

Abstract
We have shown that de novo designed peptides self-assemble in the presence of copper to create supramolecular assemblies capable of oxidation of dimethoxyphenol in the presence of dioxygen. Formation of the supramolecular assembly, akin to a protein fold, is critical for productive catalysis as the peptides possessing the same functional groups, but lacking the ability to self-assemble do not catalyze substrate oxidation. The ease with which we have discovered robust and productive oxygen activation catalysts suggests that these prion-like assemblies might have served as intermediates in evolution of enzymatic function and opens the path for development of new catalyst nanomaterials.
Keywords: peptides, de novo design, catalysis, self-assembly, supramolecular chemistry
Graphical Abstract
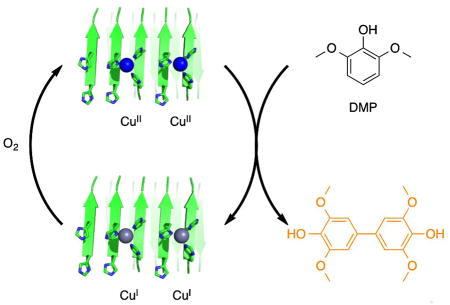
De novo designed peptides self-assemble in the presence of copper to create supramolecular assemblies that catalyze oxidation of dimethoxyphenol by dioxygen.
Proteins extensively rely on metal cofactors to achieve structural stability and function. Indeed, it is estimated that up to a third of all proteins contain various metal ions.[1] Metalloenzymes are capable of efficiently tuning the properties of a metal ion to catalyze very difficult chemical transformations such as conversion of methane to methanol or oxidative depolymerization of lignin using simple oxidants such as dioxygen and hydrogen peroxide.[2] Recently we reported that short peptides designed from the first principles self-assemble in the presence of zinc to form amyloid-like fibrils, which efficiently catalyze hydrolysis of p-nitrophenyl esters.[3] The most catalytically active fibrils showed activity that rivals that of the natural enzymes by weight. The ease with which we were able to discover such activity – the original library included only 10 peptides – suggests that self-assembly of short peptides could have been a major pathway in enzyme evolution and opens the door to utilizing this approach to a large array of chemical reactions. In this paper we ask whether self-assembly of peptides can be used to discover supramolecular catalysts for oxygen activation.
Designing efficient catalysts for oxygen activation presents several challenges: 1. The ligand in the primary coordination sphere needs to properly tune the metal ion’s redox potential; 2. The ligand has to be able to accommodate different oxidation states of the metal ion; 3. Catalysis should not involve formation of reactive oxygen species that can promote ligand decomposition. Thus it is hardly surprising that only a handful of successful protein/peptide designs capable of promoting redox reactions have been reported so far.[4]
The very simple coordination sphere produced by self-assembly of peptides previously reported by us[3] could potentially accommodate metal ions other than zinc. We set out to develop a copper based self-assembling system as copper ions are commonly employed in redox active enzymes, are hydrolytically stable at near neutral pH, and provide convenient spectroscopic handles.
As a model reaction in our studies, we have chosen oxidation of 2,6-dimethoxyphenol (DMP, Scheme 1). Oxidative dimerization of DMP produces colored product that can be easily assayed in a high throughput fashion. This reaction proceeds via CuII-mediated oxidation of the substrate; it is well benchmarked and extensively mechanistically characterized.[5] Additionally, oxidation of substituted phenols is commonly used in the polymer industry.[6]
Scheme 1.

Copper-mediated oxidation of dimethoxyphenol (DMP) by dioxygen. Fibril structures shown in this scheme are not derived from experimental data; they are based on a previously reported computational model for zinc containing peptide assemblies.[3]
In order to test whether self-assembling peptides can indeed support copper-mediated substrate oxidation, we used a small focused library of peptides previously shown to self-assemble in the presence of zinc and catalyze ester hydrolysis. The results of the initial screening are presented in Figure 1. The most active peptide (11, Ac-IHIHIQI-CONH2) showed activity that was more than an order of magnitude higher than baseline activity. The levels of activity are highly sequence dependent: the non-fibril forming control peptide (14, NH2-IHIHIQI-COOH) that has the same primary sequence but lacks the caps on the termini shows activity that is lower than that of free copper ions in buffer. Similar to our previous work, peptides containing residues that promote β-strand secondary structure (Ile and Val, 7, 8, 11 and 12) showed high activity compared to peptides with residues of lower β-sheet forming propensity (Leu and Ala, 1–5). CD spectra of 7 and 11 showed a clear β-sheet character in the presence of copper (Figure S1, Supporting Information). Replacing either His2 or His4 with alanine significantly reduces activity (Figure 1, peptides 9, 10). While both 9 and 10 form large aggregates with β-sheet character and bind copper, albeit with different stoichiometry (Figures S2, S3), they demonstrate lower activity under all conditions. This suggests that the His-X-His motif provides the most optimal functional group arrangement among the peptides studied in this work. The nature of the residue in position 6 is also important for catalytic activity, with peptides containing Gln and Tyr in this position producing the most active catalysts. Under the same conditions amyloid beta peptide (Aβ 1–40), which is known to self-assemble and bind copper,[7] does not catalyze DMP oxidation. Thus self-assembly of histidine-containing peptides alone is not sufficient for catalysis.
Figure 1.

Initial rates of product formation catalyzed by CuII (10 μM) in the absence/presence of various peptides (20 μM). All reactions are in 25 mM Hepes, pH 7.9 at room temperature, [DMP] = 500 μM. His = histidine (40 μM), Im = imidazole (40 μM), Ac-His-CONH2 = N-acetyl-histidine-amide (40 μM), Ab = Amyloid beta (10 μM).
We chose 11, the most active peptide, for in depth characterization. Based on the Job’s plot of activity (Figure S3) and copper titrations (Figure S4), we determined the ratio of CuII to 11 complex to be approximately 1:2. The rate enhancement of DMP oxidation relative to CuII in buffer is pH-dependent and reaches more than 65-fold at pH 6 (Figure S5). The catalyst undergoes multiple turnovers resulting in a complete oxidation of the substrate in less than 24 hours (Figure 2). The histidine complex of copper is completely inactive; imidazole and N-acetyl-histidine-amide show low activity, on par with that of non-self-assembling peptides 4 and 5 under the same conditions (Figure 1, Figure S6). A non-self-assembling hexahistidine peptide 16 (Figure S2), is also completely inactive, suggesting that multivalent coordination of histidines is not sufficient for activity. Thus 11 provides an appropriate coordination sphere for copper to engage in productive catalysis. Next, we performed ultracentrifugation experiment to determine the nature of the active species. Centrifugation for 1 hour at 100,000 g completely removes peptide 11 species from the solution as concluded from the absorbance of the supernatant (Figure S7). The supernatant shows no activity in DMP oxidation suggesting that large peptide aggregates are responsible for catalysis. Similarly, filtration of the copper-peptide complex through a 0.22 μm membrane (Figure S8) produces filtrate that has no activity. On the other hand, dialysis of the peptide solution fully preserves its oxygen activating activity (Figure S9). The inactive peptide 14 bound to copper, as shown by EPR data discussed below, did not show any secondary structure (Figures S1, S6) and did not precipitate in ultracentrifugation experiment under the same conditions (Figure S10).
Figure 2.

Conversion of dimethoxyphenol catalyzed by CuII in buffer, CuII in the presence of histidine (His), peptides 11 and 14. Reaction was initiated by adding DMP (200 μM). Aliquots were taken at various time points and immediately analysed by HPLC. Buffer: 25 mM Hepes, pH 8, [CuII] = 40 μM, [peptide] = 80 μM, [His] = 160 μM.
Ultracentrifugation has also provided us with an opportunity to quantitatively characterize binding of copper to the fibrils. Measurement of the equilibrium concentrations of copper in the supernatant after the centrifugation shows that under the conditions used, ~90% of the copper is bound by the peptide (Figure S10).
To probe CuII coordination environment in different peptides we used low-temperature EPR spectroscopy. The gII and AII values for CuII are commonly used to determine the composition of the copper coordination sphere. EPR spectra of CuII bound to the most active peptides (Ac-IHIHIQI-CONH2 and Ac-VHVHVYV-CONH2) are of the classic type 2 with gII = 2.27 and AII = 167 G, values that are consistent with either 3N1O or 2N2O coordination environment in the equatorial plane based on the Blumberg-Peisach plots (Figures 3, S11).[8] Peptide 14 (NH2-IHIHIQI-COOH) that does not aggregate, has no β-sheet structure at pH 8 as shown by CD and ultracentrifugation (Figures S6 and S10) and is inactive in DMP oxidation assay, has a distinctly different EPR spectrum (Figure S11). These results suggest a major change in the coordination environment of copper despite essentially identical primary peptide sequence. EPR spectra also shed light on the possible reaction mechanism. Addition of DMP to CuII – 7 results in gradual diminishing of the EPR signal (Figure 3) consistent with the reduction of CuII to CuI in line with the previously proposed mechanism.[5c, 5d]
Figure 3.

X-band EPR spectra of CuII (150 μM) in the presence of peptide 7 (300 μM) acquired in pH 7.6 buffer (25 mM Hepes, 10 % glycerol) at 10 K. Frequency 9.39 GHz, power 5.024 mW, modulation frequency 100 kHz, modulation amplitude 8 G, conversion time 40 ms, time constant 163.8 ms. After acquiring the spectrum (solid line), DMP was added to the sample to a final concentration of 1 mM (5 μL of 70 mM solution), the sample was mixed and frozen (+DMP, 30 s, dotted line). After recording another EPR spectrum, the sample was thawed and incubated at room temperature for 10 min (+DMP, 10 min, dashed line).
The contribution of self-assembly to the overall activity can be examined by comparing the properties of peptides 4, 5 and 11. The identities of metal-binding residues are the same in all cases but the residues in the “hydrophobic” positions are varied. Peptides 4 and 5 that have lower β-sheet forming propensity bind copper but show no aggregation nor β-sheet character in the presence of the metal ion (Figures S6, S7, S10). The copper coordination environments in 5 and 11 are different as judged by the EPR data (Figures S11, S12). The observed differences in activity can be explained by the high stability of the β-sheet assemblies that lock the histidines into a conformation that is more suitable for the appropriate copper coordination akin to how a protein fold modifies metal ions properties. Comparison of peptides 4 and 11 is particularly instructive. Isomerization of the side chain of amino acid residues not involved in the primary coordination sphere of the copper ion (leucine vs. isoleucine) results in self-assembly and a ca. 5-fold increase in the initial rate of phenol oxidation.
We established that oxidation of DMP promoted by CuII-peptide is oxygen-dependent, as removal of oxygen prevented formation of the product, but introduction of O2 into the reaction mixture immediately resulted in DMP oxidation (Figure S13). Oxygen, superoxide and H2O2 do not oxidize DMP on the timescale of several hours, however formation of the product occurs immediately upon mixing CuII-peptide and DMP, emphasizing the crucial role of CuII in this reaction. Addition of catalase (Figure S14) or superoxide dismutase (Figure S15) does not substantially diminish the reaction rate mechanistically supporting the CuII-promoted oxidation and effectively excluding the possibility of radical oxidation of the substrate. Among the different metal ions tested (CuII, FeIII, FeII, MnII, NiII, CoII) only CuII catalyzes DMP oxidation to any appreciable extent, suggesting that redox potential of CuII and coordination environment supported by fibril core are optimal for catalysis (Figure S16).
Supramolecular approaches to designing efficient catalysts for chemical reactions have been extremely productive.[9] Self-assembling properties of peptides have been previously used to create multidentate ligands for transition metal catalysts,[10] hydrogels with esterase activities,[11] and light-capturing materials.[12] The ability to genetically encode large peptide libraries opens the path for discovery and optimization of peptide catalysts using high-throughput techniques.[13] In this paper we show that self-assembly of short de novo designed peptides results in formation of efficient supramolecular catalysts capable of oxygen activation. Moreover, we show that supramolecular assemblies are capable of supporting oxygenation catalysis that does not rely on radicals. This finding underscores the fact that amyloid-like assemblies formed by even very short peptides can facilitate a number of various chemical transformations in a highly sequence-specific manner. Considering recent findings that amyloid-supported metal sequestration and catalysis is more likely to be a rule rather than an exception,[3, 14] we expect de novo designed self-assembled catalytic peptides to combine the power of highly controlled metal coordination sphere common in homogeneous catalysis with practical advantages of heterogeneous catalysts. Moreover, synergistic interactions observed in these systems provide additional opportunities for high throughput screening for catalytic function. Finally, the diversity of reactions catalysed by simple peptide assemblies lends further support to the amyloid-first hypothesis of emergence of enzymatic function.[15]
Supplementary Material
Acknowledgments
This work was supported in part by a grant number 1332349 from NSF-EFRI, ORAU Ralph E. Powe Junior Faculty Enhancement award and a Humboldt Fellowship to I.V.K. EPR work was supported by an NIH grant P41GM103521. We thank Dr. Boris Dzikovski for assistance with EPR, Prof. Robert Doyle for assistance with circular dichroism, and Dr. Torsten Wöllert for assistance with ultracentrifugation.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Waldron KJ, Robinson NJ. Nat Rev Microbiol. 2009;7:25–35. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- 2.Bugg TDH, Ahmad M, Hardiman EM, Singh R. Curr Opin Biotech. 2011;22:394–400. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Rufo CM, Moroz YS, Moroz OV, Stöhr J, Smith TA, Hu X, DeGrado WF, Korendovych IV. Nat Chem. 2014;6:303–309. doi: 10.1038/nchem.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Zastrow ML, Pecoraro VL. Coord Chem Rev. 2013;257:2565–2588. doi: 10.1016/j.ccr.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chino M, Maglio O, Nastri F, Pavone V, DeGrado WF, Lombardi A. Eur J Inorg Chem. 2015;2015:3352–3352. doi: 10.1002/ejic.201500470. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Watkins DW, Armstrong CT, Anderson JLR. Curr Opin Chem Biol. 2014;19:90–98. doi: 10.1016/j.cbpa.2014.01.016. [DOI] [PubMed] [Google Scholar]; d) Roy A, Madden C, Ghirlanda G. Chem Commun. 2012;48:9816–9818. doi: 10.1039/c2cc34470j. [DOI] [PubMed] [Google Scholar]; e) Yeung N, Lin YW, Gao YG, Zhao X, Russell BS, Lei LY, Miner KD, Robinson H, Lu Y. Nature. 2009;462:1079–1082. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kleingardner JG, Bren KL. Acc Chem Res. 2015;48:1845–1852. doi: 10.1021/acs.accounts.5b00106. [DOI] [PubMed] [Google Scholar]; g) Maeda Y, Makhlynets OV, Matsui H, Korendovych IV. Annu Rev Biomed Eng. 2016 doi: 10.1146/annurev-bioeng-111215-024421. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Mattinen ML, Maijala P, Nousiainen P, Smeds A, Kontro J, Sipila J, Tamminen T, Willfor S, Viikari L. J Mol Cat B Enzym. 2011;72:122–129. [Google Scholar]; b) Solano F, Lucas-Elio P, Lopez-Serrano D, Fernandez E, Sanchez-Amat A. Fems Microbiol Lett. 2001;204:175–181. doi: 10.1111/j.1574-6968.2001.tb10882.x. [DOI] [PubMed] [Google Scholar]; c) Baesjou PJ, Driessen WL, Challa G, Reedijk J. J Mol Catal A-Chem. 1998;135:273–283. [Google Scholar]; d) Baesjou PJ, Driessen WL, Challa G, Reedijk J. J Mol Catal A-Chem. 1996;110:195–210. [Google Scholar]
- 6.a) Ikeda R, Sugihara J, Uyama H, Kobayashi S. Macromolecules. 1996;29:8702–8705. [Google Scholar]; b) Hay AS. J Polym Sci, Polym Chem. 1998;36:505–517. [Google Scholar]
- 7.Cassagnes L-E, Herve V, Nepveu F, Hureau C, Faller P, Collin F. Angew Chem Int Ed. 2013;52:11110–11113. doi: 10.1002/anie.201305372. [DOI] [PubMed] [Google Scholar]
- 8.Peisach J, Blumberg WE. Arch Biochem Biophys. 1974;165:691–708. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 9.a) Duncan KL, Ulijn RV. Biocatalysis. 2015;1:67–81. [Google Scholar]; b) Raynal M, Ballester P, Vidal-Ferran A, van Leeuwen PWNM. Chem Soc Rev. 2014;43:1660–1733. doi: 10.1039/c3cs60027k. [DOI] [PubMed] [Google Scholar]
- 10.Laungani AC, Breit B. Chem Commun. 2008:844–846. doi: 10.1039/b716529c. [DOI] [PubMed] [Google Scholar]
- 11.Singh N, Conte MP, Ulijn RV, Miravet JF, Escuder B. Chem Commun. 2015;51:13213–13216. doi: 10.1039/c5cc04281j. [DOI] [PubMed] [Google Scholar]
- 12.Fry HC, Liu Y, Dimitrijevic NM, Rajh T. Nat Commun. 2014;5:4606. doi: 10.1038/ncomms5606. [DOI] [PubMed] [Google Scholar]
- 13.Maeda Y, Javid N, Duncan K, Birchall L, Gibson KF, Cannon D, Kanetsuki Y, Knapp C, Tuttle T, Ulijn RV, Matsui H. J Am Chem Soc. 2014;136:15893–15896. doi: 10.1021/ja509393p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Friedmann MP, Torbeev V, Zelenay V, Sobol A, Greenwald J, Riek R. PLoS One. 2015;10 doi: 10.1371/journal.pone.0143948. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu YQ, Wang MF, Qi W, Su RX, He ZM. Chem J Chin Univ. 2015;36:1304–1309. [Google Scholar]; c) Singh N, Tena-Solsona M, Miravet JF, Escuder B. Isr J Chem. 2015;55:711–723. [Google Scholar]; d) Zhang CQ, Xue XD, Luo Q, Li YW, Yang KN, Zhuang XX, Jiang YG, Zhang JC, Liu JQ, Zou GZ, Liang XJ. ACS Nano. 2014;8:11715–11723. doi: 10.1021/nn5051344. [DOI] [PubMed] [Google Scholar]; e) Gazit E. Nanomedicine. 2014;9:2433–2436. doi: 10.2217/nnm.14.173. [DOI] [PubMed] [Google Scholar]; f) Bolisetty S, Mezzenga R. Nat Nanotech. 2016;11:365–371. doi: 10.1038/nnano.2015.310. [DOI] [PubMed] [Google Scholar]; g) Tena-Solsona M, Nanda J, Diaz-Oltra S, Chotera A, Ashkenasy G, Escuder B. Chem Eur J. 2016;22:6687–6694. doi: 10.1002/chem.201600344. [DOI] [PubMed] [Google Scholar]
- 15.Greenwald J, Riek R. J Mol Biol. 2012;421:417–426. doi: 10.1016/j.jmb.2012.04.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.