5 mL with an SD of 20 mL.
Absolute change in LVEF of 6% vs. 1% with an SD of 5%.
The secondary efficacy endpoint is the time to cardiovascular mortality or WHF through week 52.
Tertiary (exploratory) endpoints will include, through 39 and 52 weeks, time to all‐cause mortality, time to cardiovascular mortality, WHF, admission for HF, cardiovascular death or HF admission, death or hospital admission, total days in intensive care, total outpatient days alive, total days on any i.v. positive inotropic drug, change in 6MWT distance and MLHFQ score, changes in INTERMACS class, changes in NYHA class, change in blood values (blood cell counts, C‐reactive protein, creatinine), and total cost in healthcare utilization. Change in LVESV, LV end‐diastolic volume (LVEDV), LVEF, and LV mass index at 26, 39, and 52 weeks and changes in NT‐proBNP and troponin‐T at 39 weeks, as well as total costs and healthcare utilization will also be examined.
Safety endpoints will be assessed in patients as treated and include mortality through 104 weeks, readmissions, cardiac transplantation, MI, stroke, and SAEs through weeks 52 and 104, and non‐serious AEs through week 52. Safety will also be assessed by the occurrence of aborted sudden death events, defined as resuscitated sudden death or appropriate defibrillator therapy for sustained ventricular tachyarrhythmias, through 39, 52, and 104 weeks. The CEC will adjudicate all post‐randomization deaths, MI, stroke, and aborted sudden death events.
Study oversight and events assessment
A Steering Committee (see Appendix 1), comprised of the global co‐ordinating investigators and five additional members, will oversee the study, including data collection and implementation of operational issues that may arise and that may warrant a protocol amendment or other corrective action. The Steering Committee will also approve policy regarding presentations and publications.
A blinded CEC, composed of cardiologists with expertise in HF, interventional techniques, and electrophysiology, will be responsible for reviewing and adjudicating specified clinical events collected in the trial. An unblinded, independent, interventional cardiologist will review and adjudicate the relatedness of peri‐procedural SAEs to the catheter or cell product.
An independent Data Safety Monitoring Board (DSMB; see Appendix 1), consisting of HF and interventional cardiologists, and including a clinical pharmacologist and biostatistician, will assess all SAEs (myocardial trauma, perforation, pericardial tamponade, ventricular tachyarrhythmias causing haemodynamic instability, stroke), as well as reviews of aggregate safety. Safety analysis of the C‐Cathez® catheter will be performed after 40 patients have reached the 4‐week follow‐up visit. Accumulated safety and efficacy data of the C3BS‐CQR‐1 stem cell product will be evaluated after enrolment of ∼120 patients and a minimum of 13 weeks of follow‐up, at which time a futility analysis of the primary efficacy outcome will be performed. For this unblinded examination of the primary endpoint, a two‐sided alpha of 0.0001 will be considered to have been spent. The DSMB will make recommendations to the Sponsor regarding the continuation, modification, or stoppage of the trial. The full‐cohort study will be unblinded and the primary analysis will be performed after the last patient randomized has been followed for 39 weeks.
Discussion
The late phase CHART‐1 trial builds upon findings and experience derived from its predecessor, the C‐CURE study.16 The general clinical condition, chronic ischaemic HF, and the test agent, cardiopoietic stem cells (C3BS‐CQR‐1, processed at a central facility), are consistent between studies. CHART‐1 incorporates unique elements in the design for power to validate the efficacy signals initially observed in the C‐CURE trial.
Entry criteria are designed to include patients who are symptomatic and at high risk despite traditional therapy for HF. Inclusion of high‐risk populations (HF decompensation within 12 months of screening) targets a population in need, and serves to increase the probability of detecting a treatment effect in follow‐up. As a late‐phase clinical trial, CHART‐1 incorporates randomization and treatment allocation after successful MSC expansion. Randomization is more commonly performed before any cell processing, a less than optimal approach when utilizing autologous bio‐therapeutics. Incorporating assessment of MSCs in the inclusion process reduces patient‐to‐patient variability in stem cell availability and potency among randomized patients. Delaying randomization even further, i.e. until after successful production of cardiopoietic stem cells, rather than at the MSC expansion phase, while conceivable, provides limited advantage while adding significant cost burden.
The study procedure in CHART‐1 takes advantage of a trans‐endocardial cell delivery catheter, a novel system (C‐Cathez® catheter) designed to increase tissue distribution and retention of injected stem cells.20 The Control arm as well as the Process Failure group will undergo a sham procedure, instead of receiving placebo injection, in order to achieve a reasonable balance between study risk and scientific rigour. While placebo‐controlled, double‐blinded injections have been utilized in other stem cell trials,22 the risks of intramyocardial injection are not negligible.23 Effective barriers to ensure blinding24 are intrinsic to this study in order to assess the effectiveness of the interventional therapy. Maintaining the blinding is a difficult task in studies requiring invasive therapies such as the present one.25 On the one hand, blinded (both patient and physician) assessment of the treatment effects is crucial to ensure objective assessment, but on the other hand performing a full sham procedure including intramycoardial injection was felt to add unnecessary risk for patients. Therefore, first the teams performing the efficacy assessments and those performing the procedure will be separated into two distinct firewalled teams and, secondly, patients receiving sham treatment would receive a mini‐procedure; however, no intramyocaridal injections will be attempted.
Furthermore, endpoint selection is of critical importance in the design of HF trials and continues to evolve reflective of the systemic nature of the disease.26 To this end, the CHART‐1 design combines in a hierarchical manner survival time, WHF events, quality of life, functional capacity, and parameters of LV remodelling as a primary efficacy outcome. The relevance of the specified changes in MLHFQ, 6MWT, LVESV, and LVEF merits discussion. Some parameters, including the 6MWT and MLHFQ, are subjective and may be particularly influenced by knowledge of the treatment assignment. The absolute change in MLHFQ score that would indicate a clinical meaningful outcome is not certain. The selected 10‐point change excludes chance variability, and was associated with substantially increased risks of death and re‐hospitalization in patients with advanced chronic HF followed for 18 months on average.27 Regarding the 6MWT, a 43 m improvement was found to be statistically significant in the COMPANION study and accompanied by reduced risks of death or HF rehospitalization at 6 months.28 We have therefore considered a 40 m change as meaningful. Lastly, LVESV and LVEF are more objective measures of response, which, when considered in conjunction with more subjective parameters, provide a clinically valuable readout of regenerative impact.26 However, when taken alone, in a patient population with baseline NYHA class II/III symptoms and LVESV of 200 mL, it has been suggested that a change of 10 mL is clinically meaningful,29 informing the CHART‐1 design.
Study designs based exclusively on a hard endpoint, such as mortality, disregard patient symptoms and functional capacity which, in the setting of severe HF, are vitally important to the patient themselves. Such studies would also mandate an order of magnitude larger patient cohort, typically in the thousands, which is not feasible in this type of very intensive study. A composite endpoint not only provides power above that afforded by a single endpoint, but importantly allows formal assessment of the therapy's effectiveness across multiple clinical parameters. Composite endpoints have been used, including combined death and recurrent hospitalization with the method of Finkelstein and Schoenfeld,18 in device‐based clinical trials as a comprehensive analytical approach.30 The composite endpoint method affords simultaneous insight into multiple facets of the disease, and asserts whether the intervention has an overall benefit weighting in a descending order outcomes by importance.26, 31 So the risks of a spurious false‐positive or false‐negative finding, which in small studies and single endpoints are high, are diminished in a large trial such as CHART‐1 utilizing this approach.
Summary
Cardiopoiesis offers an optimizing platform by which to generalize the regenerative potential of patient‐derived stem cells in the context of HF. CHART‐1 is a late‐phase, randomized study designed to establish the efficacy and safety of autologous cardiopoietic stem cells in the treatment of chronic ischaemic HF. In utilizing cardiac lineage‐directed cells and a delivery catheter featuring increased tissue retention, CHART‐1 will serve as the foundation for a global CHART programme.
Funding
The CHART‐1 study is sponsored by Celyad S.A. The authors acknowledge the support by the Cardiovascular Center Aalst, the Mayo Clinic Center for Regenerative Medicine, the Marriott Foundation, the Michael S. and Mary Sue Shannon Family, and the Russ and Kathy VanCleve Foundation.
Conflict of interest: J.B. is a member of an institution which is a shareholder of Celyad S.A. B.D. and G.C. are employees of Momentum Research Inc., which has provided consulting services to NovaCardia, Merck, Corthera, Novartis, Singulex, ChanRx, Laguna, Sorbent Therapeutics, Celyad S.A., Trevena, Amgen, and Anexon. W.S. is employed as Chief Medical Officer by and has stock options in Celyad S.A. T.P. is an employee of Duke Clinical Research Institute, which has received research funding from Celyad S.A. and Baxter Healthcare; and has consulting or advisory agreements with Capricor Inc. and Pluristem. R.H. is scientific co‐founder of and has equity in Celladon Corp. T.H. is on the Steering Committee of or is the Principal Investigator for studies conducted by Aastrom, Mesoblast, Capricor, Cytori, and the US National Institutes of Health. B.G. has no conflicts related to this paper. Unrelated activities: member of the Data Safety Monitoring Board for the RENEW study (sponsor: Baxter, Inc.) and NCT02032004 (sponsor TEVA Pharmaceuticals) both without financial financial or other remuneration. G.F. is a member of Committees or Principal Investigator of trials sponsored by Celyad S.A., Vifor, Bayer, Novartis, and the European Union, including CHART‐1 and CHART‐2. M.M. has received consulting incomes from Bayer, Novartis, and Servier, and honoraria for speeches from Abbott Vascular. A.B. received Mayo Clinic‐administered research grants from. The Mayo Clinic has rights to future royalties from Celyad S.A. C.H. is employed as Chief Executive Officer by and is a founder of and shareholder in Celyad S.A. M.T. has participated in research grants on cell therapy from the EU Horizon 2020 programme and from the Polish National Center for Research and Development and National Center for Science. Unrelated to study topic: Servier and Bayer, consulting fees and honoraria, lecture fees; Amgen, consulting fees. W.W. has received Institutional research grants from pharmaceutical (AstraZeneca, Therabel) and device (AbbottVascular, Biosensors, Biotronik, Boston Scientific, Cordis, Medtronic, Orbus Neich, St Jude, Terumo, Tryton) companies, and is a shareholder and non‐executive board member of Argonauts, Genae, and Celyad S.A. A.T. has received Mayo Clinic‐administered research grants from Celyad S.A. The Mayo Clinic has rights to future royalties from Celyad S.A.
Study Committees
Steering Committee: Andre Terzic, MD, PhD, Rochester, MN, USA; Jozef Bartunek, MD, PhD, Aalst, Belgium; Roger Hajjar, PhD; Guy Heyndrickx, MD, PhD, Christian Homsy, MD, Timothy Henry, MD, Thomas Povsic, MD, PhD, Marco Metra, MD, PhD, Warren Sherman, MD, Michal Tendera, MD, PhD, William Wijns, MD, PhD.
Clinical Events Committee: John R. Teerlink, MD, San Francisco, CA, USA, Chair; Tom De Potter, MD, Aalst, Belgium; Jeffrey Zimmet, MD, PhD, San Francisco, CA, USA; Gert‐Jan Laarman, MD, Tilburg, The Netherlands (peri‐procedural event adjudication)
Data Safety Monitoring Committee: Scott Waldman, MD, PhD, Philadelphia, PA, USA, Chair; Jan Tijssen, MD, Amsterdam, The Netherlands; Adriaan A. Voors, MD, PhD, Gronigen, The Netherlands; Cecilia Linde, MD, Stockholm, Sweden.
The copyright line for this article was changed on September 21, 2016 after original online publication.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després J, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 2. Braunwald E. The war against heart failure; the Lancet lecture. Lancet 2014;385:812–824. [DOI] [PubMed] [Google Scholar]
- 3. Bartunek J, Vanderheyden M, Hill J, Terzic A. Cells as biologics for cardiac repair in ischaemic heart failure. Heart 2010;96:792–800. [DOI] [PubMed] [Google Scholar]
- 4. Lin Z, Pu WT. Strategies for cardiac regeneration and repair. Sci Transl Med 2014;6:239rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sanganalmath SK, Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ Res 2013;113:810–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Telukuntla KS, Suncion VY, Schulman IH, Hare JM. The advancing field of cell‐based therapy: insights and lessons from clinical trials. J Am Heart Assoc 2013;2:e000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Menasché P. Stem cells in the management of advanced heart failure. Curr Opin Cardiol 2015;30:179–185. [DOI] [PubMed] [Google Scholar]
- 8. Terzic A, Behfar A. Regenerative heart failure therapy headed for optimization. Eur Heart J. 2014;35:1231–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jeevanantham V, Butler M, Saad A, Abdel‐Latif A, Zuba‐Surma EK, Dawn B. Adult bone marrow cell therapy improves survival and induces long‐term improvement in cardiac parameters: a systematic review and meta‐analysis. Circulation. 2012;126:551–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fisher SA, Brunskill SJ, Doree C, Mathur A, Taggart DP, Martin‐Rendon E. Stem cell therapy for chronic ischaemic heart disease and congestive heart failure. Cochrane Database Syst Rev 2014;4:CD007888. [DOI] [PubMed] [Google Scholar]
- 11. Behfar A, Crespo‐Diaz R, Terzic A, Gersh BJ. Cell therapy for cardiac repair: lessons from clinical trials. Nat Rev Cardiol 2014;11:232–246. [DOI] [PubMed] [Google Scholar]
- 12. Cogle CR, Wise E, Meacham AM, Zierold C, Traverse JH, Henry TD, Perin EC, Willerson JT, Ellis SG, Carlson M, Zhao DX, Bolli R, Cooke JP, Anwaruddin S, Bhatnagar A, da Graca Cabreira‐Hansen M, Grant MB, Lai D, Moyé L, Ebert RF, Olson RE, Sayre SL, Schulman IH, Bosse RC, Scott EW, Simari RD, Pepine CJ, Taylor DA; Cardiovascular Cell Therapy Research Network (CCTRN) . Detailed analysis of bone marrow from patients with ischemic heart disease and left ventricular dysfunction: BM CD34, CD11b, and clonogenic capacity as biomarkers for clinical outcomes. Circ Res 2014;115:867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Behfar A, Terzic A. Stem cells versus senescence: the yin and yang of cardiac health. J Am Coll Cardiol 2015;65:148–150. [DOI] [PubMed] [Google Scholar]
- 14. Behfar A, Terzic A. Stem cell in the rough: repair quotient mined out of a bone marrow niche. Circ Res 2014;115:814–816. [DOI] [PubMed] [Google Scholar]
- 15. Behfar A, Yamada S, Crespo‐Diaz R, Nesbitt JJ, Rowe LA, Perez‐Terzic C, Gaussin V, Homsy C, Bartunek J, Terzic A. Guided cardiopoiesis enhances therapeutic benefit of bone marrow human mesenchymal stem cells in chronic myocardial infarction. J Am Coll Cardiol 2010;56:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bartunek J, Behfar A, Dolatabadi D, Vanderheyden M, Ostojic M, Dens J, El Nakadi B, Banovic M, Beleslin B, Vrolix M, Legrand V, Vrints C, Vanoverschelde JL, Crespo‐Diaz R, Homsy C, Tendera M, Waldman S, Wijns W, Terzic A. Cardiopoietic stem cell therapy in heart failure: the C‐CURE (Cardiopoietic stem Cell therapy in heart failURE) multicenter randomized trial with lineage‐specified biologics. J Am Coll Cardiol 2013;61:2329–2338. [DOI] [PubMed] [Google Scholar]
- 17. Packer M. Proposal for a new clinical end point to evaluate the efficacy of drugs and devices in the treatment of chronic heart failure. J Card Fail 2001;7:176–182. [DOI] [PubMed] [Google Scholar]
- 18. Finkelstein DM, Schoenfeld DA. Combining mortality and longitudinal measures in clinical trials. Stat Med 1999;18:1341–1354. [DOI] [PubMed] [Google Scholar]
- 19. Rector TS, Cohn JN. Assessment of patient outcome with the Minnesota Living With Heart Failure Questionnaire: reliability and validity during a randomized, double‐blind, placebo‐controlled trial of pimobendan. Pimobendan multicenter research group. Am Heart J 1992;124:1017–1025. [DOI] [PubMed] [Google Scholar]
- 20. Behfar A, Latere JP, Bartunek J, Homsy C, Daro D, Crespo‐Diaz RJ, Stalboerger PG, Steenwinckel V, Seron A, Redfield MM, Terzic A. Optimized delivery system achieves enhanced endomyocardial stem cell retention. Circ Cardiovasc Interv 2013;6:710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, Verani MS. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart: a statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Circulation 2002;105:539–5422. [DOI] [PubMed] [Google Scholar]
- 22. Povsic TJ, O'Connor CM, Henry T, Taussig A, Kereiakes DJ, Fortuin FD, Niederman A, Schatz R, Spencer Rt, Owens D, Banks M, Joseph D, Roberts R, Alexander JH, Sherman W. A double‐blind, randomized, controlled, multicenter study to assess the safety and cardiovascular effects of skeletal myoblast implantation by catheter delivery in patients with chronic heart failure after myocardial infarction. Am Heart J 2011;162:654–662. [DOI] [PubMed] [Google Scholar]
- 23. Kastrup J, Jorgensen E, Ruck A, Tagil K, Glogar D, Ruzyllo W, Botker HE, Dudek D, Drvota V, Hesse B, Thuesen L, Blomberg P, Gyongyosi M, Sylven C. Direct intramyocardial plasmid vascular endothelial growth factor‐a165 gene therapy in patients with stable severe angina pectoris: a randomized double‐blind placebo‐controlled study—The Euroinject One trial. J Am Coll Cardiol 2005;45:982–988. [DOI] [PubMed] [Google Scholar]
- 24. Bhatt DL, Kandzari DE, O'Neill WW, D'Agostino R, Flack JM, Katzen BT, Leon MB, Liu M, Mauri L, Negoita M, Cohen SA, Oparil S, Rocha‐Singh K, Townsend RR, Bakris GL. A controlled trial of renal denervation for resistant hypertension. N Engl J Med 2014;370:1393–1401. [DOI] [PubMed] [Google Scholar]
- 25. Zannad F, Stough WG, Piña IL, Mehran R, Abraham WT, Anker SD, De Ferrari GM, Farb A, Geller NL, Kieval RS, Linde C, Redberg RF, Stein K, Vincent A, Woehrle H, Pocock SJ. Current challenges for clinical trials of cardiovascular medical devices. Int J Cardiol 2014;175:30–37. [DOI] [PubMed] [Google Scholar]
- 26. Zannad F, Garcia AA, Anker SD, Armstrong PW, Calvo G, Cleland JG, Cohn JN, Dickstein K, Domanski MJ, Ekman I, Filippatos GS, Gheorghiade M, Hernandez AF, Jaarsma T, Koglin J, Konstam M, Kupfer S, Maggioni AP, Mebazaa A, Metra M, Nowack C, Pieske B, Pina IL, Pocock SJ, Ponikowski P, Rosano G, Ruilope LM, Ruschitzka F, Severin T, Solomon S, Stein K, Stockbridge NL, Stough WG, Swedberg K, Tavazzi L, Voors AA, Wasserman SM, Woehrle H, Zalewski A, McMurray JJ. Clinical outcome endpoints in heart failure trials: a European Society of Cardiology Heart Failure Association consensus document. Eur J Heart Fail 2013;15:1082–1094. [DOI] [PubMed] [Google Scholar]
- 27. Alla F, Briancon S, Guillemin F, Juilliere Y, Mertes PM, Villemot JP, Zannad F. Self‐rating of quality of life provides additional prognostic information in heart failure. Insights into the Epical study. Eur J Heart Fail 2002;4:337–343. [DOI] [PubMed] [Google Scholar]
- 28. Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac‐resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004;350:2140–2150. [DOI] [PubMed] [Google Scholar]
- 29. Bellenger NG, Davies LC, Francis JM, Coats AJ, Pennell DJ. Reduction in sample size for studies of remodeling in heart failure by the use of cardiovascular magnetic resonance. J Cardiovasc Magn Res 2000;2:271–278. [DOI] [PubMed] [Google Scholar]
- 30. Lefevre T, Kappetein AP, Wolner E, Nataf P, Thomas M, Schachinger V, De Bruyne B, Eltchaninoff H, Thielmann M, Himbert D, Romano M, Serruys P, Wimmer‐Greinecker G. One year follow‐up of the multi‐centre European Partner Transcatheter Heart Valve study. Eur Heart J 2011;32:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anker SD, Agewall S, Borggrefe M, Calvert M, Jaime Caro J, Cowie MR, Ford I, Paty JA, Riley JP, Swedberg K, Tavazzi L, Wiklund I, Kirchhof P. The importance of patient‐reported outcomes: a call for their comprehensive integration in cardiovascular clinical trials. Eur Heart J 2014;35:2001–2009. [DOI] [PubMed] [Google Scholar]