

 This work is licensed under a
This work is licensed under a Abstract
Biologically active steroids are transported in the blood by albumin, sex hormone-binding globulin (SHBG), and corticosteroid-binding globulin (CBG). These plasma proteins also regulate the non-protein-bound or ‘free’ fractions of circulating steroid hormones that are considered to be biologically active; as such, they can be viewed as the ‘primary gatekeepers of steroid action’. Albumin binds steroids with limited specificity and low affinity, but its high concentration in blood buffers major fluctuations in steroid concentrations and their free fractions. By contrast, SHBG and CBG play much more dynamic roles in controlling steroid access to target tissues and cells. They bind steroids with high (~nM) affinity and specificity, with SHBG binding androgens and estrogens and CBG binding glucocorticoids and progesterone. Both are glycoproteins that are structurally unrelated, and they function in different ways that extend beyond their transportation or buffering functions in the blood. Plasma SHBG and CBG production by the liver varies during development and different physiological or pathophysiological conditions, and abnormalities in the plasma levels of SHBG and CBG or their abilities to bind steroids are associated with a variety of pathologies. Understanding how the unique structures of SHBG and CBG determine their specialized functions, how changes in their plasma levels are controlled, and how they function outside the blood circulation provides insight into how they control the freedom of steroids to act in health and disease.
Keywords: albumin, sex hormone-binding globulin, corticosteroid-binding globulin, testosterone, estradiol, glucocorticoids, progesterone, serine protease inhibitor
Introduction
Upon their release from steroidogenic cells, biologically active steroids are transported in the blood largely by albumin, sex hormone-binding globulin (SHBG), and corticosteroid-binding globulin (CBG). Additionally, these proteins regulate the non-protein-bound or ‘free’ fraction of steroid hormones in plasma, and control their ability to leave the blood vessels within tissues and to access their target cells (Siiteri et al. 1982).
Albumin binds all classes of steroids with low (µM) affinity, but its very high plasma concentrations and ligand-binding capacity allow it to buffer fluctuations in steroid levels and their distribution between other steroid-binding proteins and the free fraction in plasma. Unlike aldosterone, which is bound primarily by albumin, other steroid hormones bind to CBG and SHBG with high (nM) affinity and specificity, with SHBG binding the major androgens and estrogens, and CBG binding the glucocorticoids and progesterone, preferentially (Westphal 1986). Although CBG and SHBG are present in much lower concentrations in plasma than albumin, their high affinity and specificity for steroids enables them to play much more dynamic roles in determining the plasma concentrations of their main ligands. In addition, they control the amounts of free steroids that passively diffuse into cells, and they accomplish this in distinct and diverse ways (Hammond 2011, Perogamvros et al. 2012).
The liver is responsible for plasma SHBG and CBG production, but their genes are also expressed in several other tissues where their protein products function differently than in the blood (Hammond 2002, 2011). Programmed fluctuations in plasma SHBG and CBG levels occur throughout development (Scrocchi et al. 1993a, b, Hammond 2011), and abnormal plasma levels of both proteins have been linked to the risk of diseases and their associated pathologies (Hammond et al. 2012, Perogamvros et al. 2012). Therefore, understanding how the unique structures of SHBG and CBG determine their specialized functions, how changes in their plasma levels are controlled, and how they function outside the blood circulation is integral to understanding how they function as the ‘primary gatekeepers of steroid action’.
Free steroids are active steroids
The free hormone hypothesis provides a foundation for understanding how steroids act at the target cell level by postulating that only free steroids that are not bound by proteins passively diffuse through the plasma membranes of cells (Mendel 1989). Steroids that are loosely and non-specifically bound to albumin have also been proposed to be accessible to tissues (Pardridge 1988), but steroids still have to dissociate from albumin before they diffuse into cells and exert their activities. Numerous reports of the facilitated uptake of SHBG-bound steroids have also surfaced (Bordin & Petra 1980, Pardridge 1988, Porto et al. 1991, Hammes et al. 2005), but have never been substantiated in physiologically relevant contexts. At present, the proposition that only free steroids diffuse into cells therefore still best explains the clinical manifestations of either steroid hormone excess or deficiency, and knowledge of free steroid concentrations in plasma is critical to understanding their biological activities.
Access of plasma steroids to target tissues and cells
While measurements of free steroid concentrations remain the most robust indicator of the biological activities of plasma steroids (Vermeulen et al. 1999), adoption of the free hormone hypothesis as a universal explanation for how steroids access their target cells in different tissues and organ systems is overly simplistic (Mendel 1989). This is because steroid-target cells in multicellular organ systems are often compartmentalized and separated from the blood vasculature. Moreover, tissues and organ systems vary enormously in terms of their vascular permeability and the nature of their blood supply, including blood flow and transit time. Extreme examples include the highly fenestrated aspect of the blood vasculature in the liver, where hepatocytes are essentially bathed in blood, vs cells within the brain and testis that are separated by blood barriers. In addition, sex steroid-sensitive epithelial cells in organs such as the prostate, breast, and endometrium are separated from blood capillaries by complex basement membranes, and are compartmentalized together with other cell types (e.g., stroma and adipocytes), in which steroids may either act directly or are metabolically converted into more active hormones in intracrine or paracrine fashions. Thus, the locations of target cells in relation to the blood supply, the endothelial vascular permeability, the composition of the extravascular fluids and extracellular matrix, as well as the juxtaposition of different cell types within a tissue all dictate the ultimate ability of steroids to access their target cells. This review provides examples of how albumin, CBG, and SHBG function in concert with each other, as well as separately, to control the actions of steroid hormones in both the blood and extravascular tissue compartments.
Albumin
Albumin is the most abundant protein in the blood and it binds steroids and other small lipophilic molecules non-specifically. Its plasma concentrations are normally relatively constant and approximately 1000 times greater than those of the other major steroid-binding proteins (Dunn et al. 1981). However, albumin’s affinity for steroids is 3–4 orders of magnitude lower than those of CBG or SHBG (Dunn et al. 1981), the plasma concentrations of which undergo much greater fluctuations than albumin, as will be illustrated below. However, reductions in plasma albumin concentrations, which are often seen in patients with severe malnutrition, cirrhosis, the nephrotic syndrome, and other critical illnesses, have been predicted to alter the plasma distribution of testosterone (Dunn et al. 1981). Although the mathematical models used in latter study indicated that changes in plasma albumin levels predict only a small effect on the plasma distribution of cortisol, a recent study in critically ill patients has indicated that this introduces a bias in calculations of plasma free cortisol levels (Molenaar et al. 2015). Under most conditions, however, where plasma albumin levels are with normal ranges, its main function is to buffer changes in the plasma distribution of steroids when their concentrations increase transiently, or when the production or function of CBG or SHBG change under different physiological conditions or during disease.
Corticosteroid-binding globulin
Apart from fish, all other vertebrate classes have a plasma protein that binds glucocorticoids and progesterone with high affinity (Westphal 1986). The primary structure of CBG defines it as a clade A serine proteinase inhibitor (SERPINA) family member (Hammond et al. 1987) and examination of recent genome databases (www.ncbi.nlm.nih.gov; www.ensembl.org) reveals that SERPINA6 is in synteny with several other SERPINA genes in all mammals; thus, supporting the notion that CBG arose as a result of gene duplications within this cluster of SerpinA genes (Billingsley et al. 1993). Unlike other SERPINAs encoded by genes within this syntenic gene cluster, CBG (SERPINA6) is not known to inhibit proteases. However, as described in detail below, the specific cleavage of CBG by proteases within a distinct structural domain serves to promote the targeted delivery of CBG ligands to their sites of action.
The most obvious function of CBG in the blood is to transport glucocorticoids (Brien 1981, Perogamvros et al. 2012) and recent studies have confirmed that CBG is the primary determinant of circulating plasma cortisol levels in humans (Bolton et al. 2014). Perturbations in the plasma distribution of cortisol in patients with CBG variants with reduced affinity for cortisol, for example, CBG D367N (Emptoz-Bonneton et al. 2000), or inactive CBG, as observed for CBG G237V (Perogamvros et al. 2010) or CBG W371S (Hill et al. 2012), illustrate the key role that CBG plays in regulating the free fraction of cortisol in the blood circulation, and how albumin acts to buffer the plasma distribution and free fraction of cortisol in cases of CBG deficiencies (Fig. 1A).
Figure 1.
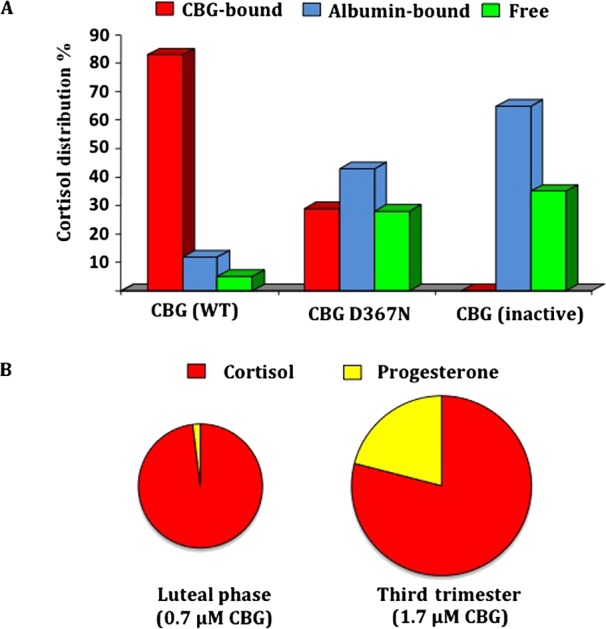
(A) Influence of CBG on the plasma distribution of cortisol. (B) Estimated proportional occupancy of plasma CBG by its major ligands, cortisol and progesterone, in blood samples taken from women before and during pregnancy, and the invervillous compartment of the placenta at term. (A) The plasma distribution of cortisol in individuals with normal CBG (WT); a CBG D367N variant with a four-fold reduction in affinity for cortisol (Emptoz-Bonneton et al. 2000), or after heat denaturation to inactivate CBG in a normal human sample (Siiteri et al. 1982), were determined by centrifugal ultrafiltration dialysis (Hammond et al. 1980). Note that the cortisol distribution in plasma with inactive CBG is expected to resemble that in patients homozygous for naturally occurring CBG variants with undetectable steroid-binding activity, for example, CBG G237V (Perogamvros et al. 2010) or CBG W371S (Hill et al. 2012). (B) Proportional occupancy of CBG in serum from women during the luteal phase of the menstrual cycle vs the third trimester of pregnancy as estimated computationally from data of serum CBG, cortisol and progesterone levels (Dunn et al. 1981).
In addition to the liver, Serpina6 is expressed at relatively high levels in several other tissues, such as the endocrine pancreas and proximal convoluted tubules of the kidney during early development in mice (Scrocchi et al. 1993a, b). These extrahepatic sites of CBG synthesis do not contribute to plasma CBG levels, and the functions of CBG in these locations appear to be distinct from that of plasma CBG. For instance, the CBG produced by the developing rodent kidney is secreted luminally into the proximal convoluted tubules (Scrocchi et al. 1993a). In this extravascular compartment, it is likely that CBG acts to control the activities of unconjugated corticosterone excreted by the kidneys of immature rodents, at a time when plasma CBG levels are very low due to a delay in their postnatal resumption of hepatic CBG production (Smith & Hammond 1991, Scrocchi et al. 1993a). Remarkably, although SERPINA6 (CBG) mRNA levels in the developing mouse kidney are greater than in any other tissue, including the liver, at any stage of development (Scrocchi et al. 1993a), there are no reports of abnormal renal development or function in Cbg-deficient mice. Moreover, while some CBG-deficient humans present with hypotension (Emptoz-Bonneton et al. 2000, Torpy et al. 2001), there is also no evidence that they suffer from renal disease.
There is information on the tissue-specific expression of CBG in other animal models during development (Smith & Hammond 1991, Berdusco et al. 1995), including baboons (Pepe et al. 1996), but changes in SERPINA6 expression in liver or any other tissues during early human development are unknown. However, as in other mammals, plasma CBG levels are relatively low in human neonates and remain so for the first month (Scott & Wells 1995). The reductions in plasma CBG levels in fetuses and neonates either just before or just after delivery may be important because this will increase the free glucocorticoid levels required for the maturation of organ, such as the lung (Smith & Hammond 1991).
It is widely recognized that CBG regulates the circulating levels and plasma distribution of glucocorticoids in mammals. However, during the second and third trimesters of human pregnancy, the large amounts of progesterone produced by the placental trophoblast are capable of displacing glucocorticoids from CBG, and under these circumstances CBG will assume the role of a major plasma progesterone transport protein at least during late gestation (Fig. 1B). In support of this, it has been reported that plasma CBG may influence circulating progesterone levels during human pregnancy and serves as a local regulator of progesterone levels and activity at the maternal–fetal interface (Benassayag et al. 2001). This was further illustrated in a study of pregnant Chinese women with a relatively common (frequency of 1:37 in Han Chinese) non-synonymous SNP (rs146744332) that results in the production of a secretion-deficient CBG A51V variant (Lin et al. 2012). In the latter study, CBG levels correlated with amounts of circulating progesterone during the first two trimesters of pregnancy, as well as the amounts of progesterone in the intervillous blood (Lei et al. 2015). As also reported previously (Benassayag et al. 2001), the CBG levels in intervillous blood samples taken from placentas at term vary by almost 10-fold in this study, while the corresponding progesterone levels varied by only three-fold (Lin et al. 2012). Moreover, plasma levels of CBG in some intervillous blood samples at term were similar to the corresponding maternal circulating levels, while in others they were as much as four times lower (Lin et al. 2012). Importantly, in those intervillous samples with very low CBG levels, the levels of progesterone were about two to three times higher than in the peripheral blood (Lin et al. 2012). When this is considered in relation to the estimates of the proportional occupancy of CBG by progesterone in maternal blood in late gestation (Fig. 1B), it can be reasonably predicted that even greater proportions of the CBG in intervillous blood will be occupied by progesterone with more displacement of cortisol, as compared with the maternal circulation, and this will be especially evident in those intervillous samples with low (~100nM) CBG levels.
Although such large differences in intervillous blood levels of CBG must also translate into large differences in the free fractions of both cortisol and progesterone at the maternal interface at term, the physiological significance of this remains obscure, especially because there were no obvious differences in pregnancy outcomes or the health of neonates in CBG-deficient pregnant women as compared with women with normal plasma CBG levels (Lei et al. 2015). However, the sex ratio of offspring delivered by women with the secretion-deficient CBG variant (CBG A51V) was significantly female skewed (Lei et al. 2015). This latter observation is of interest because stress during pregnancy causes elevations in plasma cortisol, and has been associated with female skewing of the sex ratio at birth (Navara 2010), which may be exacerbated in the offspring of CBG-deficient women. It should also be noted that CBG may act differently in regulating progesterone and or cortisol bioavailability at human fetal–maternal interface in different species, including rodents, where the placenta does not make large amounts of progesterone. In those species, there is a compensatory sustained production of high levels of progesterone by the corpus luteum until close to term, and it is possible that CBG plays a role in transporting progesterone of ovarian origin and regulating its actions during early pregnancy, at a time when progesterone has an essential role in supporting blastocyst implantation in all species (Graham & Clarke 1997).
Crystal structure analyses of human and rat CBG showed that the steroid-binding site is located close to the surface of the protein (Klieber et al. 2007, Gardill et al. 2012, Bolton et al. 2014), rather than being buried within its core as previously thought (Defaye et al. 1980, Lin et al. 2010), and revealed how different steroids interact with specific residues in the steroid-binding site (Lin et al. 2010). These experiments also demonstrate how different steroids bind to CBG with high affinity, how proteolysis of its ‘reactive center loop’ (RCL) promotes the irreversible loss of high-affinity binding activity (Lin et al. 2009, 2010), and how naturally occurring mutations of critical residues cause abnormalities in the ability of CBG to bind its ligands (Lin et al. 2010, Simard et al. 2015). As many as 15 human SERPINA6 polymorphisms have been characterized with defects in the production or steroid-binding activity of CBG (Simard et al. 2015), and these are listed in Table 1. In addition, numerous SerpinA6 mutations have been reported in animals that were used to study the actions of glucocorticoid in health and disease (Moisan 2010). Knowledge of these CBG variants is important because the algorithms used to calculate free glucocorticoid concentrations in blood samples rely on the assumption that the steroid-binding affinities of CBG in a particular species are constant, and this highlights the need to develop accurate methods to directly determine free plasma glucocorticoid concentrations.
Table 1.
Non-synonymous SERPINA6 polymorphisms linked to abnormalities in CBG production or steroid-binding activity.
| SNP (ID) | MAFa | Amino acidb | Effect on production or steroid binding | References |
|---|---|---|---|---|
| N/A | N/A | L(5)CfsX26 | Translation stop/no production | Torpy et al. (2012) |
| rs777245398 | <0.00001 | W(11)Stop | Translation stop/no production | Torpy et al. (2001) |
| rs148218218 | 0.0004 | H14R | Produced/Decreased capacity | Simard et al. (2015) |
| rs143058829 | 0.0002 | H14Q | Produced/Decreased affinity | Simard et al. (2015) |
| rs370353870 | 0.00008 | I48N | Low secretion | Simard et al. (2015) |
| rs146744332 | 0.004 | A51V | Low secretion | Lin et al. (2012) |
| rs187253929 | 0.0004 | H89Y | Produced/Decreased affinity | Simard et al. (2015) |
| rs113418909 | 0.0022 | L93H | Produced/Decreased affinity | Smith et al. (1992) |
| rs202107375 | 0.00007 | E102G | Produced/Decreased capacity | Lin et al. (2012) |
| rs754814260 | <0.00001 | G237V | Produced/No binding activity | Perogamvros et al. (2010) |
| rs201880274 | <0.00001 | P246L | Produced but not secreted | Simard et al. (2015) |
| rs267604111 | N/A | R260L | Produced/No binding activity | Simard et al. (2015) |
| rs374191911 | 0.00008 | 1279F | Produced/Decrease affinity | Simard et al. 2015 |
| rs28929488 | 0.0004 | D367N | Produced/Decreased affinity | Emptoz-Bonneton et al. (2000) |
| rs267607282 | N/A | W371S | Produced/No binding activity | Hill et al. (2012) |
Minor allele frequency (MAF) report as the highest frequency among current databases
Residue numbering is from the amino-terminus of the mature polypeptide sequence (i.e., does not include the 22 residue leader sequence). Amino acids within the leader sequence are indicated in parenthesis.
The observation that C57BL/6J mice are much more sensitive to an acute inflammatory challenge with TNFα when compared with DBA/2J mice enabled the mapping of this response to the murine Cbg gene locus in a reference panel of recombinant inbred (BXD) mouse strains derived by crossing C57BL/6J and DBA/2J mice (Libert et al. 1999). This early indication that a pathological response to acute inflammation was linked to CBG made sense because plasma CBG levels in both male and female C57BL/6 had been reported to be about 50% lower than those in DBA/2J mice (Jones et al. 1998). The link between low plasma CBG levels and this clinical phenotype was later substantiated in Cbg (SerpinA6)–/– mice that were shown to be much more sensitive to an acute inflammatory challenge than their wild-type counterparts (Petersen et al. 2006). Recently, we have also found that plasma CBG deficiencies in different colonies of Sprague–Dawley rats, which are widely used in studies of glucocorticoid-dependent stress responses and inflammation, are associated with a greater sensitivity to an acute inflammatory challenge (Bodnar et al. 2015).
A role for CBG in controlling the activities of glucocorticoids during infectious and inflammatory diseases was suspected from early studies that revealed dynamic reductions in plasma CBG levels in patients with acute infections, traumatic injuries, or severe inflammation (Savu et al. 1981, Zouaghi et al. 1985, Pugeat et al. 1989, Bernier et al. 1998), and similar changes were observed in animal studies (Savu et al. 1980, Faict et al. 1983, Garrel et al. 1993). The discovery that the gene encoding human CBG is located within a cluster of closely related SERPINA genes (Underhill & Hammond 1989, Seralini et al. 1990), many of which control proteases or other aspects of inflammatory responses, provided insight into the evolutionary origins of CBG. This also helps to understand why specific proteases target CBG and disrupt its ability to bind steroids, thereby facilitating the release of anti-inflammatory steroids at sites of tissue damage or inflammation (Hammond et al. 1987, 1990, 1991, Hammond 1990).
It is now known that several endogenous proteases, including neutrophil elastase (Hammond et al. 1990) and chymotrypsin (Lewis & Elder 2014), and a protease (LasB) produced by the opportunistic bacterial pathogen, Pseudomonas aeruginosa (Simard et al. 2014), specifically target and cleave the protease-sensitive RCL of CBG disrupting its ability to bind steroids with high affinity. While the significance of RCL cleavage by chymotrypsin is unclear, neutrophil elastase and LasB are present at sites of inflammation and infection, and their ability to specifically target and cleave the RCL of CBG is thought to promote the release of glucocorticoids at these locations.
Plasma CBG levels decrease rapidly in patients and animals undergoing acute inflammation (Savu et al. 1981, Garrel et al. 1993, Garrel 1996), and it is assumed that this reflects an initial proteolysis of the RCL that will cause a rapid plasma redistribution of glucocorticoids with increases in the albumin-bound and free fractions both locally and systemically. Furthermore, any increases in adrenal glucocorticoid production driven by adrenocorticotrophic hormone (ACTH)-mediated responses to stress can be expected to overwhelm the reduced CBG steroid-binding capacity and accentuate systemic increases in plasma free cortisol levels. It is also known that glucocorticoids (Smith & Hammond 1992) and the pro-inflammatory cytokine, interleukin (IL)-6 (Bartalena et al. 1993), downregulate hepatic CBG production, and increases in their production will act to maintain low plasma CBG levels during the course of inflammation. At some point during the recovery from inflammation, plasma CBG levels are expected to gradually rebound to maintain a normal homeostatic balance of plasma glucocorticoid levels and bioavailability.
Importantly, this model provides a framework for studies of how plasma CBG might be used as a biomarker of the severity of inflammation, and the time course of infectious or acute inflammatory diseases, as well as studies of how pre-existing deficiencies in either the production of CBG or its steroid-binding properties might contribute to poor responses to these diseases. Moreover, given the growing number of human SERPINA6 polymorphisms that compromise the production or function of CBG (Cizza et al. 2011, Simard et al. 2015), and the increased frequency of some of these mutations in specific ethnic groups/geographic locations (Cizza et al. 2011, Lin et al. 2012), the impact of CBG deficiencies on the onset of acute inflammation, as well as the recovery process, needs to be investigated in specific patient groups and animal models.
Sex hormone-binding globulin
Despite species differences in the way the SHBG gene is expressed in the liver during postnatal life, all mammals produce plasma SHBG during critical phases of gonadal and reproductive tract development (Hammond 2011). In human blood, high SHBG levels during childhood likely serve to restrict the premature actions of sex steroids until SHBG declines in both sexes as puberty advances (Hammond 2011, Hammond et al. 2012). Serum SHBG levels are generally higher in women than in men, and there is a marked difference in the relative occupancy of SHBG steroid-binding sites between the sexes, with only ~20% of the sites being occupied in women while ~80% of sites are occupied primarily by the much higher plasma testosterone concentrations in men (Dunn et al. 1981).
The steroid occupancy of SHBG is further reduced in women taking oral contraceptives that promote large increases in serum SHBG levels, while simultaneously preventing ovarian sex steroid production. It is assumed that the increases in SHBG levels in women taking oral contraceptives reflect increased hepatic SHBG production, because similar five- to ten-fold increases in serum SHBG levels occur in concert with increases in estrogen levels during human pregnancy (Anderson 1974). Increases in serum SHBG in women under these conditions undoubtedly influence the plasma distribution of both androgens and estrogens, and this property has been exploited therapeutically to reduce androgen exposures in women with symptoms of hyperandrogenism (Dewis et al. 1985). How estrogens mediate increases in hepatic SHBG expression remains to be determined but they most likely function via the estrogen receptorα(ERα (ESR1)) because their abilities to increase SHBG production in HepG2 cells, which are known to be ERα deficient, is increased in a HepG2 cell line (Hep89) engineered to overexpress ERα (Nader et al. 2006, Hammond et al. 2008). Although the significance of the large increases in maternal plasma SHBG is unclear, transient androgenization has been reported in a pregnant woman with a SHBG deficiency. The fact that androgenic symptoms resolved in this patient, postpartum, suggested that this may be due to an exposure to fetal adrenal androgens that escape placental metabolism, and which would normally be bound by elevated SHBG levels during pregnancy (Hogeveen et al. 2002).
Our understanding of the structure and function of SHBG advanced considerably after the crystal structure of the N-terminal laminin G-like domain of SHBG was resolved in complex with a variety of sex steroid ligands (Grishkovskaya et al. 2000, 2002). These high-resolution structures confirmed that androgens and estrogens interact competitively with the same steroid-binding site, but are positioned in opposite and inverted orientations (Grishkovskaya et al. 2002), and that each subunit of the SHBG homodimer contained a steroid-binding site (Avvakumov et al. 2001). The crystal structures also revealed the location of a calcium-binding site suspected as being essential for both the dimerization and steroid binding of SHBG, and provided insight into how the chelation of calcium in EDTA-treated plasma disrupts these critical structural and functional properties of the molecule (Grishkovskaya et al. 2000). In addition, they showed that a zinc atom, positioned at what appears to be an entrance to the steroid-binding site of human SHBG, reduces its affinity for estrogens specifically (Avvakumov et al. 2000). This zinc-binding site of plasma SHBG is unlikely to be fully occupied because free zinc concentrations in plasma are very low, but it may be occupied in extravascular tissue compartments, such as the prostate and the male reproductive tract, where zinc levels are exceptionally high.
Crystal structures of human SHBG have also provided unprecedented insight into its ligand-binding site, and how some naturally occurring SHBG variants (Table 2) are produced or function abnormally (Wu & Hammond 2014). Several of these naturally occurring SHBG variants exhibit differences in their relative affinities for either androgens or estrogens, and their positions within the SHBG crystal structure suggest that androgens and estrogens enter or exit the steroid-binding pocket in different ways (Wu & Hammond 2014). The latter observations may explain why androgens and estrogens are oriented so differently within the steroid-binding site (Grishkovskaya et al. 2002).
Table 2.
Non-synonymous SHBG polymorphisms linked to abnormalities in SHBG production or steroid-binding activity.
| SNP (ID) | MAFa | Amino acidb | Effect on production or steroid binding | References |
|---|---|---|---|---|
| rs373254168 | 0.00008 | T7N | Produced/Loss of O-glycosylation | Wu & Hammond (2014) |
| rs143521188 | <0.00008 | T48I | Inefficient dimerization/impaired Ca2+ binding/reduced affinity for DHT | Wu & Hammond (2014) |
| rs373769356 | 0.00008 | R123C | Reduced affinity for DHT/Increased affinity for E2 | Wu & Hammond (2014) |
| rs143269613 | 0.00008 | R123H | Reduced affinity for DHT/increased affinity for E2 | Wu & Hammond (2014) |
| rs368589266 | 0.00008 | R135C | Produced/Increased affinity for E2 | Wu & Hammond (2014) |
| rs6258 | 0.006 | P156L | Produced/Reduced affinity for T | Ohlsson et al. (2011) |
| rs145273466 | 0.0005 | L165M | Produced/Increased affinity for E2 | Wu & Hammond (2014) |
| rs372114420 | 0.00008 | E176K | Produced/Increased affinity for E2 | Wu & Hammond (2014) |
| rs146779355 | 0.00008 | G195E | Low secretion/reduced affinity for DHT | Wu & Hammond (2014) |
| N/A | N/A | G195R | No secretion | Vos et al. (2014) |
| rs6259 | 0.09 | D327N | Produced/Additional N-glycosylation site/normal steroid binding | Power et al. (1992) |
MAF report as the highest frequency among current databases
Residue numbering is from the amino-terminus of the mature polypeptide sequence (i.e., does not include the 29 residue leader sequence).
It has been proposed that SHBG leaves the blood circulation in some tissues and interacts directly with proteins on the plasma membranes of specific cell types, and that this may contribute to either the delivery of SHBG-bound sex steroids via endocytotic mechanisms or to cell membrane receptor-mediated signaling (Rosner et al. 2010). We have been unable to confirm these observations, but our studies of mice expressing human SHBG transgenes have shown that SHBG does exit the blood vessels in some tissues, and accumulates within extracellular tissue compartments, such as the stroma of the endometrium and epididymis (Ng et al. 2006). Moreover, we obtained evidence that this involves a steroid ligand-dependent interaction between SHBG and two members of the fibulin family of extracellular matrix-associated proteins, fibulin-1D and fibulin-2 (Ng et al. 2006). The biological significance of this remains to be determined, but it provides in vivo evidence that SHBG has the capacity to act in extravascular compartments, extending its functions beyond that of a transport protein that regulates free sex steroids levels in the blood.
Ever since it was realized that the proportions of free testosterone and estradiol in blood samples are inversely related to those of SHBG (Anderson 1974), serum SHBG and testosterone measurements have been used in algorithms to calculate free testosterone levels in patients with suspected hyperandrogenism or hypoandrogenism (Vermeulen et al. 1999). Similar relationships between SHBG and free estradiol levels explain how serum SHBG levels contribute to abnormal estrogen exposures in lean women who have high SHBG levels and at risk for osteoporosis (Davidson et al. 1982, Devine et al. 2005) vs obese postmenopausal women who have low SHBG levels and are at high risk for endometrial cancer (Nisker et al. 1980).
The mathematical models used to calculate free plasma androgen or estrogen levels currently rely on SHBG measurements obtained using immunoassays (Vermeulen et al. 1999), and they are based on the assumptions that all SHBG molecules react similarly immunologically and have identical steroid-binding properties. These assumptions are fallible, however, because some SHBG variants are not recognized in immunoassays, while others have abnormal affinities for sex steroids (Wu & Hammond 2014), including the SHBG P156L variant with a reduced affinity for testosterone that is present in ~1% of Caucasians, and increases the free fraction of testosterone in the blood of male carriers (Ohlsson et al. 2011). These findings highlight the pressing need for sensitive mass spectrometric methods to measure both total and free sex steroid levels in the blood.
This is emerging as an important issue because several other relatively common variations in the human SHBG coding sequence are linked to abnormal plasma SHBG levels (Table 2). They include a common non-synonymous SNP (rs6259) that causes a substitution of Asp327 with an Asn residue (D327N) and creates an extra N-linked glycosylation site (Power et al. 1992), the utilization of which retards the plasma clearance of SHBG (Cousin et al. 1998). Individuals who carry the rs6259 SNP have slightly elevated SHBG levels that have been negatively associated with the risk of developing breast cancer (Forsti et al. 2002, Cui et al. 2005) or type 2 diabetes (Ding et al. 2009). Several polymorphisms in the non-coding regions of human SHBG also appear to influence the hepatic production or blood levels of SHBG (Hogeveen et al. 2001, Cousin et al. 2004), and numerous reports have associated them with a variety of hormone-related or metabolically related diseases (Xita & Tsatsoulis 2010). There is also a growing awareness that abnormal plasma SHBG levels, and the subsequent changes in the plasma levels and distribution of sex steroids, are not only predictive of numerous clinical conditions, including low bone density (Slemenda et al. 1997) and osteoporotic fracture (Cawthon et al. 2016) in men, and risk for the metabolic syndrome and its associated diseases in both sexes (Kim & Halter 2014), but may be directly implicated in the etiology of some of these diseases (Legrand et al. 2001, Ding et al. 2009).
Despite the large number of SHBG measurements performed for diagnostic purposes, it is remarkable that there have been only two reports of complete SHBG deficiencies in humans. An early report of undetectable (<10nM) plasma SHBG levels (Ahrentsen et al. 1982) has remained unsubstantiated, but a rare missense genetic mutation that produces a secretion defective SHBG variant (SHBG G195R) was recently reported in a young man and his sister, both of whom were homozygous for the mutant SHBG allele and had no detectable SHBG in their blood (Vos et al. 2014). As expected, plasma testosterone concentrations in the male proband were well below the normal range, yet his free testosterone levels were normal. Clinical assessments indicated fatigue, overt muscle weakness, and low body weight, and other symptoms of hypoandrogenism, but gonadal development and sperm production and function appeared to be normal. The proband’s affected sibling reported a late menarche and irregular menstrual cycles, but surprisingly had no signs of hirsutism or hyperandrogenism (Vos et al. 2014). Although this report provides an indication that plasma SHBG is not essential for male reproductive development and sperm production, the proband’s clinical phenotype suggests a more direct role for SHBG in supporting the anabolic activities of androgens. However, it was noted that this phenotype might be related to possible consanguinity in this pedigree (Vos et al. 2014).
Studies of SHBG expression in human liver cells in culture (Jänne & Hammond 1998), and in a transgenic mouse model (Jänne et al. 1998) indicate that the transcription unit responsible for plasma SHBG production spans a 4.3kb region within the short arm of human chromosome 17 (Berube et al. 1990). The sequence flanking exon 1 of this transcription unit lacks a typical TATA box, but the transcription factor hepatocyte nuclear factor 4α(HNF4α) appears to substitute for the TATA-binding protein in helping to recruit the transcriptional machinery to the promoter (Jänne & Hammond 1998). These pioneering studies also demonstrated that a related orphan nuclear hormone receptor (NHR) family member, COUP transcription factor (COUP-TF), competes with HNF4α for the same site in the proximal promoter. While HNF4α actively enhances transcription from this site, COUP-TF represses it, and it is evident that this particular cis-element in the SHBG promoter acts together with these two transcription factors as the main on–off switch for SHBG transcription (Jänne & Hammond 1998). This provided the first explanation for why low plasma SHBG is a hallmark of the metabolic syndrome and its associated diseases, such as type 2 diabetes and cardiovascular disease (Selva et al. 2007). The latter study also challenged the assumption that elevated insulin levels are responsible for the downregulation of SHBG expression in hyperinsulinemic/hyperglycemic patients. Instead, this comprehensive study demonstrated that high levels of monosaccharides, especially fructose, repress SHBG transcription both in vivo and in vitro when administered exogenously, and showed that this is mediated by increasing palmitate levels in hepatocytes and a concomitant reduction in HNF4α levels (Selva et al. 2007). The mechanism responsible for the loss of HNF4α in hepatocytes under these conditions is unknown, but may somehow be related to the fact that fatty acids, including palmitate (Hertz et al. 1998), are ligands of HNF4α (Wisely et al. 2002). A direct correlation between hepatic HNF4-α and SHBG mRNA levels has been also observed in cancer patients, in whom hepatic HNF4-α and SHBG mRNA levels were reported to be inversely related to hepatic triglyceride levels and to decrease in relation to body mass index (Winters et al. 2014).
A variety of hormones and drugs, as well as metabolic and nutritional factors, influence the expression of human SHBG and plasma SHBG levels (Table 3). When considered together, the HNF4α emerges as a central mediator of human SHBG production by liver cells. For instance, it mediates the induction of SHBG expression in response to thyroid hormone (Selva & Hammond 2009a). In this regard, it is interesting that luteinizing hormone (LH) levels rise in hyperthyroxinemic men (Ruder et al. 1971) and are high in patients with spontaneous hyperthyroidism (Hudson & Edwards 1992), implying a role for SHBG in the feedback regulation of gonadotrophin-releasing hormone (GNRH) secretion. Elevated plasma levels of pro-inflammatory cytokines (TNF-α and IL-1β) in obese individuals contribute to a reduced hepatic expression of SHBG and low plasma SHBG levels by reducing HNF4-α levels in the liver (Simo et al. 2012a, b). Furthermore, adiponectin appears to increase plasma SHBG production indirectly by reducing heptic lipid content and increasing HNF4-α levels in the liver (Simo et al. 2014). Thus, the low plasma levels of adiponectin that are typically seen in overweight individuals at risk of having the metabolic syndrome (Kadowaki et al. 2006) may further contribute to the low plasma SHBG levels in obese patients. By contrast, peroxisome proliferator-activated receptor gamma (PPARγ) appears to interact with a different NHR response element in the SHBG proximal promoter to repress transcription in hepatocytes cultured in vitro (Selva & Hammond 2009b), and this may explain the lower plasma SHBG levels in subjects with a hyperactive 12 Ala PPARγ variant (Mousavinasab et al. 2006) that has been linked with low risk for type 2 diabetes and myocardial infarction (Regieli et al. 2009). Although this is counterintuitive because the use of potent synthetic PPARγ ligands such as insulin sensitizers, like pioglitazone, cause modest increases in plasma SHBG levels in individuals with the metabolic syndrome (Sridhar et al. 2013), the effects of these potent drugs may be multifactorial. Their effects on hepatic SHBG production may be influenced by increases in plasma adiponectin that in turn may increase SHBG production, as previously mentioned. Furthermore, the NHR response element in the SHBG proximal promoter that binds PPARγ is also known to bind HNF4-α (Jänne & Hammond 1998), as well as several other orphan members of the NHR family, including constitutive androstane receptor (CAR) and liver X receptor (LXR), which can all potentially compete for binding at this site. This is probably relevant, because pioglitazone and other insulin sensitizers most likely alter the hepatic complement of these orphan NHRs, and these changes may certainly contribute to the modest increases in plasma SHBG levels observed after their administration.
Table 3.
Hormonal, pharmaceutical, metabolic, and nutritional modifiers of hepatic SHBG production.
| Effector | Response | Mediator |
|---|---|---|
| Ethinyl estradiol | Increases SHBG expression | Mediated by ERα mechanism unknown |
| Increases plasma SHBG levels | ||
| Thyroid hormone | Increases SHBG expression | Indirect via increased hepatic HNF4-α |
| Increases plasma SHBG levels | ||
| Synthetic PPARγ ligands (thiazolidinediones) | Increase SHBG expression | Indirect presumably via increased hepatic HNF4-α via reduced hepatic lipids and higher adiponectin levels |
| Increase plasma SHBG levels | ||
| Monosaccharides (glucose and fructose) | Decreases SHBG expression | Indirect via increased hepatic lipid levels and decreased HNF4-α |
| Decrease plasma SHBG levels | ||
| Pro-inflammatory cytokines (TNF-α and IL-1β) | Decreases SHBG expression | Indirect via decreased hepatic HNF4-α |
| Decrease plasma SHBG levels | ||
| Adiponectin | Increases SHBG expression | Indirect via reduced hepatic lipid levels and increased HNF4-α |
| Increases plasma SHBG levels |
Conclusion
Plasma CBG and SHBG are structurally unrelated and function in very different ways that extend well beyond simple transportation or buffering functions in the blood. Their crystal structures have demonstrated how they interact with their preferred steroid ligands, as well as providing insight into other functionally important properties, including the consequences of the RCL cleavage of CBG by specific proteases in the context of infectious and inflammatory diseases, and the impact of dimerization and the binding of divalent cations (Ca2+ and Zn2+) on the steroid-binding properties of SHBG. Knowledge of the structures together with the identification of naturally occurring variants of CBG and SHBG provide additional insight into their production and functions. They also illustrate the limitations of current methods for measuring their plasma concentrations, which are used in algorithms to calculate free steroid levels, and highlight the need for more direct methods to measure plasma free steroid concentrations. Recent insight into the molecular mechanisms responsible for regulation of hepatic CBG and SHBG production explain how abnormalities in their plasma levels are linked to the risk as well as the consequences of a variety of diseases related to abnormal steroid hormone exposures, and how they may be utilized as biomarkers of disease onset, severity, or recovery. Finally, this review provides several arguments for why CBG and SHBG should be regarded as the primary gatekeepers of steroid hormone action in the blood and extravascular tissue compartments.
Footnote
This review is based on the 2015 International Medal Lecture, presented by Dr Geoffrey Hammond at the Society for Endocrinology BES 2015, Edinburgh, UK.
Declaration of interest
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This study received support from the Canada Research Chairs Program.
Acknowledgements
The author wishes to thank Lesley Hill, Marc Simard, Warren Meyers, and Phillip Round for their critical review of this manuscript. Support from the Canada Research Chairs Program is also gratefully acknowledged.
References
- Ahrentsen OD, Jensen HK, Johnsen SG. 1982. Sex-hormone-binding globulin deficiency. Lancet 2 377. [DOI] [PubMed] [Google Scholar]
- Anderson DC. 1974. Sex-hormone-binding globulin. Clinical Endocrinology 3 69–96. ( 10.1111/j.1365-2265.1974.tb03298.x) [DOI] [PubMed] [Google Scholar]
- Avvakumov GV, Muller YA, Hammond GL. 2000. Steroid-binding specificity of human sex hormone-binding globulin is influenced by occupancy of a zinc-binding site. Journal of Biological Chemistry 275 25920–25925. ( 10.1074/jbc.M004484200) [DOI] [PubMed] [Google Scholar]
- Avvakumov GV, Grishkovskaya I, Muller YA, Hammond GL. 2001. Resolution of the human sex hormone-binding globulin dimer interface and evidence for two steroid-binding sites per homodimer. Journal of Biological Chemistry 276 34453–34457. ( 10.1074/jbc.M106274200) [DOI] [PubMed] [Google Scholar]
- Bartalena L, Hammond GL, Farsetti A, Flink IL, Robbins J. 1993. Interleukin-6 inhibits corticosteroid-binding globulin synthesis by human hepatoblastoma-derived (hep G2) cells. Endocrinology 133 291–296. [DOI] [PubMed] [Google Scholar]
- Benassayag C, Souski I, Mignot TM, Robert B, Hassid J, Duc-Goiran P, Mondon F, Rebourcet R, Dehennin L, Nunez EA, et al. 2001. Corticosteroid-binding globulin status at the fetomaternal interface during human term pregnancy. Biology of Reproduction 64 812–821. ( 10.1095/biolreprod64.3.812) [DOI] [PubMed] [Google Scholar]
- Berdusco ET, Yang K, Hammond GL, Challis JR. 1995. Corticosteroid-binding globulin (CBG) production by hepatic and extra-hepatic sites in the ovine fetus; effects of CBG on glucocorticoid negative feedback on pituitary cells in vitro. Journal of Endocrinology 146 121–130. ( 10.1677/joe.0.1460121) [DOI] [PubMed] [Google Scholar]
- Bernier J, Jobin N, Emptoz-Bonneton A, Pugeat MM, Garrel DR. 1998. Decreased corticosteroid-binding globulin in burn patients: relationship with interleukin-6 and fat in nutritional support. Critical Care Medicine 26 452–460. ( 10.1097/00003246-199803000-00014) [DOI] [PubMed] [Google Scholar]
- Berube D, Seralini GE, Gagne R, Hammond GL. 1990. Localization of the human sex hormone-binding globulin gene (SHBG) to the short arm of chromosome 17 (17p12 → p13). Cytogenetics and Cell Genetics 54 65–67. ( 10.1159/000132958) [DOI] [PubMed] [Google Scholar]
- Billingsley GD, Walter MA, Hammond GL, Cox DW. 1993. Physical mapping of four serpin genes: alpha 1-antitrypsin, alpha 1-antichymotrypsin, corticosteroid-binding globulin, and protein C inhibitor, within a 280-kb region on chromosome I4q32.1. American Journal of Human Genetics 52 343–353. [PMC free article] [PubMed] [Google Scholar]
- Bodnar TS, Hill LA, Taves MD, Yu W, Soma KK, Hammond GL, Weinberg J. 2015. Colony-specific differences in endocrine and immune responses to an inflammatory challenge in female Sprague Dawley rats. Endocrinology 156 4604–4617. ( 10.1210/en.2015-1497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton JL, Hayward C, Direk N, Lewis JG, Hammond GL, Hill LA, Anderson A, Huffman J, Wilson JF, Campbell H, et al. 2014. Genome wide association identifies common variants at the SERPINA6/SERPINA1 locus influencing plasma cortisol and corticosteroid binding globulin. PLoS Genetics 10 e1004474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordin S, Petra PH. 1980. Immunocytochemical localization of the sex steroid-binding protein of plasma in tissues of the adult monkey macaca nemestrina. PNAS 77 5678–5682. ( 10.1073/pnas.77.10.5678) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien TG. 1981. Human corticosteroid binding globulin. Clinical Endocrinology 14 193–212. ( 10.1111/j.1365-2265.1981.tb00616.x) [DOI] [PubMed] [Google Scholar]
- Cawthon PM, Schousboe JT, Harrison SL, Ensrud KE, Black D, Cauley JA, Cummings SR, LeBlanc ES, Laughlin GA, Nielson CM, et al. 2016. Sex hormones, sex hormone binding globulin, and vertebral fractures in older men. Bone 84 271–278. ( 10.1016/j.bone.2016.01.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizza G, Bernardi L, Smirne N, Maletta R, Tomaino C, Costanzo A, Gallo M, Lewis JG, Geracitano S, Grasso MB, et al. 2011. Clinical manifestations of highly prevalent corticosteroid-binding globulin mutations in a village in southern Italy. Journal of Clinical Endocrinology and Metabolism 96 E1684–E1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin P, Dechaud H, Grenot C, Lejeune H, Pugeat M. 1998. Human variant sex hormone-binding globulin (SHBG) with an additional carbohydrate chain has a reduced clearance rate in rabbit. Journal of Clinical Endocrinology and Metabolism 83 235–240. [DOI] [PubMed] [Google Scholar]
- Cousin P, Calemard-Michel L, Lejeune H, Raverot G, Yessaad N, Emptoz-Bonneton A, Morel Y, Pugeat M. 2004. Influence of SHBG gene pentanucleotide TAAAA repeat and D327N polymorphism on serum sex hormone-binding globulin concentration in hirsute women. Journal of Clinical Endocrinology and Metabolism 89 917–924. ( 10.1210/jc.2002-021553) [DOI] [PubMed] [Google Scholar]
- Cui Y, Shu XO, Cai Q, Jin F, Cheng JR, Cai H, Gao YT, Zheng W. 2005. Association of breast cancer risk with a common functional polymorphism (Asp327Asn) in the sex hormone-binding globulin gene. Cancer Epidemiology, Biomarkers & Prevention 14 1096–1101. [DOI] [PubMed] [Google Scholar]
- Davidson BJ, Ross RK, Paganini-Hill A, Hammond GD, Siiteri PK, Judd HL. 1982. Total and free estrogens and androgens in postmenopausal women with hip fractures. Journal of Clinical Endocrinology and Metabolism 54 115–120. ( 10.1210/jcem-54-1-115) [DOI] [PubMed] [Google Scholar]
- Defaye G, Basset M, Monnier N, Chambaz EM. 1980. Electron spin resonance study of human transcortin: thiol groups and binding site topography. Biochimica et Biophysica Acta 623 280–294. ( 10.1016/0005-2795(80)90256-1) [DOI] [PubMed] [Google Scholar]
- Devine A, Dick IM, Dhaliwal SS, Naheed R, Beilby J, Prince RL. 2005. Prediction of incident osteoporotic fractures in elderly women using the free estradiol index. Osteoporosis International 16 216–221. ( 10.1007/s00198-004-1674-6) [DOI] [PubMed] [Google Scholar]
- Dewis P, Petsos P, Newman M, Anderson DC. 1985. The treatment of hirsutism with a combination of desogestrel and ethinyl oestradiol. Clinical Endocrinology 22 29–36. ( 10.1111/j.1365-2265.1985.tb01061.x) [DOI] [PubMed] [Google Scholar]
- Ding EL, Song Y, Manson JE, Hunter DJ, Lee CC, Rifai N, Buring JE, Gaziano JM, Liu S. 2009. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. New England Journal of Medicine 361 1152–1163. ( 10.1056/NEJMoa0804381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JF, Nisula BC, Rodbard D. 1981. Transport of steroid hormones: binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. Journal of Clinical Endocrinology and Metabolism 53 58–68. ( 10.1210/jcem-53-1-58) [DOI] [PubMed] [Google Scholar]
- Emptoz-Bonneton A, Cousin P, Seguchi K, Avvakumov GV, Bully C, Hammond GL, Pugeat M. 2000. Novel human corticosteroid-binding globulin variant with low cortisol-binding affinity. Journal of Clinical Endocrinology and Metabolism 85 361–367. [DOI] [PubMed] [Google Scholar]
- Faict D, Vandoren G, De Moor P, Lesaffre E, Verhoeven G. 1983. Response of transcortin and alpha 2u-globulin to turpentine-induced inflammation in the rat: influence of corticosteroids and prolactin. Journal of Endocrinology 99 465–468. ( 10.1677/joe.0.0990465) [DOI] [PubMed] [Google Scholar]
- Forsti A, Jin Q, Grzybowska E, Soderberg M, Zientek H, Sieminska M, Rogozinska-Szczepka J, Chmielik E, Utracka-Hutka B, Hemminki K. 2002. Sex hormone-binding globulin polymorphisms in familial and sporadic breast cancer. Carcinogenesis 23 1315–1320. ( 10.1093/carcin/23.8.1315) [DOI] [PubMed] [Google Scholar]
- Gardill BR, Vogl MR, Lin HY, Hammond GL, Muller YA. 2012. Corticosteroid-binding globulin: structure-function implications from species differences. PLoS ONE 7 e52759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrel DR. 1996. Corticosteroid-binding globulin during inflammation and burn injury: nutritional modulation and clinical implications. Hormone Research 45 245–251. ( 10.1159/000184796) [DOI] [PubMed] [Google Scholar]
- Garrel DR, Zhang L, Zhao XF, Hammond GL. 1993. Effect of burn injury on corticosteroid-binding globulin levels in plasma and wound fluid. Wound Repair and Regeneration 1 10–14. ( 10.1046/j.1524-475X.1993.10105.x) [DOI] [PubMed] [Google Scholar]
- Graham JD, Clarke CL. 1997. Physiological action of progesterone in target tissues. Endocrine Reviews 18 502–519. [DOI] [PubMed] [Google Scholar]
- Grishkovskaya I, Avvakumov GV, Sklenar G, Dales D, Hammond GL, Muller YA. 2000. Crystal structure of human sex hormone-binding globulin: steroid transport by a laminin G-like domain. EMBO Journal 19 504–512. ( 10.1093/emboj/19.4.504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishkovskaya I, Avvakumov GV, Hammond GL, Catalano MG, Muller YA. 2002. Steroid ligands bind human sex hormone-binding globulin in specific orientations and produce distinct changes in protein conformation. Journal of Biological Chemistry 277 32086–32093. ( 10.1074/jbc.M203999200) [DOI] [PubMed] [Google Scholar]
- Hammes A, Andreassen TK, Spoelgen R, Raila J, Hubner N, Schulz H, Metzger J, Schweigert FJ, Luppa PB, Nykjaer A, et al. 2005. Role of endocytosis in cellular uptake of sex steroids. Cell 122 751–762. ( 10.1016/j.cell.2005.06.032) [DOI] [PubMed] [Google Scholar]
- Hammond GL. 1990. Molecular properties of corticosteroid binding globulin and the sex-steroid binding proteins. Endocrine Reviews 11 65–79. ( 10.1210/edrv-11-1-65) [DOI] [PubMed] [Google Scholar]
- Hammond GL. 2002. Access of reproductive steroids to target tissues. Obstetrics and Gynecology Clinics of North America 29 411–423. ( 10.1016/S0889-8545(02)00008-6) [DOI] [PubMed] [Google Scholar]
- Hammond GL. 2011. Diverse roles for sex hormone-binding globulin in reproduction. Biology of Reproduction 85 431–441. ( 10.1095/biolreprod.111.092593) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GL, Smith CL, Goping IS, Underhill DA, Harley MJ, Reventos J, Musto NA, Gunsalus GL, Bardin CW. 1987. Primary structure of human corticosteroid binding globulin, deduced from hepatic and pulmonary cDNAs, exhibits homology with serine protease inhibitors. PNAS 84 5153–5157. ( 10.1073/pnas.84.15.5153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GL, Smith CL, Paterson NA, Sibbald WJ. 1990. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. Journal of Clinical Endocrinology and Metabolism 71 34–39. ( 10.1210/jcem-71-1-34) [DOI] [PubMed] [Google Scholar]
- Hammond GL, Smith CL, Underhill DA. 1991. Molecular studies of corticosteroid binding globulin structure, biosynthesis and function. Journal of Steroid Biochemistry and Molecular Biology 40 755–762. ( 10.1016/0960-0760(91)90300-T) [DOI] [PubMed] [Google Scholar]
- Hammond GL, Hogeveen KN, Visser M, Coelingh Bennink HJ. 2008. Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells. Climacteric 11 41–46. [DOI] [PubMed] [Google Scholar]
- Hammond GL, Nisker JA, Jones LA, Siiteri PK. 1980. Estimation of the percentage of free steroid in undiluted serum by centrifugal ultrafiltration-dialysis. Journal of Biological Chemistry 255 5023–5026. [PubMed] [Google Scholar]
- Hammond GL, Wu TS, Simard M. 2012. Evolving utility of sex hormone-binding globulin measurements in clinical medicine. Current Opinion in Endocrinology, Diabetes, and Obesity 19 183–189. ( 10.1097/MED.0b013e328353732f) [DOI] [PubMed] [Google Scholar]
- Hertz R, Magenheim J, Berman I, Bar-Tana J. 1998. Fatty acyl-CoA thioesters are ligands of hepatic nuclear factor-4alpha. Nature 392 512–516. ( 10.1038/33185) [DOI] [PubMed] [Google Scholar]
- Hill LA, Vassiliadi DA, Simard M, Pavlaki A, Perogamvros I, Hadjidakis D, Hammond GL. 2012. Two different corticosteroid-binding globulin variants that lack cortisol-binding activity in a Greek woman. Journal of Clinical Endocrinology and Metabolism 97 4260–4267. ( 10.1210/jc.2012-2467) [DOI] [PubMed] [Google Scholar]
- Hogeveen KN, Talikka M, Hammond GL. 2001. Human sex hormone-binding globulin promoter activity is influenced by a (TAAAA)n repeat element within an alu sequence. Journal of Biological Chemistry 276 36383–36390. ( 10.1074/jbc.M104681200) [DOI] [PubMed] [Google Scholar]
- Hogeveen KN, Cousin P, Pugeat M, Dewailly D, Soudan B, Hammond GL. 2002. Human sex hormone-binding globulin variants associated with hyperandrogenism and ovarian dysfunction. Journal of Clinical Investigation 109 973–981. ( 10.1172/Jbib214060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RW, Edwards AL. 1992. Testicular function in hyperthyroidism. Journal of Andrology 13 117–124. [PubMed] [Google Scholar]
- Jänne M, Hammond GL. 1998. Hepatocyte nuclear factor-4 controls transcription from a TATA-less human sex hormone-binding globulin gene promoter. Journal of Biological Chemistry 273 34105–34114. ( 10.1074/jbc.273.51.34105) [DOI] [PubMed] [Google Scholar]
- Jänne M, Deol HK, Power SG, Yee SP, Hammond GL. 1998. Human sex hormone-binding globulin gene expression in transgenic mice. Molecular Endocrinology 12 123–136. ( 10.1210/mend.12.1.0050) [DOI] [PubMed] [Google Scholar]
- Jones BC, Sarrieau A, Reed CL, Azar MR, Mormede P. 1998. Contribution of sex and genetics to neuroendocrine adaptation to stress in mice. Psychoneuroendocrinology 23 505–517. ( 10.1016/S0306-4530(98)00014-6) [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. 2006. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. Journal of Clinical Investigation 116 1784–1792. ( 10.1172/JCI29126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Halter JB. 2014. Endogenous sex hormones, metabolic syndrome, and diabetes in men and women. Current Cardiology Reports 16 467-014-0467-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klieber MA, Underhill C, Hammond GL, Muller YA. 2007. Corticosteroid-binding globulin, a structural basis for steroid transport and proteinase-triggered release. Journal of Biological Chemistry 282 29594–29603. ( 10.1074/jbc.M705014200) [DOI] [PubMed] [Google Scholar]
- Legrand E, Hedde C, Gallois Y, Degasne I, Boux de Casson F, Mathieu E, Basle MF, Chappard D, Audran M. 2001. Osteoporosis in men: a potential role for the sex hormone binding globulin. Bone 29 90–95. ( 10.1016/S8756-3282(01)00478-1) [DOI] [PubMed] [Google Scholar]
- Lei JH, Yang X, Peng S, Li Y, Underhill C, Zhu C, Lin HY, Wang H, Hammond GL. 2015. Impact of corticosteroid-binding globulin deficiency on pregnancy and neonatal sex. Journal of Clinical Endocrinology and Metabolism 100 1819–1827. ( 10.1210/jc.2014-4254) [DOI] [PubMed] [Google Scholar]
- Lewis JG, Elder PA. 2014. The reactive centre loop of corticosteroid-binding globulin (CBG) is a protease target for cortisol release. Molecular and Cellular Endocrinology 384 96–101. ( 10.1016/j.mce.2014.01.005) [DOI] [PubMed] [Google Scholar]
- Libert C, Wielockx B, Hammond GL, Brouckaert P, Fiers W, Elliott RW. 1999. Identification of a locus on distal mouse chromosome 12 that controls resistance to tumor necrosis factor-induced lethal shock. Genomics 55 284–289. ( 10.1006/geno.1998.5677) [DOI] [PubMed] [Google Scholar]
- Lin HY, Underhill C, Gardill BR, Muller YA, Hammond GL. 2009. Residues in the human corticosteroid-binding globulin reactive center loop that influence steroid binding before and after elastase cleavage. Journal of Biological Chemistry 284 884–896. ( 10.1074/jbc.M807376200) [DOI] [PubMed] [Google Scholar]
- Lin HY, Muller YA, Hammond GL. 2010. Molecular and structural basis of steroid hormone binding and release from corticosteroid-binding globulin. Molecular and Cellular Endocrinology 316 3–12. ( 10.1016/j.mce.2009.06.015) [DOI] [PubMed] [Google Scholar]
- Lin HY, Underhill C, Lei JH, Helander-Claesson A, Lee HY, Gardill BR, Muller YA, Wang H, Hammond GL. 2012. High frequency of SERPINA6 polymorphisms that reduce plasma corticosteroid-binding globulin activity in Chinese subjects. Journal of Clinical Endocrinology and Metabolism 97 E678–E686. [DOI] [PubMed] [Google Scholar]
- Mendel CM. 1989. The free hormone hypothesis: a physiologically based mathematical model. Endocrine Reviews 10 232–274. ( 10.1210/edrv-10-3-232) [DOI] [PubMed] [Google Scholar]
- Moisan MP. 2010. Genotype-phenotype associations in understanding the role of corticosteroid-binding globulin in health and disease animal models. Molecular and Cellular Endocrinology 316 35–41. ( 10.1016/j.mce.2009.07.017) [DOI] [PubMed] [Google Scholar]
- Molenaar N, Groeneveld AB, de Jong MF. 2015. Three calculations of free cortisol versus measured values in the critically ill. Clinical Biochemistry 48 1053–1058. ( 10.1016/j.clinbiochem.2015.07.014) [DOI] [PubMed] [Google Scholar]
- Mousavinasab F, Tahtinen T, Jokelainen J, Koskela P, Vanhala M, Oikarinen J, Laakso M, Keinanen-Kiukaanniemi S. 2006. The Pro12Ala polymorphism of the PPAR gamma 2 gene influences sex hormone-binding globulin level and its relationship to the development of the metabolic syndrome in young Finnish men. Endocrine 30 185–190. ( 10.1385/endo:30:2:185) [DOI] [PubMed] [Google Scholar]
- Nader N, Raverot G, Emptoz-Bonneton A, Déchaud H, Bonnay M, Baudin E, Pugeat M. 2006. Mitotane has an estrogenic effect on sex hormone-binding globulin and corticosteroid-binding globulin in humans. Journal of Clinical Endocrinology and Metabolism 91 2165–2170. ( 10.1210/jc.2005-2157) [DOI] [PubMed] [Google Scholar]
- Navara KJ. 2010. Programming of offspring sex ratios by maternal stress in humans: assessment of physiological mechanisms using a comparative approach. Journal of Comparative Physiology B: Biochemical, Systemic, and Environmental Physiology 180 785–796. ( 10.1007/s00360-010-0483-9) [DOI] [PubMed] [Google Scholar]
- Ng KM, Catalano MG, Pinos T, Selva DM, Avvakumov GV, Munell F, Hammond GL. 2006. Evidence that fibulin family members contribute to the steroid-dependent extravascular sequestration of sex hormone-binding globulin. Journal of Biological Chemistry 281 15853–15861. ( 10.1074/jbc.M512370200) [DOI] [PubMed] [Google Scholar]
- Nisker JA, Hammond GL, Davidson BJ, Frumar AM, Takaki NK, Judd HL, Siiteri PK. 1980. Serum sex hormone-binding globulin capacity and the percentage of free estradiol in postmenopausal women with and without endometrial carcinoma. A new biochemical basis for the association between obesity and endometrial carcinoma. American Journal of Obstetrics and Gynecology 138 637–642. ( 10.1016/0002-9378(80)90080-0) [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Wallaschofski H, Lunetta KL, Stolk L, Perry JR, Koster A, Petersen AK, Eriksson J, Lehtimaki T, Huhtaniemi IT, et al. 2011. Genetic determinants of serum testosterone concentrations in men. PLoS Genetics 7 e1002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. 1988. Selective delivery of sex steroid hormones to tissues by albumin and by sex hormone-binding globulin. Oxford Reviews of Reproductive Biology 10 237–292. [PubMed] [Google Scholar]
- Pepe GJ, Jury HH, Hammond GL, Albrecht ED. 1996. Developmental regulation of corticosteroid-binding globulin biosynthesis in the baboon fetus. Endocrinology 137 3323–3328. [DOI] [PubMed] [Google Scholar]
- Perogamvros I, Underhill C, Henley DE, Hadfield KD, Newman WG, Ray DW, Lightman SL, Hammond GL, Trainer PJ. 2010. Novel corticosteroid-binding globulin variant that lacks steroid binding activity. Journal of Clinical Endocrinology and Metabolism 95 E142–E150. [DOI] [PubMed] [Google Scholar]
- Perogamvros I, Ray DW, Trainer PJ. 2012. Regulation of cortisol bioavailability – effects on hormone measurement and action. Nature Reviews. Endocrinology 8 717–727. ( 10.1038/nrendo.2012.134) [DOI] [PubMed] [Google Scholar]
- Petersen HH, Andreassen TK, Breiderhoff T, Brasen JH, Schulz H, Gross V, Grone HJ, Nykjaer A, Willnow TE. 2006. Hyporesponsiveness to glucocorticoids in mice genetically deficient for the corticosteroid binding globulin. Molecular and Cellular Biology 26 7236–7245. ( 10.1128/MCB.00400-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto CS, Gunsalus GL, Bardin CW, Phillips DM, Musto NA. 1991. Receptor-mediated endocytosis of an extracellular steroid-binding protein (TeBG) in MCF-7 human breast cancer cells. Endocrinology 129 436–445. ( 10.1210/endo-129-1-436) [DOI] [PubMed] [Google Scholar]
- Power SG, Bocchinfuso WP, Pallesen M, Warmels-Rodenhiser S, Van Baelen H, Hammond GL. 1992. Molecular analyses of a human sex hormone-binding globulin variant: evidence for an additional carbohydrate chain. Journal of Clinical Endocrinology and Metabolism 75 1066–1070. [DOI] [PubMed] [Google Scholar]
- Pugeat M, Bonneton A, Perrot D, Rocle-Nicolas B, Lejeune H, Grenot C, Dechaud H, Brebant C, Motin J, Cuilleron CY. 1989. Decreased immunoreactivity and binding activity of corticosteroid-binding globulin in serum in septic shock. Clinical Chemistry 35 1675–1679. [PubMed] [Google Scholar]
- Regieli JJ, Jukema JW, Doevendans PA, Zwinderman AH, van der Graaf Y, Kastelein JJ, Grobbee DE. 2009. PPAR gamma variant influences angiographic outcome and 10-year cardiovascular risk in male symptomatic coronary artery disease patients. Diabetes Care 32 839–844. ( 10.2337/dbib8-1819) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner W, Hryb DJ, Kahn SM, Nakhla AM, Romas NA. 2010. Interactions of sex hormone-binding globulin with target cells. Molecular and Cellular Endocrinology 316 79–85. ( 10.1016/j.mce.2009.08.009) [DOI] [PubMed] [Google Scholar]
- Ruder H, Corvol P, Mahoudeau JA, Ross GT, Lipsett MB. 1971. Effects of induced hyperthyroidism on steroid metabolism in man. Journal of Clinical Endocrinology and Metabolism 33 382–387. ( 10.1210/jcem-33-3-382) [DOI] [PubMed] [Google Scholar]
- Savu L, Lombart C, Nunez EA. 1980. Corticosterone binding globulin: an acute phase ‘negative’ protein in the rat. FEBS Letters 113 102–106. ( 10.1016/0014-5793(80)80505-9) [DOI] [PubMed] [Google Scholar]
- Savu L, Zouaghi H, Carli A, Nunez EA. 1981. Serum depletion of corticosteroid binding activities, an early marker of human septic shock. Biochemical and Biophysical Research Communications 102 411–419. ( 10.1016/0006-291X(81)91536-9) [DOI] [PubMed] [Google Scholar]
- Scott SM, Wells L. 1995. Corticosteroid-binding globulin in preterm infants in an intensive care unit. Hormone Research 44 218–221. ( 10.1159/000184629) [DOI] [PubMed] [Google Scholar]
- Scrocchi LA, Hearn SA, Han VK, Hammond GL. 1993a. Corticosteroid-binding globulin biosynthesis in the mouse liver and kidney during postnatal development. Endocrinology 132 910–916. [DOI] [PubMed] [Google Scholar]
- Scrocchi LA, Orava M, Smith CL, Han VK, Hammond GL. 1993b. Spatial and temporal distribution of corticosteroid-binding globulin and its messenger ribonucleic acid in embryonic and fetal mice. Endocrinology 132 903–909. [DOI] [PubMed] [Google Scholar]
- Selva DM, Hammond GL. 2009a. Thyroid hormones act indirectly to increase sex hormone-binding globulin production by liver via hepatocyte nuclear factor-4alpha. Journal of Molecular Endocrinology 43 19–27. [DOI] [PubMed] [Google Scholar]
- Selva DM, Hammond GL. 2009b. Peroxisome-proliferator receptor gamma represses hepatic sex hormone-binding globulin expression. Endocrinology 150 2183–2189. [DOI] [PubMed] [Google Scholar]
- Selva DM, Hogeveen KN, Innis SM, Hammond GL. 2007. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. Journal of Clinical Investigation 117 3979–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seralini GE, Berube D, Gagne R, Hammond GL. 1990. The human corticosteroid binding globulin gene is located on chromosome 14q31-q32.1 near two other serine protease inhibitor genes. Human Genetics 86 73–75. [DOI] [PubMed] [Google Scholar]
- Siiteri PK, Murai JT, Hammond GL, Nisker JA, Raymoure WJ, Kuhn RW. 1982. The serum transport of steroid hormones. Recent Progress in Hormone Research 38 457–510. [DOI] [PubMed] [Google Scholar]
- Simard M, Hill LA, Underhill CM, Keller BO, Villanueva I, Hancock RE, Hammond GL. 2014. Pseudomonas aeruginosa elastase disrupts the cortisol-binding activity of corticosteroid-binding globulin. Endocrinology 155 2900–2908. ( 10.1210/en.2014-1055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Hill LA, Lewis JG, Hammond GL. 2015. Naturally occurring mutations of human corticosteroid-binding globulin. Journal of Clinical Endocrinology and Metabolism 100 E129–E139. [DOI] [PubMed] [Google Scholar]
- Simo R, Barbosa-Desongles A, Hernandez C, Selva DM. 2012a IL1beta down-regulation of sex hormone-binding globulin production by decreasing HNF-4alpha via MEK-1/2 and JNK MAPK pathways. Molecular Endocrinology 26 1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simo R, Barbosa-Desongles A, Saez-Lopez C, Lecube A, Hernandez C, Selva DM. 2012b. Molecular mechanism of TNFalpha-induced down-regulation of SHBG expression. Molecular Endocrinology 26 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simo R, Saez-Lopez C, Lecube A, Hernandez C, Fort JM, Selva DM. 2014. Adiponectin upregulates SHBG production: molecular mechanisms and potential implications. Endocrinology 155 2820–2830. ( 10.1210/en.2014-1072) [DOI] [PubMed] [Google Scholar]
- Slemenda CW, Longcope C, Zhou L, Hui SL, Peacock M, Johnston CC. 1997. Sex steroids and bone mass in older men. Positive associations with serum estrogens and negative associations with androgens. Journal of Clinical Investigation 100 1755–1759. ( 10.1172/JCI119701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Hammond GL. 1991. Ontogeny of corticosteroid-binding globulin biosynthesis in the rat. Endocrinology 128 983–988. ( 10.1210/endo-128-2-983) [DOI] [PubMed] [Google Scholar]
- Smith CL, Hammond GL. 1992. Hormonal regulation of corticosteroid-binding globulin biosynthesis in the male rat. Endocrinology 130 2245–2251. [DOI] [PubMed] [Google Scholar]
- Smith CL, Power SG, Hammond GL. 1992. A leu → his substitution at residue 93 in human corticosteroid binding globulin results in reduced affinity for cortisol. Journal of Steroid Biochemistry and Molecular Biology 42 671–676. ( 10.1016/0960-0760(92)90107-T) [DOI] [PubMed] [Google Scholar]
- Sridhar S, Walia R, Sachdeva N, Bhansali A. 2013. Effect of pioglitazone on testosterone in eugonadal men with type 2 diabetes mellitus: a randomized double-blind placebo-controlled study. Clinical Endocrinology 78 454–459. ( 10.1111/j.1365-2265.2012.04510.x) [DOI] [PubMed] [Google Scholar]
- Torpy DJ, Bachmann AW, Grice JE, Fitzgerald SP, Phillips PJ, Whitworth JA, Jackson RV. 2001. Familial corticosteroid-binding globulin deficiency due to a novel null mutation: association with fatigue and relative hypotension. Journal of Clinical Endocrinology and Metabolism 86 3692–3700. ( 10.1210/jcem.86.8.7724) [DOI] [PubMed] [Google Scholar]
- Torpy DJ, Lundgren BA, Ho JT, Lewis JG, Scott HS, Mericq V. 2012. CBG santiago: a novel CBG mutation. Journal of Clinical Endocrinology and Metabolism 97 E151–E155. [DOI] [PubMed] [Google Scholar]
- Underhill DA, Hammond GL. 1989. Organization of the human corticosteroid binding globulin gene and analysis of its 5-flanking region. Molecular Endocrinology 3 1448–1454. ( 10.1210/mend-3-9-1448) [DOI] [PubMed] [Google Scholar]
- Vermeulen A, Verdonck L, Kaufman JM. 1999. A critical evaluation of simple methods for the estimation of free testosterone in serum. Journal of Clinical Endocrinology and Metabolism 84 3666–3672. ( 10.1210/jcem.84.10.6079) [DOI] [PubMed] [Google Scholar]
- Vos MJ, Mijnhout GS, Rondeel JM, Baron W, Groeneveld PH. 2014. Sex hormone binding globulin deficiency due to a homozygous missense mutation. Journal of Clinical Endocrinology and Metabolism 99 E1798–E1802. [DOI] [PubMed] [Google Scholar]
- Westphal U. 1986. Steroid-protein interactions II. In Monographs on Endocrinology, pp 59–83. Berlin, Germany: Springer Verlag. [PubMed] [Google Scholar]
- Winters SJ, Gogineni J, Karegar M, Scoggins C, Wunderlich CA, Baumgartner R, Ghooray DT. 2014. Sex hormone-binding globulin gene expression and insulin resistance. Journal of Clinical Endocrinology and Metabolism 99 E2780–E2788. [DOI] [PubMed] [Google Scholar]
- Wisely GB, Miller AB, Davis RG, Thornquest AD Jr, Johnson R, Spitzer T, Sefler A, Shearer B, Moore JT, Miller AB, et al. 2002. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure 10 1225–1234. ( 10.1016/S0969-2126(02)00829-8) [DOI] [PubMed] [Google Scholar]
- Wu TS, Hammond GL. 2014. Naturally occurring mutants inform SHBG structure and function. Molecular Endocrinology 28 1026–1038. ( 10.1210/me.2014-1058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xita N, Tsatsoulis A. 2010. Genetic variants of sex hormone-binding globulin and their biological consequences. Molecular and Cellular Endocrinology 316 60–65. ( 10.1016/j.mce.2009.08.025) [DOI] [PubMed] [Google Scholar]
- Zouaghi H, Savu L, Guerot C, Gryman R, Coulon A, Nunez EA. 1985. Total and unbound cortisol-, progesterone-, oestrone- and transcortin-binding activities in sera from patients with myocardial infarction: evidence for differential responses of good and bad prognostic cases. European Journal of Clinical Investigation 15 365–370. ( 10.1111/j.1365-2362.1985.tb00286.x) [DOI] [PubMed] [Google Scholar]