

Abstract
The oral route is a preferred method of drug administration, though achieving effective drug delivery and minimizing off-target side effects is often challenging. Formulation into nanoparticles can improve drug stability in the harsh gastrointestinal (GI) tract environment, providing opportunities for targeting specific sites in the GI tract, increasing drug solubility and bioavailability, and providing sustained release in the GI tract. However, the unique and diverse physiology throughout the GI tract, including wide variation in pH, mucus that varies in thickness and structure, numerous cell types, and various physiological functions are both a barrier to effective delivery and an opportunity for nanoparticle design. Here, nanoparticle design aspects to improve delivery to particular sites in the GI tract are discussed. We then review new methods for evaluating oral nanoparticle formulations, including a short commentary on data interpretation and translation. Finally, the state-of-the-art in preclinical targeted nanoparticle design is reviewed.
Keywords: inflammatory bowel disease, colon targeting, intestinal lymphatic system, targeted delivery, in vitro-in vivo correlation
Graphical Abstract
1. Introduction
Oral ingestion remains the preferred mode of delivery for most drugs owing largely to simplicity. The oral route is associated with the greatest degree of patient compliance (especially for chronic conditions) as it ensures convenience, enables self-administration, and offers great flexibility in dosage regimen. Oral products do not require sterile conditions for their manufacture, which reduces production costs. According to the drug delivery market analysis, oral drug products accounted for 38% of the North American drug delivery market in 2012 [1]. The oral drug delivery market was valued at $64.3 billion in 2013 and is expected to cross $100 billion by 2018 [2]. Thus, oral drug delivery will continue to dominate the pharmaceutical market and drug delivery research.
The oral route is also of interest for physiological reasons. The gastrointestinal (GI) tract offers extensive surface area (300–400 m2) for drug absorption by absorptive epithelial cells (enterocytes) [3–6]. The GI tract contains many other types of cells that may participate in drug absorption, including mucin-secreting goblet cells, endocrine cells, Paneth cells and specialized M cells associated with Peyer’s patches that are responsible for antigen transportation through dendritic cells [3–6]. However, many hydrophobic and hydrophilic drugs (taxanes, aminoglycosides, polyene antibiotics etc.) have poor bioavailability when administered via the oral route due to their inadequate physicochemical (solubility, stability) and/or biopharmaceutical (permeability, metabolic stability) properties [7–9]. Furthermore, a majority of the new chemical entities generated through drug discovery screening exhibit poor aqueous solubility and/or poor permeability. It has been reported that nearly 70% of new chemical entities are dropped during pre-clinical development due to poor oral bioavailability [10]. Oral delivery is even more challenging for biologics (e.g. peptides, proteins and nucleic acids) due to their hydrophilicity (leading to low permeability), high molecular weight and poor chemical/enzymatic stability in the GI tract. Figure 1 summarizes the various challenges/barriers to effective oral drug delivery. The reader is also referred to several excellent reviews discussing the various barriers to developing drugs for effective oral delivery [3–6, 11–13].
Figure 1.

Summary of barriers that affect oral drug delivery
Numerous studies have shown that nanoparticles can improve the oral bioavailability of hydrophobic, hydrophilic and biologic drugs via various mechanisms [3–6, 14–16]. In fact, several oral nanosuspension-based products that improve drug dissolution and absorption are on the market [10]. Nanoparticle formulations with more sophisticated design aspects are in preclinical development, including those designed to target a particular region in the GI tract, only diseased regions of the GI tract, or specific cells within the GI tract. Targeting approaches aim to enable better drug absorption and/or localized treatment of various disease conditions, such as gastric ulcers, Helicobacter pylori (H. pylori) infections and ulcerative colitis. An important aspect of preclinical development of oral nanoparticle systems is the choice of experimental models. In this review, we first describe nanoparticle design considerations for targeting particular regions in the GI tract. We then discuss recent advances in in vitro and ex vivo experimental techniques for the evaluation of oral nanoparticle formulations, as well as a perspective on correlation of preclinical results with clinical translation. Finally, we highlight recent developments in the design and preclinical evaluation of targeted oral nanoparticle formulations.
2. Design considerations for targeting nanoparticles to a specific region of the GI tract
One benefit of nanoparticle formulations is the potential for providing targeted and/or localized drug delivery. Although the term “targeted” brings to mind the vision of nanoparticles that actively seek out their delivery target and selectively accumulate there, “targeting” in the GI tract is generally a more passive process. Here, we use the term “target” to refer to various strategies used for increasing residence time, increasing the relative amount of degradation/drug release that occurs, and/or facilitating interaction of nanoparticle formulations with tissues and cells in a particular section of the GI tract. In the context of the rational design of targeted oral nanoparticle formulations, we discuss diseases and delivery goals that would benefit from targeted delivery, as well as the physiological barriers and targeting opportunities (specific cell types or receptors) associated with various regions of the GI tract. Here, we broadly categorize approaches for stomach targeting, small intestine targeting, intestinal lymphatic targeting, and colon targeting. In Section 4, we further describe recent innovations in nanoparticle platforms designed to target specific regions of the GI tract.
2.1. Design considerations for stomach targeting
Targeting of therapeutic agents to the stomach has received attention for the effective treatment and management of Helicobacter pylori (H. pylori) infections. H. pylori infections affect around 50% of the global population [17–19]. Around 20% of the H. pylori infected population develop gastric disorders, such as chronic gastritis and/or gastric ulcers, and around 1–2% of H. pylori infected individuals present with gastric cancer [17–19]. Additionally, stomach targeting (and/or gastric retention) can also be useful for: (i) drugs that are primarily absorbed in the stomach (e.g. metronidazole), (ii) drugs that are poorly soluble in the intestinal milieu due to pH dependent solubility (e.g. verapamil), (iii) drugs with a narrow absorption window in the stomach or in the upper small intestine (e.g. furosemide), and (iv) drugs that degrade in the intestinal milieu (e.g. captopril) [20]. Gastric retention of the therapeutic agent is of utmost importance, yet is difficult to achieve. For effective stomach targeting, a nanoparticle delivery system must overcome various physiological hurdles, including gastric motility, gastric pH and gastric mucus.
The GI tract is in a state of continuous motility in the fasted and fed states. GI motility is categorized into inter-digestive and digestive motility. During the fasted state, the inter-digestive motility pattern is activated to empty the stomach of the residual contents of the upper GI tract [19–23]. In the inter-digestive mode, motility comprises of four phases (total duration for 4 phases: 90–120 min), and each phase involves cycles of (peristaltic) activity and quiescence. Phase III of the inter-digestive mode involves intense contractions to empty the undigested stomach contents through maximal pyloric opening. Digestive mode is initiated within 5–10 min after the ingestion of food and lasts until the food in the stomach is completely processed [19–23]. Gastric retention of nanoparticles will depend upon the motility phase that is active at the time of ingestion. For effective stomach targeting, nanoparticles should be able to withstand the peristaltic activity of the stomach. Gastric retention of the nanoparticles can also depend upon the fed or fasted state of the individual and also on the type of the ingested food (Figure 2). Additionally, the pH of stomach is highly acidic, varying between 1 and 3 depending upon the fasted or fed state of the individual [19–23]. Many drugs used for peptic ulcer and antibiotics like clarithromycin that are used for H. pylori treatment are susceptible to degradation at acidic pH and are rendered ineffective in the stomach [24, 25]. Similarly, it is important to construct nanoparticles from materials that can provide protection of acid labile therapeutic agents.
Figure 2.

In vivo distribution of the NIR dye (P2) labeled solid lipid nanoparticles following oral gavage administration in mice under fasted and fed conditions. Mice fed with a high fat diet showed fluoresence in the stomach for longer duration, indicating that the composition of the diet and fed state can affect gastroretention. Figures reproduced from reference [150] with permission.
Gastric mucus poses a significant barrier to effective stomach targeting of nanoparticles. GI mucus protects the epithelium from exposure to foreign particulates and pathogens, including nanoparticles [3]. Effective gastric mucus penetration may be of particular importance for eradication of H. pylori infection, as the bacteria is situated deep inside the gastric mucosa and attaches itself to the gastric epithelial cells with the help of adhesin-like proteins [17–18]. The total thickness of mucus (loosely adherent plus firmly adherent layers) in the human GIT ranges from 50–450 μm depending upon the location, and is thickest in the stomach and colon [3,18,19]. Nanoparticles must penetrate the mucus barrier to be retained in the stomach and locally deliver drugs eradicate H. pylori infections. For more extensive discussions of the barrier properties of GI tract mucus, the reader is referred to other papers and reviews [3, 26–28].
More generally, macroscopic delivery systems, such as tablets, microparticles, or hydrogels, are employed for increasing gastric retention [17–20, 29, 30]. The general principle is to utilize a property like geometry or density to prevent gastric emptying, allowing for prolonged drug release into the gastric milieu [17–20, 29, 30]. Although these macroscopic dosage forms increase residence time of the therapeutic agent in the stomach, they may not be optimal for the delivery drugs that have poor penetration in gastric mucosa. Studies have shown that drugs like amoxicillin, an antibiotic used for the treatment of H. pylori infections, were unable to achieve sufficient penetration in human gastric mucosal biopsies [31]. Moreover, a clinical trial demonstrated that amoxicillin- and clarithromycin-loaded mucoadhesive hydrogel did not provide an improvement in H. pylori eradication when compared to the standard antibiotic therapy [32]. Nanoparticle formulations could be useful for improving the current limitations of gastro-retentive delivery systems. Some of the design principles of gastro-retentive dosage forms can also apply to nanoparticles, such as designing floating nanoparticles (hollow or gas generating) or nanoparticles incorporated into gastro-retentive dosage forms, like tablets or clinically available raft-forming gastro-retentive systems [30]. In Section 4, we describe some recent developments in stomach-targeted nanoparticle formulations.
Currently, stomach targeted nanoparticle formulations are largely in pre-clinical development. Most reported investigations on the development of stomach-targeted nanoparticles are focused on achieving increased adhesion to the gastric mucosa (discussed in the Section 4). However, a nanoparticle designed to adhesively interact with the gastric mucosal epithelium will also interact with, and be coated by, the loosely adherent mucus layer in the lumen of the stomach, which it encounters first. Mucus secretion is continuous and rapid in the stomach to protect the epithelium from the acidity of the luminal contents [19], thus, mucoadhesive nanoparticles are likely to interact with and get cleared during the renewal of the loosely adherent mucus layer in the stomach. Hence, it may be necessary to design mucus penetrating nanoparticle formulations, especially for the treatment of H. pylori infections. Research is needed to gain a better understanding of the role that mucoadhesion, as opposed to mucus-penetration, plays in gastric retention of nanoparticle formulations.
2.2. Design considerations for targeting to small intestine
The small intestine plays a major role in the absorption of drugs, nutrients, electrolytes, vitamins and many other molecules [3–6]. Thus, when “targeting” the small intestine, the goal is generally to achieve increased nanoparticle uptake or local drug release and absorption to improve systemic drug delivery. In order to absorb molecules with diverse chemical structures, the small intestine is equipped with specialized cells, cell membrane-bound transporters, receptors, and transport mechanisms, and it is important to consider these physiological aspects while designing systems for intestinal targeting [3–6,15,16]. In addition, nanoparticles must withstand the harsh gastric milieu (described in the previous section) in order to reach the intestine unless they are formulated in a capsule that bypasses the stomach. In order to maintain particle integrity in the stomach and thus allow nanoparticles to reach the small intestine, polyanionic polymers that are insensitive to acidic pH (enteric polymers) but labile in more neutral pH have been developed [24]. Nanoparticles coated with enteric polymers can remain intact in the stomach and, as the pH increases upon reaching the intestine, the enteric polymer coating gradually dissolves and enables drug release. In this way, nanoparticles coated with enteric polymers are used to passively target drug to the small intestine. We refer the reader to more comprehensive reviews of enteric polymer formulations [24,33,34]. In the section 4, we discuss recent works for oral nanoparticles based on enteric polymers for improved oral delivery of various drugs.
“Active” targeting of nanoparticles to the small intestine includes the use of surface functionalization intended to increase interactions with specific cell types in the small intestine to increase residence time and/or uptake. There are several cell types with various characteristics and receptors present in the small intestine. Enterocytes, specialized absorptive columnar epithelial cells, constitute a major portion of the cells present in the small intestine [4–6,16,35]. The second most abundant cell type is the goblet cell, which produces mucin to form a mucus layer that coats the epithelial surface. The small intestine also contains microfold cells (M cells), which are specialized epithelial cells situated in the Peyer’s patches. The Peyer’s patches are a part of the gut associated lymphoid tissues (GALT), and in humans, they are found in the lowest part of the ileum [4–6,16,35]. Particulates, such as immunogens, bacteria, and viruses are transcytosed from the mucosal surface of the Peyer’s patches to the sub-epithelial dome through the M cells. Thus, M cells in the Peyer’s patches have a special role in the potential transport and absorption of nanoparticles. Furthermore, M cells are thought to be relatively less protected by mucus secretions and drug efflux transporters like P-glycoprotein, and thus, are a common delivery target for nanoparticles [4–6,16,35]. However, nanoparticles that adhere to and become coated in mucus may have limited access to M cells.
Enterocytes are often used to target for maximizing delivery of nanoparticles to the small intestine and absorption into systemic circulation because they are the dominant cell type in the small intestine [3–6]. However, mucus poses a significant obstacle to reaching the cell surface [3–6]. When targeting specific cells, the ability to penetrate through mucus and reach the cell surface is an important design criterion for nanoparticles. Recently, we reported that 200 nm fluorescent polystyrene nanoparticles densely coated with hydrophilic polyethylene glycol (PEG) chains penetrate through the mucus layers and uniformly coat freshly excised mouse small intestine tissue, whereas unmodified fluorescent polystyrene nanoparticles were trapped in the mucus layers [36]. Furthermore, orally-administered PEG-coated nanoparticles were found in closer proximity to the small intestine villi and provided more uniform coverage of the small intestine epithelial surface compared to unmodified nanoparticles [36]. Other investigators have used nanoparticles decorated with enzymes, like papain, or thiols to improve mucus penetration [37–40]. Enzymes like papain actively degrade mucus, while the presence of thiols on the surface of the nanoparticles reduces the degree of mucus cross-linking to increase nanoparticle penetration [37–40]. Degrading the vital barrier properties of the mucus barrier may have other unintended consequences, which must be carefully studied. In case of thiol modified nanoparticles, it is important to ascertain that the thiol groups on the surface of nanoparticles do not form disulfide bonds with mucin network, thereby immobilizing the nanoparticles in mucus. We and others have also shown that the size, surface charge, PEG density and material of the nanoparticles also govern their ability to penetrate through mucus, and these aspects should also be understood while designing nanoparticles [41–46].
Nanoparticles that penetrate the mucus barriers come into contact with the cell surfaces. Ligands for various receptors expressed in the intestine have been used to decorate the surface of nanoparticles to achieve increased residence time and/or increased uptake. Vitamin B12 (cobalamin) functionalization of nanoparticles has been of interest for targeted delivery [47,48]. The process of oral absorption of vitamin B12 involves binding of vitamin B12 to intrinsic factor (IF) in the small intestine and receptor mediated uptake of the B12-IF complex into the intestinal epithelia [4,47,48]. Receptors for the B12-IF complex are expressed on enterocytes of the duodenum, jejunum and ileum, though the majority are located in the ileum. During transport, vitamin B12-IF complex is trafficked through the lysosomal pathway, resulting in the degradation of IF [4,47,48]. Interestingly, studies of B-12 decorated nanoparticles using Caco-2 cell monolayers have shown that the conjugation of vitamin B12 on the surface of the nanoparticles does not affect the uptake [49]. However, the vitamin B12 bound nanoparticles showed different intracellular trafficking, including endolysosomal escape [49]. No studies were carried out to elucidate the reasons for different intracellular trafficking observed in case of the vitamin B12 bound nanoparticles. The potential of vitamin B12 mediated targeting of nanoparticles has also been evaluated with the help of rat, dog and pig intestinal loop models [48]. Fluorescent nanoparticles decorated with vitamin B12 showed greater uptake in the rat, dog and pig intestines as compared to unmodified nanoparticles [48].
Recently, targeting to neonatal Fc receptors (FcRn) has been explored to improve oral delivery of nanoparticles [4,48,50]. FcRn are present throughout the intestine and enable trafficking of IgG across the intestinal epithelium. FcRn bind to the Fc portion of IgG in a pH dependent manner. At pH below 6.5 (luminal pH in the duodenum is typically 5.5–6.5), FcRn have high affinity to the Fc region of IgG, whereas the affinity is minimal at pH ~7.4 (similar to the basolateral side of the duodenum). The pH sensitive interactions between IgG and FcRn are thought to enable unilateral transport of IgG across the intestinal epithelium and into the systemic circulation (further discussed in section 4) [4,48,50]. Nanoparticles decorated with the Fc portion of IgG may allow improved targeting and delivery across the duodenum and jejunum of the small intestine.
Due to the high transcytotic potential and ability to traffic particulate substances, M cells have been of great interest as a target for orally administered nanoparticles. As mentioned earlier, the relative lack of a continuous mucus coating, reduced membrane hydrolase activity, scarce glyclocalyx, and relatively sparse drug efflux transporters, offer additional advantages for nanoparticle absorption [4,15,16,35]. Extensive research has been carried out on the targeting of M cells with particulate delivery systems for oral vaccination and other immunotherapies. Numerous research studies and reviews have described the influence of nanoparticle properties such as size, surface charge, material composition and hydrophobicity on M cell uptake and targeting [15,16,35,51–53]. However, the experimental methods used may influence the extent of uptake, as discussed later in Section 3.
Lectins are perhaps the most studied ligands to improve M cell targeting by nanoparticles. Lectins are a diverse group of proteins and glycoproteins of plant or bacterial origin, and they bind with high affinity, but in a reversible manner, to specific carbohydrate residues expressed on cell surface proteins or lipids. Lectins such as wheat germ agglutinin (binding to N-acetyl-D-glucosamine and sialic acid), concanvalin A (α-D-mannose, α-D-glucose binding), tomato lectin (binding to N-acetyl-D-glucosamine), Ulex europaeus 1 lectin (L-α-fucose binding), and Aleuria aurantia lectin (L-α-fucose binding) have been used as ligand to target the apical surface of M cells [16,35,54,55]. Studies have shown that wheat germ agglutinin (WGA) and tomato lectins can withstand abnormally high amounts of pepsin, trypsin, pancreatin, and elastase without losing their cell-binding properties [55–57]. In vivo studies have shown that orally administered WGA underwent 5% and 40% degradation in mice and rats respectively [55–57].
Lectins specific to the human follicular associated endothelium (galectin 9 and sialyl Lewis A Antigen) have also been identified [16], though no information is available on their stability to proteolytic degradation. The immunogenicity, and potential toxicity limit the use of lectins as a targeting ligand [4,16,35,54,55]. Studies have also been carried out to develop/identify lectin mimetics to overcome drawbacks associated with lectins [58]. Furthermore, the loosely adherent mucus layer can act as a cell surface “decoy” that presents some of the same functionalities as the cell surface, which is another mechanism for protection from infection [59,60]. It would be beneficial to ensure that the targeting lectin or lectin decoy of choice does not interact with the mucus barrier.
M cells have a crucial role in processing of bacterial and viral pathogens and various antigens. Hence, targeting of receptors involved in pathogen-host interactions and antigen processing can also be useful for increasing M cell binding [16,51,52]. Various pattern recognition receptors (PRR) expressed on M cells and other cells in Peyer’s patches are involved in the process of invasion and colonization of enteric pathogens. PRR, such as Toll-like receptors (TLR), NOD-like receptors, C-type lectin receptors and α5β1 integrin receptors, are of interest for targeting [16,51,52]. The TLR family and α5β1 integrin receptors are specifically expressed by human and mouse M cells [16,51,52]. TLR ligands, like monophosphoryl lipid A and bacterial flagellin, and α5β1 integrin receptor ligand RGD peptide and RGD peptidomimetics, have been utilized to functionalize nanoparticles to increase interaction with M cells [16,61]. Claudin 4 is a tight junction transmembrane protein that is highly expressed on human and mouse M cells [16,62]. Claudin 4 is a receptor for Clostridium perfringens enterotoxin (CPE) that binds to the C-terminal 30 amino acids of CPE (CPE30) [63]. Hence, CPE30 has been used as a ligand for targeting nanoparticles to M cells.
Although goblet cells constitute the second largest population of intestinal epithelial cells, very few studies have been carried out to target goblet cells for improved oral delivery [16]. This is perhaps due to the relatively low phagocytic and absorptive behavior of goblet cells, as well as active mucus secretion in the opposing direction to effective drug delivery and cell uptake. Recently, in vivo phage display identified CSKSSDYQC (CSK) as a goblet cell specific targeting ligand [64], and its potential to target nanoparticles has been studied (see section 4).
2.3. Design considerations for intestinal lymphatic targeting
The lymphatic system is a complex network of conduits composed of lymphatic vessels, lymph nodes, spleen, thymus, Peyer’s patches and tonsils. “Targeting” the lymphatic system, or more generally, increasing the relative amount of drug that reaches the lymphatic system, is important in the prognosis and management of diseases like filariasis, tuberculosis, AIDS, metastatic cancers, and several chronic inflammatory diseases [65–71]. Furthermore, recent studies have demonstrated that lymph and lymphoid tissue (in particular GALT) act as a reservoir for HIV, and that the majority of currently used antiretroviral drugs have poor and/or variable penetration in the GALT [72]. Intestinal lymphatic targeting can also help to circumvent first-pass metabolism; the mesenteric lymph bypasses the liver and enters directly into the systemic circulation. Hence, intestinal lymphatic targeting can be useful for improving oral bioavailability of drugs that are susceptible to high-degree of first-pass metabolism, such as steroids [65–71]. In order to understand the attributes for intestinal lymph targeted nanoparticles, understanding the inherent function of the intestinal lymphatic system is crucial. Additionally, various studies involving design and evaluation of lymph-targeted prodrugs have been useful for the development of the lipid nanocarriers targeted to the intestinal lymphatic systems.
The intestinal lymphatic system plays a major role in the oral absorption of dietary lipids such as triglycerides and fat-soluble vitamins. After consumption of dietary lipids, triglycerides are hydrolyzed to fatty acid and monoglyceride in the gastrointestinal lumen, and then absorbed by enterocytes (Figure 3).
Figure 3.
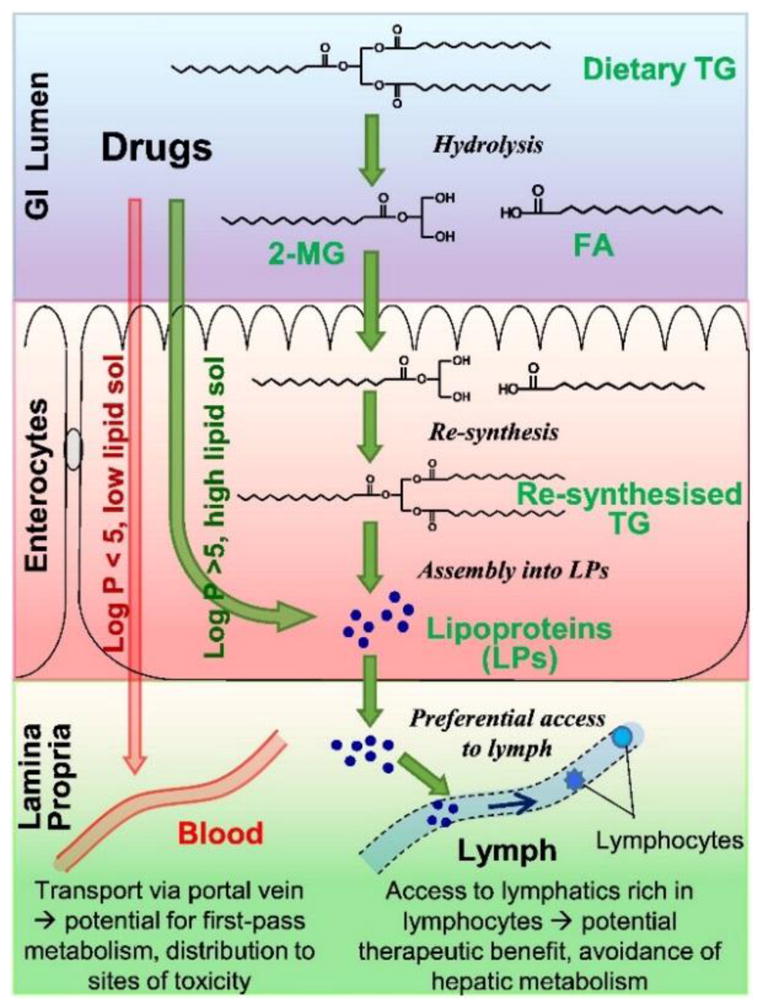
Schematic representation of triglyceride (TG) transport into the intestinal lymphatics and drug transport into the lymph via association with lymph lipoproteins. Adapted from reference [73] with permission.
Enterocytes resynthesize triglycerides from fatty acid and monoglyceride via 2-monoglyceride and/or glycerol-3-phosphate pathway. These resynthesized triglycerides are transformed into colloidal lipoproteins within the endoplasmic reticulum and undergo exocytosis from the enterocytes (Figure 3). Lymphatic capillaries are much more permeable and amenable to the particle transport than the neighboring blood capillaries [65,66,71,73]. Hence, exocytosed lipoproteins are preferentially shuttled to the mesenteric lymph vessels and, finally, into the systemic circulation. It has been observed that the highly lipophilic drugs (exhibiting log P value > 5 and solubility in long-chain triglycerides > 50 mg/g) can associate with lipoproteins during their assembly [65,66,71,73] (Figure 3). Such drugs, due to their association with lipoproteins, avoid first-pass metabolism and are transported to the systemic circulation via intestinal lymphatic transport. Intestinal lymphatic transport is involved in the oral absorption of various lipophilic drugs, like halofantrine, penclomedine, ontazolast, and cyclosporine, especially when these drugs are administered with lipids [65,66]. Lipid prodrugs have been designed for improving oral absorption of hydrophilic drugs and/or drugs with low oral bioavailability due to high first-pass metabolism. Studies have also been carried out to understand the structural requirements of various lipids for augmenting intestinal lymphatic transport; fatty acids of 14 (or greater) carbon chain length were predominantly transported via intestinal lymphatic transport [65,66,71]. Various prodrug based design studies have shown that lipid prodrugs mimicking triglycerides exhibit significantly more lymphatic transport as compared to the prodrugs mimicking monoacylglycerols [65,66,71,73]. Mono- and poly-unsaturated fatty acids promoted greater intestinal lymphatic transport as compared to the saturated fatty acids. Furthermore, phospholipids (phosphatidylcholine and lyso-phosphatidylcholine) can also enhance the intestinal lymphatic transport of drugs and vitamins [65,66]. These studies have informed the design principles for development of lipid nanocarriers, namely fatty acid glyceride and/or phospholipid containing nanocarriers, targeted to the intestinal lymphatic system. Details of recent advancements in nanoparticle targeting to the intestinal lymphatics are discussed in section 4.
2.4. Design considerations for colon targeting
Colon “targeting” generally refers to strategies for minimizing degradation and absorption in the GI tract prior to reaching the colon, which can improve local delivery to colorectal tissues. Targeting the colon has been of great interest for the treatment of various bowel ailments, such as inflammatory bowel disease (IBD), colon cancer, irritable bowel syndrome, diverticulitis, colonic dismotility, and parasitic diseases (amebiasis) [74–79]. Targeting the colon is also a strategy for improving systemic absorption of macromolecules due to the lower proteolytic activity, decreased CYP3A4 activity, diminished P-gp expression, and increased transit time compared to the small intestine [74–79]. However, the fact that the colon is the distal end of the GI tract poses significant challenges for oral nanoparticle targeting [74–79]. Nanoparticles should be able to resist (or minimize) drug release in the stomach and small intestine, despite the wide pH range encountered and presence of various digestive enzymes. Further, drug release should be triggered once the nanoparticles enter the colon.
pH sensitive polymers have been widely employed for designing nanoparticles to target the colon. The pH of the colon ranges from 6.2 to 7.2 depending upon location [74–79]. Various pH-sensitive methacrylic acid copolymers (Eudragit®) that dissolve between pH 6 to 7 have been employed for colon targeting. Eudragit® L100, Eudragit® S100 and Eudragit® FS 30D are commonly used for developing colon targeted delivery systems [24,34,74,79]. However, it should be noted that colonic disorders such as IBD can be associated with changes in the colonic pH [74,79]. Patients with ulcerative colitis can have colonic pH ranging from 2.7–5.5, and patients with Crohn’s disease have average colonic pH of 5.3 [74,79]. Interestingly, Eudragit polymers that can dissolve at pH 5.5 (Eudragit® L100-55) and at pH < 5 (Eudragit® E100) are also available [24,34]. Hence, it may be necessary to employ alternate coatings and suitable ratios of these polymers in order to prevent premature drug release in the stomach and small intestine prior to reaching colon.
The colon is responsible for maintaining the fluid and electrolyte balance via resorption of water and sodium and chloride ions. In order to achieve this, the colon is equipped with absorptive columnar enterocytes. The mucus producing goblet cells, GALT, and M cells are also present in the colon [74–79]. Hence, the design considerations for “active” targeting of nanoparticles to the colon are similar to that discussed for small intestine targeting. Additionally, mucus is a significant barrier to nanoparticles that reach the colon. The mucus layer is thickest in the colon, which reflects the importance of its barrier function. It is composed of two distinct layers: a loosely adherent mucus layer and a firmly adherent mucus layer [3]. Studies in mice, rats and humans have shown that, unlike the loosely adherent layer, the firmly adherent mucus layer is devoid of commensal bacteria in the colon [80–82]. These studies indicated that the firmly adherent mucus layer acts as an additional barrier that prevents the host from infection by the commensal bacteria [80–82]. Recently, we have shown that fluorescent polystyrene nanoparticles densely coated with PEG and ≤ 200 nm in diameter can penetrate through the colonic mucosal barrier ex vivo and in vivo, whereas unmodified nanoparticles were trapped in the colonic mucus and/or lumen [36]. Our studies also showed that the PEG coated nanoparticles demonstrated improved mucus penetration and in vivo distribution, as well as penetration into ulcerated tissue regions, in the colon in a mouse model of IBD [36]. Other studies have shown that negatively charged nanoparticles can selectively target the inflamed region of the colonic mucosa by interacting with the cationic proteins (e.g. eosinophil cationic proteins) in the inflamed region [83]. Nanoparticle size has also been shown to have an effect on targeting in the colon in the context of colitis. Particles showed significantly higher accumulation in the inflamed colonic mucosa of mice as compared to the normal mucosa in healthy mice, and 100 nm particles showed highest accumulation in the inflamed colonic mucosa [84,85]. Taken together, size, charge and surface hydrophilicity of the nanoparticles have an impact on colon targeting, which is further impacted by the presence of disease. In addition to pH, other environmental factors in the colon have been utilized for targeting. The majority of colonic pathologies involve inflammation, resulting in relatively high levels of reactive oxygen species (ROS) at the diseased tissue. For instance, individuals suffering from ulcerative colitis showed 10- to 100-fold higher mucosal levels of ROS in the biopsies of the diseased tissue [86,87]. Attempts have been made to design nanoparticles composed of ROS sensitive materials to specifically target inflamed areas of the colon in IBD. Additionally, the human colon harbors the most abundant and diverse microflora (over 400 distinct bacterial species) in the GIT, and the typical microbial load in the colon is 1010–1012 CFU/ml [76,78,88]. The bacteria are predominantly anaerobic and produce a variety of enzymes (glucuronidase, xylosidase, arabinosidase, galactosidase, nitroreductase, azoreductase, deaminase, urea hydroxylase, etc.) to accomplish fermentation of undigested contents [76,78,88]. Materials that can act as a substrate for these enzymes may find use for colon targeted delivery. In view of this, prodrugs based on azo bonds (sulfasalazine, olsalazine and balsalazide) and glucosides (dexamethasone-21-fglucosides) have been developed to achieve colon targeting. Similarly, polymers based on natural carbohydrates (pectin, guar gum, alginic acid), or azo containing monomers (azo-polyurethanes) have been studied for colon targeted nanoparticles [88,89]. However, it is important to consider the differences in the microbiota in the healthy and diseased colon, and between different animal species, in the design of nanoparticles based on microbial enzyme substrates. The normal colonic microflora is altered in the individuals suffering from IBD [90]. Various studies have shown that IBD is associated with reduced bacterial diversity, a reduction in bacteria producing anti-inflammatory substances (Faecalibacterium sp.) and increase in Enterobacteriaceae sp. (such as E. coli) [90,91]. Murine colitis models show similar alterations in the colonic microflora as found in human IBD studies [91]. Studies have also shown that enzymes, such as azoreductases, and glucuronidases, are present in the murine colon, suggesting that it may be possible to evaluate nanoparticles based on the microbial enzyme substrates in murine colitis models [92–95].
Finally, studies have also been carried out to ascertain the potential of lectins as a ligand to improve colon targeting of nanoparticles [96,97]. Additionally, aforementioned (Section 2.2) ligands for targeting enterocytes, M cells and goblets cells can also be used to improve colon targeting of nanoparticles. Because of the redundancy with the small intestine, some sort of protective coating strategy that degrades in the colon must be used to ensure maximal delivery to the colon. Specific examples of nanoparticles encompassing some of the design criteria described here are further discussed in section 4.
3. Techniques for characterizing nanoparticle systems designed for oral administration
Proper selection of characterization techniques, nanoparticle administration techniques and interpretation of results is critical in the assessment of nanoparticles designed for oral administration. A wide range of in vitro, ex vivo, and in vivo techniques are described in the literature. The driving force behind the evolution of experimental techniques used to evaluate the potential of nanoparticles for oral delivery, is the disparity in results that is often seen between methods, e.g. in vitro versus in vivo, as well as disparities in results reported by various investigators for similar nanoparticle formulations. Thus, an important challenge is to develop a framework for characterization that reliably predicts outcomes in humans. Techniques commonly used to evaluate oral nanoparticle formulations have been reviewed extensively elsewhere [98–100], so our focus is on new developments over the past several years. We describe recently reported models of the intestinal environment, followed by a review of studies that used some combination of in vitro, ex vivo, and in vivo methods to characterize nanoparticle systems. We end this section with a brief discussion of what these methods may be able to predict about the fate of nanoparticles in the ultimate in vivo model: humans.
3.1. New models of the intestinal environment
3.1.1. Cell monolayer cultures
In vitro models are more accessible and have lower cost, and higher throughput compared to in vivo models. In addition, better in vitro models may reduce of animals used for experimental purposes [101]. Caco-2 is the most widely used cell line for mimicking epithelial barrier of the intestines. Caco-2 cells were derived from human colorectal adenocarcinoma cells. Despite their colorectal origin, Caco-2 cells can be differentiated and polarized to resemble enterocytes lining the small intestine [102]. Strong correlations have been observed for passive transcellular and paracellular permeability across Caco-2 cell monolayers and oral bioavailability in humans for many drugs, though the caveats and limitations of using Caco-2 cell monolayers for drug absorption are well-reviewed elsewhere [98,103,104]. It is likely that the utility for predicting small molecule absorption, and the ease of culture and handling of these cells, led to their prolific use in evaluating nanoparticle uptake and translocation. However, it is worth noting that some researchers caution against assuming that the endocytosis events observed for nanoparticles in cell culture also occur in the GI tract in vivo [105]. For these reasons, improving in vitro cell culture models to better recapitulate intestinal barriers and transport mechanisms for a wider range of therapeutics and delivery vehicles, including nanoparticles, remains an active area of research.
Kauffman and coworkers sought to develop absorptive intestinal epithelial cell models that more fully recapitulate in vivo intestinal phenotypes [106]. They compared two sources of human intestinal epithelial cells, primary human intestinal epithelial cells (hInEpCs) and induced pluripotent stem cell (iPSC)-derived intestinal cells, to Caco-2 cells. Monolayers were assessed for marker expression, monolayer integrity, and small molecule permeability. They observed that hInEpC monolayers demonstrated more uniform expression of intestinal epithelial markers (E-cadherin, CDX2, Villin), in contrast to the known heterogeneity of Caco-2 monolayers. The overall expression of many markers by the hInEpCs and iPSC-derived cell monolayers was similar to or higher than Caco-2 monolayers. Interestingly, the trans-epithelial electrical resistance (TEER) for the hInEpCs and iPSC-derived monolayers was several-fold higher than what is typically reported for Caco-2 monolayers. Interestingly, the trans-epithelial electrical resistance (TEER) for the hInEpCs and iPSC-derived monolayers was several-fold higher than what is typically reported for Caco-2 monolayers. These results suggest that the hINEpCs and IPSC-derived models may be useful for specific types of drugs or delivery systems reliant on specific cell characteristics better recapitulated by these systems, but the use of these cells is limited by the viability and the extensive differentiation time, as reported [106]. It is possible that the increased TEER may indicate more resistance to passive absorption, so it may be interesting to test nanoparticle translocation using these cell monolayers.
One major limitation of Caco-2 monolayers is that although the cells differentiate into a heterogeneous population, the cells are entirely enterocytes. Thus, these cultures lack mucus secreting goblet cells interspersed between enterocytes. Mucus acts as a protective barrier in the intestines that is encountered by orally administered nanoparticles [3]. For this reason, many have studied and further optimized Caco-2 and goblet-like HT29 cell co-cultures in an attempt to achieve a mucus coating on the monolayer [107–109]. In the case of nanoparticles, though, many have suggested that M cells play a significant role in nanoparticle absorption. This has led others to develop co-cultures of Caco-2 cells with cells with similar characteristics as M cells, Raji B cells. It was reported that nanoparticle translocation in Caco-2/Raji co-cultures was increased compared to cultures with Caco-2 alone [110]. More recently, Schimpel and coworkers described an in vitro triple culture model to study the transport of nanoparticles [111]. Using 50 and 200 nm polystyrene nanoparticles, they demonstrated increased nanoparticle translocation in co-cultures of Caco-2/M cells vs. Caco-2 cells alone. However, the presence of HT29-MTX cells, either in double (Caco-2/HT29-MTX double culture compared to Caco-2 only) or triple culture (Caco-2/HT29-MTX/M cell triple culture compared to Caco-2/M cell double culture), significantly reduced nanoparticle translocation (Figure 4).
Figure 4.

Fluorescence microscopic z-scans of 200 nm fluorescent red polystyrene nanoparticles incubated with (a) Caco-2 monolayer, (b) Caco-2/M cell double culture, (c) Caco-2/HT29-MTX double culture, and (d) Caco-2/HT29-MTX/M cell triple culture. The arrows depict particle aggregates caught on the luminal side, and circles indicate nanoparticle uptake/penetration. The presence of mucus dramatically decreased nanoparticle uptake, while M cells facilitate nanoparticle translocation. Adapted from reference [111] with permission.
Furthermore, confocal microscopy revealed nanoparticles aggregated and became entrapped above the cell monolayers when HT29-MTX cells and mucus were present. They compared these observations to nanoparticle absorption when incubated with porcine intestinal mucosa in Ussing chambers, where it appeared the nanoparticles, particularly the 50 nm nanoparticles, more effectively permeated the mucus barrier compared to the entrapment observed in the cell cultures with mucus [111]. Considering that other reports have suggested that mucus from porcine intestines is penetrable to even very large polystyrene microparticles, additional characterization of porcine intestines and comparison to other models is needed. When obtaining porcine intestines from a butcher, post-mortem degradation of the mucus by bacterial products, the process of washing away the luminal digesta which includes the loosely adherent mucus layer, and the impact of common farming practices on the integrity and health of the intestines may all affect the barrier properties of the mucosa [112,113].
3.1.2. 3-dimensional (3D) cell and tissue cultures
New 3-dimensional (3D) culture systems aim to incorporate features of the gastrointestinal tract that are absent in monolayer cultures, such as the topography of the intestinal villi and the resident immune cells lying below the epithelium. Leonard and coworkers described the development of an inflamed 3D cell culture model for evaluating nanoformulations loaded with budesonide for potential use in patients with IBD [114,115]. It has been suggested that the permeability of the intestine and the uptake of nanoparticles in the context of inflammation is increased, which is important in the context of IBD treatment [36, 80–82]. To mimic these effects in vitro, macrophages and dendritic cells were embedded in a collagen layer on a Transwell filter insert and Caco-2 cells were seeded on top to form a monolayer with underlying immune cells [114]. Upon stimulation with IL-1β, the Caco-2 layer TEER was reduced by 20%, and release of the pro-inflammatory cytokine IL-8 increased by ~30-fold on the basolateral side. They then incubated various sized polystyrene nanoparticles (50 nm – 500 nm) with the co-cultures, and found most of the nanoparticles adhered to the top of the monolayer. However, they observed nanoparticle uptake by immunocompetent cells, which increased when the cells were stimulated with IL-1β and mobilized to the apical side of the Caco-2 monolayer [114]. This suggests that any translocation that occurred was due to internalization by activated immune cells. Leonard and coworkers then went on to test the efficacy and deposition of budesonide loaded into nanoparticles (PLGA) and liposomes [115]. Free budesonide solution and budesonide administered in PLGA nanoparticles rapidly restored the TEER to normal levels after the initial 20% drop after IL-1β stimulation. In contrast, the liposomal-budesonide worsened the inflammation and further reduced the TEER (70% of initial value), which corresponded with increased IL-8 secretion. The sustained release of budesonide from the PLGA nanoparticles appeared to provide more sustained suppression of IL-8 secretion compared to free budesonide [115]. Thus, it appears the advantages of this culture system include determining potential pro-inflammatory effects of carriers, as well as confirming the anti-inflammatory effects of loaded cargo. The increase in nanoparticle phagocytosis by immune cells that would be expected in a pro-inflammatory environment also occurred in this system. The co-culture model also appeared to elucidate a benefit of sustained release, though the impact of GI transit and residence time are not represented in a static culture.
Huang and coworkers described a similar strategy for modeling the pro-inflammatory environment of IBD in a high-throughput 24-well plate format [116]. THP-1 macrophage cells were seeded in a Matrigel solution matrix, and Caco-2 cells were placed on top in media. For the inflamed condition, Caco-2 cells were added along with THP-1 cells in media containing IL-1β and lipopolysaccharide (LPS). They then compared the uptake of quantum dots (QDs), polyetherimide/antisense oligonucleotide (ASO) nanoparticles, and galactosylated low molecular weight chitosan (G-LMWC)/ASO nanoparticles in the model system to uptake in vivo in the 2,4,6-trinitrobenzenesulfonic acid (TNBS) colitis mouse model. They generally found that the increased nanoparticle uptake observed in the inflamed vs. control culture model was similar to that observed in the TNBS model compared to healthy controls, though the extent of uptake was higher in the cell culture model compared to in vivo. They observed decreased levels of IL-6 and TNF-α mRNA and protein in vivo and in the cell culture due to antisense oligonucleotide delivery [116].
Chen and coworkers developed a 3D porous silk protein scaffolding system that included various additional features of the intestinal environment in vivo, including a hollow lumen, formation of a double mucus layer on the epithelial surface, and a progressive reduction in oxygen tension through the intestinal lumen [117]. Silk protein scaffolds surrounding non-patterned (Teflon-coated wire) and patterned (threaded nylon rod) lumens were seeded with Caco-2, HT29-MTX, and human intestinal myofibroblasts (H-InMyoFibs). The support in growth, differentiation, and expansion provided by the H-InMyoFibs extended the growth and differentiation of the epithelial cells in the Caco-2/HT29-MTX co-culture. They demonstrated that various measures were improved for the 3D culture compared to typical 2D culture, and even further improved for the patterned 3D culture compared to the non-patterned 3D culture, including: mucus layer thickness, oxygen gradient, adhesion and colonization of bacteria on the epithelial cells, and expression of various epithelial cell markers (ZO-1, E-cadherin, Villin, sucrose-isomaltase) [117]. One limitation of this system for characterizing nanoparticle systems is that the culture does not include immune cells, which as described, are considered to be a significant contributor to nanoparticle translocation and uptake. However, this system could be used to evaluate potential toxic effects of nanoparticle systems to the GI epithelium and non-pathogenic microbiota, as well as delivery of antibiotics.
Another class of 3D cultures is tissue-derived organoids and spheroids. Organoid/spheroid cultures produced from gastrointestinal tissues are widely used to study interactions with bacteria, pathogenic infections, inflammation and oxidative stress, and toxicity to cells/cell death [118–122]. Recent reports include: the work of Grabinger and coworkers toward generation of ex vivo cultures of intestinal crypt organoids and methods for monitoring and scoring organoid survival and death [123]; the work of McCracken and coworkers toward generating pluripotent stem-cell-derived gastric organoids that include gastric gland and pit-like domains, proliferative zones, surface and antral mucus cells, and the capability of supporting Helicobacter pylori infection [124]; and the work of Pastula and coworkers toward development of murine GI epithelial organoids cultured with myofibroblasts and neurons to support stem cell growth [125]. In the case of evaluating oral nanoparticle formulations, organoid systems can be used to assess pro-/anti-inflammatory properties and, in some organoid systems, impact on pathogenic/non-pathogenic bacteria. In recent work, Chia and coworkers used a 3D gastrointestinal spheroid culture model to assess the toxicity and pro-inflammatory characteristics of zinc oxide (ZnO) nanoparticles, and to compare to previous results reported using 2D cell cultures [126]. Using NCM460 (normal epithelium) and SW480 (adenocarcinoma) colorectal cells, they allowed spheroids to self-assemble in non-fouling agarose micro-molds. They found that the highest concentration of ZnO nanoparticles (1000 μM) caused a significant increase in the percentage of NCM460 cells producing reactive oxygen species (ROS) after 6 h in the 2D and 3D cultures, with a higher percentage of ROS-producing cells in the 2D cell culture. For the SW480 cancer cells, a much higher percentage of untreated cells were ROS positive in the 3D cultures compared to the 2D cultures. Interestingly, treatment with ZnO did not produce an increase in the percentage of ROS positive SW480 cells in 3D culture, but there was a significant increase in the 2D culture 6 h after exposure to 125–1000 μM ZnO nanoparticles. They went on to further demonstrate additional differences in the response of the 2D and 3D cultures to the ZnO nanoparticles, including differential inflammatory gene expression, greater resistance to DNA damage in 3D cultures, and lower levels of cytotoxicity in 3D cultures. Their results suggest that the cells supported by a 3D tissue structure experience additional cell-cell interactions and protection of inner cell layers from nanoparticle access, leading to a less severe response to ZnO nanoparticle exposure. This further implies that prior studies using 2D cultures may have overestimated the potential toxicity of ZnO nanoparticles in the GI tract in vivo [126]. Indeed, this further highlights the care that must be taken when selecting appropriate experimental methods to evaluate specific aspects of a nanoparticle formulation.
3.1.3 “Gut-on-a-chip” devices
“Gut-on-a-chip” devices combine 3D cell culture models with microfabrication technologies and microfluidics to better recapitulate mechanical microenvironments, spatiotemporal chemical gradients, and tissue-tissue interfaces [127]. In the context of evaluating oral nanoparticle formulations, these systems incorporate flow, which is an important limiting factor in nanoparticle residence time and uptake. Esch and coworkers described a porous membrane dried onto silicon pillars of defined height and width as a template for growth of Caco-2 cells into villi-like structures [128]. Taking advantage of micropatterning and microfluidics, the design evolved to include a co-culture of Caco-2 and HT29-MTX cells (intestine), HepG2/C3A cells (liver), and chambers representing the liquid portion of other organs in the human body [129]. Thus, by flowing nanoparticles through the apical side of the “intestine”, they could quantify the percentage of 50 nm polystyrene nanoparticles that crossed the Caco-2/HT29-MTX co-culture monolayer, and flow these “translocated” particles through chambers containing liver cells. Using relatively high particle concentrations (120–480 ×1011 particles/mL), they found that <10% of the nanoparticles crossed their “intestine” after 24 h of device operation. They found the nanoparticles on the apical side to largely be in aggregates, and nanoparticles recovered from the basolateral side suggested that the polystyrene nanoparticles that did cross the Caco-2/HT29-MTX monolayers were coated with proteins and lipids from the cell culture medium. They then measured the aspartate aminotransferase (AST) concentration as a measure of liver cell toxicity from nanoparticle exposure. Only the highest dose of nanoparticles caused an increase in AST levels when the HepG2/C3A cells were exposed alone. When the Caco-2/HT29-MTX cultures and HepG2/C3A cultures were combined on the device, the AST levels increased with increasing nanoparticle exposure. However, this did not result in any measurable increase in cell permeability or cell death [129]. The advantage of this microfluidic system is that it allowed communication and feedback between different cell types, though it is not clear whether the specific effect described may be recapitulated in vivo. This work also highlighted an often overlooked fact that nanoparticles with sticky surfaces typically have different physiochemical characteristics after entering biological environments, which can affect the nanoparticle behavior.
Kim and coworkers developed a human gut-on-a-chip that simulated intestinal peristalsis-type motions and flow [130]. Using microfluidic channels, the low shear stress and cyclic strain of peristalsis-like flow could be exerted on the Caco-2 cells, leading to the development of a columnar epithelium that polarized rapidly, grew villi-like folds, and allowed for co-culture of Lactobacillus rhamnosus for at least 1 week without compromising cell viability. They went on to further characterize the Caco-2 monolayers that developed in the gut-on-a-chip, describing cells forming villi with tight junctions, brush borders, mucus, and the formation of absorptive, mucus-secreting, enteroendocrine, and Paneth epithelial cell types [131]. In more recent work, they describe the incorporation of immune cells and commensal microbes that can be stably cultured for many days to weeks [132]. Their system can be used to model the pro-inflammatory environment of IBD, including bacterial overgrowth and slowing of GI motility. Although they have not yet reported characterization of nanoparticles using the gut-on-a-chip, it would be interesting to see how nanoparticle uptake differs in this cell culture system, in the context of peristaltic flow and increased barrier properties in the presence of probiotic bacteria, compared to the other 2D and 3D systems described.
3.2. Comparisons between in vitro, ex vivo, and in vivo models
Models describing the relationship between an in vitro property and an in vivo response, also called an in vitro-in vivo correlation (IVIVC), have been in development since the 1950s for oral dosage forms, where the in vitro property is drug dissolution and the in vivo property is systemic pharmacokinetics [104,133,134]. Developing IVIVC models for oral nanoparticle formulations adds an additional layer of complexity in predictive modeling of drug pharmacokinetics. Barzegar and coworkers described the development of an IVIVC model for oral nanoparticle formulations with release rate-limited absorption; the area under the curve for the in vitro drug release was correlated to the in vivo area under the curve for systemic drug levels [135]. The model parameters obtained based on experimental results varied for different formulations loading the same drug (estradiol), depending on molecular weight of the polymer used, solvents and particle size, highlighting the added complexity [135]. Predicting in vivo properties for even more complicated systems, such as nanoparticles that are intended to be taken up intact, nanoparticles that are unstable in the GI environment, or nanoparticles that are cleared rapidly, would be increasingly challenging. Further, the choice of animal model to obtain in vivo data for developing correlation models is not an obvious one. The correlation between drug bioavailability in various animal models (mice, rats, dogs, non-human primates) and humans has been extensively studied, and clearly depends on drug properties and physiological characteristics [136,137]. Thus, adding an additional layer of complexity by packaging the drug in a nanoparticle delivery system makes the issue of defining correlation between animals and humans even more nuanced. Differences in the GI physiology between animals and humans, such as mucus production and thickness, density of Peyer’s patches, pH, emptying and transit times, bacterial colonization and administration method differences between animals and humans must all be considered [3,138].
In order to build confidence in the observations of the behavior of an oral nanoparticle formulation, many researchers have taken to comparing their platform using a variety of in vitro, ex vivo, and in vivo methods. A theme we have observed commonly is to demonstrate stability of the nanoparticles in vitro (i.e. slow or incomplete drug release over a 12–24 h period), show uptake of nanoparticles loaded with fluorophores in Caco-2 cell monolayers in vitro, and then reason that increased systemic exposure of the drug cargo delivered by nanoparticles in vivo is due to nanoparticle uptake in the GI (see section 3.3 for additional discussion). Zou and Gu demonstrated that emulsified nanoparticles containing daidzin provided increased cellular uptake and transport through Caco-2 monolayers and 2.4-fold increased AUC compared to daidzin solution [139]. Yuan and coworkers developed PEGylated solid lipid nanoparticles (pSLN) and evaluated the system in Caco-2/HT29 cultures, everted rat gut sacs, and oral gavage to rats [140]. In the cell culture model, they showed increased uptake of pSLN compared to solid lipid nanoparticles (SLN) in the presence of increasing proportions of HT29 cells (and thus, increased mucus). In the everted gut sac, pSLN showed a minor increase in absorption compared to SLN, though it has been demonstrated that the mucus barrier may be compromised in ex vivo/in situ models [36]. In vivo, they found that pSLN delivering doxorubicin (Dox) provided a 2-fold increase in the AUC compared to SLN, and a 7.5-fold increase in AUC compared to oral Dox [140]. This suggests that nanoencapsulation provided a significant increase in systemic Dox absorption (3.7-fold increase), and that PEGylation of the SLN provided an additional increase in Dox delivery. Similarly, Tariq and coworkers found that PLGA nanoparticles loaded with epirubicin provided increased epirubicin delivery across Caco-2 layers and across non-everted intestinal rat sacs compared to epirubicin solution [141]. They then found that the AUC for epirubicin administered orally in a nanoparticle to rats was ~3.9-fold higher than epirubicin solution.
Also focusing on the mucus barrier, Zhu and coworkers developed self-assembled nanoparticles containing insulin for oral delivery [142]. Nanocomplexes (NCs) of the cell-penetrating peptide, penatrin, and insulin were formed and coated with N-(2-hydroxypropyl) methacrylamide copolymer (pHPMA) derivatives to form nanoparticles (NPs) with a hydrophilic, mucoinert coating. In contrast to the positively charged NCs, more neutrally-charged NPs were more stable and diffused more freely in mucin solutions in vitro. They also demonstrated that nanocomplexation improved insulin permeability across HT29-MTX-E12 cell monolayers, which was further improved by coating the NCs to make mucoinert NPs; the apparent permeability (Papp) measurements suggested that removal of mucus from the cultures provided a small improvement in insulin translocation for NCs, and employing NPs provided an additional increase in the Papp whether mucus was present in the culture or the cultures were washed to remove mucus produced by the HT29 cells. They went on to demonstrate increased insulin bioavailability and corresponding hypoglycemic effect for NPs compared to NCs. Although required oral dose of insulin delivered by NPs (75 IU/kg) was much higher than the dose delivered subcutaneously (5 IU/kg), the hypoglycemia induced by the insulin delivered as NPs was more mild and prolonged compared to the subcutaneous insulin dose. The effect of the NCs was similar to oral free insulin, presumably from lack of penetration through mucus [142]. Maisel and coworkers described similar in vitro/ex vivo evaluation of PEGylated polystyrene nanoparticles for penetration through GI mucus [36]. The PEGylated nanoparticles (mucus-penetrating particles, MPP) diffused rapidly in mucus on freshly excised mouse intestinal tissue, whereas uncoated mucoadhesive particles (MAP) were adhesively trapped in the mucus layers. When administered orally to mice, the MPP distributed uniformly over the surface of the mouse jejunum and ileum, whereas MAP remained aggregated in the intestinal lumen. They then used the nanoparticles to investigate the effect of various experimental techniques on nanoparticle distribution in the GI tract. They found that administering the particles to mice in the fed state (whereas most animals used for oral gavage experiments are fasted) led to an even more striking difference in the distribution of MPP compared to MAP, presumably because MAP also interact with and stick to digesta in the intestinal lumen. They further described that the distribution of MAP in the mouse GI tract could be artificially improved by either administering to an ileal loop ex vivo or by administering MAP in a higher volume of gavage fluid in vivo, reinforcing the importance of ensuring that experimental methods closely recapitulate the intended delivery mode (e.g. swallowing a pill) (Figure 5) [36]. It has yet to be determined whether the improved distribution and access to the absorptive epithelium by the MPP platform can also improve the oral bioavailability of drugs compared to MAP. Careful, direct comparisons using particles with similar stability, size, core composition, and drug loading and release are needed.
Figure 5.

Distribution of the same 200 nm fluorescent red mucoadhesive polystyrene nanoparticles (MAP) in the mouse ileum after administration by various routes, (A) low volume (50 μL) oral gavage, (B) high volume (250 μL) oral gavage, and (C) direct infusion into an in intestinal loop. The mode of administration significantly impacted the intestinal distribution. Adapted from reference [36] with permission.
3.3. A brief perspective on data interpretation and potential translation
When considering the value of any experimental technique for characterizing nanoparticle systems designed for drug and gene delivery, the most important aspect is how well the technique can recapitulate the environment and biological barriers that the nanoparticle would encounter when administered to the target population (e.g. a particular disease state) via the intended route (e.g. oral versus pulmonary). For oral administration, there are additional considerations, such as if a particular location in the GI tract or cell type is targeted for delivery, and if delivery should be locally to the tissue or requires absorption into the systemic circulation or lymph. There is a vast array of potential applications for oral nanoparticle delivery, and this section does not intend to exhaustively discuss all possibilities. In many cases, though, the goal is to use nanoparticles to increase systemic bioavailability. Perhaps one of the more controversial issues in this area is the question of whether nanoparticles are taken up by the absorptive epithelium and enter the systemic circulation. Cell culture models suggest that nanoparticles of optimal size and surface characteristics translocate easily, even passively [109, 143]. There are also reports of significant nanoparticle accumulation in the systemic circulation when administered to ligated intestinal loops [144]. However, when considering whether 15–30% or more of an orally ingested nanoparticle dose could make it to the systemic circulation, physiology must be considered.
The primary purpose of the GI tract is digestion and assimilation of nutrients, and while fulfilling this purpose, the GI tract must also act as a barrier and perform surveillance to keep out pathogens. Nutrient absorption is dependent on digestion into small peptides and monosaccharides, with the larger lipid/bile micelles and intact proteins being a few nanometers in size [145]. In contrast, the GI tract has defenses for larger viral and bacterial pathogens and environmental particles, including particulates added to food like titanium dioxide (TiO2) and silicon dioxide (SiO2). Indeed, studies in the toxicology field investigating the unintentional systemic exposure of these types of additive particulates have shown much less systemic accumulation than what has been described for drug delivery studies [146]. However, Cho and coworkers found that, in comparison to TiO2 nanoparticles (26 nm), systemic zinc exposure from zinc oxide (ZnO) nanoparticles (89 nm) was much higher, presumably because the ZnO particles were degraded in the GI tract and subsequently absorbed [147]. Similarly, it was found that orally-administered PEG-coated gold nanoparticles (45 nm) were absorbed at significantly higher levels than uncoated gold nanoparticles (23 nm), though both coated and uncoated particles had overall low bioavailability of <1% [148]. The advantage of these analyses is that unlike measurements of fluorophores or radiolabels passively loaded inside particles as a surrogate for particle uptake, or particles made of materials that can potentially be degraded in the GI tract prior to absorption, quantification of systemic uptake and organ accumulation should be more accurate. It is possible that nanoparticles composed of polymeric or lipid materials just behave differently in the GI tract than inorganic materials, but the apparent disparity in the extent of uptake of nanoparticles after oral administration in the toxicology literature is worth noting. Further, Sinnecker and coworkers recently reported minimal absorption of the same type of polystyrene nanoparticles studied in numerous other reports citing high systemic absorption [149]. Using an isolated perfused rat small intestine, essentially no 20 and 200 nm carboxylate-modified polystyrene nanoparticles were detected in the vascular and lymph systems. In contrast, over 95% of the nanoparticles were recovered in the perfused luminal samples and dissolved mucus [149]. Similarly, our own work has found that carboxylate-modified polystyrene nanoparticles can be found associated with mucus in the GI tract (oral, ex vivo tracking), which would preclude cell uptake that may occur. However, gavaging large volumes of fluid and administering to intestinal loops led to increased exposure of the polystyrene nanoparticles to the epithelium [36], which would potentially allow for more systemic absorption. Thus, some controversy over the extent of uptake of intact nanoparticles may derive from differences in experimental methods. Hu and coworkers also recently commented on whether SLN are absorbed intact after oral administration, as many other reports suggested [150]. By co-loading a water-quenchable dye and a standard dye in the SLN, they were able to differentiate between intact SLN and dye that had been released from degraded SLN. Through cell culture and in vivo studies, they concluded that SLN are not absorbed from the intestines intact, and that the presence of mucus limits interactions between the nanocarriers and cell membranes [150]. What the authors describe makes sense from the physiological viewpoint of how lipid-based substances are handled for nutrient absorption in the GI tract.
For many types of nanoparticles and microparticles, it is generally thought that translocation via M cells in the Peyer’s patches is a primary mechanism for uptake. This characterization is consistent with the role of the Peyer’s patches in sampling the luminal contents for bacteria and other pathogens, as generating mucosal immune responses is thought to be the main protective function for the vast majority of human pathogenic microorganisms [151]. Many reports describing microparticle translocation via M cells were performed using intestinal loop models [152–155], a method that was initially employed for investigating the mechanisms of bacterial infection in the GI tract and assessing drug permeability in the intestine [156,157]. However, the minimal extent to which bacterial infection occurs in the healthy GI tract necessitates the use of loops and forced, artificial exposure to the GI, which also likely leads to an artificial increase in the systemic uptake of particles in these models. Man and coworkers describe exposing the follicle associated epithelium (FAE) of the Peyer’s patch to pathogenic bacterial components to subsequently increase the translocation of microparticles by the FAE [155,158]. However, this was also studied using intestinal loops, the extent of activation by bacterial exposure and subsequent access of the microparticles to the FAE that would occur in vivo is unclear. It seems to us that if large doses of microparticles were able to access the systemic circulation via the Peyer’s patches in the GI tract, this would signal a larger susceptibility to infection, or in other words, a breach in mucosal defenses. Thus far, we have not discussed formulations with various targeting moieties, permeation enhancers, or mucus degrading capabilities, which if properly exploited, would also be considered a breach in normal mucosal defenses. Situations where the benefits outweigh the risk for potential harm likely exist, but would have to be evaluated on a case-by-case basis.
A greater question is whether one actually wants or needs nanoparticles to be absorbed intact after oral administration. Of course, this depends on the properties of the cargo and the delivery goal. In the cases of compounds that are highly degradable in the GI tract, packaging within a nanoparticle can improve bioavailability just by protecting the cargo. Peptides/proteins, nucleic acids, and other macromolecules can be protected and stabilized within the nanoparticle core or complex. For insoluble drugs, improving the dissolution can make a tremendous difference in oral absorption. Inherently, drugs that are typically amenable to formulation as nanoparticles have solubility issues that lead to limited GI absorption of the unencapsulated drug. Essentially, all oral nanoparticle products on the market are nanosuspension formulations, which are insoluble drug particles that have been milled down to nanosized pieces. Just by increasing the surface area for dissolution, more of the drug is able to be absorbed by the normal route of absorption [159]. The beauty of the approach is the simplicity, which aids in the feasibility of clinical implementation. Formulation of drugs in nanoparticles could also potentially provide more sustained absorption as drug is released from the nanoparticles during transit through the GI tract. Similarly, mucoadhesive strategies aim to take advantage of increased residence time in the GI tract to provide sustained drug release and absorption [10]. In all of these cases, the particle does not need to be absorbed intact to provide improved systemic drug exposure. Furthermore, it seems it would be generally preferable if the nanoparticle components remained in the GI tract where they could be easily excreted as waste, rather than requiring processing by the mononuclear phagocyte system (MPS) and potentially causing systemic toxicity. Thus, we hypothesize that a combination of increased residence time and increased proximity to and coverage of the absorptive epithelium by nanoparticles could maximize systemic drug bioavailability [36]. Regardless, the field has much more to learn through preclinical experimentation and controlled clinical trials.
4. Recent developments in targeting oral nanoparticles to specific sites in the GI tract
Building off of our discussion of approaches for “targeting” oral nanoparticle formulations to specific tissues and/or cells in the GI tract in section 2, we next discuss recent advancements in nanoparticle technologies for targeting in the GI tract. Again, “targeting” refers to formulation properties intended to increase residence time, increase the relative amount of nanoparticle degradation/drug release, and/or facilitate interactions of nanoparticles with particular tissues and cells in particular sections of the GI tract. Following our emphasis on the state-of-the-art, we focus here on works published since the year 2010.
4.1. Nanoparticles targeted to the stomach
The majority of formulations designed to target the stomach are composed of materials that are considered mucoadhesive to increase gastric retention. Among these, the most commonly used mucoadhesive polymer is chitosan, a naturally occurring polysaccharide composed of β-(1–4)-linked D-glucosamine and N-acetyl-D-glucosamine. Chitosan contains primary amines that interact with the negatively charged sugars on mucin glycoproteins [160], and thus is used widely as a mucoadhesive for delivery to mucosal surfaces. However, chitosan ionizes and rapidly dissolves in the acidic pH of the stomach. Thus, for stomach targeting, chitosan is complexed with polyanions to form nanoparticles that do not rapidly dissolve at acidic pH. Chang and coworkers described amoxicillin-loaded nanoparticles based on chitosan and poly-glutamic acid polyelectrolyte complexes for the treatment of H. pylori infections [161]. They also developed a cross-linked hydrogel composed of alginate, gelatin and calcium ions to incorporate the amoxicillin nanoparticles. Interestingly, release of amoxicillin from chitosan-polyglutamic acid complex nanoparticles was significantly lower at acidic pH (pH 1.2) after their incorporation in the hydrogel. To determine the therapeutic potential of the nanoparticles, fluorescently labeled nanoparticles were incubated with gastric adenocarcinoma (AGS) cell monolayers infected with H. pylori. Confocal microscopy studies revealed that the nanoparticles were able to interact with the AGS cells at the site of H. pylori infection [161]. This investigation discussed the importance of incorporating nanoparticles in a suitable gastroretentive dosage form to sustain the release of the drug at an acidic pH of stomach, though no ex vivo or in vivo gastric retention studies or efficacy studies were described.
Lin and colleagues designed polyelectrolyte nanoparticles based on fucose substituted chitosan and heparin (a polyanion) for targeted treatment of H. pylori infection [162]. The fucose was intended to target the fucose receptors present on the H. pylori. The complexed nanoparticles were loaded with amoxicillin. In vitro release studies at pH 1.2 showed that 40% of encapsulated amoxicillin released in the first 2 h. Thus, the authors used genipin to induce crosslinks in the chitosan-heparin nanoparticles, which led to slower release of amoxicillin in vitro. Electron microscopy studies showed that amoxicillin-loaded, genipin-crosslinked chitosan-heparin nanoparticles attached and distorted the shape of H. pylori. In vitro antimicrobial efficacy studies revealed that the amoxicillin-loaded, genipin-crosslinked fucose-chitosan-heparin nanoparticles were more active against H. pylori compared to non-fucosylated, amoxicillin-loaded nanoparticles and amoxicillin solution. Finally, they demonstrated in mice that orally delivered amoxicillin-loaded, genipin crosslinked fucose-chitosan-heparin nanoparticles were almost 4-fold more effective in clearing H. pylori infection compared to amoxicillin solution [162]. It would have been interesting to also compare the in vivo efficacy of the non-fucosylated, amoxicillin-loaded nanoparticles in clearing H. pylori infection to support the benefit of fucose-mediated targeting in vivo. It would have also been interesting to visualize the duration of in vivo gastric retention of the fucosylated chitosan-heparin complex nanoparticles and chitosan-heparin complex nanoparticles. In a second investigation, Lin and coworkers demonstrated the potential of berberine-loaded, genipin crosslinked fucose-chitosan-heparin nanoparticles in the treatment of H. pylori infection and found similar results [163].
Recently, Lin and colleagues described stomach targeted nanoparticles for improved oral treatment of gastric cancer [164]. Several cancers, including gastric cancer, display carbohydrate antigens and/or increased expression of fusocyl transferase at the cell membrane. Thus, they designed nanoparticles containing a combination of fucosylated chitosan, PEGylated chitosan, gelatin, and epigallocatechin gallate (EGCG-PFCH-NPs), a natural polyphenol with anticancer activity. PEGylated chitosan was employed to improve the intermolecular crosslinking and protect the gelatin in the nanoparticles from pepsin. In vitro release studies showed that the presence of PEGylated chitosan significantly reduced the release of epigallocatechin gallate from the nanoparticles in pH 1.2 buffer. In vivo studies in mice showed that orally administered Vivo-Tag 750-labeled EGCG-PFCH-NPs demonstrated strong fluorescence signal in the stomach for 6 h, and the fluorescence signal in stomach diminished somewhat by 9 h (Figure 6) [164].
Figure 6.

In vivo gastric retention of orally administered, NIR dye labeled, nanoparticles composed of fucosylated chitosan, PEGylated chitosan, gelatin, and epigallocatechin gallate (FCS/PCS/Gel/EGCG-NPs). FCS/PCS/Gel/EGCG-NPs were localized in the stomach for the first 6 h and started spreading further in the GI tract at 9 h. This is Adapted from reference [164] with permission.
It would have been interesting to also compare the in vivo gastric retention of the EGCG-chitosan-gelatin NPs, EGCG-PEGylated chitosan-gelatin NPs, and EGCG-fucosylated chitosan-gelatin NPs to ascertain the roles of PEGylation and fucosylation of the chitosan in increasing gastric retention. Finally, Lin and coworkers studied the in vivo efficacy of the EGCG-PFCH-NPs in an orthotopic gastric tumor mouse model. Oral administration of EGCG-PFCH-NPs (equivalent to 40.0 mg/kg epigallocatechin gallate) showed significantly higher anti-tumor activity compared to 40 mg/kg epigallocatechin gallate solution (Figure 7) [164]. It would have been interesting to demonstrate the benefit of the fucose-mediated targeting of the tumor cells, in addition to increasing the duration of EGCG delivery in the proximity of the tumor, in vivo by also evaluating the anti-tumor activity of the non-fucosylated EGCG-PEGylated chitosan-gelatin NPs.
Figure 7.

In vivo efficacy of orally delivered fucosyl chitosan (FCS)-PEGylated Chitosan (PCS)-gelatin (Gel)-epigallocatechin gallate (EGCG)-nanoparticles (NPs) (equivalent to 40 mg/kg EGCG), EGCG 40 mg/kg solution and FCS/PCS/Gel solution (negative control) in mice bearing orthotopic Luc MKN45 xenografts (gastric carcinoma). FCS/PCS/Gel/EGCG-NPs showed significantly higher anti-tumor activity compared to EGCG solution, presumably due to enhanced gastroretention leading to localized delivery of EGCG in the vicinity of gastric cancer. Adapted from reference [164] with permission from American Chemical Society.
Deng and coworkers designed polyelectrolyte complex nanoparticles composed of chitosan and PEGylated-poly glutamic acid-doxorubicin conjugate [165]. The in vitro doxorubicin release studies showed that chitosan-PEGylated-poly glutamic acid-doxorubicin conjugate showed significantly lower release of doxorubicin (< 30% over 50 h) compared to the PEGylated-poly glutamic acid-doxorubicin conjugate (~ 80% DOX release over 50 h). Although the nanoparticles were developed for improving oral absorption of the doxorubicin for cancer therapy and not for stomach targeting, the biodistribution studies showed that orally administered chitosan-PEGylated polyglutamic acid-doxorubicin conjugate nanoparticles localized in the stomach for up to 12 h [165]. It would have been interesting to carefully study the contributions of PEG and chitosan in the gastric nanoparticle retention. In vivo efficacy studies in Ehrlich ascites tumor-bearing Balb/c mice showed equivalent anti-tumor activity, better survival and lower toxicity of orally administered doxorubicin nanoparticles as compared to the systemic doxorubicin [165]. Although increased gastric retention was not the intention for the nanoparticle formulation, it would have been interesting to determine whether there was an added advantage in increasing residence time in the stomach, i.e. whether doxorubicin absorption occurred primarily through the stomach or the intestine.
Suwannateep and colleagues fabricated curcumin loaded nanoparticles for improved oral bioavailability [166]. They used either ethyl cellulose or a mixture of ethyl cellulose and methyl cellulose to encapsulate curcumin in the nanoparticles. In vitro release studies showed less than 10% release of the curcumin in the pH 1.2 buffer even after 24 h, though studies were not carried out in the presence of gastric enzymes. Ethyl cellulose-curcumin nanoparticles and ethyl cellulose-methyl cellulose-curcumin nanoparticles showed 8.5- and 3.6-fold increase in the curcumin bioavailability, and greater maximum plasma concentration (Cmax), compared to curcumin in solution. Moreover, the Cmax of curcumin was observed within 100 min for the nanoparticles as well as curcumin solution [166], indicating that the stomach likely served as the primary site for systemic absorption of curcumin. In vivo biodistribution studies revealed that curcumin levels in the stomach of the mice treated with ethyl cellulose-curcumin nanoparticles were 4-fold higher than those treated with curcumin solution at 2 h post-administration [166]. To verify improved gastric retention of the ethyl cellulose nanoparticles, Suwannateep and colleagues excised the stomachs of mice treated with nanoparticles 2 h post-administration and imaged them using scanning electron microscopy (SEM). SEM studies showed the presence of intact ethyl cellulose nanoparticles adhered to the stomach [166]. In a subsequent study, the same group evaluated the potential of clarithromycin-loaded ethyl cellulose nanoparticles for the treatment of H. pylori infection [167]. As anticipated, clarithromycin-loaded ethyl cellulose nanoparticles showed greater efficacy in clearing H. pylori infection in mice compared to clarithromycin suspension. Similar results were also obtained for Garcinia mangostana extract loaded into the ethyl cellulose nanoparticles [168].
Recently, polyethylene oxide-acrylate (PEO-Acr) substituted ethyl cellulose has been synthesized to develop nanoparticles that may modulate the mucus microstructure [169]. The investigators hypothesized that the acrylate-terminated ethyl cellulose would react via thiol-ene click reaction with the thiols generated from the treatment of mucin with biocompatible reducing agents. The investigators demonstrated that the treatment of mucin with tris (2-carboxyethyl) phosphine hydrochloride (TCEP) or vitamin C could generate free thiols that subsequently react with the PEO-Acr terminated ethyl cellulose [169]. Furthermore, ex vivo studies on porcine stomach showed that PEO-Acr terminated fluorescent ethyl cellulose nanoparticles could bind to the mucosal surface in the presence of reducing agent TCEP, and the extent of binding was greater than that of non-modified fluorescent ethyl cellulose nanoparticles. Finally, the in vivo efficacy of clarithromycin-loaded, PEO-Acr modified, ethyl cellulose nanoparticles co-administered with vitamin C was evaluated in H. pylori infected mice. Interestingly, these nanoparticles in combination with vitamin C showed greater eradication of H. pylori than non-modified clarithromycin-ethyl cellulose nanoparticles and clarithromycin suspension [169]. It would be interesting to further establish the ability of vitamin C to generate free thiols in vivo, and use fluorescence imaging to assess whether the reaction with the acrylate terminated nanocarriers increases gastric retention in vivo.
As described, most of the studies using mucoadhesive nanoparticles to increase gastric retention have focused on the treatment of H. pylori infections. A recent study by Navabi and coworkers showed that acute and chronic H. pylori infection attenuated the rate of mucin production and turnover in the murine gastric mucosa [170]. The normal mucus turnover rate in mice was around 6 h, the turnover rate was greater than 12 h in H. pylori infected mice [170]. This may contribute to the benefits observed for mucoadhesive nanoparticles in the treatment of murine H. pylori infection. Accordingly, Navabi and coworkers suggested that nanoparticles adhering to gastric mucus may not be retained for more than 6 h or 12 h in healthy and H. pylori infected mice, respectively. In humans, H. pylori infection has been associated with an increase in mucosal permeability and disruption of the mucosal barrier [18,19]. Clinical trials have shown that antibiotic-loaded mucoadhesive hydrogels did not offer any advantages with respect to efficacy compared to conventional treatment regimen [32]. Hence, it is difficult to predict the success of mucoadhesive nanoparticles in the treatment of H. pylori infected individuals. More clinical studies are needed to evaluate the use of mucoadhesive formulations for H. pylori infection and other gastric ailments, such as cancer.
To date, we are unaware of studies that have employed muco-inert nanoparticle formulations for stomach targeting. It is not yet clear whether muco-inert nanomaterials would be useful for improved gastric retention or for the treatment of H. pylori infection and gastric cancer. Future studies to compare the gastro-retentive potential of mucoadhesive nanoparticles in comparison with muco-inert nanoparticles are needed.
4.2. Nanoparticles targeted to the small intestine
Targeting of nanoparticles to the small intestine has been extensively studied as a method to potentially enhance for improving systemic absorption of drugs, peptides, vaccines and other macromolecules. The use of nanoparticles based on enteric polymers, either alone or in combination with other polymers, to prevent drug degradation in the stomach and/or to achieve site-specific release in the small intestine has been discussed in recent reviews [24,33,34]. Thus, we focus our discussion on recently published investigations that describe nanoparticles functionalized with ligands to improve their uptake in enterocytes, M cells and goblet cells.
He and colleagues developed insulin-loaded SLN using a vitamin B12 stearate conjugate for targeting [171]. Co-administration of vitamin B12 with vitamin B12 functionalized insulin SLN (B12-insulin-SLN) significantly reduced the blood glucose lowering capability of B12-insulin-SLN in diabetic rats. Furthermore, intra-jejunal administration of the B12-insulin-SLN was carried out to bypass association of vitamin B12 on the SLN with the intrinsic factor in the stomach that is required for absorption of vitamin B12. As expected, intra-jejunal administration of B12-insulin-SLN significantly decreased the efficacy of insulin compared to orally delivered B12-insulin-SLN. In comparison to subcutaneously administered insulin, the oral B12-insulin-SLN administration showed approximately 10% bioavailability. However, B12-insulin-SLN provided a 60% reduction in blood glucose that was stably maintained from 6 to 12 h. Furthermore, B12-insulin-SLN showed almost 2-fold more bioavailability compared to non-modified insulin SLN [171].
Verma and colleagues synthesized insulin-loaded calcium phosphate nanoparticles (Insu-Cap NPs) that were coated with alternating layers of sodium alginate and chitosan-vitamin B12 conjugate (B12-LbL-Insu-Cap NPs) [172]. Insu-Cap NPs coated with alternate layers of chitosan and sodium alginate were also developed (LbL-Insu-Cap NPs) to assess the effect of vitamin B12 on targeted oral delivery. In vitro studies showed that B12-LbL- Insu-Cap NPs and LbL-Insu-Cap NPs offered protection from proteolytic enzymes, such as trypsin and chymotrypsin, and also maintained the bioactivity of insulin. In vitro Caco-2 cell culture studies showed that B12-LbL-Insu-Cap NPs had at least 2-fold increased uptake compared to LbL-Insu-Cap NPs. Furthermore, Caco-2 cell uptake of B12-LbL-Insu-Cap NPs was reduced in the absence of intrinsic factor in the cell culture media. In vivo studies in rats showed increased localization of fluorescently labeled B12-LbL-Insu-Cap NPs in the ileum compared to LbL-Insu-Cap NPs. In comparison to subcutaneous insulin (5 IU/kg), oral administration of B12-LbL-Insu-Cap NPs and LbL-Insu-Cap NPs (both equivalent to 50 IU/kg insulin) showed relative bioavailability of ~ 27% and ~ 6%, respectively [172]. B12-LbL-Insu-Cap NPs achieved similar glucose lowering effects to that of subcutaneous insulin, whereas LbL-Insu-Cap NPs did not cause a measurable hypoglycemic effect after oral administration (Figure 8) [172]. The investigation demonstrated a potential benefit for vitamin B12-mediated absorption of insulin.
Figure 8.

(A) Serum insulin and (B) blood glucose levels in diabetic rats after oral delivery of 50 IU/kg insulin solution (
 ), 50 IU/kg vitamin B12 functionalized chitosan-alginate-calcium phosphate insulin nanoparticles (
), 50 IU/kg vitamin B12 functionalized chitosan-alginate-calcium phosphate insulin nanoparticles (
 ), and 50 IU/kg chitosan-alginate-calcium phosphate insulin nanoparticles (
), and 50 IU/kg chitosan-alginate-calcium phosphate insulin nanoparticles (
 ), compared to 5 IU/kg insulin subcutaneous injection (●). Vitamin B12 functionalized chitosan-alginate-calcium phosphate insulin nanoparticles showed significantly higher serum insulin levels (p < 0.001) and significantly lower blood glucose levels (p < 0.001) as compared to chitosan-alginate-calcium phosphate insulin nanoparticles. Adapted from [172] with permission.
), compared to 5 IU/kg insulin subcutaneous injection (●). Vitamin B12 functionalized chitosan-alginate-calcium phosphate insulin nanoparticles showed significantly higher serum insulin levels (p < 0.001) and significantly lower blood glucose levels (p < 0.001) as compared to chitosan-alginate-calcium phosphate insulin nanoparticles. Adapted from [172] with permission.
Pridgen and coworkers described the use of FcRn receptor targeting to improve oral delivery of insulin [50]. The investigators fabricated PLA-PEG nanoparticles and decorated the surface of the nanoparticles with the Fc portion of IgG. Fc-functionalized nanoparticles (Fc-NPs) had significantly increased uptake by Caco-2 cells compared to unmodified nanoparticles, and the cell uptake of Fc-decorated nanoparticles was inhibited by the presence of the Fc portion of human IgG in the cell culture media. In vivo biodistribution studies of orally administered fluorescent Fc-NPs showed localization on the basolateral side of the duodenal epithelial cells. On the contrary, unmodified fluorescent NPs did not enter the villi [50]. Biodistribution studies using 14C labeled Fc-NPs also showed significantly higher levels of 14C in various other organs compared to unmodified NPs, suggesting increased absorption. However, the plasma concentration of 14C over time was not evaluated. In vivo efficacy studies showed that insulin-Fc-NPs (equivalent to 1.1 U/kg insulin) significantly reduced blood glucose levels for 10 h, whereas unmodified insulin nanoparticles (equivalent to 1.1 U/kg insulin) and i.v. injection of insulin (3.3 U/kg) were not as effective [50]. The authors did not compare the efficacy of orally delivered insulin-Fc-NPs with subcutaneously delivered insulin, which is commonly used as the positive control in animal and human studies. However, they did demonstrate that increased insulin delivery was not seen in FcRn knockout mice, suggesting that FcRn functionalization increased nanoparticle-mediated insulin delivery. Similar results were observed when insulin-Fc-NPs were co-administered with IgG in wild type mice [50]. To date, this is the only study demonstrating the use of FcRn functionalization of nanoparticles for improved oral absorption. It would be interesting to study whether FcRn functionalized nanoparticles are able to improve oral absorption of drugs with poor solubility and permeability like docetaxel.
Lectins have been widely used as a ligand to improve M cell mediated uptake of nanoparticles, particularly for vaccination. Ma and coworkers developed polymer-lipid hybrid nanoparticles containing PLGA, phospholipids, and monophosphoryl lipid A (MPLA) [173]. The hybrid nanoparticles were functionalized with UEA-1 lectin. In vitro studies using a human M cell culture model showed 3-fold higher uptake of the lectin-functionalized fluorescent hybrid nanoparticles compared to unmodified nanoparticles. Ex vivo studies using a ligated mouse ileal loop confirmed higher uptake of the lectin-functionalized nanoparticles in the Peyer’s patches compared to unmodified nanoparticles. Fluorescence imaging studies showed that orally delivered lectin-functionalized nanoparticles showed significantly higher localization in the intestine for up to 10 h, whereas unmodified nanoparticles showed negligible fluorescence in the intestine [173]. It should be noted that the MPLA coating of the nanoparticles could also help to target TLR receptors on M cells (see section 2.2). It would have been interesting to study in vitro and ex vivo uptake of hybrid nanoparticles containing only MPLA to assess the contribution of the MPLA coating on the particle uptake. In vivo murine immunization studies showed that orally delivered lectin-functionalized nanoparticles containing ovalbumin generated significantly higher mucosal immune response compared to unmodified hybrid nanoparticles.
Xia and coworkers developed itraconazole nanocrystals (ITZ-NCs) stabilized with poly(acrylic acid)-b-poly(methyl acrylate) (PAA-b-PMA) copolymer [174]. Wheat-germ agglutinin (WGA) was conjugated to the carboxylic acid groups of the polymeric stabilizer present on the surface of nanocrystals. Surface crosslinks were introduced to enhance association of the WGA to ITZ-NCs (WGA-crosslinked ITZ-NCs). Biodistribution studies showed significantly higher localization of the fluorescent WGA crosslinked NCs in the duodenum and ileum after 2 h compared to WGA functionalized NCs and unmodified NCs. Pharmacokinetic studies in rats showed that orally administered WGA-crosslinked ITZ-NCs resulted in 2-fold increase in the plasma concentration (Cmax) and AUC of ITZ compared to non-crosslinked WGA-ITZ-NCs, and around 3-fold increase in Cmax and AUC of ITZ compared to ITZ-NCs [174]. The study suggested that crosslinking of WGA functionalized polymer increased the association of WGA with ITZ-NCs, leading to an increase in the oral absorption as compared to the non-crosslinked WGA-ITZ-NCs. The increased proteolytic stability of WGA in rats (see section 2.2) likely also contributed to improved ITZ delivery.
The role of integrin receptor targeting in oral delivery and targeting of nanocarriers has been evaluated by Liu and coworkers [175,176]. The investigators used FQSIYPpIK (FQS) peptide as a ligand to target α5β3 integrin receptors. PLGA-PEG micelles loaded with insulin were fabricated, and the surface of the micelles was decorated with the FQS peptide conjugated to trimethyl chitosan. Functionalization of the nanoparticles with FQS peptide resulted in significantly higher uptake of the FQS-trimethyl chitosan functionalized NPs (FQS-NPs) in Caco-2 cells in vitro compared to trimethyl chitosan coated NPs (TMC-NPs), and unmodified NPs. In vivo biodistribution studies showed significantly higher localization of FITC-insulin in the intestinal villi in rats treated with FQS-NPs compared to TMC-NPs and unmodified NPs [175]. In vivo efficacy studies in diabetic rats showed that oral administration of FQS-NPs yielded significantly increased hypoglycemic effect compared to TMC-NPs and unmodified NPs. Pharmacokinetic studies on insulin delivered via various oral nanoformulations corroborated the results of in vivo efficacy studies [175]. It would also be interesting to evaluate the effect of the FQS peptide-targeting without the combined effect of chitosan, which is known to increase permeability and uptake [160].
Rajapaksa and colleagues established the potential of Claudin-4 targeting to improve uptake of nanoparticles in the Peyer’s patch M cells [63]. They used CPE30 (see section 2.2) peptide as a ligand to increase interactions between nanoparticles and M cells. They demonstrated increased localization of CPE30-functionalized fluorescent nanoparticles in the Peyer’s patch M cells both in vitro and in vivo [63]. To date, no further studies have been reported on ability of CPE30-functionalized nanoparticles to improve oral delivery and targeting of therapeutic agents. CPE30 is derived from Clostridium perfringens enterotoxin and no information is available on its toxicity, pathogenicity or immunogenicity, which may limit the use of CPE30.
Few studies have been carried out to target goblet cells to improve oral delivery of therapeutic agents and/or nanoparticles. As goblet cells secrete mucus, a protective barrier function, their role in absorption of therapeutic agents is relatively unexplored. Nevertheless, a random phage display identified CSKSSDYQC (CSK) peptide as a ligand with high affinity to goblet cells [64]. Jin and coworkers explored the potential of CSK peptide functionalized TMC NPs (CSK-TMC-NPs) for oral delivery of insulin [177]. An in vitro Caco-2/HT-29-MTX co-culture model was used to evaluate uptake of fluorescent CSK-TMC-NPs compared to unmodified TMC NPs. CSK-TMC-NPs showed significantly increased cell uptake compared to unmodified TMC NPs. However, the presence of mucus produced by HT29-MTX cells reduced the uptake of both peptide-functionalized and unmodified nanoparticles. Using ligated ileum loop model, fluorescent CSK-TMC-NPs were shown to localize more in goblet cells compared to unmodified TMC NPs. Furthermore, FITC-insulin encapsulated in CSK-TMC-NPs permeated deeply into the villi in the loop model. In vivo efficacy studies in diabetic rats demonstrated that insulin-loaded CSK-TMC-NPs had significantly higher hypoglycemic effect than achieved with unmodified TMC NPs [177]. Similar observations have been reported for Exendin-4-loaded CSK-TMC-NPs [178]. Although these studies demonstrated that goblet cell targeting ligands could improve oral absorption of insulin in rodents, more studies need to be carried out to critically evaluate the potential of goblet cell targeting. It is important to compare the potential of CSK peptide to other targeting ligands, such as lectins, for their ability to improve enterocyte targeting and oral delivery of model fluorophores. Zhang and coworkers showed that the ability of TMC to modulate tight junction proteins was one of the mechanisms responsible for the increased in vitro uptake of CSK-TMC-NPs and TMC-NPs in Caco-2/HT-29-MTX co-culture model [179]. Chitosan or chitosan derivatives have been used for many aforementioned investigations for fabricating nanoparticles. In order to understand the potential of various peptide ligands in improving oral absorption, it is essential to study in vivo biodistribution of nanoparticles composed of materials other than chitosan, after functionalization with CSK peptide or other peptide ligands. It should also be noted that functionalization of nanoparticles with peptides can alter their surface characteristics, including charge and surface hydrophobicity/hydrophilicity. Moreover, stability of the peptide in the GI tract could be positively or negatively affected after conjugation to the nanoparticles. It may be necessary to use nanoparticles functionalized with scrambled peptide sequences (but with similar physicochemical properties as peptide ligands) to ascertain the effect of peptide functionalization of nanoparticles on oral absorption. These comparative studies will give additional insight into the potential of peptide functionalized nanoparticles for targeting of the small intestine.
4.3. Nanoparticles targeted to the intestinal lymphatic system
Targeting the intestinal lymphatic system is useful for improving oral bioavailability of the drugs that undergo extensive hepatic first-pass metabolism and/or enzymatic degradation in the GI tract. Additionally, lymphatic targeting may be useful to deliver antiretroviral drugs to the GALT, which acts as a reservoir for HIV infection. For intestinal lymphatic targeting, nanoparticles composed of various fatty acid mono-, di- and triglycerides were found to be useful. Fatty acid glycerides, such as glyceryl behenate (Compritol ATO), glyceryl palmitosterate (Precirol ATO), medium chain triglycerides, and long chain triglycerides are used in the pharmaceutical industry for various oral delivery applications [180]. Nanoparticles composed of such lipid materials are categorized into solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLCs) depending upon their composition. A detailed description of the composition, advantages and applications of SLN and NLCs can be found in several published reviews [180–183]. Here, we focus on recent reports that describe the potential of SLN/NLC for targeting the intestinal lymphatic system.
Alex and coworkers developed lopinavir-loaded glyceryl behenate SLN to improve oral bioavailability of lopinavir [184]. Lopinavir is an integral part of the highly active antiretroviral therapy (HAART), but its oral bioavailability is limited due to low water solubility and high metabolism. Lopinavir SLN provided a 4-fold increased lopinavir plasma concentration (Cmax) as compared to the lopinavir suspension in rats, and the relative oral bioavailability of lopinavir delivered in SLN was 200-fold higher than that of lopinavir suspension. Furthermore, intestinal lymphatic transport of lopinavir was found to be 4.91-fold higher for SLN compared to lopinavir suspension [184]. It is interesting that they observed a 200-fold increase in oral bioavailability with only a 5-fold increase in lymphatic transport with SLN, though this was not discussed. Lopinavir is highly susceptible to intestinal metabolism, and thus, is usually co-administered with ritonavir in clinical settings to reduce its metabolism. The dramatic increase in the oral bioavailability of lopinavir after encapsulation in SLN could potentially be due to reduction in the metabolism of lopinavir in the small intestine. It would have been interesting to study pharmacokinetics of lopinavir SLN co-administered with an intestinal lymphatic transport inhibitor, such as cycloheximide. It would also have been interesting to compare the oral bioavailability of lopinavir SLN with lopinavir suspension co-administered with ritonavir.
Candesartan cilexetil is an anti-hypertensive drug with low oral bioavailability due to rapid hydrolysis in the GI tract. Zhang and coworkers formulated candesartan cilexetil-loaded glyceryl monostearate SLN and found that SLN resulted in 12-fold improvement in oral bioavailability of candesartan cilexetil in rats as compared to the drug suspension [185]. Pharmacokinetic studies revealed that the values of plasma concentration and area under the curve (AUC0-t) were reduced by 40% and 30% respectively when cycloheximide, a lymphatic transport inhibitor, was co-administered with candersartan cilexetil SLN [185]. It is likely that the reduced GI hydrolysis of candesartan cilexetil due to encapsulation in SLN also contributed to the improvement in oral bioavailability, though this was not addressed. Intestinal lymphatic transport mediated improvement in oral bioavailability after encapsulation in SLN or NLCs was also observed for raloxifene, tripterine, diethyl carbamazine, silymarin, docetaxel and lacidipine [186–191].
Nassar and coworkers focused on development of oral nanoparticles for delivery of docetaxel, a hydrophobic anticancer agent with poor solubility and permeability. In addition to low solubility and permeability, P-gp mediated efflux and CYP3A4 metabolism contribute to the poor oral bioavailability of docetaxel [7]. Nassar and coworkers developed PLGA nanocapsules to encapsulate docetaxel. The docetaxel-loaded PLGA nanocapsules contained oleoyl macrogol glycerides (Labrafil 1944 CS) in the core of the nanocapsules [192]. The docetaxel PLGA nanocapsules were further encapsulated into an enteric polymer (Eudragit L100-55) to generate enteric microparticles containing docetaxel PLGA nanocaspules (MPs-DTX-NCs). Oral delivery of MPs-DTX-NCs (equivalent to 5 mg/kg docetaxel) yielded more than 2.5-fold increase in the AUC compared to the intravenous injection of Taxotere, a commercial docetaxel (5 mg/kg) formulation. Fluorescent imaging studies showed that orally delivered fluorescent MPs-DTX- NCs accumulated in the mesenteric lymph nodes for up to 8 h [192]. The use of C18 unsaturated fatty acid glycerides (Labrafil 1944 CS) in the nanocapsule core may have contributed to intestinal lymphatic targeting (See section 2.3), though this effect was not explored. In another study [193] they found that docetaxel was not systemically absorbed upon oral administration of MPs-DTX-NCs (equivalent to 10 mg/kg of docetaxel) when the rats were pre-treated with cycloheximide 1.5 h before the oral administration of MPs-DTX-NCs (Figure 9), thus suggesting oral bioavailability is due to lymphatic transport.
Figure 9.

Effect of administration of cycloheximide (intestinal lymphatic transporter inhibitor) on the pharmacokinetics of docetaxel administered in enteric microparticles containing PLGA nanocapsules (MPs-DTX-NCs) orally administered to rats. Adapted from [193] with permission.
The potential of MPs-DTX-NCs to improve oral bioavailability of docetaxel was also established in minipigs (Table 1); oral administration of MPs-DTX-NCs led to almost 10-fold increased docetaxel plasma concentration and AUC compared to docetaxel solution.
Table 1.
Pharmacokinetics of orally delivered Taxotere® and enteric microparticles containing docetaxel PLGA nanocaspules (both equivalent to 1.25 mg/kg of docetaxel) in minipigs (n=3). Enteric microparticles containing docetaxel PLGA nanocaspules showed significantly higher Cmax and AUC as compared to Taxotere® (p < 0.05). Adapted from reference [193] with permission
| Oral Formulation | Cmax (ng/mL) | AUC (h.ng/mL) | Clearance (L/h/kg) |
|---|---|---|---|
| Docetaxel solution | 97.6 ± 221 | 797.7 ± 1141 | 4 ± 2.8 |
| Enteric microparticles containing docetaxel PLGA nanocaspules | 827.9 ± 558 | 7923.1 ± 5644 | 0.4 ± 0.5 |
Finally, the investigators compared the efficacy of oral MPs-DTX-NCs (equivalent to 10 mg/kg of docetaxel) and intraperitoneal injection of Taxotere (equivalent to 10 mg/kg of docetaxel) in reducing metastatic lung tumors in severe combined immune deficiency-beige (SCID-bg) mice. Orally delivered MPs-DTX-NCs showed significantly higher anti-metastatic activity compared to the i.p. injection of Taxotere. Furthermore, the survival rate for mice treated with MPs-DTX-NCs was 100% at 75 days, whereas there was 0% survival in the Taxotere treated mice at day 75. Mice receiving MPs-DTX-NCs showed the first sign of mortality on day 90, and all the animals died by day 129 [193]. MPs-DTX-NCs showed potential for oral cancer therapy and clearly demonstrated the potential for intestinal lymphatic targeting of drugs.
4.4. Nanoparticles targeted to the colon
The most common application for targeting orally administered nanoparticles to the colon is local treatment of inflammatory bowel disease (IBD). Again, “targeting” the colon refers to nanoparticle design strategies for maintaining particle integrity and protecting the drug cargo during transit through the stomach and small intestines to maximize delivery to the colon. As described in section 2.4, colon targeting of oral drug delivery systems largely depends on the use of pH sensitive polymers, either alone or in combination with other approaches for colon targeting, including polysaccharides and azo containing moieties.
Gugulothu and coworkers developed pH sensitive Eudragit S100 nanoparticles containing curcumin, celecoxib, or a combination of curcumin and celecoxib, for the treatment of IBD [194]. In a TNBS-induced rat colitis model, orally administered curcumin nanoparticles and celecoxib nanoparticles showed significantly lower myeloperoxidase (MPO) activity (a measure of inflammatory cell activity) in rat colon homogenates compared to curcumin suspension, celecoxib suspension, and a combination drug suspension. Furthermore, curcumin-celecoxib combination nanoparticles were more effective than curcumin nanoparticles, celecoxib nanoparticles or curcumin-celecoxib suspension. Naeem and colleagues designed nanoparticles based on a combination of Eudragit FS 30D (a pH sensitive polymer) and Eudragit RSPO (a water swellable polymer) [195]. The use of water-swellable Eudragit RSPO in combination with Eudragit FS 30D prolonged the release of budesonide at colonic pH, though the authors did not explain the mechanism. The investigators encapsulated budesonide into Eudragit FS 30D/RSPO combination nanoparticles for the treatment of IBD. They also fabricated fluorescent combination Eudragit FS 30D/RSPO nanoparticles and fluorescent Eudragit FS 30D nanoparticles and studied their distribution in mice with dextran sulfate sodium (DSS) induced colitis. Interestingly, orally administered Eudragit FS 30D/RSPO combination nanoparticles showed presence of fluorophore in the colon for at least 10 h, and the fluorescence intensity at all time points was significantly greater than that of the Eudragit FS 30D nanoparticles. Finally, efficacy studies using budesonide-loaded combination Eudragit FS 30D/RSPO nanoparticles also showed significantly increased amelioration of IBD symptoms (disease activity index, body weight, colon length, colon weight to length ratio and histology) compared to Eudragit FS 30D nanoparticles and untreated controls [195]. In a second study, Naeem and coworkers developed Eudragit S100 and azo-polyurethane combination nanoparticles to investigate the role of pH and enzyme sensitive materials in colon targeting [196]. The authors carried out in vivo GI tract distribution studies on orally delivered fluorescent Eudragit/azo-polyurethane nanoparticles and fluorescent Eudragit S100 nanoparticles in rats. At the end of 8 h, orally administered Eudragit/azo-polyurethane nanoparticles had delivered 17% and 42% of the total fluorophore quantity to the cecum and colon, respectively, whereas Eudragit S100 nanoparticles delivered less than 10% of the fluorophore to the cecum and colon [196]. This suggests that the introduction of polymers that degrade in the presence of colonic enzymes, like azoreductase, could be useful to increase nanoparticle-mediated drug delivery to the colon. \
Ali and coworkers developed budesonide-loaded PLGA nanoparticles coated with Eudragit S100 for oral IBD therapy [197]. In vitro release studies showed that the Eudragit S100 coating on PLGA nanoparticles dramatically reduced the release rate of budesonide in pH 1.2 buffer as compared to the uncoated PLGA nanoparticles, suggesting the coating would provide increased stability in the stomach environment. Ali and coworkers used murine TNBS-, oxazolone- and DSS-induced colitis models to evaluate in vivo efficacy of orally administered budesonide solution, budesonide-loaded PLGA nanoparticles, and Eudragit S100-coated PLGA nanoparticles containing budesonide. Efficacy was assessed by mini-endoscopy in live mice, MPO activity measurement using immunohistochemistry and histology scores. Eudragit S100-coated PLGA nanoparticles showed better efficacy compared to uncoated nanoparticles and budesonide solution in ameliorating symptoms of IBD in mice (Figure 10) [197]. Further, fluorescence imaging demonstrated that Eudragit S100-coated PLGA nanoparticles showed better localization in the colon compared to uncoated PLGA nanoparticles, though the difference was not quantified. A pharmacokinetic study investigating the colon tissue and plasma levels of budesonide would have been informative. In a similar study, Beloqui and colleagues demonstrated superior efficacy of Eudragit S100-coated PLGA nanoparticles containing curcumin compared to curcumin suspension in a murine DSS induced colitis model [198].
Figure 10.

Efficacy of orally administered budesonide solution, budesonide PLGA nanoparticles and Eudragit S100-coated budesonide PLGA nanoparticles in various murine colitis models. (A) In vivo high resolution mini-endoscopy of mice intestines, (B) colitis scores after treatment with different budesonide formulations and untreated controls, and (C) combined colitis scores obtained from various murine colitis models. Eudragit S100-coated budesonide PLGA nanoparticles showed significantly higher efficacy (p < 0.001) than budesonide PLGA nanoparticles. Adapted from [197] with permission.
As mentioned in section 2.4, IBD is characterized by a dramatic increase in mucosal reactive oxygen species (ROS) concentrations in the mucosa of diseased tissues. Hence, materials that degrade in the presence of ROS to release drug at the sites of inflammation and/or scavenge ROS to decrease inflammation could be useful as orally administered nanoparticles targeted to the colon for treatment of IBD. Vong and coworkers have carried out a series of investigations to explore the potential of ROS sensitive nanoparticles for the treatment of IBD [199–201]. They explored the use of nitroxide radical-based nanoparticles to reduce oxidative stress and inflammation in colonic tissue for the treatment of IBD. They synthesized a self-assembling redox polymer (PEG-b-PMOT) containing PEG 5000 shell and non-biodegradable oxymethylstyrene core attached to a nitroxide radical [199,200]. Fluorescently-labeled and radioisotope-labeled redox nanoparticles were synthesized, and their ability to target the colon was tested using a murine DSS induced colitis model. Interestingly, the redox nanoparticles showed significant accumulation in the inflamed colon and the presence of nitroxide radicals in colon also confirmed the targeting capability of redox nanoparticles [199,200]. Despite the small size (40 nm) of the orally delivered redox nanoparticles, there was no presence of nitroxide radicals in the blood even after 24 h, whereas mice dosed orally with free nitroxide had an average concentration of 20 ng/ml nitroxide radicals in the blood [199]. In vivo efficacy of orally administered redox nanoparticles (dosed at 50 mg/kg, 100 mg/kg and 300 mg/kg) as compared to oral mesalamine suspension (35 mg/kg) was evaluated in a murine DSS-induced colitis model. Redox nanoparticles administered orally at 300 mg/kg provided significantly increased amelioration of IBD symptoms (disease activity index, colon length, and IL-1β levels) compared to orally administered mesalamine suspension (Figure 11) [200].
Figure 11.

Efficacy of orally delivered 100 mg/kg or 300 mg/kg redox nanoparticles (RNPo) and 35 mg/kg mesalamine (5-ASA) in a murine DSS-induced colitis model. Efficacy evaluated by measuring (A) disease activity index, (B) colon length, and (C) secretion of inflammatory cytokine IL-1β. Orally delivered RNPo at the dose of 300 mg/kg showed better efficacy than mesalamine at 35 mg/kg (p < 0.05). Adapted from reference [200] with permission.
The investigators also showed that the orally administered redox nanoparticles did not have any harmful effects on the total number of aerobic and anaerobic bacteria in the colon, but the specific bacterial species were not reported [201]. It would be interesting to further explore the efficacy of redox nanoparticles loaded with standard IBD drugs, such as mesalamine or budesonide, for the synergistic treatment of IBD. More recently, the redox nanoparticles have been evaluated for the treatment of colitis associated colon cancer (CAC) [202]. To evaluate the efficacy, azoxymethane (AOM)/DSS-induced CAC was established in mice. The redox nanoparticles were orally delivered (200 mg/kg/day as oral gavage or 5 mg/ml in drinking water) for 50 days. Analysis of various parameters, such as disease activity index, endoscopy, tumor score and histology, showed that the redox nanoparticles had significant activity against CAC in mice [202]. However, the authors did not compare the efficacy of redox nanoparticles with standard chemotherapeutic agents like 5-fluorouracil or oxaliplatin. The safety profile of the redox nanoparticles, administered through drinking water (5 mg/ml), was evaluated for 1 month. No changes in the hematological parameters and GI histology were observed. Utility of the redox nanoparticles in combination with chemotherapeutic agents, such as doxorubicin and irinotecan, was also evaluated for the treatment of CAC, which showed significantly improved anticancer activity (Figure 12) [203]. In addition, the cardiotoxic side effects of doxorubicin are mediated through generation of ROS in cardiomyocytes, so the ability of the redox nanoparticles to scavenge ROS led to a significant reduction in cardiotoxicity [203].
Figure 12.

Efficacy of oral redox nanoparticles (RNPo) in combination with intravenous doxorubicin (DOX) in azoxymethane-dextran sulfate sodium (AOM/DSS) induced colitis associated cancer in mice evaluated by (A) colonic tumor score, (B) survival, and (C) superoxide production in the heart. DOX (5 mg/kg) was injected i.p. in mice once a week, while RNPo (2.5 mg/mL) were administered to mice in free drinking water starting on day 35, and the treatments were stopped on day 70. DOX + RNPo combination treatment showed enhanced anti-cancer efficacy and less cardiotoxicity as compared to DOX (*p < 0.05). Adapted from [203] with permission.
Coco and coworkers compared the colon targeting potential of nanoparticles functionalized with GSQSHPRH peptide (a peptide ligand that targets inflamed murine mucosa identified using phage display technique) or mannose (targets mannose receptors present on immune cells) [204]. Biodegradable nanoparticles functionalized with mannose or GSQSHPRH peptide, and containing radiolabeled ovalbumin, were prepared. The targeting potential of ligands was compared by measuring ex vivo uptake of ovalbumin in colon tissue obtained from either healthy mice or mice with DSS-included colitis. Mannose-functionalized PLGA nanoparticles showed at least 3-fold higher uptake of ovalbumin in the inflamed colon tissue compared to normal colonic tissue, whereas no selectivity was noted for peptide-functionalized nanoparticles. Furthermore, the uptake of peptide-functionalized nanoparticles was around 2-fold less in the inflamed colon tissue compared to mannose-decorated nanoparticles [204]. It is well known that IBD and inflammatory disorders are characterized by immune cell recruitment in diseased tissues, which was the justification for the apparent increased selectivity of mannose-functionalized nanoparticles. Functionalization of nanoparticles with sugar molecules would also likely change the surface properties, including charge and hydrophobicity. It would be interesting to functionalize nanoparticles with non-specific sugar molecules, such as glucose, to further demonstrate the specific targeting effect of mannose upon oral nanoparticle administration. Additionally, these observations would be further supported by in vivo studies in different murine colitis models.
Laroui and colleagues described development of poly-lactic acid-polyethylene glycol (PLA-PEG) nanoparticles containing tumor necrosis factor (TNF)-α siRNA [205]. Anti-TNF-α antibodies, such as infliximab, are commonly used for the treatment of IBD [206]. In view of this, strategies that can reduce TNF-α expression in colitis could be useful for the treatment of IBD. The investigators used polyethyleneimine (PEI) to complex the TNF-α siRNA, and the complexes were loaded inside PLA-PEG nanoparticles. The nanoparticle surfaces were functionalized with the Fab′ portion of the F4/80 antibody, a monoclonal antibody against murine macrophages. In vitro cell uptake studies using murine macrophages showed greater uptake and TNF-α siRNA silencing activity by Fab′-F4/80-functionalized nanoparticles compared to naked siRNA and non-targeted nanoparticles [205]. The targeted and non-targeted nanoparticles containing TNF-α siRNA or scrambled siRNA were dispersed in a chitosan-alginate hydrogel and delivered orally to mice with DSS-induced colitis. The efficacy of the nanoparticles was measured using MPO expression, accumulation of IKβα protein (an inhibitor of NFκb activity and indicator for TNF-α downregulation), and histology. As compared to the non-targeted TNF-α siRNA nanoparticles, the Fab′-F4/80-functionalized nanoparticles containing TNF-α siRNA provided a significant reduction in MPO levels, reduction in the accumulation of IKβα protein, and decreased neutrophil infiltration, crypt destruction and mucosal ulceration [205]. The scrambled siRNA-containing nanoparticles did not show any therapeutic activity. The authors also demonstrated that, as compared to the non-targeted nanoparticles, the targeted nanoparticles delivered fluorescent siRNA in vitro to a greater population of murine macrophages obtained from mice with colitis [205]. In addition to the use of Fab′ portion as a targeting ligand, the use of the chitosan-alginate hydrogel was an interesting aspect of the investigation. The dispersion of targeted nanoparticles in the chitosan-alginate hydrogel may have helped to maintain the integrity of the antibody-based targeting ligand in the GI tract. However, the authors did not explore this aspect. It would be interesting to know the difference in the distribution and efficacy of the targeted nanoparticles delivered with or without hydrogels. In conclusion, colon targeted nanoparticles have potential in the treatment of IBD and other colonic disorders.
5. Future perspective
Achieving efficient and effective delivery via the oral route is one of the “holy grails” of drug delivery. This perspective is due to the simplicity and acceptability of oral administration, as well as the challenge in achieving effective delivery to target cells or tissues with minimal off-target side effects. Nanoparticle formulations are employed for oral drug delivery for various reasons, including protection of drug cargo, targeting of particular delivery sites in the GI tract, and improving oral bioavailability. Here, we aimed to discuss important aspects of oral nanoparticle formulation, including design criteria and the state-of-the-art in experimental methods and nanoparticle platforms. One important design challenge that is often not considered is maintaining simplicity in formulation. Complexity can often introduce variability and, even more importantly from the perspective of helping patients, complexity can limit clinical translation. The GI tract performs numerous tasks and embodies a wide array of functionalities, and oftentimes, designing formulations that take advantage of, or work in concert with, natural biological processes leads to favorable outcomes. Furthermore, proper selection of experimental methods is critical for preclinical validation. As methods continue to evolve, more clear pathways to clinical studies will emerge. What is clear, however, is the tremendous potential for nanomedicine to positively impact the effectiveness of oral drug delivery.
Acknowledgments
This work was supported by National Institutes of Health grants U19AI133127, R21/R33AI094519, and R01DK107806; the Johns Hopkins University Center for AIDS Research P30AI094189; and the 2015 Burroughs Wellcome Fund Preterm Birth Initiative. We thank Dr. Richard Cone for helpful discussions. We also thank Mr. Jeffrey Chang for his help with reference formatting.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1. [accessed 12.1.2016]; http://www.americanpharmaceuticalreview.com/Featured-Articles/148747-Major-Advances-in-Oral-Drug-Delivery-over-the-Past-15-Years/
- 2. [accessed 12.1.2016]; http://www.micromarketmonitor.com/market-report/oral-drug-delivery-reports-6701101662.html.
- 3.Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64:557–570. doi: 10.1016/j.addr.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pridgen EM, Alexis F, Farokhzad OC. Polymeric nanoparticle drug delivery technologies for oral delivery applications. Expert Opin Drug Deliv. 2015;12:1459–1473. doi: 10.1517/17425247.2015.1018175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter AC, Elsom J, Wibroe PP, Moghimi SM. Polymeric particulate technologies for oral drug delivery and targeting: a pathophysiological perspective. Nanomedicine. 2012;8(Suppl 1):S5–S20. doi: 10.1016/j.nano.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Pawar VK, Meher JG, Singh Y, Chaurasia M, Reddy BS, Chourasia MK. Targeting of gastrointestinal tract for amended delivery of protein/peptide therapeutics: strategies and industrial perspectives. J Control Release. 2014;196:168–183. doi: 10.1016/j.jconrel.2014.09.031. [DOI] [PubMed] [Google Scholar]
- 7.Malingré MM, Beijnen JH, Schellens JH. Oral delivery of taxanes. Invest New Drugs. 2001;19:155–162. doi: 10.1023/a:1010635000879. [DOI] [PubMed] [Google Scholar]
- 8.Thornton SJ, Wasan KM. The reformulation of amphotericin B for oral administration to treat systemic fungal infections and visceral leishmaniasis. Expert Opin Drug Deliv. 2009;6:271–284. doi: 10.1517/17425240902802861. [DOI] [PubMed] [Google Scholar]
- 9.De Leo L, Di Toro N, Decorti G, Malusà N, Ventura A, Not T. Fasting increases tobramycin oral absorption in mice. Antimicrob Agents Chemother. 2010;54:1644–1646. doi: 10.1128/AAC.01172-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao L, Liu G, Ma J, Wang X, Zhou L, Li X, Wang F. Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs. Pharm Res. 2013;30:307–324. doi: 10.1007/s11095-012-0889-z. [DOI] [PubMed] [Google Scholar]
- 11.Smart AL, Gaisford S, Basit AW. Oral peptide and protein delivery: intestinal obstacles and commercial prospects. Expert Opin Drug Deliv. 2014;11:1323–1335. doi: 10.1517/17425247.2014.917077. [DOI] [PubMed] [Google Scholar]
- 12.Renukuntla J, Vadlapudi AD, Patel A, Boddu SH, Mitra AK. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447:75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choonara BF, Choonara YE, Kumar P, Bijukumar D, du Toit LC, Pillay V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol Adv. 2014;32:1269–1282. doi: 10.1016/j.biotechadv.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Roger E, Lagarce F, Garcion E, Benoit JP. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. 2010;5:287–306. doi: 10.2217/nnm.09.110. [DOI] [PubMed] [Google Scholar]
- 15.Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65:822–832. doi: 10.1016/j.addr.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116:1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Bardonnet PL, Faivre V, Pugh WJ, Piffaretti JC, Falson F. Gastroretentive dosage forms: overview and special case of Helicobacter pylori. J Control Release. 2006;111:1–18. doi: 10.1016/j.jconrel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 18.Adebisi AO, Conway BR. Modification of drug delivery to improve antibiotic targeting to the stomach. Ther Deliv. 2015;6:741–762. doi: 10.4155/tde.15.35. [DOI] [PubMed] [Google Scholar]
- 19.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61:75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Adebisi A, Conway BR. Gastroretentive microparticles for drug delivery applications. J Microencapsul. 2011;28:689–708. doi: 10.3109/02652048.2011.590613. [DOI] [PubMed] [Google Scholar]
- 21.Soybel DI. Anatomy and physiology of the stomach. Surg Clin North Am. 2005;85:875–894. doi: 10.1016/j.suc.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Gelberg HB. Comparative anatomy, physiology, and mechanisms of disease production of the esophagus, stomach, and small intestine. Toxicol Pathol. 2014;42:54–66. doi: 10.1177/0192623313518113. [DOI] [PubMed] [Google Scholar]
- 23.Kong F, Singh RP. Disintegration of solid foods in human stomach. J Food Sci. 2008;73:R67–R80. doi: 10.1111/j.1750-3841.2008.00766.x. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida T, Lai TC, Kwon GS, Sako K. pH- and ion-sensitive polymers for drug delivery. Expert Opin Drug Deliv. 2013;10:1497–1513. doi: 10.1517/17425247.2013.821978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erah PO, Goddard AF, Barrett DA, Shaw PN, Spiller RC. The stability of amoxycillin, clarithromycin and metronidazole in gastric juice: relevance to the treatment of Helicobacter pylori infection. J Antimicrob Chemother. 1997;39:5–12. doi: 10.1093/jac/39.1.5. [DOI] [PubMed] [Google Scholar]
- 26.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai SK, Wang YY, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61:86–100. doi: 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61:75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Garg T, Kumar A, Rath G, Goyal AK. Gastroretentive drug delivery systems for therapeutic management of peptic ulcer. Crit Rev Ther Drug Carrier Syst. 2014;31:531–557. doi: 10.1615/critrevtherdrugcarriersyst.2014011104. [DOI] [PubMed] [Google Scholar]
- 30.Prajapati VD, Jani GK, Khutliwala TA, Zala BS. Raft forming system-an upcoming approach of gastroretentive drug delivery system. J Control Release. 2013;168:151–165. doi: 10.1016/j.jconrel.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura M, Spiller RC, Barrett DA, Wibawa JI, Kumagai N, Tsuchimoto K, Tanaka T. Gastric juice, gastric tissue and blood antibiotic concentrations following omeprazole, amoxicillin and clarithromycin triple therapy. Helicobacter. 2003;8:294–299. doi: 10.1046/j.1523-5378.2003.00156.x. [DOI] [PubMed] [Google Scholar]
- 32.Gisbert JP, Torrado G, Torrado S, Olivares D, Pajares JM. Clinical trial evaluating amoxicillin and clarithromycin hydrogels (Chitosan-polyacrylic acid polyionic complex) for H. pylori eradication. J Clin Gastroenterol. 2006;40:618–622. doi: 10.1097/00004836-200608000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Wang XQ, Zhang Q. pH-sensitive polymeric nanoparticles to improve oral bioavailability of peptide/protein drugs and poorly water-soluble drugs. Eur J Pharm Biopharm. 2012;82:219–229. doi: 10.1016/j.ejpb.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Thakral S, Thakral NK, Majumdar DK. Eudragit: a technology evaluation. Expert Opin Drug Deliv. 2013;10:131–149. doi: 10.1517/17425247.2013.736962. [DOI] [PubMed] [Google Scholar]
- 35.Bakhru SH, Furtado S, Morello AP, Mathiowitz E. Oral delivery of proteins by biodegradable nanoparticles. Adv Drug Deliv Rev. 2013;65:811–821. doi: 10.1016/j.addr.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 36.Maisel K, Ensign L, Reddy M, Cone R, Hanes J. Effect of surface chemistry on nanoparticle interaction with gastrointestinal mucus and distribution in the gastrointestinal tract following oral and rectal administration in the mouse. J Control Release. 2015;197:48–57. doi: 10.1016/j.jconrel.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller C, Perera G, König V, Bernkop-Schnürch A. Development and in vivo evaluation of papain-functionalized nanoparticles. Eur J Pharm Biopharm. 2014;87:125–131. doi: 10.1016/j.ejpb.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Wilcox MD, Van Rooij LK, Chater PI, Pereira de Sousa I, Pearson JP. The effect of nanoparticle permeation on the bulk rheological properties of mucus from the small intestine. Eur J Pharm Biopharm. 2015;96:484–487. doi: 10.1016/j.ejpb.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 39.Pereira de Sousa I, Cattoz B, Wilcox MD, Griffiths PC, Dalgliesh R, Rogers S, Bernkop-Schnürch A. Nanoparticles decorated with proteolytic enzymes, a promising strategy to overcome the mucus barrier. Eur J Pharm Biopharm. 2015;97:257–264. doi: 10.1016/j.ejpb.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Köllner S, Dünnhaupt S, Waldner C, Hauptstein S, Pereira de Sousa I, Bernkop-Schnürch A. Mucus permeating thiomer nanoparticles. Eur J Pharm Biopharm. 2015;97:265–272. doi: 10.1016/j.ejpb.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Dawson M, Krauland E, Wirtz D, Hanes J. Transport of polymeric nanoparticle gene carriers in gastric mucus. Biotechnol Prog. 2004;20:851–857. doi: 10.1021/bp0342553. [DOI] [PubMed] [Google Scholar]
- 42.Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang YY, Cone R, Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A. 2007;104:1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ensign LM, Henning A, Schneider CS, Maisel K, Wang YY, Porosoff MD, Cone R, Hanes J. Ex vivo characterization of particle transport in mucus secretions coating freshly excised mucosal tissues. Mol Pharm. 2013;10:2176–2182. doi: 10.1021/mp400087y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yildiz HM, McKelvey CA, Marsac PJ, Carrier RL. Size selectivity of intestinal mucus to diffusing particulates is dependent on surface chemistry and exposure to lipids. J Drug Target. 2015;23:768–774. doi: 10.3109/1061186X.2015.1086359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdulkarim M, Agulló N, Cattoz B, Griffiths P, Bernkop-Schnürch A, Borros SG, Gumbleton M. Nanoparticle diffusion within intestinal mucus: Three-dimensional response analysis dissecting the impact of particle surface charge, size and heterogeneity across polyelectrolyte, pegylated and viral particles. Eur J Pharm Biopharm. 2015;97:230–238. doi: 10.1016/j.ejpb.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 46.Pereira de Sousa I, Moser T, Steiner C, Fichtl B, Bernkop-Schnürch A. Insulin loaded mucus permeating nanoparticles: Addressing the surface characteristics as feature to improve mucus permeation. Int J Pharm. 2016;500:236–244. doi: 10.1016/j.ijpharm.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 47.Clardy SM, Allis DG, Fairchild TJ, Doyle RP. Vitamin B12 in drug delivery: breaking through the barriers to a B12 bioconjugate pharmaceutical. Expert Opin Drug Deliv. 2011;8:127–140. doi: 10.1517/17425247.2011.539200. [DOI] [PubMed] [Google Scholar]
- 48.Russell-Jones G. Intestinal receptor targeting for peptide delivery: an expert’s personal perspective on reasons for failure and new opportunities. Ther Deliv. 2011;2:1575–1593. doi: 10.4155/tde.11.129. [DOI] [PubMed] [Google Scholar]
- 49.Fowler R, Vllasaliu D, Trillo FF, Garnett M, Alexander C, Horsley H, Smith B, Whitcombe I, Eaton M, Stolnik S. Nanoparticle transport in epithelial cells: pathway switching through bioconjugation. Small. 2013;9:3282–3294. doi: 10.1002/smll.201202623. [DOI] [PubMed] [Google Scholar]
- 50.Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS, Langer R, Farokhzad OC. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci Transl Med. 2013;5:213ra167. doi: 10.1126/scitranslmed.3007049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devriendt B, De Geest BG, Goddeeris BM, Cox E. Crossing the barrier: Targeting epithelial receptors for enhanced oral vaccine delivery. J Control Release. 2012;160:431–439. doi: 10.1016/j.jconrel.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 52.Wang M, Gao Z, Zhang Z, Pan L, Zhang Y. Roles of M cells in infection and mucosal vaccines. Hum Vaccin Immunother. 2014;10:3544–3551. doi: 10.4161/hv.36174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116:1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 54.Jepson MA, Clark MA, Hirst BH. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv Drug Deliv Rev. 2004;56:511–525. doi: 10.1016/j.addr.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 55.Gabor F, Bogner E, Weissenboeck A, Wirth M. The lectin-cell interaction and its implications to intestinal lectin-mediated drug delivery. Adv Drug Deliv Rev. 2004;56:459–480. doi: 10.1016/j.addr.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 56.Pusztai A, Ewen SW, Grant G, Peumans WJ, van Damme EJ, Rubio L, Bardocz S. Relationship between survival and binding of plant lectins during small intestinal passage and their effectiveness as growth factors. Digestion. 1990;46:308–316. doi: 10.1159/000200402. [DOI] [PubMed] [Google Scholar]
- 57.Lavelle EC. Targeted delivery of drugs to the gastrointestinal tract. Crit Rev Ther Drug Carrier Syst. 2001;18:341–386. [PubMed] [Google Scholar]
- 58.Lambkin I, Pinilla C, Hamashin C, Spindler L, Russell S, Schink A, Moya-Castro R, Allicotti G, Higgins L, Smith M, Dee J, Wilson C, Houghten R, O’Mahony D. Toward targeted oral vaccine delivery systems: selection of lectin mimetics from combinatorial libraries. Pharm Res. 2003;20:1258–1266. doi: 10.1023/a:1025061317400. [DOI] [PubMed] [Google Scholar]
- 59.Crouzier T, Beckwitt CH, Ribbeck K. Mucin multilayers assembled through sugar-lectin interactions. Biomacromolecules. 2012;13:3401–3408. doi: 10.1021/bm301222f. [DOI] [PubMed] [Google Scholar]
- 60.Adebisi AO, Conway BR. Lectin-conjugated microspheres for eradication of Helicobacter pylori infection and interaction with mucus. Int J Pharm. 2014;470:28–40. doi: 10.1016/j.ijpharm.2014.04.070. [DOI] [PubMed] [Google Scholar]
- 61.Fievez V, Plapied L, des Rieux A, Pourcelle V, Freichels H, Wascotte V, Vanderhaeghen ML, Jerôme C, Vanderplasschen A, Marchand-Brynaert J, Schneider YJ, Préat V. Targeting nanoparticles to M cells with non-peptidic ligands for oral vaccination. Eur J Pharm Biopharm. 2009;73:16–24. doi: 10.1016/j.ejpb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 62.Tamagawa H, Takahashi I, Furuse M, Yoshitake-Kitano Y, Tsukita S, Ito T, Matsuda H, Kiyono H. Characteristics of claudin expression in follicle-associated epithelium of Peyer’s patches: preferential localization of claudin-4 at the apex of the dome region. Lab Invest. 2003;83:1045–1053. doi: 10.1097/01.lab.0000078741.55670.6e. [DOI] [PubMed] [Google Scholar]
- 63.Rajapaksa TE, Stover-Hamer M, Fernandez X, Eckelhoefer HA, Lo DD. Claudin 4-targeted protein incorporated into PLGA nanoparticles can mediate M cell targeted delivery. J Control Release. 2010;142:196–205. doi: 10.1016/j.jconrel.2009.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang SK, Woo JH, Kim MK, Woo SS, Choi JH, Lee HG, Lee NK, Choi YJ. Identification of a peptide sequence that improves transport of macromolecules across the intestinal mucosal barrier targeting goblet cells. J Biotechnol. 2008;135:210–216. doi: 10.1016/j.jbiotec.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 65.Porter CJ, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 2001;50:61–80. doi: 10.1016/s0169-409x(01)00151-x. [DOI] [PubMed] [Google Scholar]
- 66.Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008;60:702–716. doi: 10.1016/j.addr.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cai S, Yang Q, Bagby TR, Forrest ML. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv Drug Deliv Rev. 2011;63:901–908. doi: 10.1016/j.addr.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chaudhary S, Garg T, Murthy RS, Rath G, Goyal AK. Recent approaches of lipid-based delivery system for lymphatic targeting via oral route. J Drug Target. 2014;22:871–882. doi: 10.3109/1061186X.2014.950664. [DOI] [PubMed] [Google Scholar]
- 69.Singh I, Swami R, Khan W, Sistla R. Lymphatic system: a prospective area for advanced targeting of particulate drug carriers. Expert Opin Drug Deliv. 2014;11:211–229. doi: 10.1517/17425247.2014.866088. [DOI] [PubMed] [Google Scholar]
- 70.Khan AA, Mudassir J, Mohtar N, Darwis Y. Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. Int J Nanomedicine. 2013;8:2733–2744. doi: 10.2147/IJN.S41521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trevaskis NL, Kaminskas LM, Porter CJ. From sewer to saviour-targeting the lymphatic system to promote drug exposure and activity. Nat Rev Drug Discov. 2015;14:781–803. doi: 10.1038/nrd4608. [DOI] [PubMed] [Google Scholar]
- 72.van Marle G, Gill MJ, Kolodka D, McManus L, Grant T, Church DL. Compartmentalization of the gut viral reservoir in HIV-1 infected patients. Retrovirology. 2007;4:87. doi: 10.1186/1742-4690-4-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han S, Quach T, Hu L, Wahab A, Charman WN, Stella VJ, Trevaskis NL, Simpson JS, Porter CJ. Targeted delivery of a model immunomodulator to the lymphatic system: comparison of alkyl ester versus triglyceride mimetic lipid prodrug strategies. J Control Release. 2014;177:1–10. doi: 10.1016/j.jconrel.2013.12.031. [DOI] [PubMed] [Google Scholar]
- 74.Collnot EM, Ali H, Lehr CM. Nano- and microparticulate drug carriers for targeting of the inflamed intestinal mucosa. J Control Release. 2012;161:235–246. doi: 10.1016/j.jconrel.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 75.Wolk O, Epstein S, Ioffe-Dahan V, Ben-Shabat S, Dahan A. New targeting strategies in drug therapy of inflammatory bowel disease: mechanistic approaches and opportunities. Expert Opin Drug Deliv. 2013;10:1275–1286. doi: 10.1517/17425247.2013.800480. [DOI] [PubMed] [Google Scholar]
- 76.Esseku F, Adeyeye MC. Bacteria and pH-sensitive polysaccharide-polymer films for colon targeted delivery. Crit Rev Ther Drug Carrier Syst. 2011;28:395–445. doi: 10.1615/critrevtherdrugcarriersyst.v28.i5.10. [DOI] [PubMed] [Google Scholar]
- 77.Patel M, Shah T, Amin A. Therapeutic opportunities in colon-specific drug-delivery systems. Crit Rev Ther Drug Carrier Syst. 2007;24:147–202. doi: 10.1615/critrevtherdrugcarriersyst.v24.i2.20. [DOI] [PubMed] [Google Scholar]
- 78.Sinha VR, Kumria R. Microbially triggered drug delivery to the colon. Eur J Pharm Sci. 2003;18:3–18. doi: 10.1016/s0928-0987(02)00221-x. [DOI] [PubMed] [Google Scholar]
- 79.Hua S, Marks E, Schneider JJ, Keely S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: selective targeting to diseased versus healthy tissue. Nanomedicine. 2015;11:1117–1132. doi: 10.1016/j.nano.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 80.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011;108:4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johansson ME, Gustafsson JK, Holmén-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjövall H, Hansson GC. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63:281–291. doi: 10.1136/gutjnl-2012-303207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jakobsson HE, Rodríguez-Piñeiro AM, Schütte A, Ermund A, Boysen P, Bemark M, Sommer F, Bäckhed F, Hansson GC, Johansson ME. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015;16:164–177. doi: 10.15252/embr.201439263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jubeh TT, Barenholz Y, Rubinstein A. Differential adhesion of normal and inflamed rat colonic mucosa by charged liposomes. Pharm Res. 2004;21:447–453. doi: 10.1023/B:PHAM.0000019298.29561.cd. [DOI] [PubMed] [Google Scholar]
- 84.Lamprecht A, Schäfer U, Lehr CM. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18:788–793. doi: 10.1023/a:1011032328064. [DOI] [PubMed] [Google Scholar]
- 85.Youshia J, Lamprecht A. Size-dependent nanoparticulate drug delivery in inflammatory bowel diseases. Expert Opin Drug Deliv. 2016;13:281–294. doi: 10.1517/17425247.2016.1114604. [DOI] [PubMed] [Google Scholar]
- 86.Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Weissman GS, Katz S, Floyd RA, McKinley MJ, Fisher SE, Mullin GE. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41:2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 87.Sedghi S, Fields JZ, Klamut M, Urban G, Durkin M, Winship D, Fretland D, Olyaee M, Keshavarzian A. Increased production of luminol enhanced chemiluminescence by the inflamed colonic mucosa in patients with ulcerative colitis. Gut. 1993;34:1191–1197. doi: 10.1136/gut.34.9.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang L, Sang Y, Feng J, Li Z, Zhao A. Polysaccharide-based micro/nanocarriers for oral colon-targeted drug delivery. J Drug Target. 2016:1–11. doi: 10.3109/1061186X.2015.1128941. [DOI] [PubMed] [Google Scholar]
- 89.Van den Mooter G, Maris B, Samyn C, Augustijns P, Kinget R. Use of azo polymers for colon-specific drug delivery. J Pharm Sci. 1997;86:1321–1327. doi: 10.1021/js9702630. [DOI] [PubMed] [Google Scholar]
- 90.Schippa S, Conte MP. Dysbiotic events in gut microbiota: impact on human health. Nutrients. 2014;6:5786–5805. doi: 10.3390/nu6125786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nguyen TL, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech. 2015;8:1–16. doi: 10.1242/dmm.017400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Canevari M, Castagliuolo I, Brun P, Cardin M, Schiavon M, Pasut G, Veronese FM. Poly(ethylene glycol)-mesalazine conjugate for colon specific delivery. Int J Pharm. 2009;368:171–177. doi: 10.1016/j.ijpharm.2008.09.058. [DOI] [PubMed] [Google Scholar]
- 93.Ruiz JF, Kedziora K, Windle H, Kelleher DP, Gilmer JF. Investigation into drug release from colon-specific azoreductase-activated steroid prodrugs using in-vitro models. J Pharm Pharmacol. 2011;63:806–816. doi: 10.1111/j.2042-7158.2011.01289.x. [DOI] [PubMed] [Google Scholar]
- 94.de Moreno de LeBlanc A, Perdigón G. Reduction of beta-glucuronidase and nitroreductase activity by yoghurt in a murine colon cancer model. Biocell. 2005;29:15–24. [PubMed] [Google Scholar]
- 95.Cardon ME, Norin E, Midtvedt T. β-Glucuronidase activity in germ-free, monoassociated and conventional mice. Microb Ecol Health Dis. 2006;18:38–41. [Google Scholar]
- 96.Minko T. Drug targeting to the colon with lectins and neoglycoconjugates. Adv Drug Deliv Rev. 2004;56:491–509. doi: 10.1016/j.addr.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 97.Moulari B, Béduneau A, Pellequer Y, Lamprecht A. Lectin-decorated nanoparticles enhance binding to the inflamed tissue in experimental colitis. J Control Release. 2014;188:9–17. doi: 10.1016/j.jconrel.2014.05.046. [DOI] [PubMed] [Google Scholar]
- 98.Gamboa JM, Leong KW. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv Drug Deliv Rev. 2013;65:800–810. doi: 10.1016/j.addr.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shahbazi MA, Santos HA. Improving oral absorption via drug-loaded nanocarriers: absorption mechanisms, intestinal models and rational fabrication. Curr Drug Metab. 2013;14:28–56. [PubMed] [Google Scholar]
- 100.Lefebvre DE, Venema K, Gombau L, Valerio LG, Jr, Raju J, Bondy GS, Bouwmeester H, Singh RP, Clippinger AJ, Collnot EM, Mehta R, Stone V. Utility of models of the gastrointestinal tract for assessment of the digestion and absorption of engineered nanomaterials released from food matrices. Nanotoxicology. 2015;9:523–542. doi: 10.3109/17435390.2014.948091. [DOI] [PubMed] [Google Scholar]
- 101.Kura AU, Fakurazi S, Hussein MZ, Arulselvan P. Nanotechnology in drug delivery: the need for more cell culture based studies in screening. Chem Cent J. 2014;8:46. doi: 10.1186/1752-153X-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- 103.Ungell AL, Karlsson J. Cell cultures in drug discovery: an industrial perspective. In: Waterbeemd H, Lennernäs H, Artursson P, editors. Drug Bioavailability: Estimation of Solubility, Permeability, Absorption and Bioavailability. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2004. pp. 90–131. [Google Scholar]
- 104.Volpe DA. Application of method suitability for drug permeability classification. AAPS J. 2010;12:670–678. doi: 10.1208/s12248-010-9227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Powell JJ, Faria N, Thomas-McKay E, Pele LC. Origin and fate of dietary nanoparticles and microparticles in the gastrointestinal tract. J Autoimmun. 2010;34:226–233. doi: 10.1016/j.jaut.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 106.Kauffman AL, Gyurdieva AV, Mabus JR, Ferguson C, Yan Z, Hornby PJ. Alternative functional in vitro models of human intestinal epithelia. Front Pharmacol. 2013;4:79. doi: 10.3389/fphar.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wikman-Larhed A, Artursson P. Co-cultures of human intestinal goblet (HT29-H) and absorptive (Caco-2) cells for studies of drug and peptide absorption. Eur J Pharm Sci. 1995;3:171–183. [Google Scholar]
- 108.Walter E, Janich S, Roessler BJ, Hilfinger JM, Amidon GL. HT29-MTX/Caco-2 cocultures as an in vitro model for the intestinal epithelium: in vitro-in vivo correlation with permeability data from rats and humans. J Pharm Sci. 1996;85:1070–1076. doi: 10.1021/js960110x. [DOI] [PubMed] [Google Scholar]
- 109.Mahler GJ, Shuler ML, Glahn RP. Characterization of Caco-2 and HT29-MTX cocultures in an in vitro digestion/cell culture model used to predict iron bioavailability. J Nutr Biochem. 2009;20:494–502. doi: 10.1016/j.jnutbio.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 110.des Rieux A, Fievez V, Théate I, Mast J, Préat V, Schneider YJ. An improved in vitro model of human intestinal follicle-associated epithelium to study nanoparticle transport by M cells. Eur J Pharm Sci. 2007;30:380–391. doi: 10.1016/j.ejps.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 111.Schimpel C, Teubl B, Absenger M, Meindl C, Fröhlich E, Leitinger G, Zimmer A, Roblegg E. Development of an advanced intestinal in vitro triple culture permeability model to study transport of nanoparticles. Mol Pharm. 2014;11:808–818. doi: 10.1021/mp400507g. [DOI] [PubMed] [Google Scholar]
- 112.Wijtten PJ, van der Meulen J, Verstegen MW. Intestinal barrier function and absorption in pigs after weaning: a review. Br J Nutr. 2011;105:967–981. doi: 10.1017/S0007114510005660. [DOI] [PubMed] [Google Scholar]
- 113.Lindberg JE. Fiber effects in nutrition and gut health in pigs. J Anim Sci Biotechnol. 2014;5:15. doi: 10.1186/2049-1891-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leonard F, Collnot EM, Lehr CM. A three-dimensional coculture of enterocytes, monocytes and dendritic cells to model inflamed intestinal mucosa in vitro. Mol Pharm. 2010;7:2103–2119. doi: 10.1021/mp1000795. [DOI] [PubMed] [Google Scholar]
- 115.Leonard F, Ali H, Collnot EM, Crielaard BJ, Lammers T, Storm G, Lehr CM. Screening of budesonide nanoformulations for treatment of inflammatory bowel disease in an inflamed, 3D cell-culture model. ALTEX. 2012;29:275–285. doi: 10.14573/altex.2012.3.275. [DOI] [PubMed] [Google Scholar]
- 116.Huang Z, Wang Z, Long S, Jiang H, Chen J, Zhang J, Dong L. A 3-D artificial colon tissue mimic for the evaluation of nanoparticle-based drug delivery system. Mol Pharm. 2014;11:2051–2061. doi: 10.1021/mp400723j. [DOI] [PubMed] [Google Scholar]
- 117.Chen Y, Lin Y, Davis KM, Wang Q, Rnjak-Kovacina J, Li C, Isberg RR, Kumamoto CA, Mecsas J, Kaplan DL. Robust bioengineered 3D functional human intestinal epithelium. Sci Rep. 2015;5:13708. doi: 10.1038/srep13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bermudez-Brito M, Plaza-Díaz J, Fontana L, Muñoz-Quezada S, Gil A. In vitro cell and tissue models for studying host-microbe interactions: a review. Br J Nutr. 2013;109:S27–34. doi: 10.1017/S0007114512004023. [DOI] [PubMed] [Google Scholar]
- 119.Fang SB, Schüller S, Phillips AD. Human Intestinal In Vitro Organ Culture as a Model for Investigation of Bacteria-Host Interactions. J Exp Clin Med. 2013;5:43e50. [Google Scholar]
- 120.Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Ettayebi K, Blutt SE, Hyser JM, Zeng XL, Crawford SE, Broughman JR, Estes MK, Donowitz M. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 2014;239:1124–1134. doi: 10.1177/1535370214529398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hartman KG, Bortner JD, Jr, Falk GW, Ginsberg GG, Jhala N, Yu J, Martín MG, Rustgi AK, Lynch JP. Modeling human gastrointestinal inflammatory diseases using microphysiological culture systems. Exp Biol Med (Maywood) 2014;239:1108–1123. doi: 10.1177/1535370214529388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vu LT, Less RR, Rajagopalan P. The promise of organotypic hepatic and gastrointestinal models. Trends Biotechnol. 2014;32:406–413. doi: 10.1016/j.tibtech.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 123.Grabinger T, Luks L, Kostadinova F, Zimberlin C, Medema JP, Leist M, Brunner T. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis. 2014;5:e1228. doi: 10.1038/cddis.2014.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McCracken KW, Catá EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, Wells JM. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516:400–404. doi: 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pastuła A, Middelhoff M, Brandtner A, Tobiasch M, Höhl B, Nuber AH, Demir IE, Neupert S, Kollmann P, Mazzuoli-Weber G, Quante M. Three-Dimensional Gastrointestinal Organoid Culture in Combination with Nerves or Fibroblasts: A Method to Characterize the Gastrointestinal Stem Cell Niche. Stem Cells Int. 2016;2016:3710836. doi: 10.1155/2016/3710836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chia SL, Tay CY, Setyawati MI, Leong DT. Biomimicry 3D gastrointestinal spheroid platform for the assessment of toxicity and inflammatory effects of zinc oxide nanoparticles. Small. 2015;11:702–712. doi: 10.1002/smll.201401915. [DOI] [PubMed] [Google Scholar]
- 127.Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21:745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Esch MB, Sung JH, Yang J, Yu C, Yu J, March JC, Shuler ML. On chip porous polymer membranes for integration of gastrointestinal tract epithelium with microfluidic ‘body-on-a-chip’ devices. Biomed Microdevices. 2012;14:895–906. doi: 10.1007/s10544-012-9669-0. [DOI] [PubMed] [Google Scholar]
- 129.Esch MB, Mahler GJ, Stokol T, Shuler ML. Body-on-a-chip simulation with gastrointestinal tract and liver tissues suggests that ingested nanoparticles have the potential to cause liver injury. Lab Chip. 2014;14:3081–3092. doi: 10.1039/c4lc00371c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim HJ, Huh D, Hamilton G, Ingber DE. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip. 2012;12:2165–2174. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- 131.Kim HJ, Ingber DE. Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr Biol (Camb) 2013;5:1130–1140. doi: 10.1039/c3ib40126j. [DOI] [PubMed] [Google Scholar]
- 132.Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci U S A. 2016;113:E7–E15. doi: 10.1073/pnas.1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen J, Burgess DJ. In vitro-in vivo correlation for complex non-oral drug products: Where do we stand? J Control Release. 2015;219:644–651. doi: 10.1016/j.jconrel.2015.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S, Dickinson PA, Dressman J, Holm R, Klein S, Mann J, McAllister M, Minekus M, Muenster U, Müllertz A, Verwei M, Vertzoni M, Weitschies W, Augustijns P. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:342–366. doi: 10.1016/j.ejps.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 135.Barzegar-Jalali M, Mohammadi K, Mohammadi G, Valizadeh H, Barzegar-Jalali A, Adibkia K, Nokhodchi A. A correlative model to predict in vivo AUC for nanosystem drug delivery with release rate-limited absorption. J Pharm Sci. 2012;15:583–591. doi: 10.18433/j3np5n. [DOI] [PubMed] [Google Scholar]
- 136.Musther H, Olivares-Morales A, Hatley OJ, Liu B, Rostami Hodjegan A. Animal versus human oral drug bioavailability: do they correlate? Eur J Pharm Sci. 2014;57:280–291. doi: 10.1016/j.ejps.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Akabane T, Tabata K, Kadono K, Sakuda S, Terashita S, Teramura T. A comparison of pharmacokinetics between humans and monkeys. Drug Metab Dispos. 2010;38:308–316. doi: 10.1124/dmd.109.028829. [DOI] [PubMed] [Google Scholar]
- 138.Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos. 1995;16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 139.Zou T, Gu L. TPGS emulsified zein nanoparticles enhanced oral bioavailability of daidzin: in vitro characteristics and in vivo performance. Mol Pharm. 2013;10:2062–2070. doi: 10.1021/mp400086n. [DOI] [PubMed] [Google Scholar]
- 140.Yuan H, Chen CY, Chai GH, Du YZ, Hu FQ. Improved transport and absorption through gastrointestinal tract by PEGylated solid lipid nanoparticles. Mol Pharm. 2013;10:1865–1873. doi: 10.1021/mp300649z. [DOI] [PubMed] [Google Scholar]
- 141.Tariq M, Alam MA, Singh AT, Iqbal Z, Panda AK, Talegaonkar S. Biodegradable polymeric nanoparticles for oral delivery of epirubicin: In vitro, ex vivo, and in vivo investigations. Colloids Surf, B. 2015;128:448–456. doi: 10.1016/j.colsurfb.2015.02.043. [DOI] [PubMed] [Google Scholar]
- 142.Zhu X, Shan W, Zhang P, Jin Y, Guan S, Fan T, Yang Y, Zhou Z, Huang Y. Penetratin derivative-based nanocomplexes for enhanced intestinal insulin delivery. Mol Pharm. 2014;11:317–328. doi: 10.1021/mp400493b. [DOI] [PubMed] [Google Scholar]
- 143.Braakhuis HM, Kloet SK, Kezic S, Kuper F, Park MV, Bellmann S, van der Zande M, Le Gac S, Krystek P, Peters RJ, Rietjens IM, Bouwmeester H. Progress and future of in vitro models to study translocation of nanoparticles. Arch Toxicol. 2015;89:1469–1495. doi: 10.1007/s00204-015-1518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Reineke JJ, Cho DY, Dingle YT, Morello AP, Jacob J, Thanos CG, Mathiowitz E. Unique insights into the intestinal absorption, transit, and subsequent biodistribution of polymer-derived microspheres. Proc Natl Acad Sci U S A. 2013;110:13803–13808. doi: 10.1073/pnas.1305882110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gardner ML. Gastrointestinal absorption of intact proteins. Annu Rev Nutr. 1988;8:329–350. doi: 10.1146/annurev.nu.08.070188.001553. [DOI] [PubMed] [Google Scholar]
- 146.Bellmann S, Carlander D, Fasano A, Momcilovic D, Scimeca JA, Waldman WJ, Gombau L, Tsytsikova L, Canady R, Pereira DI, Lefebvre DE. Mammalian gastrointestinal tract parameters modulating the integrity, surface properties, and absorption of food-relevant nanomaterials. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7:609–622. doi: 10.1002/wnan.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cho WS, Kang BC, Lee JK, Jeong J, Che JH, Seok SH. Comparative absorption, distribution, and excretion of titanium dioxide and zinc oxide nanoparticles after repeated oral administration. Part Fibre Toxicol. 2013;10:9. doi: 10.1186/1743-8977-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hinkley GK, Carpinone P, Munson JW, Powers KW, Roberts SM. Oral absorption of PEG-coated versus uncoated gold nanospheres: does agglomeration matter? Part Fibre Toxicol. 2015;12:9. doi: 10.1186/s12989-015-0085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sinnecker H, Krause T, Koelling S, Lautenschläger I, Frey A. The gut wall provides an effective barrier against nanoparticle uptake. Beilstein J Nanotechnol. 2014;5:2092–2101. doi: 10.3762/bjnano.5.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hu X, Fan W, Yu Z, Lu Y, Qi J, Zhang J, Dong X, Zhao W, Wu W. Evidence does not support absorption of intact solid lipid nanoparticles via oral delivery. Nanoscale. 2016 doi: 10.1039/c5nr07474f. [DOI] [PubMed] [Google Scholar]
- 151.Holmgren J, Czerkinsky C, Eriksson K, Mharandi A. Mucosal immunisation and adjuvants: a brief overview of recent advances and challenges. Vaccine. 2003;21:S89–95. doi: 10.1016/s0264-410x(03)00206-8. [DOI] [PubMed] [Google Scholar]
- 152.Jepson MA, Clark MA, Foster N, Mason CM, Bennett MK, Simmons NL, Hirst BH. Targeting to intestinal M cells. J Anat. 1996;189:507–516. [PMC free article] [PubMed] [Google Scholar]
- 153.Pappo J, Ermak TH. Uptake and translocation of fluorescent latex particles by rabbit Peyer’s patch follicle epithelium: a quantitative model for M cell uptake. Clin Exp Immunol. 1989;76:144–148. [PMC free article] [PubMed] [Google Scholar]
- 154.Thomas NW, Jenkins PG, Howard KA, Smith MW, Lavelle EC, Holland J, Davis SS. Particle uptake and translocation across epithelial membranes. J Anat. 1996;189:487–490. [PMC free article] [PubMed] [Google Scholar]
- 155.Meynell HM, Thomas NW, James PS, Holland J, Taussig MJ, Nicoletti C. Up-regulation of microsphere transport across the follicle-associated epithelium of Peyer’s patch by exposure to Streptococcus pneumoniae R36a. FASEB J. 1999;13:611–619. doi: 10.1096/fasebj.13.6.611. [DOI] [PubMed] [Google Scholar]
- 156.De SN, Chatterje DN. An experimental study of the mechanism of action of Vibriod cholerae on the intestinal mucous membrane. J Pathol Bacteriol. 1953;66:559–562. doi: 10.1002/path.1700660228. [DOI] [PubMed] [Google Scholar]
- 157.Schanker LS, Tocco DJ, Brodie BB, Hogben CA. Absorption of drugs from the rat small intestine. J Pharmacol Exp Ther. 1958;123:81–88. [PubMed] [Google Scholar]
- 158.Man AL, Prieto-Garcia ME, Nicoletti C. Improving M cell mediated transport across mucosal barriers: do certain bacteria hold the keys? Immunology. 2004;113:15–22. doi: 10.1111/j.1365-2567.2004.01964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fox CB, Kim J, Le LV, Nemeth CL, Chirra HD, Desai TA. Micro/nanofabricated platforms for oral drug delivery. J Control Release. 2015;219:431–444. doi: 10.1016/j.jconrel.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.George M, Abraham TE. Polyionic hydrocolloids for the intestinal delivery of protein drugs: alginate and chitosan--a review. J Control Release. 2006;114:1–14. doi: 10.1016/j.jconrel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 161.Chang CH, Lin YH, Yeh CL, Chen YC, Chiou SF, Hsu YM, Chen YS, Wang CC. Nanoparticles incorporated in pH-sensitive hydrogels as amoxicillin delivery for eradication of Helicobacter pylori. Biomacromolecules. 2010;11:133–142. doi: 10.1021/bm900985h. [DOI] [PubMed] [Google Scholar]
- 162.Lin YH, Tsai SC, Lai CH, Lee CH, He ZS, Tseng GC. Genipin-cross-linked fucose-chitosan/heparin nanoparticles for the eradication of Helicobacter pylori. Biomaterials. 2013;34:4466–4479. doi: 10.1016/j.biomaterials.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 163.Lin YH, Lin JH, Chou SC, Chang SJ, Chung CC, Chen YS, Chang CH. Berberine-loaded targeted nanoparticles as specific Helicobacter pylori eradication therapy: in vitro and in vivo study. Nanomedicine. 2015;10:57–71. doi: 10.2217/nnm.14.76. [DOI] [PubMed] [Google Scholar]
- 164.Lin YH, Chen ZR, Lai CH, Hsieh CH, Feng CL. Active targeted nanoparticles for oral administration of gastric cancer therapy. Biomacromolecules. 2015;16:3021–3032. doi: 10.1021/acs.biomac.5b00907. [DOI] [PubMed] [Google Scholar]
- 165.Deng L, Dong H, Dong A, Zhang J. A strategy for oral chemotherapy via dual pH-sensitive polyelectrolyte complex nanoparticles to achieve gastric survivability, intestinal permeability, hemodynamic stability and intracellular activity. Eur J Pharm Biopharm. 2015;97:107–117. doi: 10.1016/j.ejpb.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 166.Suwannateep N, Banlunara W, Wanichwecharungruang SP, Chiablaem K, Lirdprapamongkol K, Svasti J. Mucoadhesive curcumin nanospheres: biological activity, adhesion to stomach mucosa and release of curcumin into the circulation. J Control Release. 2011;151:176–182. doi: 10.1016/j.jconrel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 167.Pan-In P, Banlunara W, Chaichanawongsaroj N, Wanichwecharungruang S. Ethyl cellulose nanoparticles: clarithomycin encapsulation and eradication of H. pylori. Carbohydr Polym. 2014;109:22–27. doi: 10.1016/j.carbpol.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 168.Pan-in P, Tachapruetinun A, Chaichanawongsaroj N, Banlunara W, Suksamrarn S, Wanichwecharungruang S. Combating Helicobacter pylori infections with mucoadhesive nanoparticles loaded with Garcinia mangostana extract. Nanomedicine. 2014;9:457–468. doi: 10.2217/nnm.13.30. [DOI] [PubMed] [Google Scholar]
- 169.Tachaprutinun A, Pan-In P, Samutprasert P, Banlunara W, Chaichanawongsaroj N, Wanichwecharungruang S. Acrylate-tethering drug carrier: covalently linking carrier to biological surface and application in the treatment of Helicobacter pylori infection. Biomacromolecules. 2014;15:4239–4248. doi: 10.1021/bm5012618. [DOI] [PubMed] [Google Scholar]
- 170.Navabi N, Johansson ME, Raghavan S, Lindén SK. Helicobacter pylori infection impairs the mucin production rate and turnover in the murine gastric mucosa. Infect Immun. 2013;81:829–837. doi: 10.1128/IAI.01000-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.He H, Wang P, Cai C, Yang R, Tang X. VB12-coated Gel-Core-SLN containing insulin: Another way to improve oral absorption. Int J Pharm. 2015;493:451–459. doi: 10.1016/j.ijpharm.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 172.Verma A, Sharma S, Gupta PK, Singh A, Teja BV, Dwivedi P, Gupta GK, Trivedi R, Mishra PR. Vitamin B12 functionalized layer by layer calcium phosphate nanoparticles: A mucoadhesive and pH responsive carrier for improved oral delivery of insulin. Acta Biomater. 2016;31:288–300. doi: 10.1016/j.actbio.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 173.Ma T, Wang L, Yang T, Ma G, Wang S. M-cell targeted polymeric lipid nanoparticles containing a Toll-like receptor agonist to boost oral immunity. Int J Pharm. 2014;473:296–303. doi: 10.1016/j.ijpharm.2014.06.052. [DOI] [PubMed] [Google Scholar]
- 174.Xia D, Tao J, He Y, Zhu Q, Chen D, Yu M, Cui F, Gan Y. Enhanced transport of nanocage stabilized pure nanodrug across intestinal epithelial barrier mimicking Listeria monocytogenes. Biomaterials. 2015;37:320–332. doi: 10.1016/j.biomaterials.2014.10.038. [DOI] [PubMed] [Google Scholar]
- 175.Liu C, Shan W, Liu M, Zhu X, Xu J, Xu Y, Huang Y. A novel ligand conjugated nanoparticles for oral insulin delivery. Drug Deliv. 2015;27:1–11. doi: 10.3109/10717544.2015.1058433. [DOI] [PubMed] [Google Scholar]
- 176.Xu Y, Xu J, Shan W, Liu M, Cui Y, Li L, Liu C, Huang Y. The transport mechanism of integrin αvβ3 receptor targeting nanoparticles in Caco-2 cells. Int J Pharm. 2016;500:42–53. doi: 10.1016/j.ijpharm.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 177.Jin Y, Song Y, Zhu X, Zhou D, Chen C, Zhang Z, Huang Y. Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials. 2012;33:1573–1582. doi: 10.1016/j.biomaterials.2011.10.075. [DOI] [PubMed] [Google Scholar]
- 178.Li X, Wang C, Liang R, Sun F, Shi Y, Wang A, Liu W, Sun K, Li Y. The glucose-lowering potential of exenatide delivered orally via goblet cell-targeting nanoparticles. Pharm Res. 2015;32:1017–1027. doi: 10.1007/s11095-014-1513-1. [DOI] [PubMed] [Google Scholar]
- 179.Zhang J, Zhu X, Jin Y, Shan W, Huang Y. Mechanism study of cellular uptake and tight junction opening mediated by goblet cell-specific trimethyl chitosan nanoparticles. Mol Pharm. 2014;11:1520–1532. doi: 10.1021/mp400685v. [DOI] [PubMed] [Google Scholar]
- 180.Muchow M, Maincent P, Muller RH. Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev Ind Pharm. 2008;34:1394–1405. doi: 10.1080/03639040802130061. [DOI] [PubMed] [Google Scholar]
- 181.Date AA, Joshi MD, Patravale VB. Parasitic diseases: Liposomes and polymeric nanoparticles versus lipid nanoparticles. Adv Drug Deliv Rev. 2007;59:505–521. doi: 10.1016/j.addr.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 182.Puglia C, Bonina F. Lipid nanoparticles as novel delivery systems for cosmetics and dermal pharmaceuticals. Expert Opin Drug Deliv. 2012;9:429–441. doi: 10.1517/17425247.2012.666967. [DOI] [PubMed] [Google Scholar]
- 183.Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine. 2016;12:143–161. doi: 10.1016/j.nano.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 184.Aji MRA, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42:11–18. doi: 10.1016/j.ejps.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 185.Zhang Z, Gao F, Bu H, Xiao J, Li Y. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in rats. Nanomedicine. 2012;8:740–747. doi: 10.1016/j.nano.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 186.Chaudhary S, Garg T, Murthy RS, Rath G, Goyal AK. Development, optimization and evaluation of long chain nanolipid carrier for hepatic delivery of silymarin through lymphatic transport pathway. Int J Pharm. 2015;485:108–121. doi: 10.1016/j.ijpharm.2015.02.070. [DOI] [PubMed] [Google Scholar]
- 187.ElKasabgy NA, Elsayed I, Elshafeey AH. Design of lipotomes as a novel dual functioning nanocarrier for bioavailability enhancement of lacidipine: in-vitro and in-vivo characterization. Int J Pharm. 2014;472:369–379. doi: 10.1016/j.ijpharm.2014.06.048. [DOI] [PubMed] [Google Scholar]
- 188.Siram K, Chellan VR, Natarajan T, Krishnamoorthy B, Ebrahim HRM, Karanam V, Muthuswamy SS, Ranganathan HP. Solid lipid nanoparticles of diethylcarbamazine citrate for enhanced delivery to the lymphatics: in vitro and in vivo evaluation. Expert Opin Drug Deliv. 2014;11:1351–1365. doi: 10.1517/17425247.2014.915310. [DOI] [PubMed] [Google Scholar]
- 189.Zhang X, Zhang T, Zhou X, Liu H, Sun H, Ma Z, Wu B. Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation. J Pharm Sci. 2014;103:1711–1719. doi: 10.1002/jps.23967. [DOI] [PubMed] [Google Scholar]
- 190.Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomedicine. 2014;9:495–504. doi: 10.2147/IJN.S56648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ravi PR, Aditya N, Kathuria H, Malekar S, Vats R. Lipid nanoparticles for oral delivery of raloxifene: optimization, stability, in vivo evaluation and uptake mechanism. Eur J Pharm Biopharm. 2014;87:114–124. doi: 10.1016/j.ejpb.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 192.Nassar T, Attili-Qadri S, Harush-Frenkel O, Farber S, Lecht S, Lazarovici P, Benita S. High plasma levels and effective lymphatic uptake of docetaxel in an orally available nanotransporter formulation. Cancer Res. 2011;71:3018–3028. doi: 10.1158/0008-5472.CAN-10-3118. [DOI] [PubMed] [Google Scholar]
- 193.Attili-Qadri S, Karra N, Nemirovski A, Schwob O, Talmon Y, Nassar T, Benita S. Oral delivery system prolongs blood circulation of docetaxel nanocapsules via lymphatic absorption. Proc Natl Acad Sci U S A. 2013;110:17498–17503. doi: 10.1073/pnas.1313839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gugulothu D, Kulkarni A, Patravale V, Dandekar P. pH-sensitive nanoparticles of curcumin-celecoxib combination: evaluating drug synergy in ulcerative colitis model. J Pharm Sci. 2014;103:687–696. doi: 10.1002/jps.23828. [DOI] [PubMed] [Google Scholar]
- 195.Naeem M, Choi M, Cao J, Lee Y, Ikram M, Yoon S, Lee J, Moon HR, Kim MS, Jung Y, Yoo JW. Colon-targeted delivery of budesonide using dual pH- and time-dependent polymeric nanoparticles for colitis therapy. Drug Des Devel Ther. 2015;9:3789–3799. doi: 10.2147/DDDT.S88672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Naeem M, Kim W, Cao J, Jung Y, Yoo JW. Enzyme/pH dual sensitive polymeric nanoparticles for targeted drug delivery to the inflamed colon. Colloids Surf B Biointerfaces. 2014;123:271–278. doi: 10.1016/j.colsurfb.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 197.Ali H, Weigmann B, Neurath MF, Collnot EM, Windbergs M, Lehr CM. Budesonide loaded nanoparticles with pH-sensitive coating for improved mucosal targeting in mouse models of inflammatory bowel diseases. J Control Release. 2014;183:167–177. doi: 10.1016/j.jconrel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 198.Beloqui A, Coco R, Memvanga PB, Ucakar B, des Rieux A, Préat V. pH-sensitive nanoparticles for colonic delivery of curcumin in inflammatory bowel disease. Int J Pharm. 2014;473:203–212. doi: 10.1016/j.ijpharm.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 199.Vong LB, Tomita T, Yoshitomi T, Matsui H, Nagasaki Y. An orally administered redox nanoparticle that accumulates in the colonic mucosa and reduces colitis in mice. Gastroenterology. 2012;143:1027–1036. doi: 10.1053/j.gastro.2012.06.043. [DOI] [PubMed] [Google Scholar]
- 200.Vong LB, Mo J, Abrahamsson B, Nagasaki Y. Specific accumulation of orally administered redox nanotherapeutics in the inflamed colon reducing inflammation with dose-response efficacy. J Control Release. 2015;210:19–25. doi: 10.1016/j.jconrel.2015.05.275. [DOI] [PubMed] [Google Scholar]
- 201.Vong LB, Yoshitomi T, Morikawa K, Saito S, Matsui H, Nagasaki Y. Oral nanotherapeutics: effect of redox nanoparticle on microflora in mice with dextran sodium sulfate-induced colitis. J Gastroenterol. 2014;49:806–813. doi: 10.1007/s00535-013-0836-8. [DOI] [PubMed] [Google Scholar]
- 202.Vong LB, Yoshitomi T, Matsui H, Nagasaki Y. Development of an oral nanotherapeutics using redox nanoparticles for treatment of colitis-associated colon cancer. Biomaterials. 2015;55:54–63. doi: 10.1016/j.biomaterials.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 203.Vong LB, Nagasaki Y. Combination treatment of murine colon cancer with doxorubicin and redox nanoparticles. Mol Pharm. 2016 doi: 10.1021/acs.molpharmaceut.5b00676. In press. [DOI] [PubMed] [Google Scholar]
- 204.Coco R, Plapied L, Pourcelle V, Jérôme C, Brayden DJ, Schneider YJ, Préat V. Drug delivery to inflamed colon by nanoparticles: comparison of different strategies. Int J Pharm. 2013;440:3–12. doi: 10.1016/j.ijpharm.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 205.Arora Z, Shen B. Biological therapy for ulcerative colitis. Gastroenterol Rep. 2015;3:103–109.H. doi: 10.1093/gastro/gou070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Laroui, Viennois E, Xiao B, Canup BS, Geem D, Denning TL, Merlin D. Fab′-bearing siRNA TNFα-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J Control Release. 2014;186:41–53. doi: 10.1016/j.jconrel.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]