

Abstract
The objective of this study was to assess the relationship between short‐term and long‐term treatment effects measured by the American College of Rheumatology (ACR) 50 responses and to assess the feasibility of predicting 6‐month efficacy from short‐term data. A rheumatoid arthritis (RA) database was constructed from 68 reported trials. We focused on the relationship between 3‐ and 6‐month ACR50 treatment effects and developed a generalized nonlinear model to quantify the relationship and test the impact of covariates. The ΔACR50 at 6 months strongly correlated with that at 3 months, moderately correlated with that at 2 months, and only weakly correlated with results obtained at <2 months. A scaling factor that reflected the ratio of 6‐ to 3‐month treatment effects was estimated to be 0.997, suggesting that the treatment effects at 3 months are approaching a “plateau.” Drug classes, baseline Disease Activity Score in 28 Joints, and the magnitude of control arm response did not show significant impacts on the scaling factor. This work quantitatively supports the empirical clinical development paradigm of using 3‐month efficacy data to predict long‐term efficacy and to inform the probability of clinical success based on early efficacy readout.
Keywords: rheumatoid arthritis, ACR50, meta‐analysis, efficacy
Rheumatoid arthritis (RA), an autoimmune disease, affects about 1.3 million adults in the United States alone.1 The strategy for treating RA has evolved from reducing joint pain toward altering disease progression and improving both radiographic and functional outcomes. Corticosteroids and nonsteroidal anti‐inflammatory drugs (NSAIDs) that reduce acute inflammation have a short onset of action, but they do not significantly change disease progression. In contrast, disease‐modifying antirheumatic drugs (DMARDs) not only treat symptoms but also slow progressive joint destruction. Methotrexate (MTX) is the most commonly used nonbiological DMARD. Nine biologic DMARDs approved for RA treatment have provided additional treatment options for patients who show an inadequate response to traditional DMARD agents.2 They are classified into different classes based on their mechanisms of action. They include anti‐TNFs (tumor necrosis factors) (adalimumab, certolizumab, etanercept, golimumab, and infliximab), anti‐interleukin (IL)‐1 (anakinra), anti‐IL‐6 receptor (tocilizumab), T‐cell costimulatory blocking agents (abatacept), and B cell–depleting agents (rituximab). More recently, tofacitinib, a Janus‐associated kinase (JAK) inhibitor, became the first approved orally active small‐molecule DMARD for RA.3, 4 Other oral kinase inhibitors, such as spleen tyrosine kinase (SYK) inhibitors, are also under clinical development for the treatment of RA.5 Aiming for higher efficacies and better safety profiles, many other efforts are under way to explore additional drug targets for RA therapies.6
The development of a new therapy for the treatment of RA is costly. Generally, in early‐stage proof‐of‐concept (PoC) RA trials, the 3‐month efficacy is used as the primary endpoint, whereas the 6‐month efficacy result is relied on as the primary endpoint for late‐stage clinical trials. By leveraging the information from a large RA database derived from multiple therapeutic intervention trials, we aimed to quantitatively test such an empirical practice and to provide insight into the ability to predict long‐term clinical efficacy based on short‐term data. A model‐based meta‐analysis was conducted to explore the treatment‐effect relationships between early time points (≤3 months) and late time points (6 to 12 months). The ratio between the treatment effects obtained at the 2 time points was quantified. Given the observed differences in the onset time of drug response among different drug classes, the impact of drug class was evaluated and tested as a covariate of the scaling factor.
The American College of Rheumatology (ACR) has defined response criteria for RA treatments, and they are termed ACR scores. They include ACR20, ACR50, and ACR70, representing 20%, 50%, and 70% improvements in disease activity after treatment.7 Currently, ACR20 is one of the most commonly used primary efficacy endpoints in RA clinical trials leading to approval. A model‐based meta‐analysis was developed to examine longitudinal ACR20 for currently approved biologics, and it appeared that ACR20 reached a maximum treatment effect at approximately 3 months for most compounds.8 Given the relatively high efficacy of the more effective biological DMARDs, ACR20 may not represent the optimal clinical response outcome measure for rheumatologists.9 In contrast, ACR50 appears to be a better endpoint to distinguish clinically significant treatment effects between treatment arms for contemporary RA trials.10 Therefore, ACR50 was evaluated in our meta‐analysis.
The Disease Activity Score in 28 joints (DAS28) is another clinical efficacy endpoint that provides a quantitative measure of disease activity. In contrast to categorical ACR scores, it is a continuous variable. It is calculated using a formula based on the number of tender and swollen joints within a group of 28 joints and the level of acute‐phase reactants (eg, erythrocyte sedimentation rate [ESR] or C‐reactive protein [CRP]). Because the DAS28 is a continuous variable, the sample size can be greatly reduced for a desired power for efficacy assessment. Thus, for extended interest, DAS28 was also included as one of the exploratory analysis endpoints.
To our knowledge, a model‐based meta‐analysis of the relationship between short‐term and long‐term clinical efficacies in RA trials has not been previously undertaken. Our meta‐analysis provides a quantitative assessment based on a large pool of clinical data and demonstrates a robust correlation between 3‐month and 6‐month (or longer) ACR50 readouts.
Methods
Data Sources and Data Sets Used in the Analysis
The primary data sources were controlled clinical trials for the treatment of RA. The data were published in the medical literature or were available from the FDA or accessible as EMA drug labeling information published between 1994 and 2012. Other data sources included meeting abstracts and presentations. The objective was to include all the information on biologic DMARDs as well as synthetic DMARDs currently approved or in development for RA. Only randomized controlled RA trials with at least 12 weeks of treatment were included. Collected clinical efficacy endpoints included ACR20, ACR50, ACR70, and DAS28 (DAS28‐ESR). A master data set was generated by filtering the database for trials reporting ACR50 efficacy for at least 2 time points (2‐week, 1‐, 2‐, 3‐, 6‐, or 12‐month points) as well as trials reporting the corresponding placebo efficacies. This resulted in a total of 68 trials comprised of 13 drug classes and 28 drugs with which the exploratory analysis was conducted (Table 2).
Table 2.
Data Sets Used in Different Analysis Scenarios*
| Data Set | Analysis Scenarios | Number of Trials | Number of Arms | Number of Patients | Number of Drug Classes | Number of Drugs |
|---|---|---|---|---|---|---|
| 1 | ACR50 exploratory assessment | 68 | 257 | 27 176 | 13 | 28 |
| 2 | 3‐ to 6‐ month ACR50 | 40 | 117 | 17 330 | 7 | 12 |
| 3 | 3‐ to 12‐ month ACR50 | 11 | 27 | 5606 | 5 | 7 |
| 4 | 2‐ to 6‐ month ACR50 | 32 | 87 | 15 152 | 6 | 11 |
| 5 | DAS28 exploratory assessment | 22 | 69 | 6560 | 6 | 10 |
| 6 | 1‐ to 6‐ month DAS28 | 12 | 29 | 4551 | 6 | 7 |
| 7 | Additional data for extended analysis | 4 | 9 | 901 | 2 | 2 |
*Data were included only if the trial had ACR50 or DAS28 reported at both time points of interest for each scenario.
A final data set was further filtered for trials with both 3‐month and 6‐month ACR50 outcomes. Trials with placebo patients switched to treatment at 3 months were excluded from the data set. Seven drug classes and 12 drugs were encompassed by this analysis, including (1) anti‐TNF: etanercept, infliximab, adalimumab, certolizumab, golimumab; (2) anti‐IL‐1: anakinra; (3) T‐cell costimulatory blocking agents: abatacept; (4) B‐cell‐depleting agents: rituximab; (5) anti‐IL‐6 receptor: tocilizumab; (6) JAK kinase inhibitor: tofacitinib; and (7) SYK kinase inhibitor: fostamatinib. Model‐based analysis was performed on the final data set (Table 1). Similar analyses were also performed on relationships between 3‐ and 12‐month ACR50 values as well as 2‐ and 6‐month ACR50 values with separate filtered data sets specified in Table 2.
Table 1.
Summary of the Final Data Set Used to Model the Relationship of the ACR50 Treatment Effect Between 3 Months and 6 Months
| Name of Drug | Class of Drug | Number of Trials | Number of Arms | Number of Patients | References |
|---|---|---|---|---|---|
| Adalimumab | anti‐TNF | 6 | 20 | 2729 | 14, 15, 16, 17, 18, 19 |
| Certolizumab | anti‐TNF | 5 | 13 | 1977 | 20, 21, 22, 23, 24 |
| Etanercept | anti‐TNF | 6 | 13 | 1553 | 25, 26, 27, 28, 29, 30 |
| Golimumab | anti‐TNF | 3 | 10 | 1308 | 31, 32, 33 |
| Anakinra | anti‐IL‐1 | 1 | 6 | 419 | 34 |
| Tocilizumab (Actemra) | anti‐IL‐6R | 7 | 16 | 4326 | 35, 36, 37, 38, 39, 40, 41 |
| Ocrelizumab | anti‐CD20 | 1 | 4 | 145 | 42 |
| Ofatumumab | anti‐CD20 | 1 | 2 | 260 | 43 |
| Rituximab | anti‐CD20 | 2 | 5 | 1247 | 44, 45 |
| Abatacept | anti‐CD28 | 5 | 12 | 2069 | 46, 47, 48, 49, 50, 51 |
| Fostamatinib | SYK kinase inhibitor | 1 | 3 | 457 | 52 |
| Tofacitinib | JAK kinase inhibitor | 2 | 13 | 840 | 53, 54 |
| (MTX) | (DMARD) | 28 | |||
| TOTAL | 40 | 117 | 17 330 |
Analysis Methodology
The clinical response ACR50 is a summary level result derived from individual patient binary responses (yes or no) in each trial arm. The number of patients (X) achieving ACR50 responses for the jth arm of the ith trial at time point t with sample size N (Nij) was assumed to follow a binomial distribution at the 3‐month or 6‐month time point:
The probability of a patient achieving ACR50 (Pijt) was restricted to the range of 0 to 1, and a logit transformation was applied to the ACR50 value. Because the sample size (Nij) in each trial arm was sufficiently large, a large sample normal approximation to the binomial likelihood was used to fit the ACR50 response with the following model with variance P(1 – P)/N for the mean incidence:
where the function f{x} is the inverse logit transformation, f{x} = exp(x)/[1 + exp(x)], to constrain probabilities between 0 and 1. Parameters θit are the logit‐transformed probabilities of ACR50 for the control arm in the ith trial at time point t, and trtij are the treatment effects for the jth treated arm from the ith trial. Parameter λt is the scaling factor that represents the ratio of the treatment effects, and we set λ at 3 months, ie, λ3 = 1. The 3‐month and 6‐month relationships can also be expressed in a log odds ratio (OR) format:
where OR3 and OR6 are the odds ratios of the treated arm to the control arm in achieving ACR50 at the 3‐month point and the 6‐month point, respectively. Although ΔACR50 is more frequently used clinically by rheumatologists, the treatment effect expressed here in term of odds ratio to the control is found to be more accurately analyzed for the binomially distributed ACR50. Results of ACR50 were analyzed using nonlinear regression implemented in the generalized nonlinear least‐squares (gnls) and nonlinear mixed‐effect (nlme) routines in S‐PLUS 8.2.
Due to the large variability of the ACR50 response in the control arm and because of the primary interest in the scaling factor of the treatment effect, a different fixed effect was estimated for each control group in every trial and time point. The scaling factor was estimated with both a fixed effect and a random intertrial variability. During the model development, the intertrial variability was not statistically significant; therefore, the intertrial variability was removed from the final model, and only a fixed effect was estimated for the scaling factor. There was no significant between‐trial heterogeneity in the scaling factor. The interarm variability was accounted for by the binomial distribution of the response variable.
The dependencies of the scaling factor λ on various covariates were evaluated in the model. These covariates included the following: (1) drug classes, 7 drug classes were tested individually or as either “TNF blockers” or “non‐TNF blockers”; (2) mean value of patient baseline DAS28 for each arm, a continuous variable measured at predose to reflect the severity of the disease; (3) the patient population, a categorical variable that separated trials into “MTX naive (N = 4),” “DMARDs‐IR (inadequate responder) or MTX‐IR (N = 33),” or “TNF‐IR (N = 3)” groups; (4) the mean value of the disease duration in years for each arm; (5) the magnitude of the control arm response; (6) other demographic variables such as mean body weights and sex. Model selection was based on a log‐likelihood ratio test at an acceptance level of P = 0.05.
Additional data sets from 4 Roche RA clinical trials (including data from 901 patients treated with 2 drug classes: MAb1 and MAb2, unpublished data) were obtained after the meta‐analysis. They were combined and used as an external validation data set to examine the relationship between treatment effects at 3‐month and 6‐month data points.
We also explored the relationship between DAS28 readouts at early and late time points. DAS28‐ESR results were included. Our interest stemmed from the fact that using it for efficacy assessment permitted a smaller sample size for a desired statistical power. A collection of 22 trials that reported DAS28 at either of 2 time points (2 weeks, 1, 2, 3, or 6 months) was included in this exploratory analysis (Table 2).
Results
Overview of the Short‐Term to Long‐Term Efficacy Relationships
The initial exploratory analysis utilized a master database that contained a total of 68 randomized trials in RA, representing data from more than 27 000 patients treated with 13 different drug classes. These trials reported ACR50 data from at least 2 of the following time points: 2 weeks, 1 month, 2 months, 3 months, 6 months, or 12 months. To explore the relationship between ACR50 treatment effects at early and later time points, a matrix of pairwise scatter plots was made for ΔACR50 (difference from control) (Figure 1a) or log OR (treated vs control) (Figure 1b) among these time points. The treatment effect after 3 months was observed to be strongly correlated with that seen after 6 months (with a correlation coefficient R2 = 0.90) or 12 months (R2 = 0.95). The treatment effect after 2 months was moderately correlated with that at 6 months (R2 = 0.61) or 12 months (R2 = 0.76), whereas treatment effects measured earlier than 2 months had much weaker correlations with either 6‐ or 12‐month efficacy (R2 ≤ 0.32). These observations encouraged us to further characterize the correlation between 3‐month and 6‐month treatment effects in ACR50.
Figure 1.
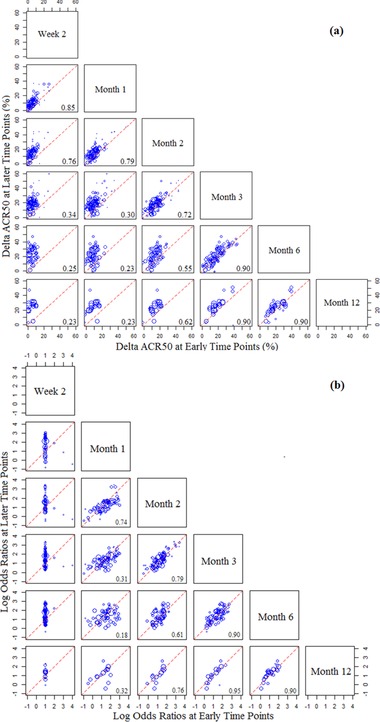
Pairwise correlations of the treatment effects (ACR50) between early and late time points. The treatment effect for each treated arm is expressed either in linear format (ΔACR50, panel A) or in log OR format (odds ratio of treated vs control; panel B). In each scatter plot the symbols represent the treatment effect at a later time point (specified at the right end of each row) vs that at an early time point (specified at the top of each column). Symbol size reflects the sample size for each treatment arm. The value of R2 for paired values is indicated at the right bottom corner. Unity (dashed) lines represent equal efficacies of the early and late time points, whereas points above the lines suggest higher efficacies at the late time point, and points below the lines suggest higher efficacies at the early time point.
Observed Relationship Between 3‐Month and 6‐Month ACR50 Treatment Effects
A data set that included only 3‐month (12–14 weeks) and 6‐month (24–26 weeks) ACR50 values was filtered from the master data set. A total of 40 randomized controlled trials were included in the final data set, representing data from 17,330 patients who were treated with 7 different drug classes (Table 1).
The trials included in the database ranged from phase 1 to phase 3, and the control arms differed from placebo control to active comparator control. In that database we observed a strong correlation of either ΔACR50 (Figure 2a) or log OR values (Figure 2b) between readouts at 3‐month points and 6‐month points, and the variability was moderate. Most data points were distributed around and centered along the unity line, indicating that treatment effects were similar at both time points.
Figure 2.
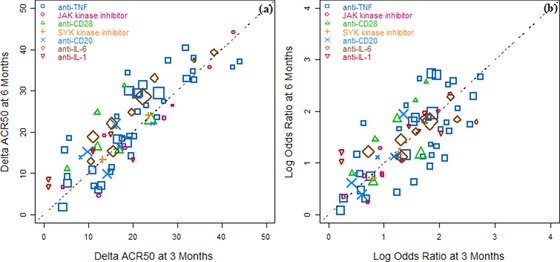
Strong correlations were observed between the treatment effects at 3 months and 6 months. (a) Scatter plot of the linear form of treatment effect, ΔACR50, at 6 months vs 3 months. (b) Scatter plot of log odds ratios of treated vs control at 6 months vs 3 months. The symbols represent different drug classes. The symbol size reflects the sample size for each treatment arm. The unity line represents equal efficacies at the 2 time points. Both plots suggest a similar mean effect after 3 months and 6 months for the trials tested in the database. With this data set the scaling factor was estimated to be 0.997 (95%CI: 0.903, 1.09). ACR50, American College of Rheumatology score 50; CI, confidence interval.
Model Development and Covariate Tests
A generalized nonlinear model was used to quantify the relationship between early time points (2 or 3 months) and late time points (6‐ or 12‐month) based on ACR50 data. Both the control arm and the treated arm were fitted simultaneously in the model. A scaling factor (λ) was used in the model to correlate the 3‐month efficacy to the 6‐month efficacy. The scaling factor was estimated to be 0.997 with 95%CI of (0.903, 1.09), indicating a consistently strong correlation between 3‐month and 6‐month ACR50 treatment effects in these clinical trials.
We tested covariates that might have impacted the relationship. These included drug class, specific patient populations, patient baseline disease severity (indicated by baseline DAS28 level), average disease duration, the magnitude of the control arm response, as well as various demographic variables such as age and sex. The observed “scaling factor,” which is the ratio of 6‐month to 3‐month log OR of “treated to control,” was calculated in each case. The ratios are plotted in Figure 3 for different covariates such as drug classes (Figure 3a), baseline DAS28 levels (Figure 3b), magnitude of ACR50 response of the control arm (Figure 3c), and disease duration prior to the treatment (Figure 3d). These plots indicate that the scaling factor was independent of any of the covariates evaluated. Thus, none of the evaluated covariates showed a strong effect.
Figure 3.
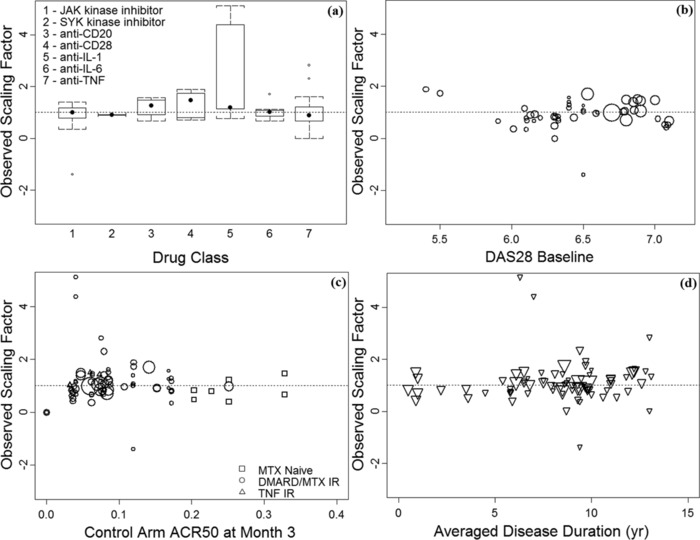
Observed 3‐ to 6‐month treatment scaling factor is independent of various covariates tested. Scaling factor λ was not statistically significantly impacted by the various covariates tested, including (a) drug class, (b) baseline DAS28 score, (c) the magnitude of control arm ACR50 at 3 months, and (d) averaged patient disease duration. Dotted line shows the model‐predicted scaling factor (λ = 0.997). Symbol size in scatter plots reflects the sample size in each treatment arm.
Extended Analysis
To validate findings from this meta‐analysis, we obtained 4 additional unpublished data sets (N = 901, Table 2) from Roche clinical trials studying treatments of RA. The objective of the external validation was to further confirm the consistent trend of mean scaling factor that is independent of drug class. Consistent with previous findings, the ACR50 treatment effects observed in these 4 trials at 3 months and 6 months were highly correlated (correlation coefficient R2 = 0.893). The scatter plots in Figure 4 demonstrated a consistent relationship between the 3‐month and 6‐month ACR50 responses for these 4 trials as compared with the rest of the data points from the final database. When the data from these 4 trials were pooled with the meta‐analysis final database, the scaling factor was reestimated to be 1.01 with 95%CI (0.91, 1.10), very similar to the initial estimate based on published data.
Figure 4.
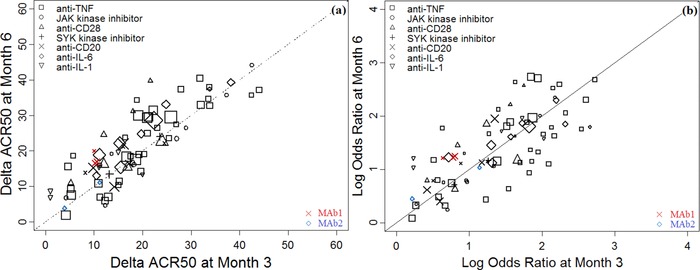
Additional data sets further validated a consistent relationship between treatment effects determined after 3 months and 6 months. Scatter plots overlay the additional data (MAb1 and MAb2) with the model‐evaluated data set 2 (Table 2) for (a) the ΔACR50 readouts at 6 months vs 3 months and (b) the log odds ratios of treated vs control at 6 months vs 3 months. The unity line represents equal efficacies at the 2 time points. The symbols represent different drug classes, and solid symbols represent the data points from the external data set. The symbol size reflects the sample size for each treatment arm. With the additional data set, the scaling factor was reestimated to be 1.01 (95%CI: 0.91, 1.10), very similar to the estimated values shown in Figure 2.
As stated in the Methods section, an exploratory analysis of DAS28 readouts was also conducted using a similar approach to that for ACR50. We constructed a scatter plot of pairwise ΔΔDAS28 (differences between the treatment arm and the control arm of baseline‐adjusted DAS28 values) at early time points and late time points (Figure 5). In contrast to ΔACR50, a strong relationship between early and late ΔΔDAS28 readouts was evident as early as 1 month, suggesting that a 1‐month ΔΔDAS28 may be indicative of the 3‐ or 6‐month ΔΔDAS28 outcomes. Due to the small number of trials (n = 12) reporting DAS28 at both the 1‐month and 6‐month readouts, further covariate testing was not performed.
Figure 5.
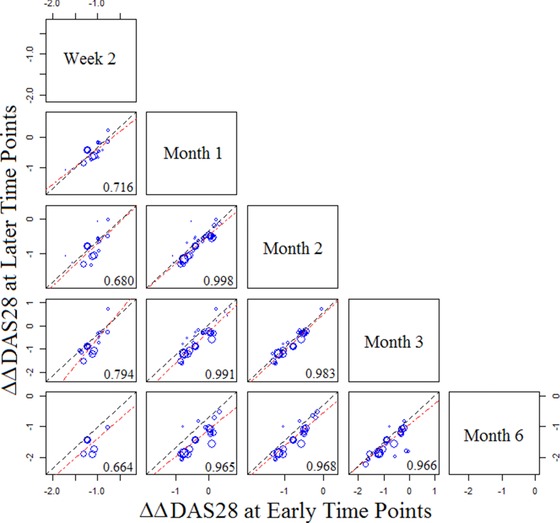
Pairwise scatter plots of ΔΔDAS28 between early and late time points. In each plot the symbols represent the observed ΔΔDAS28 at a later time point (specified at the far right end of each row) vs an early time point (specified at the top of each column). Symbol size reflects the sample size of each treatment arm. Unity (dashed) lines represent equal efficacies at the early and late time points; points above suggest a higher efficacy at a late time point, and points below suggest a higher efficacy at an early time point. DAS28, disease activity score in 28 joints.
Discussion
Meta‐analyses have been widely used to facilitate the development of drugs for many diseases.11 These analyses are useful for interpreting safety and efficacy results from many trials. They also allow indirect comparisons of different treatment options as well as the characterization of the relationships between different endpoints. Mandema et al.2 applied a nonlinear regression model to compare the dose‐response relationship among anti‐TNFs and other biologics for ACR20, 50, and 70 efficacy endpoints. Demin et al used a longitudinal model to quantify ACR20 responses and to support the decision‐making process for the development of an in‐house biologic drug for RA.8 Other analyses compared efficacy and safety data between treatments using relative risk ratios or odds ratios to a common comparator.12, 13 The focus of our analysis was to quantitatively assess the relationship between short‐term and long‐term treatment effects based on a large RA database.
Based on 40 controlled randomized clinical trials for treatment of RA, we demonstrated that the 3‐month readout of ACR50 is a reasonable predictor of a drug's long‐term efficacy. With diverse mechanisms of action for various biological DMARDs, different treatment durations might be required to achieve a maximal effect. Thus, one would assume that a longer time point might be needed to demonstrate maximal efficacy. However, the results of our meta‐analysis suggested that the treatment effects measured by the ACR50 at 3 months were highly correlated with those at 6 months, and the scaling factor for the treatment effect (in term of odds ratios to the control) was close to 1. Even though the absolute value of ACR50 may be higher at 6 months than at 3 months, the treatment effect after adjusting for the control arm effects was very similar at both time points (Figure 2), and this result held true for all drug classes evaluated. Four unpublished data sets from additional RA trials were used as an external validation, and they further confirmed a consistent trend regardless of drug classes. Therefore, such a trend can be widely used to guide drug development of new drugs.
The fact that the 3‐ to 6‐month efficacy scaling factor was close to 1 implies that efficacy is approaching a “plateau” after 3 months of treatment. To further confirm this conclusion, a similar approach was applied to quantify the relationship between 3‐ and 12‐month ACR50 readouts. Consistent with earlier findings, a high correlation was also observed between 3‐ and 12‐month readouts, and the estimated scaling factor (λ2) was also centered close to 1 (λ2 = 1.19 with 95%CI values of [0.971, 1.410]). Although there was noise for the scaling factor for some individual trials, the trend of the estimated scaling factors suggested that 3‐month efficacy in general is highly representative of the long‐term efficacy. Hence, this outcome offers a quantitatively robust rationale for decision making based on short‐term clinical outcome in the development of therapeutics for RA treatment.
Our analysis supports the empirical practice seen in many PoC studies in which 3‐month efficacy results were heavily relied on to make “GO/NO GO” decisions for molecules under development. Such a decision entails risk in cases when short‐term efficacy readouts do not translate well into robust long‐term clinical efficacy. Our meta‐analysis lends credibility to the industry approach by demonstrating a highly correlated relationship between ACR50 treatment effects at 3‐ and 6‐month time points using a model‐based approach.
Efforts were also made to determine whether readouts at even earlier time points, such as 1 or 2 months, could robustly project long‐term outcomes, hence allowing companies to accelerate drug development. Results showed that after 1 month treatment effects measured by ACR50 correlated only weakly with that seen at 6 months. In contrast, a moderate correlation was observed between ΔACR50 at 2 months and 6 months (Figure 1b). The scaling factor was estimated to be 1.05 based on 32 trials in 5 drug classes (that had both 2‐ and 6‐month readouts available), and it was not affected by the drug class. Therefore, the ACR50 efficacy measured as early as 2 months may provide an insightful projection regarding whether the treatment has any meaningful benefit compared with the control arm.
In contrast to the ACR50 as a categorical endpoint, the DAS28 is a continuous variable. DAS28 is becoming more attractive in early‐stage clinical trials because the trial sample size can be greatly reduced for the desired power level. Our exploratory analysis of the DAS28 readout at different times suggested that DAS28 readouts as early as 1 month appeared to correlate strongly with long‐term efficacy readouts (Figure 5). In addition, DAS28 demonstrated faster response kinetics than ACR50. It is speculated that key components of the DAS28 score, ie, the CRP or ESR, contribute to the rapid demonstration of the treatment effect because they reflect acute‐phase responses to inflammation. It should be noted that DAS28 data are not as rich as ACR50 data in the database, so the outcome of this assessment needs to be interpreted with caution.
There are some caveats to the analysis. First, in order to focus on quantifying the relationship between treatment effects of 2 readout time points and to avoid any impact of control arm effects, a nonparametric approach was used for every control group in every trial and time point. The advantage is that no arbitrary assumption had to be made on the distribution of the control arm response; hence, the within‐trial relationship between 3‐ and 6‐month responses can be analyzed in the most unbiased manner. Second, the model is more readily applicable to predictions of a later time point response using earlier readouts within the same trial rather than across trials. In cases of cross‐trial prediction, intertrial variability should be taken into account. The present meta‐analysis included 3 trials of small‐molecule nonconventional DMARDs, including tofacitinib and fostamatinib. The limited data from the available trials showed no significant differences in the relationship between 3‐ and 6‐month treatment readouts compared with biologics. Future robust analyses may be carried out when sufficient clinical data are available.
In conclusion, our study is the first that quantitatively demonstrated a strong relationship between 3‐month and 6‐month clinical efficacy readouts in RA clinical trials for different classes of therapies using model‐based meta‐analysis. A scaling factor that represented ratios of the treatment effect from 3 months to 6 months was estimated to be 0.997 (95%CI 0.903, 1.09), suggesting that the treatment effect at 3 months was reaching an “efficacy plateau” and could well predict the 6‐month outcome. It is beneficial to reduce the duration of early clinical trials assessing proof of concept for a particular mechanism of action. Thus, our findings support empirical practices seen in most PoC studies in which 3‐month results are utilized. Furthermore, even earlier time points (eg, 2‐month outcome) can also be considered to provide insight into drug efficacy and be used for an early evaluation of the clinical success rate. Finally, it is recognized that early efficacy readouts may be helpful at the PoC stage, but this does not supplant the need for longer‐term safety monitoring that must be accrued in the later stages of clinical development.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
supplementary material
Acknowledgments
The authors would like to thank Stuart Lacey, Senior Statistical Scientist from the Department of Statistics at Roche Pharmaceuticals, Welwyn, UK, for his assistance in obtaining the clinical trial data as external validation data sets. The authors also thank Williams Dexter Kennedy, Senior Medical Director from Clinical Sciences at Genentech, Inc, South San Francisco, CA, for providing comments during the development of this manuscript. The authors thank Anshin Biosolutions Corp for providing editing and formatting assistance.
Disclosures
Yehong Wang, Rui Zhu, Jim Xiao, Jin Y. Jin, and Meina T. Tang are all employees of Genentech (a member of the Roche Group) and also hold stock in F. Hoffmann‐La Roche. Jim Xiao and John C. Davis Jr are former employees of Genentech and have no financial connections. Jaap W. Mandema has received consulting fees from Genentech.
References
- 1. Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58(1):15–25. [DOI] [PubMed] [Google Scholar]
- 2. Mandema JW, Salinger DH, Baumgartner SW, Gibbs MA. A dose‐response meta‐analysis for quantifying relative efficacy of biologics in rheumatoid arthritis. Clin Pharmacol Ther. 2011;90(6):828–835. [DOI] [PubMed] [Google Scholar]
- 3. Rakieh C, Conaghan PG. Tofacitinib for treatment of rheumatoid arthritis. Adv Ther. 2013;30(8):713–726. [DOI] [PubMed] [Google Scholar]
- 4. van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve‐month data from a twenty‐four‐month phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):559–570. [DOI] [PubMed] [Google Scholar]
- 5. Simmons DL. Targeting kinases: a new approach to treating inflammatory rheumatic diseases. Curr Opin Pharmacol. 2013;13(3):426–434. [DOI] [PubMed] [Google Scholar]
- 6. Malviya G, Salemi S, Lagana B, Diamanti AP, D'Amelio R, Signore A. Biological therapies for rheumatoid arthritis: progress to date. BioDrugs. 2013;27(4):329–345. [DOI] [PubMed] [Google Scholar]
- 7. Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–735. [DOI] [PubMed] [Google Scholar]
- 8. Demin I, Hamren B, Luttringer O, Pillai G, Jung T. Longitudinal model‐based meta‐analysis in rheumatoid arthritis: an application toward model‐based drug development. Clin Pharmacol Ther. 2012;92(3):352–359. [DOI] [PubMed] [Google Scholar]
- 9. Pincus T, Stein CM. ACR 20: clinical or statistical significance? Arthritis Rheum. 1999;42(8):1572–1576. [DOI] [PubMed] [Google Scholar]
- 10. Chung CP, Thompson JL, Koch GG, Amara I, Strand V, Pincus T. Are American College of Rheumatology 50% response criteria superior to 20% criteria in distinguishing active aggressive treatment in rheumatoid arthritis clinical trials reported since 1997? A meta‐analysis of discriminant capacities. Ann Rheum Dis. 2006;65(12):1602–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mould DR. Model‐based meta‐analysis: an important tool for making quantitative decisions during drug development. Clin Pharmacol Ther. 2012;92(3):283–286. [DOI] [PubMed] [Google Scholar]
- 12. Schoels M, Aletaha D, Smolen JS, Wong JB. Comparative effectiveness and safety of biological treatment options after tumour necrosis factor α inhibitor failure in rheumatoid arthritis: systematic review and indirect pairwise meta‐analysis. Ann Rheum Dis. 2012;71(8):1303–1308. [DOI] [PubMed] [Google Scholar]
- 13. Aaltonen KJ, Virkki LM, Malmivaara A, Konttinen YT, Nordstrom DC, Blom M. Systematic review and meta‐analysis of the efficacy and safety of existing TNF blocking agents in treatment of rheumatoid arthritis. PLoS One. 2012;7(1):e30275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Furst DE, Schiff MH, Fleischmann RM, et al. Adalimumab, a fully human anti tumor necrosis factor‐alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis). J Rheumatol. 2003;30(12):2563–2571. [PubMed] [Google Scholar]
- 15. Weinblatt ME, Keystone EC, Furst DE, et al. Adalimumab, a fully human anti‐tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45. [DOI] [PubMed] [Google Scholar]
- 16. Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti‐tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo‐controlled, 52‐week trial. Arthritis Rheum. 2004;50(5):1400–1411. [DOI] [PubMed] [Google Scholar]
- 17. van de Putte LB, Atkins C, Malaise M, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mease PJ, Gladman DD, Ritchlin CT, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double‐blind, randomized, placebo‐controlled trial. Arthritis Rheum. 2005;52(10):3279–3289. [DOI] [PubMed] [Google Scholar]
- 19. Miyasaka N, CHANGE Study Investigators. Clinical investigation in highly disease‐affected rheumatoid arthritis patients in Japan with adalimumab applying standard and general evaluation: the CHANGE study. Mod Rheumatol. 2008;18(3):252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamamoto K, Takeuchi T, Yamanaka H, et al. Efficacy and safety of certolizumab pegol plus methotrexate in Japanese rheumatoid arthritis patients with an inadequate response to methotrexate: the J‐RAPID randomized, placebo‐controlled trial. Mod Rheumatol. 2014;24(5):715–724. [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto K, Takeuchi T, Yamanaka H, et al. Efficacy and safety of certolizumab pegol without methotrexate co‐administration in Japanese patients with native rheumatoid arthritis: the HIKARI randomized, placebo‐controlled trial. Mod Rheumatol. 2014;24(4):552–560. [DOI] [PubMed] [Google Scholar]
- 22. Smolen J, Landewe RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6):797–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fleischmann R, Vencovsky J, van Vollenhoven RF, et al. Efficacy and safety of certolizumab pegol monotherapy every 4 weeks in patients with rheumatoid arthritis failing previous disease‐modifying antirheumatic therapy: the FAST4WARD study. Ann Rheum Dis. 2009;68(6):805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keystone E, van der Heijde D, Mason D, et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty‐two‐week, phase III, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study. Arthritis Rheum. 2008;58(11):3319–3329. [DOI] [PubMed] [Google Scholar]
- 25. Combe B, Codreanu C, Fiocco U, et al. Etanercept and sulfasalazine, alone and combined, in patients with active rheumatoid arthritis despite receiving sulfasalazine: a double‐blind comparison. Ann Rheum Dis. 2006;65(10):1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mease PJ, Kivitz AJ, Burch FX, et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum. 2004;50(7):2264–2272. [DOI] [PubMed] [Google Scholar]
- 27. Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double‐blind randomised controlled trial. Lancet. 2004;363(9410):675–681. [DOI] [PubMed] [Google Scholar]
- 28. Bathon JM, Martin RW, Fleischmann RM, et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med. 2000;343(22):1586–1593. [DOI] [PubMed] [Google Scholar]
- 29. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–259. [DOI] [PubMed] [Google Scholar]
- 30. Moreland LW, Schiff MH, Baumgartner SW, et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130(6):478–486. [DOI] [PubMed] [Google Scholar]
- 31. Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti‐tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate‐naive patients with active rheumatoid arthritis: twenty‐four‐week results of a Phase III, multicenter, randomized, double‐blind, placebo‐controlled study of golimumab before methotrexate as first‐line therapy for early‐onset rheumatoid arthritis. Arthritis Rheum. 2009;60(8):2272–2283. [DOI] [PubMed] [Google Scholar]
- 32. Keystone EC, Genovese MC, Klareskog L, et al. Golimumab, a human antibody to tumour necrosis factor {alpha} given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO‐FORWARD Study. Ann Rheum Dis. 2009;68(6):789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kremer J, Ritchlin C, Mendelsohn A, et al. Golimumab, a new human anti‐tumor necrosis factor alpha antibody, administered Intravenously in patients with active rheumatoid arthritis: forty‐eight‐week efficacy and safety results of a phase III randomized, double‐blind, placebo‐controlled study. Arthritis Rheum. 2010;62(4):917–928. [DOI] [PubMed] [Google Scholar]
- 34. Cohen S, Hurd E, Cush J, et al. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin‐1 receptor antagonist, in combination with methotrexate: results of a twenty‐four‐week, multicenter, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum. 2002;46(3):614–624. [DOI] [PubMed] [Google Scholar]
- 35. Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL‐6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader‐blinded randomised controlled trial of tocilizumab. Ann Rheum Dis. 2007;66(9):1162–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emery P, Keystone E, Tony HP, et al. IL‐6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti‐tumour necrosis factor biologicals: results from a 24‐week multicentre randomised placebo‐controlled trial. Ann Rheum Dis. 2008;67(11):1516–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Genovese MC, Mckay JD, Nasonov EL, et al. Interleukin‐6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease‐modifying antirheumatic dugs the tocilizumab in combination with traditional disease‐modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58(10):2968–2980. [DOI] [PubMed] [Google Scholar]
- 38. Smolen JS, Beaulieu A, Rubbert‐Roth A, et al. Effect of interleukin‐6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double‐blind, placebo‐controlled, randomised trial. Lancet. 2008;371(9617):987–997. [DOI] [PubMed] [Google Scholar]
- 39. Nishimoto N, Miyasaka N, Yamamoto K, et al. Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth factor by IL‐6 receptor inhibition therapy. Mod Rheumatol. 2009;19(1):12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kremer JM, Blanco R, Brzosko M, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate results from the double‐blind treatment phase of a randomized placebo‐controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011;63(3):609–621. [DOI] [PubMed] [Google Scholar]
- 41. Yazici Y, Curtis JR, Ince A, et al. Efficacy of tocilizumab in patients with moderate to severe active rheumatoid arthritis and a previous inadequate response to disease‐modifying antirheumatic drugs: the ROSE study. Ann Rheum Dis. 2012;71(2):198–205. [DOI] [PubMed] [Google Scholar]
- 42. Harigai M, Tanaka Y, Maisawa S, JA21963 Study Group . Safety and efficacy of various dosages of ocrelizumab in Japanese patients with rheumatoid arthritis with an inadequate response to methotrexate therapy: a placebo‐controlled double‐blind parallel‐group study. J Rheumatol. 2012;39(3):486–495. [DOI] [PubMed] [Google Scholar]
- 43. Taylor PC, Quattrocchi E, Mallett S, Kurrasch R, Petersen J, Chang DJ. Ofatumumab, a fully human anti‐CD20 monoclonal antibody, in biological‐naive, rheumatoid arthritis patients with an inadequate response to methotrexate: a randomised, double‐blind, placebo‐controlled clinical trial. Ann Rheum Dis. 2011;70(12):2119–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti‐tumor necrosis factor therapy: results of a multicenter, randomized, double‐blind, placebo‐controlled, phase III trial evaluating primary efficacy and safety at twenty‐four weeks. Arthritis Rheum. 2006;54(9):2793–2806. [DOI] [PubMed] [Google Scholar]
- 45. Tak PP, Rigby W, Rubbert‐Roth A, et al. Sustained inhibition of progressive joint damage with rituximab plus methotrexate in early active rheumatoid arthritis: 2‐year results from the randomised controlled trial IMAGE. Ann Rheum Dis. 2012;71(3):351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kremer JM, Westhovens R, Leon M, et al. Treatment of rheumatoid arthritis by selective inhibition of T‐cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349(20):1907–1915. [DOI] [PubMed] [Google Scholar]
- 47. Genovese MC, Becker J, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005;353(11):1114–1123. [DOI] [PubMed] [Google Scholar]
- 48. Kremer JM, Dougados M, Emery P, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve‐month results of a phase iib, double‐blind, randomized, placebo‐controlled trial. Arthritis Rheum. 2005;52(8):2263–2271. [DOI] [PubMed] [Google Scholar]
- 49. Kremer JM, Genant HK, Moreland LW, et al. Effects of abatacept in patients with methotrexate‐resistant active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2006;144(12):865–876. [DOI] [PubMed] [Google Scholar]
- 50. Westhovens R, Robles M, Ximenes AC, et al. Clinical efficacy and safety of abatacept in methotrexate‐naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68(12):1870–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takeuchi T, Matsubara T, Nitobe T, et al. Phase II dose‐response study of abatacept in Japanese patients with active rheumatoid arthritis with an inadequate response to methotrexate. Mod Rheumatol. 2013;23(2):226–235. [DOI] [PubMed] [Google Scholar]
- 52. Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363(14):1303–1312. [DOI] [PubMed] [Google Scholar]
- 53. Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose‐ranging study of the oral JAK inhibitor tofacitinib (CP‐690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease‐modifying antirheumatic drugs. Arthritis Rheum. 2012;64(3):617–629. [DOI] [PubMed] [Google Scholar]
- 54. Kremer JM, Cohen S, Wilkinson BE, et al. A phase IIb dose‐ranging study of the oral JAK inhibitor tofacitinib (CP‐690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64(4):970–981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
supplementary material