

Summary
Nanodiscs and isotropic bicelles are promising membrane mimetics in the field of solution NMR spectroscopy of integral membrane proteins (IMPs). Despite varied challenges to solution NMR studies of IMPs, we attribute the paucity of solution NMR structures in these environments to the inability of diverse IMPs to withstand detergent treatment during standard nanodisc and bicelle preparations. Here, we present a strategy that creates small isotropic bicelles from IMPs co-translationally embedded in large nanodiscs using cell-free expression. Our results demonstrate appreciable gains in NMR spectral quality while preserving lipid-IMP contacts. We validate the approach on the detergent sensitive LspA, which finally allowed us to perform high quality triple resonance NMR experiments for structural studies. Our strategy of producing bicelles from nanodiscs comprehensively avoids detergent during expression and preparation and is suitable for solution NMR spectroscopy of lipid-IMPs complexes.
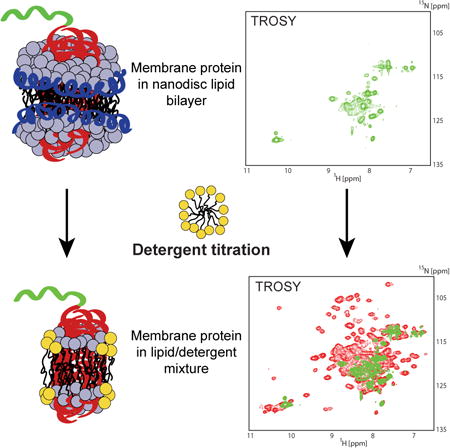
Graphical abstract
Laguerre et al. show that nanodisc bilayers can be peeled away from embedded membrane proteins by detergent titration to make bicelles. Avoiding initial detergent solubilization, this method preserves lipid contacts and functional folds of detergent-sensitive membrane proteins. The resulting improvements in spectral intensity facilitate high resolution NMR spectroscopy for structure determination.
Introduction
Solution NMR is an important tool for the biophysical characterization of integral membrane proteins (IMPs). A significant stumbling block to its commonplace use is the process of selecting a membrane mimetic that preserves the structure and function of an IMP suitable for solution NMR studies (Warschawski et al., 2011). Many successful NMR studies are conducted in detergent solutions on well-behaved targets that are impervious to a variety of conditions (Kang and Li, 2011). However, the often deleterious effects of detergents on IMP stability, function and structure are widely acknowledged. Efforts to address this have witnessed the development of a range of membrane mimetics that provide or preserve lipids for IMP complexes (Raschle et al., 2010).
Nanodiscs and bicelles are popular membrane mimetics with lipid bilayers suitable for solution NMR studies of IMPs. Nanodiscs (NDs) are water soluble nanoparticles consisting of lipid bilayers encapsulated by two membrane spanning proteins (MSPs) (Denisov et al., 2004) whose lengths can be modified to generate NDs of various sizes (Hagn et al., 2013). The well-established isotropic bicelle, consisting of a detergent stabilized lipid bilayer, also presents an attractive option for solution NMR studies (Dürr et al., 2012; Kim et al., 2009). Unfortunately, there are few structures of IMPs solved by solution NMR in either of these conditions. This may not be surprising for the recently introduced ND. However, this scarcity is conspicuous for the long established isotropic bicelle, although limited recipe formulation, which predominantly relies on the use of phosphocholine lipids, has certainly contributed to their sporadic use. The paucity of solution NMR structures in these systems can, in part, be attributed to the slow tumbling rate of large IMP complexes, which exacerbates the effects of the characteristic spectral overlap and broad line widths suffered by NMR spectra of IMPs. However, there are various strategies that address the size restrictions and spectral overlap of large complexes, such as selective labeling strategies (Sobhanifar et al., 2010), perdeuteration and the TROSY effect (Pervushin et al., 1997). Further, advances that tackle the problem that high levels of deuteration pose to obtaining structural information via 1H interactions, such as special isotopic labels (Kerfah et al., 2015; Tugarinov and Kay, 2003) and methods to attain distance information that forego the NOE (Hass and Ubbink, 2014; Iwahara et al., 2007) have made the pursuit of large IMP complexes possible. Hence, technological stumbling blocks posed by solution NMR cannot fully account for the lack of solution NMR structures of IMPs in NDs and bicelles.
Standard ND and bicelle preparations begin with IMPs solubilized in detergent, relying on the expectation that lost function or structure can be restored upon re-introduction into the lipid bilayer as typified by model membrane proteins (MPs) (Lau and Bowie, 1997; London and Khorana, 1982). However, model MPs exhibiting robust resistance to detergent effects and well dispersed NMR spectra (Nietlispach and Gautier, 2011), are most likely the exception as opposed to the rule. Strategies that eliminate detergent exposure during crucial initial IMP preparation steps, maintaining the presence of lipids, while addressing the spectral overlap of IMPs, would reduce the time-consuming screening that characterizes the preliminary stages of NMR solution studies of non-model MPs.
With this in mind, we have pursued cell-free (CF) protein synthesis to prepare IMPs expressed directly into ND lipid bilayers (Katzen et al., 2008). The CF system offers numerous advantages to structural biologists focusing on IMPs. Attractive features include high expression yields and access to the protein synthesis machinery during expression making the use of sophisticated labeling strategies to address the overlapped spectra relatively routine (Sobhanifar et al., 2010). However, it is the co-translational expression of IMPs into preformed, easy-to-handle, water-soluble bilayers that crucially avoids the use of harsh detergents for solubilization and extraction (Lyukmanova et al., 2012; Proverbio et al., 2013). Thereafter, bicelles or mixed micelles can then be generated by quantitatively titrating the IMP-ND complexes with suitable detergents. As detergent exposure is the last step, this process would be of interest for those IMP targets where lipid contact is obligatory for stability and/or function. The main benefit of this procedure is the drastic improvement in spectral intensity of the IMPs over MP-ND samples, which makes triple resonance NMR experiments in the pursuit of structural characterization feasible.
We demonstrate and validate our method on four MPs, including the detergent sensitive pharmacological drug target, lipoprotein signal peptidase II (LspA). We also present the first structural analysis of LspA by solution NMR in conditions suitable for performing triple resonance NMR experiments that, in combination with a combinatorial labeling strategy, led to its full backbone assignment and secondary structure prediction.
Results
Detergent screening of LspA for solution NMR
Initially, we assessed E.coli expressed LspA in n-dodecyl β-D-maltoside (DDM), however, 15N-TROSY spectra (Figure 1A) exhibited poor sensitivity common to IMPs in DDM (Sanders and Sönnichsen, 2006) and suggested that selective labeling would be necessary. Furthermore, LspA DDM samples were unstable at high temperature making it unsuitable for NMR experiments. Exchange from DDM to other detergents, such as fos-choline 12 (DPC), led to monumental loss of protein. The expected requirement for labeling strategies, deuteration and the possibility to express into different membrane mimetics made CF expression a logical step. We expressed 2H/15N-labeled LspA in precipitate, detergent and lipid CF modes (P-CF, D-CF and L-CF respectively). We assessed the functionality of LspA solubilized in various detergents by incubating LspA with a lipid modified substrate, a fusion protein of maltose binding protein and lipoprotein (MBP-LPP). Cleavage by active LspA at the lipobox site between the MBP and LPP is apparent by the presence of the free MBP at approximately 40 kDa and cleaved LPP at 10 kDa (Figure S1). Incubation of LspA with its specific inhibitor, globomycin, prevents cleavage. LspA expressed in P-CF mode was functional in a range of detergents (Figure S1B). DPC was selected as the most suitable for solution NMR, however LspA DPC samples demonstrated very poor TROSY quality (Figure 1B), attributed to aggregation and low concentrations of LspA. Unfortunately, LspA proved to be highly unstable in many detergents and was unamenable to a variety of concentration and exchange techniques, necessary to produce high concentrated samples for NMR spectroscopy. After extensive screening to improve LspA stability with various additives and lipid and detergent mixtures, P-CF produced LspA samples were abandoned. High concentrations of LspA in predominantly detergent solutions necessary to obtain a sufficient NMR sample were never achieved and never exceeded 50 μM. LspA expressed poorly in D-CF and L-CF modes and was insufficient for solution NMR samples or standard bicelle preparation from liposomes.
Figure 1.
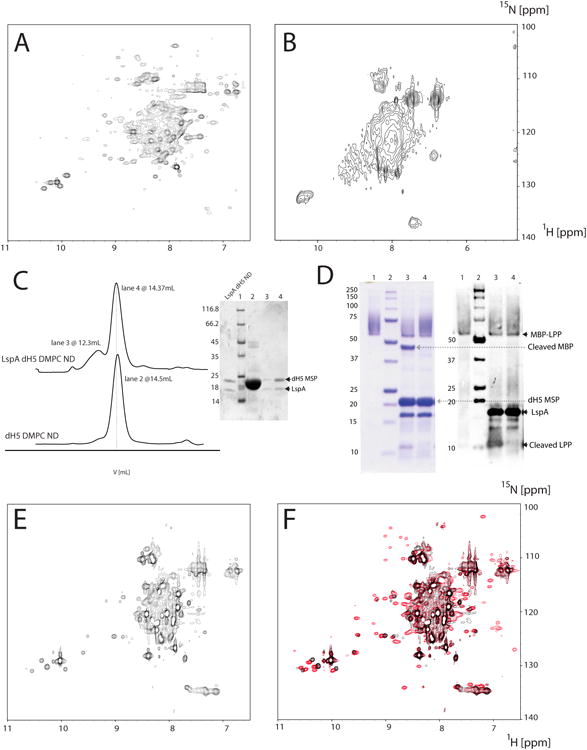
Sample optimization and characterization of LspA. 15N-labeled LspA in DDM from E.coli (A) and 2H/15N-labeled LspA in DPC from P-CF expression (B). (C) Size exclusion chromatograms of empty and LspA dH5 DMPC NDs. The molecular weight obtained by calibration of an analytical Superdex 200 is 95kDa for empty dH5 DMPC NDs and 102kDa for LspA embedded dH5 DMPC NDs. Buffer conditions were 20mM TrisHCl pH 8, 100mM NaCl. The majority of LspA in dH5 DMPC NDs are in a single homogenous peak (lane 4 of the Coomassie stained gel) that is slightly larger than empty dH5 DMPC NDs (lane 2). A small amount of LspA is also present in larger aggregates (lane 3). (D) Functional assay of LspA in dH5 DMPC NDs. Active LspA (lane 3) cleaves at the lipobox of the substrate fusion protein MBP-LPP (lane 1) and free MBP and free LPP are visible on SDS-PAGE gel and the western blot respectively. Lane 4 contains LspA in dH5 DMPC ND incubated with globomycin and does not cleave MBP-LPP. Free MBP and dH5 MSP are absent in lanes 3 and 4 of the western blot due to their consistently poor visualization with the anti-his antibody. (E) [15N-1H] TROSY of LspA in dH5 DMPC ND without globomycin. (F) Overlay of a [15N-1H] TROSY of LspA in dH5 DMPC NDs with globomycin (red) and the spectrum shown in (E) without the inhibitor.
Cell-free expression and optimization of IMPs in NDs
Assuming that LspA requires lipids to maintain a stable fold, we trialed the direct expression into preformed DMPC NDs that has been successfully established with other MPs (Katzen et al., 2008; Proverbio et al., 2013; Roos et al., 2014). After testing different versions of MSP, we chose the dH5 version due to highest stability and small size. Using size-exclusion chromatography, the insertion of LspA into the dH5 DMPC NDs was assessed and confirmed by its similar retention volume as empty NDs (Figure 1C). The functionality of LspA in dH5 DMPC NDs was verified by incubation and inhibition with MBP-LPP and globomycin respectively (Figure 1D). LspA dH5 DMPC ND samples were stable for longer than 1 week and concentrations > 200 μM could be produced. Unfortunately, TROSY spectra of 2H/15N-labeled LspA in NDs displayed poor chemical shift dispersion and a lack of uniformity of line shape and intensity (Figure 1E). We assessed whether globomycin would result in improved spectra by adding globomycin to the CF reaction mixture to produce holo-LspA samples. Indeed, TROSY spectra of holo-LspA demonstrate a substantial improvement in the spectral quality (Figure 1F), suggesting globomycin stabilizes the structure of LspA by suppressing conformational exchange processes attributed to high flexibility. Despite significant improvement in the spectral dispersion of holo-LspA, the rotational correlation time of LspA in dH5 DMPC NDs remained quite long (82 ns at 318 K) preventing the measurement of high resolution NMR spectra even at elevated temperatures. However, during our sample optimization efforts, we fortuitously discovered that titration of LspA ND samples with small quantities of detergent, including DPC and 1,2-diheptanoyl.sn-glycero-3-phoscholine (diC7PC) significantly improved the quality of the TROSY spectra (Figure 2A).
Figure 2.
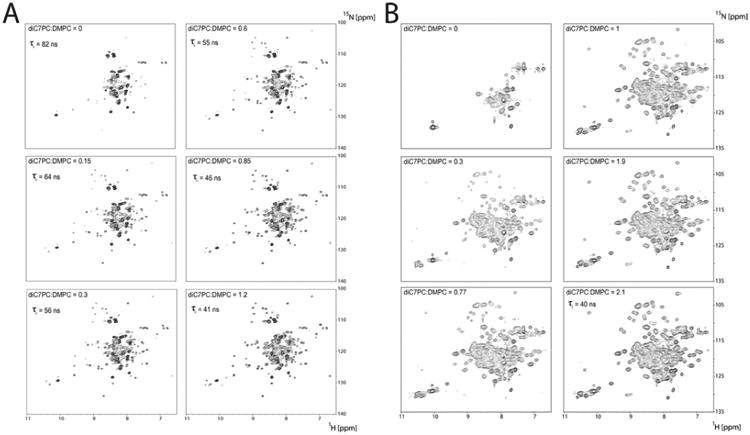
Effects of diC7PC titration on detergent sensitive IMPs in dH5 DMPC NDs. [15N,1H]-BEST-TROSY spectra (800 MHz) of (A) 2H/15N-labeled LspA and (B) EmrE in dH5 DMPC ND during diC7PC titration. Rotational correlation times as determined by TRACT are indicated.
Validating the effects of detergent on MP in NDs
To characterize the effect of detergent on our MP-ND preparations, we performed titrations of DPC or diC7PC to LspA NDs samples in a stepwise fashion and measured two dimensional [15N,1H]-TRACT experiments at each titration point. Progressive addition of diC7PC to LspA NDs samples increased signal intensity without changes in peak position (Figure 2A and S2A). The optimum for LspA is reached at a diC7PC:DMPC ratio of 1.2. At higher concentrations, signals begin to broaden (Figure S2A). To exclude the protein specificity of this effect, we expressed EmrE bound to its substrate tetraphenoylphosphonium (TPP+), bacterial proteorhodopsin (PR) and the voltage sensing domain of the phosphoinositide phosphatase from Danio rerio (DrVSD) into dH5 DMPC and DMPG NDs and repeated the detergent titration to ratios that achieve maximum spectral enhancement (Figure 2B, S2B and S3). In each case, addition of detergent resulted in an appreciable enhancement of spectral resolution and signal intensities with minimal change in the resonance positions.
The titration of EmrE in dH5 DMPC NDs with diC7PC had the most pronounced effect and is attributed to its dimeric state and concomitant larger size (Ubarretxena-Belandia et al., 2003). EmrE requires bicellar conditions to produce adequate TROSY spectra and is detergent sensitive (Morrison and Henzler-Wildman, 2012). Our ratios are the inverse of the standard q ratio which reports long-chain:short-chain lipid ratio, however our [15N, 1H]-TROSY spectrum of EmrE is very similar to published spectra in isotropic bicelles (Morrison et al., 2012) suggesting similar sample conditions despite different q ratios This suggests that EmrE is in contact with DMPC lipid in our titrated mixtures at a diC7PC:DMPC ratio of 2.1. In contrast to EmrE, PR is monomeric, stable and functional in a predominately micellar environment (Reckel et al., 2011). This detergent insensitivity is reflected in that spectra at the final titration point in DMPG ND (ratio of 2.8) are very similar to PR in diC7PC micelles (Figure S4C and S4D). We also performed titration experiments with DrVSD in DMPG ND which is assumed to form dimers in lipid bilayers (Li et al., 2014, 2015). Addition of DPC resulted in an appreciable enhancement of spectral resolution and signal intensities, however, the spectra did not reach the quality obtained with the other three proteins. This is presumably due to DrVSD conformational flexibility in the absence of an inhibitor. Nevertheless, the general trend seen with LspA, PR and EmrE is present as demonstrated with proton-carbon correlation spectra of selectively 13C-labeled DrVSD (Figure S3). The results with the different MPs demonstrate that the improvement in spectra is not protein specific.
To assess whether the improvement in [15N, 1H]-TROSY spectra is due to a decrease in transverse relaxation rates of the overall MP particle, we measured the rotational correlation time during the diC7PC titration for LspA, EmrE and PR using a two dimensional version of the TRACT experiment. The τc values for LspA decreased as the titration progressed and at the optimal diC7PC:DMPC ratio of 1.2 yielded values slightly above 40 ns at a temperature of 45°C, corresponding to a molecular weight of approximately 100 kDa. Both EmrE and PR demonstrated a decrease in their initial τc values from > 100 ns to approximately 40 ns for EmrE and 32 ns for PR at their final and optimal ratio. Despite similar τc values at their final titration points, each MP-ND has a different optimal diC7PC ratio indicating that different quantities of detergent are required to optimize the spectral quality of different MP samples. This result suggests that while the overall effect of the titration is not protein dependent, the optimal detergent lipid ratio is. This might reflect differences in the affinity of certain MPs for lipids and/or the different number of lipid molecules in the NDs depending on the size of the MP. These results also demonstrate the mildness of this method in that it preserves MP-lipid complexes necessary to maintain functional folds.
Detergent effects on ND bilayers and lipid-IMP contacts
To investigate the effect of the detergent on the lipid, we measured 31P NMR spectra on ND samples with and without MP during the titration (Figure 3). The 31P chemical shifts of DMPC and diC7PC reflect overall particle size, organization and homogeneity with broader, more upfield shifts indicating larger particles (liposomes, bicelles and macrodiscs) (Park et al., 2011). The chemical shift of the 31P peak of DMPC in NDs has been shown to correspond to the isotropic value of the phosphodiester group (Nolandt et al., 2012). The 31P chemical shift of DMPC in our NDs was recorded at -0.69 ppm. As the titration progresses, the diC7PC signal appears at -0.63 ppm. The DMPC peak at -0.69 ppm sharpens and displays a peak doubling at detergent-to-lipid ratios between 0.6 and 1.0. At higher ratios, the second DMPC peak collapses leaving a DMPC peak at -0.67 ppm. The splitting of the DMPC signal is most likely due to a transition of larger ND bilayers to smaller bicelle-like structures. The effects of diC7PC on DMPC in NDs is consistent irrespective of whether the NDs contain a MP (Figure S5). This supports our assertion that the detergent affects the lipids of NDs in a protein independent manner.
Figure 3.
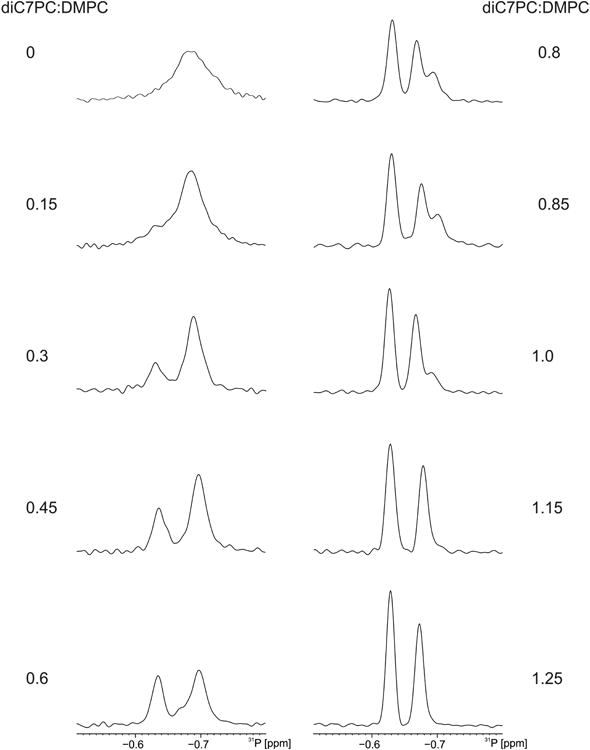
Effect of detergent on ND bilayer. LspA containing dH5 DMPC ND titrated with diC7PC as monitored by 31P NMR at 202 MHz. The highfield and lowfield signals are assigned to DMPC and diC7PC, respectively. The DMPC signals exhibit a splitting at detergent-lipid ratios between approximately 0.6 and 1.0, what might be interpreted as a decrease in particle size from ND to smaller bilayers. Lipid:detergent ratios were calculated from the integrated peak volumes when peaks are resolved. For conditions without resolved peaks the ratios were back-calculated based on the amount of detergent added.
The [15N, 1H]-TROSY spectra of all the IMPs display a lack of significant amide chemical shift changes during the titration, which led us to investigate whether this is attributed to the IMPs remaining in contact with lipids. To investigate this, we measured 3D 15N-resolved [1H,1H]-NOESY spectra of 15N-labeled LspA after titration with deuterated diC7PC. We could observe intermolecular NOEs between amide protons of transmembrane helices of LspA and the fatty acid methylene groups of DMPC (at 1.25 ppm) (Figure 4). Repeating these experiments with deuterated DMPC but protonated diC7PC resulted in the disappearance of all intermolecular NOEs, demonstrating that the transmembrane helices of LspA are surrounded by lipid molecules.
Figure 4.

Projections along the 15N dimensions of 3D NOESY-[15N,1H]-TROSY spectra (950 MHz) of 2H/15N-labeled LspA at diC7PC-DMPC ratios of 1.2. Samples employed for recording the spectra were prepared with protonated DMPC/d26-diC7PC (left) and d54-DMPC/protonated diC7PC (right). Indicated in red are the NOEs to the DMPC fatty acid methylene at 1.25ppm which are noticeably absent on the right. DMPC NOEs in the left spectrum are missing for certain residues (around 9.5ppm for V40, N47 and N53) due to their location in the β-sheets which are not embedded within the lipid bilayer. Peaks appearing in the spectrum on the right hand side are due to incomplete deuteration of that sample.
Destruction of the ND
We investigated the role of MSP and its ability to maintain the integrity of the ND during detergent titration. We repeated the diC7PC titration to empty 13C15N dH5 NDs and to free dH5 MSP (Figure S6A and S6B). At the end point of the titration, ND made with dH5 MSP have a very similar spectrum to dH5 MSP directly titrated with diC7PC (Figure 5A and S6). These results are independent whether the NDs contain a MP or not (Figure S6B and S6C), suggesting that dH5 MSP becomes detached from the MP-DMPC complex during diC7PC titration.
Figure 5.

Effects of dH5 MSP to detergent and destruction of the ND. (A) Comparison of 13C15N dH5 MSP in DMPC NDs (left) and free in solution (right) after titration with diC7PC which display marked similarity. (B) [15N,1H]-BEST-TROSY spectra of EmrE (B) and LspA (C) in dH5 MSP/DMPC/detergent mixtures (left) and after the removal of dH5 MSP (right).
To further demonstrate that dH5 MSP is detached from the ND during the titration, we removed dH5 MSP from the EmrE and LspA titrated samples by purification. This analysis showed a clear separation of the MPs and MSP (Figure S7A). Reanalyzing the eluted fractions by NMR spectroscopy showed a very similar spectra as obtained by titrating LspA with DPC or EmrE with diC7PC (Figure 5B and 5C) suggesting these complexes retain lipids and further supporting the suggestion that detergent titration to the MP-NDs fragments the ND components. This interpretation is also consistent with the effect of the detergent titration on PR dH5 DMPG ND resulting in detergent solubilized monomers as demonstrated by very similar correlation times at the end of the titration and in diC7PC micelles (32 ns vs 28ns) (Figure S4A and S4B). Interestingly, removal of MSP from LspA samples had differing outcomes depending on whether DPC or diC7PC was used (Figure S7B). Comparison of the 31P NMR spectra of DPC and diC7PC re-purified samples following titration suggests that all lipids were lost from samples purified with diC7PC (Figure S7C). Overall, diC7PC and DPC titration on MP in NDs have the identical effect of reducing the correlational tumbling time by formation of smaller bicelles/mixed micelles. However, these re-purification experiments demonstrate the differential abilities of different detergents in preserving MP-lipid complexes.
NMR spectroscopy, combinatorial labeling and assignment of LspA
Subsequent to confirming and validating the detergent titration to NDs, we used this sample preparation technique to pursue the structural investigation of LspA. Despite the improvement in dispersion upon binding of globomycin, the spectral overlap necessitated the use of a combinatorial labeling strategy as previously published (Löhr et al., 2012). Unlike previous combinatorial labeling strategies, the labeling scheme (Table 1) chosen for LspA required the use of selective labels in a deuterated background. Details about the combinatorial strategy are described in the supplemental information. Briefly, a suite of 2D HSQC, HN(CO), HN(CA), HN(COCA), DQ-HN(CA) and Hα-filtered HN(CA) experiments on 3 samples were collected and analyzed to obtain the backbone assignment of LspA (Figure 6 and 7A). In addition, we prepared uniformly 2H,13C, 15N-labeled LspA in dH5 DMPC ND and recorded 3D BEST-[15N,1H]-TROSY- versions of HNCA, HNCO, HNCACB and HN(CO)CA (Figure 7B) which in combination with the backbone assignment from the combinatorial labeling allowed us to ascertain 13Cα and 13Cβ chemical shifts and facilitated the resonance assignment of 98% of all non-proline amide 1H and 15N, 98% of 13Cα and 84% of 13Cβ resonances (Biological Magnetic Resonance Bank (BMRB) accession code 26857).
Table 1. Combinatorial selective labelling scheme employed for backbone assignment of LspA.
| labelling type a | sample # | ||
|---|---|---|---|
|
|
|||
| 1 | 2 | 3 | |
| 2H/15N | Thr | Thr | Thr |
| Lys | Lys | Phe | |
| Trp | Tyr | Trp | |
| Leu | Arg | Arg | |
| 2H/13C/15N b | Ala | Ile | Val |
| 1-13C c | Pro | Pro | Ser |
| Asp | Ser | Asp | |
| 2-13C d | Phe | Phe | Leu |
| Tyr | Leu | Tyr | |
amino acids not listed were uniformly 2H labelled.
fully 13C and 15N labelled.
selectively 13C labelled at the carbonyl position.
selectively 13C-labeled at the α-position.
Figure 6.
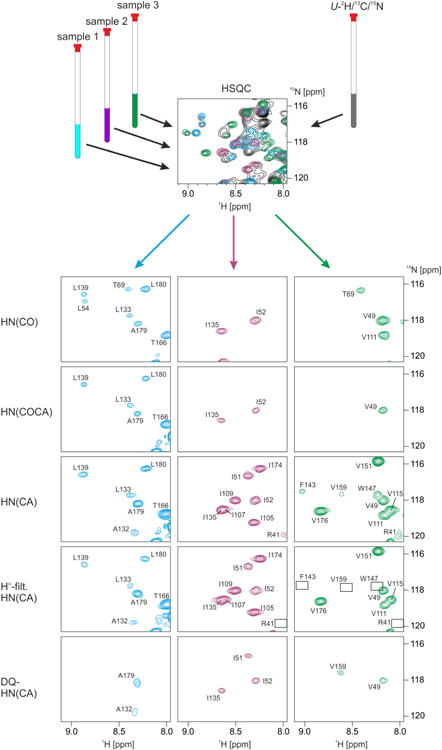
Application of combinatorial 15N, 13Cα, 13C' labeling to LspA. An overlay of regions from 15N-HSQC spectra of a uniformly (black contours) and the three selectively labelled (blue, magenta and green contours) samples is shown at the top. Spectra analysis for the uniformly labelled protein is severely hampered by overlap, whereas almost all signals (exception: I107/I135 in the HN(CA) of sample 2) are well-resolved in the series of 2D triple-resonance spectra of the three samples of the combinatorial scheme. Signals that are missing in the Hα-filtered HN(CA) but present in the reference HN(CA), which indicates that they arise from 2H/15N-labelled amino acids preceded by 2-13C-labelled ones (see table S1 of the supplemental information), are highlighted by empty rectangles.
Figure 7.
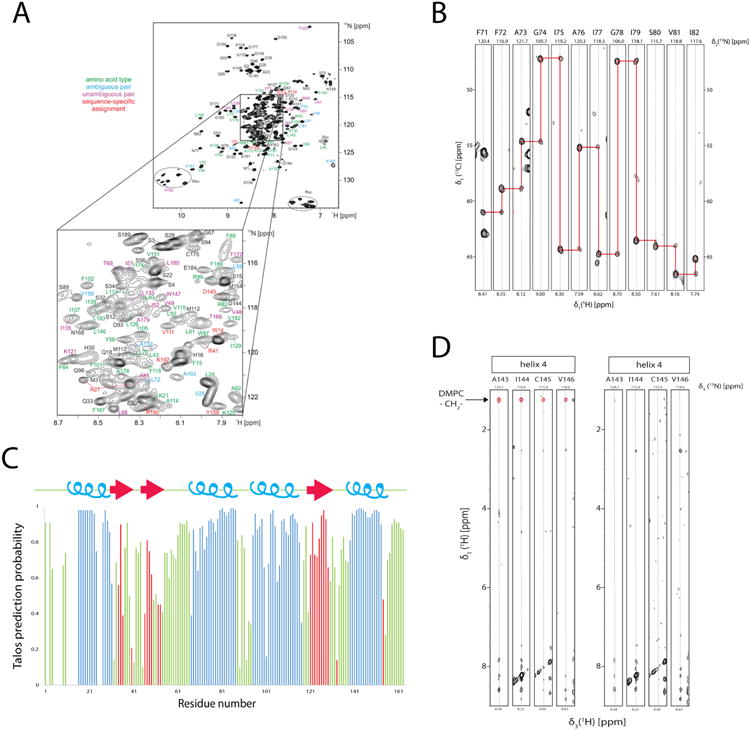
Backbone amide and 13Cα assignment and secondary structure determination of LspA in dH5 DMPC ND titrated with diC7PC. (A) Overview of assignment of backbone amide signal of LspA. For residues with colored annotations the following information was obtained from combinatorial 15N, 13Cα, and 13C' labeling: green, only the amino acid was determined, blue, ambiguous sequential pairs (two possibilities for the amino acid type preceding the detected amide), magenta, unambiguous pairs (both amino acid types of a i-1, i pair determined, and red, unique pairs (i.e. the i-1 / i amino-acid type combination occurs only once in the sequence. (B) Example strips taken from 3D HNCA experiments show the spectral quality although without the backbone assignment from the combinatorial labeling strategy would have been very difficult due to degeneracy in 13Cα chemical shifts and prior knowledge of preceding residues gained from the combinatorial labeling strategy. (C) Secondary structure prediction taken from the program Talos+ indicates 4 helices (blue) and 3 β-strands (red) in the loop regions. (D) Strips from two 3D 15N-resolved 1H,1H NOESY spectra for residues in TM4. Two samples of 2H/15N-labeled LspA in deuterated diC7PC:protonated DMPC (left) and protonated diC7PC :deuterated DMPC (right) titration, measured at 45° at 950 MHz are shown. Residues in helix 4 have NOEs to the DMPC fatty acid methylene chain at 1.25 ppm (red) only when in protonated DMPC samples. Protonated diC7PC methylene signals are not apparent in either sample.
The chemical shift data obtained were used to calculate the secondary structure of LspA in complex with globomycin with Talos+ (Shen et al., 2009) and Dangle (Cheung et al., 2010) (Figure 7C). We identified four α-helices and three β-strands in the loop regions between helix 1 and 2, and helix 3 and 4. The fourth helix (TM4) which harbors a proposed catalytic aspartate is four amino acids shorter than the other helices. Our backbone assignments allowed us to confirm specific DMPC contacts to residues in TM4 by comparing the 3D 15N-resolved 1H,1H NOESY spectra in deuterated and protonated DMPC samples titrated with diC7PC. Residues in TM4 show NOEs to the DMPC fatty acid methylene chain at 1.25 ppm only when in protonated DMPC samples (Figure 7D). Comparison of our secondary structure of LspA in complex with globomycin with the recently published crystal structure (Vogeley et al., 2016) confirms the presence of these β-strands which lie on the periplasmic side of the membrane and the shorter TM4. In addition, the backbone assignment allows us to confirm that many of the residues that become visible upon forming a complex with globomycin (Figure 1E and 1F) are located in and around the globomycin binding site of LspA such as A51 and N53 in the ‘pH region’. The assignments will assist in future investigations into the dynamics of LspA substrate recognition and binding.
Discussion
Although not necessarily intuitive, creating isotropic bicelles or mixed micelles from NDs presents hitherto uncharacterized advantages for solution NMR studies of lipid-IMP complexes. The general principle arises from the process of ND formation, which occurs by the removal of sodium cholate from specific mixtures of MSP and lipids (Bayburt and Sligar, 2003). Hence, subsequent addition of detergents should trigger the dissolution of the ND bilayer leaving each component surrounded by detergent particles. The dramatic improvement in NMR spectra quality and the ability of detergent sensitive IMPs to preserve lipid contacts during this process motivated our efforts to validate its use for solution NMR spectroscopy.
We sought to demonstrate that the overall effect on the IMP is due to the gradual destruction of the ND as previous reports have shown the co-existence of NDs and diC6PC bicelles (Lai et al., 2015). Similarity between the NMR spectra of IMPs in titrated ND samples and in samples where MSP has been removed, confirms that the titration creates a smaller and more micellar environment for the IMP. Essentially, this process of making bicelles by titrating detergent to NDs peels the lipid away from the MP and MSP in a controlled fashion. Incremental detergent titration ensures that the balance between preserving functionally relevant lipid-IMP complexes while producing high quality NMR spectra can be identified for individual IMP in a ND. Our results with LspA, EmrE, PR and DrVSD demonstrate that the overall effect is protein and lipid independent. This broadens the potential application of our method to include a greater range of lipids and more complicated mixed micelles and isotropic bicelles that can be catered to individual IMPs. The most important feature of this strategy as demonstrated with LspA and EmrE, is that it is mild enough to allow detergent sensitive IMPs to retain lipid contacts necessary for stability and function. The difference in the detergent:lipid ratios required to result in optimal spectra for the different MPs suggests that individual lipid affinities of IMPs dictates the presence of lipid in the final complexes. Usually, the role of lipids is probed by comparing different membrane mimetic environments to ascertain the most native-like in which to proceed with functionally relevant MP studies (Etzkorn et al., 2013; Kucharska et al., 2015). We expect our method to, in the first instance, ease these types of labor intensive investigations and in the second, to contribute to studies into the lipid specificity of individual IMPs. Our method will also be applicable for MP-ND samples prepared in the conventional method without co-translational CF insertion.
Common for solution NMR studies of IMPs, as exemplified by LspA, a significant initial effort is made to address the poor tumbling rate of IMPs by exhaustive detergent screening. Unfortunately, the effects of detergents on LspA stability renders them unsuitable for solution NMR experiments, much less subsequent standard preparations to form bicelles or NDs. The strategy outlined here neatly circumvents the need for highly concentrated IMP samples in detergent solutions and is on a par with styrene malic acid copolymers, one of the few membrane mimetics that allows purification from lipid membranes without the use of detergent (Dörr et al., 2014). While the mechanism of co-insertion into ND lipid bilayers is not fully explained, models where MP folding and insertion may not strictly require translocon machinery have been proposed (Cymer et al., 2015). Unassisted helical insertion has been demonstrated to have similar free energies as MP insertion by the Sec61 translocon (Ulmschneider et al., 2014). Furthermore, direct expression into bilayers is gaining recognition as a method to produce functional MPs in lipid bilayers that have never been in contact with detergent (Goren et al., 2009; Henrich et al., 2016; Kalmbach et al., 2007; Long et al., 2012; Yildiz et al., 2012).
Finally, it should be noted that despite the solubility and function of LspA in DPC and DDM, the recent crystal structure of LspA in complex with globomycin was solved using lipid cubic phase (Vogeley et al., 2016). At this stage, the relevance of each LspA subunit containing six monoolein molecules is pure speculation. However, it is indisputable that this method acknowledges the lipid requirement of IMPs for their stability and function (Caffrey, 2015). Similarly, our method preserves lipid-IMP complexes for solution NMR based structure determination, allowing the recording of NMR spectra with the necessary chemical shift dispersion and sensitivity.
Experimental Procedures
Cell-free expression of MPs
LspA and EmrE in pET28, PR in piVEX2.3d (Roche Applied Science) and DrVSD in pET15b vectors with N-terminal 6× histag and streptag were expressed in a S30-based continuous exchange cell-free system in P-CF, D-CF and L-CF according to previously published protocols (Schwarz et al., 2007). For 15N/2H labeled samples, stable-isotope-labeled amino acid mixtures (Cambridge Isotope Laboratories) and algal amino acid mixtures (Sigma Aldrich) in total concentration of 15 mg/mL were added to the reaction mixture. For selectively labeled DrVSD samples, 13C-ε Met and 13C/15N Ile (Sigma Aldrich) were added directly to the reaction mixture. LspA, EmrE and PR were expressed in the presence of 0.05 mM globomycin (in ethanol), 0.1 mM tetraphenylphosphonium (TPP+) and 0.6 mM retinal respectively which were added directly to reaction mixture.
Detergent-MP sample preparation
P-CF LspA pellets from 3mL reactions were collected by centrifugation at 13,000 rpm, washed with S30 buffer and dissolved in 0.5% DPC in 20 mM HEPES pH 8, 20 mM Imidazole, 300 mM NaCl buffer. Samples were applied to Ni-affinity columns (Fast Flow 6 Sepharose, GE Healthcare), washed with 10 CV with 20 mM HEPES pH 8, 30 mM imidazole 300 mM NaCl buffer and eluted in 5 ml fractions with 20 mM HEPES pH 8, 400 mM imidazole, 300 mM NaCl buffer. LspA samples were concentrated and buffer exchanged in centrifugal concentration devices (10,000 MWCO, Amicon) and were prone to high amount of precipitation. Final sample concentrations never exceeded 50 μM and were unstable after 1 day. PR was expressed in D-CF mode according to previously published protocols (Reckel et al., 2011) with minor modifications. GDN (Anatrace) replaced digitonin in the original protocol. PR in diC7PC micelles was purified on StrepTactin Sepharose (GE Healthcare) with 0.1% diC7PC
MSP was removed from titrated NMR samples by purifying on StrepTactin Sepharose (GE Healthcare) with either 0.04% DPC or 0.09% diC7PC in washing and elution buffers. Tricine-PAGE gels were run to confirm that elution fractions contained only the strep-tagged MPs. Incidentally, running the re-purifications in detergent concentrations lower than their corresponding critical micellar concentration was insufficient to remove dH5 MSP from the titrated samples irrespective of final titration concentration. Protein concentration of MPs extracted from titrated ND samples did not exceed 20 μM due to massive protein loss during the concentration steps.
Nanodisc-MP sample preparation
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Avanti Polar Lipids, Inc.) and 1,2-dimyristolsn-glycero-3-phosphoglycerol (DMPG) (Avanti Polar Lipids, Inc.) MSP and NDs were prepared according to previously published protocols (Denisov et al., 2004; Roos et al., 2012). dH5 MSP version was chosen due to its stability and small size (Hagn et al., 2013). A final concentration of 100 μM NDs were added to the reaction mixture of LspA and EmrE and PR and DrVSD respectively (Roos et al., 2014). The samples were purified with Ni-affinity purification on Fast Flow 6 Sepharose (GE Healthcare) and subsequently streptag purified on StrepTactin Sepharose (GE Healthcare) to remove empty his-tagged NDs from the samples. The sample were concentrated and buffer exchanged in centrifugal concentration devices (10,000 MWCO, Amicon). LspA dH5 DMPC ND were applied to a calibrated analytical gel filtration S200 column (GE Healthcare). All purification buffers for EmrE contained 0.5 -1 mM TPP. Final concentrations of NMR samples ranged from 0.1-0.3 mM. The final NMR buffer conditions were 20mM sodium acetate buffer pH 4 for LspA and 20mM potassium phosphate pH 7 20mM NaCl 1mM DTT 3mM TPP+ for EmrE. DPC and diC7PC were titrated to MP-ND samples at 45°. Detergent-lipid ratios were determined by integrating 31P DMPC, diC7PC and DPC peaks. NOESY experiments required the use of deuterated diC7PC and deuterated DMPC (CortecNet, France).
NMR spectroscopy
Multidimensional NMR experiments were conducted at a sample temperature of 318 K on Bruker Avance 600, 700, 800, 900 and 950 MHz spectrometers equipped with cryogenic 13C/15N triple-resonance probes. Two dimensional 15N-1H correlations were of the TROSY type (Pervushin et al., 1997) and employed sensitivity-enhanced gradient echo/antiecho selection (Kay et al., 1992). Acceleration of longitudinal 1H relaxation between scans was achieved by the Band-selective Excitation Short-Transient (BEST) technique (Farjon et al., 2009; Schanda et al., 2006). Proton-carbon correlations of methyl groups employed a gradient-selected version of the [13C,1H]-SOFAST-HMQC pulse sequence (Schanda et al., 2005). Three-dimensional 15N-resolved NOESY spectra were recorded with a mixing time of 250 ms using a [15N,1H]-TROSY based detection scheme.
Rotational correlation times were determined with the [15N,1H]-TRACT method (Lee et al., 2006). Since intensities of the full amide proton envelopes in 1D spectra were dominated by very strong signals of tag or other very mobile residues, a 2D 15N-resolved version was employed in order to avoid severe underestimation of correlation times. A total of 15-20 cross peaks known to belong to structured regions of the proteins were chosen for evaluation. Rate constants for cross correlation between 15N CSA and 1H-15N dipole-dipole interactions (2ηxy) were measured essentially as described by Palmer and co-workers (Wang et al., 2003), by interleaved recording of pairs of 2D TROSY spectra with additional 15N Hahn echo periods (Δ) during which either the broad or the narrow doublet component relaxes. Durations of the Δ periods were adjusted to 5-10 ms. To account for intrinsic differences in signal intensities between the two versions of the pulse sequence a pair of reference spectra was recorded with a short relaxation period (Δref = 0.5 ms) and cross correlation rates were calculated according to 2ηxy = In [Iα(Δ) Iβ(Δref) / Iα(Δ) Iβ(Δref)] / (Δ − Δref), where Iαand Iβ are the signal intensities observed in spectra that select the slow and fast relaxing 15N doublet component, respectively during Δ.
One-dimensional 31P spectra were recorded at a sample temperature of 318 K on a Bruker DRX500 spectrometer equipped with a broadband inverse probe. The recycle delay was set to 4 s and WALTZ-16 proton decoupling was applied during an acquisition period of 0.6 s.
A set of [15N,1H]-BEST-TROSY type 2D HSQC, HN(CO), HN(COCA), HN(CA), Hα-filtered HN(CA), and DQ-HN(CA) spectra on the combinatorial selectively labeled samples listed in Table 1 was recorded at 600 MHz. The pulse sequence employed for the Hα-filtered HN(CA) is shown in the Supplemental Information (Figure S8) while those of the remaining experiments were published previously (Löhr et al., 2012). All samples had a protein concentration of 0.3 mM, and the diC7PC:DMPC ratio was adjusted to ca. 1.0. Spectral widths were 13 and 50 ppm in the 1H and 15N dimensions, respectively. Acquisition times were 41 ms in the 1H dimension and 63 ms in the 15N dimension, corresponding to 320 and 192 complex data points. Measurement times of individual experiments ranged from 3h (32 scans/FID) for HSQCs to 48 h (512 scans/FID) for DQ-HN(CA) spectra. Three-dimensional HNCA, HNCO, HN(CO)CA, HN(CA)CO, HNCACB and HN(CO)CACB experiments on LspA employed standard [15N,1H]-TROSY-type pulse sequences (Salzmann et al., 1998, 1999) modified to include BEST longitudinal relaxation enhancement (Farjon et al., 2009; Favier and Brutscher, 2011). Experiments were carried out at 600, 800, 900 or 950 MHz using a 0.4 mM uniformly 2H/13C/15N-labeled sample with a diC7PC: DMPC ratio of ca. 0.8.
Supplementary Material
Highlights.
A strategy to produce bicelles from integral membrane proteins in nanodisc bilayers
Suitable for solution NMR studies of detergent sensitive membrane proteins
Vast improvements in signal intensity allow high resolution NMR spectroscopy
Acknowledgments
The plasmid of MSP1D1ΔH5 was kindly provided by Gerhard Wagner (Harvard Medical School). We thank Vertex Pharmaceuticals Inc. for the MBP-LPP substrate and the gift of globomycin in the initial stages of this project when it was not commercially available. This work was supported by the state of Hesse (Center for Biomolecular Magnetic Resonance), the German Research Foundation (D0545/11), NIH (U54GM087519), Vertex Pharmaceuticals Inc. and the Cluster of Excellence Frankfurt (Macromolecular Complexes).
Footnotes
Author Contributions: Conceptualization, A.L., F.L and V.D.; Methodology, A.L. and F.L.; Investigation, A.L., F.L., E.H. and B.H.; Writing, A.L., F.L., V.D.; Funding Acquisition, V.D., J.M.M., E.P., F.B.; Resources, N.A.M., P.J.C., J.M.M. and E.P.; Supervision, V.D.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bayburt TH, Sligar SG. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci Publ Protein Soc. 2003;12:2476–2481. doi: 10.1110/ps.03267503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr Sect F Struct Biol Commun. 2015;71:3–18. doi: 10.1107/S2053230X14026843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung MS, Maguire ML, Stevens TJ, Broadhurst RW. DANGLE: A Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J Magn Reson San Diego Calif 1997. 2010;202:223–233. doi: 10.1016/j.jmr.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Cymer F, von Heijne G, White SH. Mechanisms of Integral Membrane Protein Insertion and Folding. J Mol Biol. 2015;427:999–1022. doi: 10.1016/j.jmb.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis AA, Opella SJ. Bicelle samples for solid-state NMR of membrane proteins. Nat Protoc. 2007;2:2332–2338. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- Dörr JM, Koorengevel MC, Schäfer M, Prokofyev AV, Scheidelaar S, van der Cruijsen EAW, Dafforn TR, Baldus M, Killian JA. Detergent-free isolation, characterization, and functional reconstitution of a tetrameric K+ channel: the power of native nanodiscs. Proc Natl Acad Sci U S A. 2014;111:18607–18612. doi: 10.1073/pnas.1416205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr UHN, Gildenberg M, Ramamoorthy A. The Magic of Bicelles Lights Up Membrane Protein Structure. Chem Rev. 2012;112:6054–6074. doi: 10.1021/cr300061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr UHN, Soong R, Ramamoorthy A. When detergent meets bilayer: Birth and coming of age of lipid bicelles. Prog Nucl Magn Reson Spectrosc. 2013;69:1–22. doi: 10.1016/j.pnmrs.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, Wagner G. Cell-free expressed bacteriorhodopsin in different soluble membrane mimetics: biophysical properties and NMR accessibility. Struct Lond Engl 1993. 2013;21:394–401. doi: 10.1016/j.str.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farjon J, Boisbouvier J, Schanda P, Pardi A, Simorre JP, Brutscher B. Longitudinal-Relaxation-Enhanced NMR Experiments for the Study of Nucleic Acids in Solution. J Am Chem Soc. 2009;131:8571–8577. doi: 10.1021/ja901633y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier A, Brutscher B. Recovering lost magnetization: polarization enhancement in biomolecular NMR. J Biomol NMR. 2011;49:9–15. doi: 10.1007/s10858-010-9461-5. [DOI] [PubMed] [Google Scholar]
- Goren MA, Nozawa A, Makino S, Wrobel RL, Fox BG. Cell-free translation of integral membrane proteins into unilamelar liposomes. Methods Enzymol. 2009;463:647–673. doi: 10.1016/S0076-6879(09)63037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Etzkorn M, Raschle T, Wagner G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass MAS, Ubbink M. Structure determination of protein-protein complexes with long-range anisotropic paramagnetic NMR restraints. Curr Opin Struct Biol. 2014;24:45–53. doi: 10.1016/j.sbi.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Henrich E, Ma Y, Engels I, Münch D, Otten C, Schneider T, Henrichfreise B, Sahl HG, Dötsch V, Bernhard F. Lipid Requirements for the Enzymatic Activity of MraY Translocases and in Vitro Reconstitution of the Lipid II Synthesis Pathway. J Biol Chem. 2016;291:2535–2546. doi: 10.1074/jbc.M115.664292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara J, Tang C, Marius Clore G. Practical aspects of (1)H transverse paramagnetic relaxation enhancement measurements on macromolecules. J Magn Reson San Diego Calif 1997. 2007;184:185–195. doi: 10.1016/j.jmr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach R, Chizhov I, Schumacher MC, Friedrich T, Bamberg E, Engelhard M. Functional cell-free synthesis of a seven helix membrane protein: in situ insertion of bacteriorhodopsin into liposomes. J Mol Biol. 2007;371:639–648. doi: 10.1016/j.jmb.2007.05.087. [DOI] [PubMed] [Google Scholar]
- Kang C, Li Q. Solution NMR study of integral membrane proteins. Curr Opin Chem Biol. 2011;15:560–569. doi: 10.1016/j.cbpa.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Katzen F, Fletcher JE, Yang JP, Kang D, Peterson TC, Cappuccio JA, Blanchette CD, Sulchek T, Chromy BA, Hoeprich PD, et al. Insertion of membrane proteins into discoidal membranes using a cell-free protein expression approach. J Proteome Res. 2008;7:3535–3542. doi: 10.1021/pr800265f. [DOI] [PubMed] [Google Scholar]
- Kerfah R, Plevin MJ, Sounier R, Gans P, Boisbouvier J. Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins. Curr Opin Struct Biol. 2015;32:113–122. doi: 10.1016/j.sbi.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent Advances in the Application of Solution NMR Spectroscopy to Multi-Span Integral Membrane Proteins. Prog Nucl Magn Reson Spectrosc. 2009;55:335–360. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharska I, Edrington TC, Liang B, Tamm LK. J Biomol NMR. 2015;61:261–274. doi: 10.1007/s10858-015-9905-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai G, Forti KM, Renthal R. Kinetics of lipid mixing between bicelles and nanolipoprotein particles. Biophys Chem. 2015;197:47–52. doi: 10.1016/j.bpc.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry (Mosc) 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- Lee D, Hilty C, Wider G, Wüthrich K. Effective rotational correlation times of proteins from NMR relaxation interference. J Magn Reson. 2006;178:72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Li Q, Wanderling S, Paduch M, Medovoy D, Singharoy A, McGreevy R, Villalba-Galea CA, Hulse RE, Roux B, Schulten K, et al. Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat Struct Mol Biol. 2014;21:244–252. doi: 10.1038/nsmb.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Shen R, Treger JS, Wanderling SS, Milewski W, Siwowska K, Bezanilla F, Perozo E. Resting state of the human proton channel dimer in a lipid bilayer. Proc Natl Acad Sci U S A. 2015;112:E5926–5935. doi: 10.1073/pnas.1515043112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhr F, Reckel S, Karbyshev M, Connolly PJ, Abdul-Manan N, Bernhard F, Moore JM, Dötsch V. Combinatorial triple-selective labeling as a tool to assist membrane protein backbone resonance assignment. J Biomol NMR. 2012;52:197–210. doi: 10.1007/s10858-012-9601-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhr F, Tumulka F, Bock C, Abele R, Dötsch V. An extended combinatorial 15N, 13Cα, and 13C′ labeling approach to protein backbone resonance assignment. J Biomol NMR. 2015;62:263–279. doi: 10.1007/s10858-015-9941-8. [DOI] [PubMed] [Google Scholar]
- London E, Khorana HG. Denaturation and renaturation of bacteriorhodopsin in detergents and lipid-detergent mixtures. J Biol Chem. 1982;257:7003–7011. [PubMed] [Google Scholar]
- Long AR, O'Brien CC, Alder NN. The cell-free integration of a polytopic mitochondrial membrane protein into liposomes occurs cotranslationally and in a lipid-dependent manner. PloS One. 2012;7:e46332. doi: 10.1371/journal.pone.0046332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyukmanova EN, Shenkarev ZO, Khabibullina NF, Kopeina GS, Shulepko MA, Paramonov AS, Mineev KS, Tikhonov RV, Shingarova LN, Petrovskaya LE, et al. Lipid-protein nanodiscs for cell-free production of integral membrane proteins in a soluble and folded state: comparison with detergent micelles, bicelles and liposomes. Biochim Biophys Acta. 2012;1818:349–358. doi: 10.1016/j.bbamem.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Morrison EA, Henzler-Wildman KA. Reconstitution of integral membrane proteins into isotropic bicelles with improved sample stability and expanded lipid composition profile. Biochim Biophys Acta. 2012;1818:814–820. doi: 10.1016/j.bbamem.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2012;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nietlispach D, Gautier A. Solution NMR studies of polytopic α-helical membrane proteins. Curr Opin Struct Biol. 2011;21:497–508. doi: 10.1016/j.sbi.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Nolandt OV, Walther TH, Grage SL, Ulrich AS. Magnetically oriented dodecylphosphocholine bicelles for solid-state NMR structure analysis. Biochim Biophys Acta BBA - Biomembr. 2012;1818:1142–1147. doi: 10.1016/j.bbamem.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Park SH, Berkamp S, Cook GA, Chan MK, Viadiu H, Opella SJ. Nanodiscs vs. Macrodiscs for NMR of Membrane Proteins. Biochemistry (Mosc) 2011;50:8983–8985. doi: 10.1021/bi201289c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proverbio D, Roos C, Beyermann M, Orbán E, Dötsch V, Bernhard F. Functional properties of cell-free expressed human endothelin A and endothelin B receptors in artificial membrane environments. Biochim Biophys Acta. 2013;1828:2182–2192. doi: 10.1016/j.bbamem.2013.05.031. [DOI] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Etzkorn M, Wagner G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr Opin Struct Biol. 2010;20:471–479. doi: 10.1016/j.sbi.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reckel S, Gottstein D, Stehle J, Löhr F, Verhoefen MK, Takeda M, Silvers R, Kainosho M, Glaubitz C, Wachtveitl J, et al. Solution NMR structure of proteorhodopsin. Angew Chem Int Ed Engl. 2011;50:11942–11946. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos C, Zocher M, Müller D, Münch D, Schneider T, Sahl HG, Scholz F, Wachtveitl J, Ma Y, Proverbio D, et al. Characterization of co-translationally formed nanodisc complexes with small multidrug transporters, proteorhodopsin and with the E. coli MraY translocase. Biochim Biophys Acta. 2012;1818:3098–3106. doi: 10.1016/j.bbamem.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Roos C, Kai L, Haberstock S, Proverbio D, Ghoshdastider U, Ma Y, Filipek S, Wang X, Dötsch V, Bernhard F. High-Level Cell-Free Production of Membrane Proteins with Nanodiscs. In: Alexandrov K, Johnston WA, editors. Cell-Free Protein Synthesis. Humana Press; 2014. pp. 109–130. [DOI] [PubMed] [Google Scholar]
- Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci U S A. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann M, Wider G, Pervushin K, Senn H, Wüthrich K. TROSY-type Triple-Resonance Experiments for Sequential NMR Assignments of Large Proteins. J Am Chem Soc. 1999;121:844–848. [Google Scholar]
- Sanders CR, Sönnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem MRC. 2006;44 Spec No:S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- Schanda P, Kupce E, Brutscher B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR. 2005;33:199–211. doi: 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- Schanda P, Van Melckebeke H, Brutscher B. Speeding Up Three-Dimensional Protein NMR Experiments to a Few Minutes. J Am Chem Soc. 2006;128:9042–9043. doi: 10.1021/ja062025p. [DOI] [PubMed] [Google Scholar]
- Schwarz D, Junge F, Durst F, Frölich N, Schneider B, Reckel S, Sobhanifar S, Dötsch V, Bernhard F. Preparative scale expression of membrane proteins in Escherichia coli-based continuous exchange cell-free systems. Nat Protoc. 2007;2:2945–2957. doi: 10.1038/nprot.2007.426. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhanifar S, Reckel S, Junge F, Schwarz D, Kai L, Karbyshev M, Löhr F, Bernhard F, Dötsch V. Cell-free expression and stable isotope labelling strategies for membrane proteins. J Biomol NMR. 2010;46:33–43. doi: 10.1007/s10858-009-9364-5. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003;22:6175–6181. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider MB, Ulmschneider JP, Schiller N, Wallace BA, von Heijne G, White SH. Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat Commun. 2014;5:4863. doi: 10.1038/ncomms5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogeley L, El Arnaout T, Bailey J, Stansfeld PJ, Boland C, Caffrey M. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science. 2016;351:876–880. doi: 10.1126/science.aad3747. [DOI] [PubMed] [Google Scholar]
- Wang C, Rance M, Palmer AG. Mapping chemical exchange in proteins with MW > 50 kD. J Am Chem Soc. 2003;125:8968–8969. doi: 10.1021/ja035139z. [DOI] [PubMed] [Google Scholar]
- Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand É, Marcotte I. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta. 2011;1808:1957–1974. doi: 10.1016/j.bbamem.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Yildiz AA, Knoll W, Gennis RB, Sinner EK. Cell-free synthesis of cytochrome bo(3) ubiquinol oxidase in artificial membranes. Anal Biochem. 2012;423:39–45. doi: 10.1016/j.ab.2012.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.