

Abstract
The marine alphaproteobacterium strain O3.65 was isolated from an enrichment culture of surface seawater contaminated with weathered oil (slicks) from the Deepwater Horizon (DWH) oil spill and belongs to the ubiquitous, diverse and ecological relevant Roseobacter group within the Rhodobacteraceae. Here, we present a preliminary set of physiological features of strain O3.65 and a description and annotation of its draft genome sequence. Based on our data we suggest potential ecological roles of the isolate in the degradation of crude oil within the network of the oil-enriched microbial community. The draft genome comprises 4,852,484 bp with 4,591 protein-coding genes and 63 RNA genes. Strain O3.65 utilizes pentoses, hexoses, disaccharides and amino acids as carbon and energy source and is able to grow on several hydroxylated and substituted aromatic compounds. Based on 16S rRNA gene comparison the closest described and validated strain is Phaeobacter inhibens DSM 17395, however, strain O3.65 is lacking several phenotypic and genomic characteristics specific for the genus Phaeobacter. Phylogenomic analyses based on the whole genome support extensive genetic exchange of strain O3.65 with members of the genus Ruegeria, potentially by using the secretion system type IV. Our physiological observations are consistent with the genomic and phylogenomic analyses and support that strain O3.65 is a novel species of a new genus within the Rhodobacteraceae.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-016-0201-7) contains supplementary material, which is available to authorized users.
Keywords: Rhodobacterales, Rhodobacteraceae, Roseobacter, Oil spill, Deepwater Horizon, Oil degradation, Hydrocarbon, Marine bacteria
Introduction
The Roseobacter clade is a major marine bacterial group, often associated with phytoplankton blooms [1–3], and accounts for up to 35 % of the bacterioplankton in coastal waters and the Southern Ocean [4–6]. The Roseobacter clade belongs to the family Rhodobacteraceae within the order Rhodobacterales, among the Alphaproteobacteria [7]; organisms of this group show a highly diversified range of physiological adaptations to various marine ecosystems [4, 5, 8]. Several taxa of this group are stimulated by different hydrocarbon compounds in laboratory experiments or in situ, suggesting a function in aerobic hydrocarbon degradation. Furthermore, pathways for oxygenic degradation of aromatic compounds and genes encoding for enzymes in alkane degradation were described for these bacteria [9]. Contributions of Roseobacter-related phylotypes to oil degradation were indicated by surveys using 16S rRNA gene based molecular biological techniques [10–14], but only a few studies were based on cultivation approaches [15, 16].
With this study, we fill this gap by specific isolation, genomic and physiological analysis of a bacterium of the Roseobacter clade isolated from seawater contaminated with weathered oil slicks from the Deepwater Horizon oil spill, one of the worst anthropogenic disasters in maritime petroleum production. Within 84 days (20th April to 15th July 2010) over 4.1 million barrels (~6.5x108 L) of crude oil burst out into the Gulf of Mexico in a water depth of 1500 m [17]. Massive microbial community shifts were observed in the deep hydrocarbon plume at about 1,100 m depth, and in surface waters contaminated with slicks of weathered oil [12, 18–21].
Approximately two weeks after the beginning of the discharge, the first samples of oil slick-contaminated surficial seawater were collected, and were dominated by aromatic hydrocarbon degrading Cycloclasticus spp. and heterotrophic members of the Alteromonadales (Pseudoalteromonas, Alteromonas and Colwellia spp.) as well as members of the Rhodobacteraceae [20]. Passow and colleagues [22] reported that weathered crude oil slicks at the air-water interface were transformed into water-in-oil emulsions. These emulsions promoted the formation exopolymeric substances, mostly composed of polysaccharides; these coalesced into huge mucus-rich marine snow aggregates acting as hot spots for oil-specialized or -associated microbes, in which emulsified oil and EPS served as diverse food source for the highly active bacterial community [12, 23]. The bacterial communities associated with these aggregates included diverse phyla of Gammaproteobacteria, Bacteroidetes, and different organisms of the Roseobacter clade [12], and were distinctly different compared to those in the oil-contaminated water column [20].
The succession of different microbial taxa being abundant at distinct time points or steps during degradation of oil-derived hydrocarbons suggests a metabolic network comprising i) primary hydrocarbon-degrading and specialized microbes (involved in consumption, hydrolysis, oxidation of distinct hydrocarbons), ii) emulsifying microbes increasing the hydrocarbon bioavailability for the networkers, and iii) a very diverse group of secondary hydrocarbon consumers. All together form a complex assemblage of microbes involved in degradation of a wide spectrum of oil-derived hydrocarbons [12, 24].
Strain O3.65 was isolated from contaminated seawater of the DWH oil spill. Subsequent comparative analysis of the 16S rRNA gene sequences revealed that strain O3.65 belongs to the Roseobacter group, with Phaeobacter and Ruegeria species as closest described relatives. Here, we present a set of features and physiological characteristics of strain O3.65, and a description of the draft and annotated genome sequence of this organism. Furthermore, we partially elucidate its contribution in oil degradation and classify strain O3.65 into the above mentioned microbial oil degradation network based on the genomic and physiological analyses.
Organism information
Classification and features
Strain O3.65 was isolated from an enrichment culture of surface seawater sample contaminated with weathered oil from the DWH oil spill (Table 1). The sample was collected on June 1st in 2010, and was subsequently stored undisturbed in a 50 ml Falcon tube for four years at 4 °C in the dark. The inoculum for isolation was taken from the underlying water–oil phase, directly below the oil layer (Additional file 1: Figure S1), and streaked out on agar plates (1.5 % w/v) containing 10 % marine broth (MB 2216, Difco) diluted with artificial seawater [25]. Plates were incubated at 20 °C in the dark until colonies were visible (2–5 days). For purification single colonies were picked and transferred at least three times to fresh plates with the same medium. Tests for purity of the culture, extraction of chromosomal DNA and sequencing of the 16S rRNA gene sequence were performed after Giebel et al. [26].
Table 1.
Classification and general features of Rhodobacteraceae strain O3.65 according to the MIGS recommendations [92]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [93] | |
| Phylum Proteobacteria | TAS [94] | ||
| Class Alphaproteobacteria | TAS [95, 96] | ||
| Order Rhodobacterales | TAS [95] | ||
| Family Rhodobacteraceae | TAS [7, 95, 97] | ||
| Genus not specified | |||
| Species not specified | |||
| (Type) strain: O3.65 (LPUY00000000.1) | |||
| Gram stain | negative | IDA | |
| Cell shape | rod shaped | IDA | |
| Motility | motile | IDA | |
| Sporulation | none | NAS | |
| Temperature range | mesophile | IDA | |
| Optimum temperature | 30 °C | IDA | |
| pH range; Optimum | not specified | ||
| Carbon source | oligo-, di-saccharides, organic acids, amino acids, hydroxylated aromatic hydrocarbons | IDA | |
| Energy metabolism | heterotrophic | IDA | |
| MIGS-6 | Habitat | marine | IDA |
| MIGS 6.3 | Salinity | 1- < 8 %, optimum 3.5 % | IDA |
| MIGS-22 | Oxygen requirement | aerobic | IDA |
| MIGS-15 | Biotic relationship | unknown | NAS |
| MIGS-14 | Pathogenicity | none | NAS |
| Biosafety level | 1 | TAS [98] | |
| MIGS-4 | Geographic location | Gulf of Mexico | IDA |
| MIGS-5 | Sample collection | June 1, 2010 | IDA |
| MIGS-4.1 | Latitude | 28°43.967 N | IDA |
| MIGS-4.2 | Longitude | 88°22.993 W | IDA |
| MIGS-4.4 | Altitude | not specified |
aEvidence codes - IDA inferred from direct assay, TAS traceable author statement (i.e., a direct report exists in the literature), NAS non-traceable author statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [99]
Comparison of the 16S rRNA gene sequence of strain O3.65 with those of type strains of the Rhodobacteraceae was performed using the Blast search tool of the National Center for Biotechnology Information [27]. For phylogenetic analysis and similarity matrix calculation we used the ARB software [28]. The tree in Fig. 1 comprises all currently available genome sequenced Phaeobacter, Pseudophaeobacter, Leisingera and Ruegeria strains, covering most of the type strains and species of those groups and additional genome-sequenced species of the Roseobacter group.
Fig. 1.
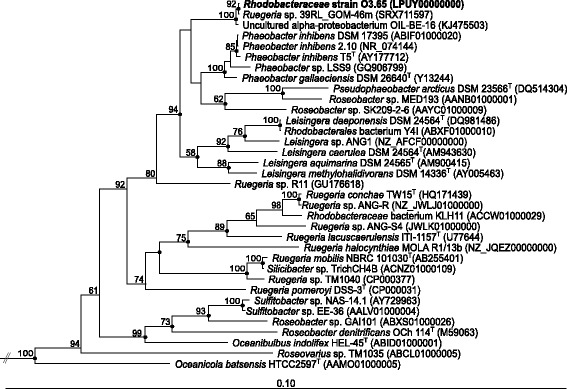
Phylogenetic tree highlighting the position of Rhodobacteraceae strain O3.65 relative to other genome sequenced and type strains within the genera Phaeobacter, Pseudophaeobacter, Ruegeria, Leisingera and additional strains of the Rhodobacteraceae. The tree was inferred from nearly full-length 16S rRNA gene sequences (≥1300 bp) using the neighbour joining tool of the ARB software [28]. Only bootstrap values ≥50 % (derived from 1000 replicates) are shown. Filled circles indicate nodes also recovered reproducibly with maximum-likelihood (RAxML) calculation. Strains and their corresponding GenBank accession numbers are listed in Additional file 1: Table S1. All strains in the tree are genome sequenced, except clone Oil-BE-016 (KJ475503). Type strains are designated by T. Three Synechococcus strains (AY946243, CP000951, AF448073) served as outgroup (not shown)
Based on the 16S rRNA gene sequences a greater monophyletic cluster, supported by a high bootstrap value of 94 %, was obtained encompassing strain O3.65 and related sequences, as well as the genera Phaeobacter, Pseudophaeobacter and Leisingera. Strain O3.65 forms a subcluster together with the undescribed strain Ruegeria sp. 39RL_GOM-46 m (SRX711597) isolated from an oil-amended biotrap, and the clone Oil-BE-016 (KJ475503, [12]) obtained from an oil slick sample after lab incubation, both from the DWH oil spill and having a sequence similarity of 100 and 99 %, respectively. The 16S rRNA gene sequence of strain O3.65 shows minimal dissimilarities to those of its closest described and validated relatives, i.e. 1.6 % to Phaeobacter inhibens DSM 17395 and 1.7 % to both type strains P. gallaeciensis DSM 26640T and Phaeobacter inhibens T5T. Dissimilarity values increased up to 1.9 or higher for type species of the genera Ruegeria, Leisingera and Pseudophaeobacter (Additional file 1: Table S1; [12, 29–58]). Despite these low dissimilarity values, classification of strain O3.65 as a new Phaeobacter species was not supported by phylogenetic analysis only on 16S rRNA gene level (Fig. 1). The clearly separated subcluster of strain O3.65 leads to the assumption that this organism represents a new phylogenetic lineage at the species and genus level. Comparative analysis of genomic data (see below, Fig. 2) supports a classification as a new genus within the Rhodobacteraceae. The multitude of recent reclassifications of species within the Phaeobacter-Leisingera group [29–31, 59] shows the difficulty of accurate classification of (new) species related to these closely related genera. Furthermore, we suggest reclassification of strain Ruegeria sp. 39RL_GOM-46 m based on a coherent description and validation of strain O3.65 as member of a new genus in the future.
Fig. 2.
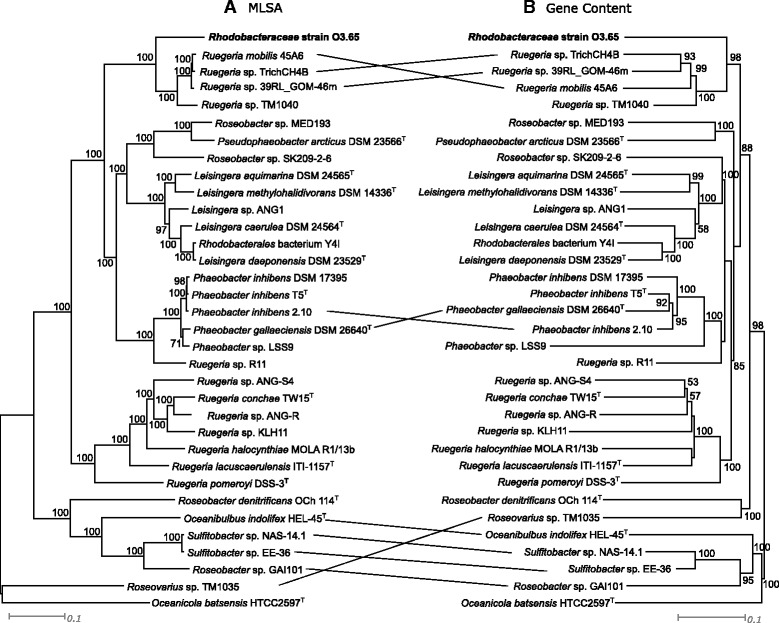
Tanglegram of genome based trees. a Maximum likelihood tree based on genomic data of organisms affiliated with the genera Phaeobacter, Pseudophaeobacter, Ruegeria, Leisingera and additional strains of the Roseobacter clade inferred with 500 bootstraps (BS) with RAxML after Stamatakis (2014) [100]. The alignment was created from 684 orthologous single-copy genes present in all genomes (Multilocus Sequence Analysis; MLSA) after total protein sequences of the genomes were extracted from the corresponding GenBank files and used for the downstream analysis with an in house pipeline at the Goettingen Genomics Laboratory (J. Vollmers, unpubl.). In brief, clusters of orthologs were generated using proteinortho version 5 [101], inparalogs were removed, the remaining sequences were aligned with MUSCLE [102] and poorly aligned positions automatically filtered from the alignments using Gblocks [103]. b Gene content tree including singletons of the same organisms as in A based on an orthologs-content matrix representing presence or absence of a gene in a certain genome, inferred with Neighbour Joining (1000 BS). Both scripts for this pipeline, PO_2_MLSA.py and PO_2_GENECONTENT.py, are available at github. Numbers at the nodes specify BS values ≥50 %. Scale bars represent 10 % sequence divergence. For Genbank accession numbers see Additional file 1: Table S1. For a clear view only lines were given linking the same species at different positions
Besides strain O3.65, we isolated similar organisms with the same 16S rRNA gene sequence from agar plates inoculated with oil-polluted seawater from another sample taken at a different station after the DWH oil spill (data not shown). Furthermore, two independent studies found previously the same phylotype of strain O3.65 (SRX711597) and a second phylotype very similar (1382/1383 identities, [12]) to strain O3.65 in the Gulf of Mexico (see above). Therefore we conclude that strain O3.65 represents a physiologically and ecologically relevant ecotype for the DWH oil spill.
Cells of strain O3.65 are ovoid rods, with a length of 1.3–2.2 μm and a width of 0.6–1.0 μm (Fig. 3). Cells are motile by means of a polar flagellum. O3.65 is a Gram-negative, marine, aerobic, mesophilic bacterium with an optimal growth temperature between 30 and 35 °C and an optimal salinity between 2.5 and 5 %. On Difco Marine Broth (MB) 2216 agar (Becton Dickinson, MD, USA) strain O3.65 forms smooth, shiny and convex colonies with regular edges of white to light beige color. Strain O3.65 utilizes pentoses, hexoses and disaccharides [(+)-L-arabinose, (+)-D-xylose, (−)-D-ribose, (+)-D-glucose (−)-L-fucose, (−)-D-fructose, (+)-D-cellobiose, (+)-D-sucrose;1 g/l final] as well as most amino acids (L-forms of alanine, aspartic acid, glutamic acid, histidine, arginine, threonine, tryptophane, phenylalanine, proline, leucine, valine; 1 mM final) as carbon and energy sources. Strain O3.65 is able to grow on several aromatic compounds, i.e., 4-hydroxy-benzoic acid, 3,4-dihydroxy-benzoic acid, p-coumarin, ferulic acid, tryptophan and vanillin.
Fig. 3.
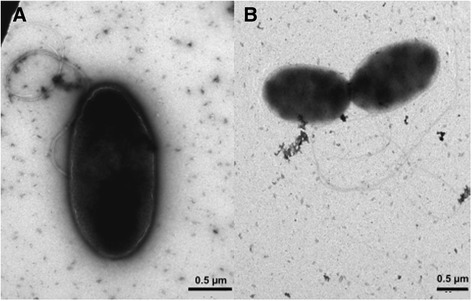
Transmission electron micrographs of Rhodobacteraceae strain O3.65. a The typical rod-shaped morphology of a single cell with intact bundle of flagella, and (b) two cells by binary fission and their flagella. Cells were negatively stained. Scale bars 0.5 μm
Genome sequencing information
Genome project history
The genome of strain O3.65 was selected for sequencing based on its phylogenetic affiliation with the ecologically important and worldwide distributed Roseobacter clade and the lack of roseobacteral genomes in the course of studies on oil degradation of the DWH oil spill. The genome sequence was completed on February 18th, 2015, and presented for public access on January 19th, 2016. The genome project was deposited in the Genomes OnLine Database (GOLD) as project Gp0111538. The Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number LPUY00000000.1. The version described in this paper is version 1. Table 2 presents a summary of the project information.
Table 2.
Project information for Rhodobacteraceae strain O3.65
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Draft |
| MIGS-28 | Libraries used | Nextera xt |
| MIGS-29 | Sequencing platforms | Illumina GAiix |
| MIGS-31.2 | Fold coverage | 71.5x |
| MIGS-30 | Assemblers | SPAdes v3.5 |
| MIGS-32 | Gene calling method | Prodigal v2.50 |
| Genome Database release | IMG; 2608642179 | |
| Genbank ID | LPUY00000000.1 | |
| Genbank Date of Release | January 19th, 2016 | |
| GOLD ID | Gp0111538 | |
| BIOPROJECT | PRJNA305382 | |
| MIGS-13 | Source Material Identifier | O3.65 |
| Project relevance | environmental |
Growth conditions and DNA preparation
Strain O3.65 was grown at 20 °C in marine broth (MB2216, Difco) in the dark to the late exponential phase. Cells were harvested by centrifugation (10 000 g at 4 °C for 20 min) and subsequent DNA extraction was performed using a Power Soil DNA Isolation Kit (MoBio) according to the manufacturer’s specifications. The protocol includes bead beating for mechanical as well as chemical methods for cell lysis. A total of 1.3 μg of DNA was obtained.
Genome sequencing and assembly
Whole-genome sequencing was performed using Illumina technology. Preparation of a paired-end sequencing library with the Illumina Nextera XT library preparation kit and sequencing of the library using the Genome Analyzer IIx were performed as described by the manufacturer (Illumina, San Diego, CA, USA). A total of 4.6 million paired-end reads were derived from sequencing and trimmed using Trimmomatic version 0.32 [60]. De novo assembly of all trimmed reads with SPAdes version 3.5.0 [61] resulted in 125 contigs and 71.5-fold coverage.
Genome annotation
Protein-coding genes were identified as part of the genome annotation pipeline of the Integrated Microbial Genomes platform using Prodigal v2.50. The predicted CDS were translated and used to search the CDD, KEGG, UniProt, TIGRFam, Pfam and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Non-coding genes and miscellaneous features were predicted using tRNAscan-SE [62], RNAmmer [63], Rfam [64], TMHMM [65] and SignalP [66]. Additional gene prediction analyses and functional annotation were performed within the IMG-ER platform [67].
Genome properties
The genome statistics are provided in Table 3. The draft genome of strain O3.65 consists of 125 scaffolds with a total length of 4,852,484 bp and an overall G + C content of 61.50 %. Of the 4,654 predicted genes, 4591 (98.65 %) are protein-coding, and 63 are RNA genes. No pseudogenes or CRISPR counts were found. Most of the protein-coding genes (71 %) were assigned to putative functions. Besides the chromosome we assume strain O3.65 is carrying at least five extrachromosomal elements derived from five different typical plasmid repABC-type replication modules, commonly found within the Rhodobacteraceae [68]. The number and length of scaffolds of this draft genome did not allow a detailed view on plasmid organization. The distribution of genes into COGs functional categories is listed in Table 4.
Table 3.
Nucleotide content and gene count levels of the draft genome of Rhodobacteraceae strain O3.65
| Attribute | Genome (total) | |
|---|---|---|
| Value | % of total | |
| Genome size (bp) | 4,852,484 | 100.00 |
| DNA coding (bp) | 4,330,569 | 89.25 |
| DNA G + C (bp) | 2,984,418 | 61.50 |
| DNA scaffolds | 125 | |
| Total genes | 4,654 | 100.00 |
| Protein-coding genes | 4,591 | 98.65 |
| RNA genes | 63 | 1.35 |
| Pseudo genes | 0 | |
| Genes in internal clusters | ||
| Genes with function prediction | 3,868 | 83.11 |
| Genes assigned to COGs | 3,308 | 71.08 |
| Genes assigned to pfam domains | 3,953 | 84.94 |
| Genes with signal peptides | 390 | 8.38 |
| Genes with transmembrane helices | 991 | 21.29 |
| CRISPR repeats | 0 | |
Table 4.
Number of genes associated with the 25 general COG functional categories of Rhodobacteraceae strain O3.65
| Code | Value | %age | Description |
|---|---|---|---|
| J | 193 | 5.19 | Translation, ribosomal structure and biogenesis |
| A | n.a. | n.a. | RNA processing and modification |
| K | 300 | 8.07 | Transcription |
| L | 110 | 2.96 | Replication, recombination and repair |
| B | 3 | 0.08 | Chromatin structure and dynamics |
| D | 39 | 1.05 | Cell cycle control, Cell division, chromosome partitioning |
| V | n.a. | n.a. | Defense mechanisms |
| T | 58 | 1.56 | Signal transduction mechanisms |
| M | 135 | 3.63 | Cell wall/membrane biogenesis |
| N | 183 | 4.92 | Cell motility |
| U | 64 | 1.72 | Intracellular trafficking and secretion |
| O | 1 | 0.03 | Posttranslational modification, protein turnover, chaperones |
| C | 11 | 0.3 | Energy production and conversion |
| G | 83 | 2.23 | Carbohydrate transport and metabolism |
| E | 156 | 4.19 | Amino acid transport and metabolism |
| F | 249 | 6.7 | Nucleotide transport and metabolism |
| H | 346 | 9.3 | Coenzyme transport and metabolism |
| I | 393 | 10.57 | Lipid transport and metabolism |
| P | 91 | 2.45 | Inorganic ion transport and metabolism |
| Q | 183 | 4.92 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 216 | 5.81 | General function prediction only |
| S | 218 | 5.86 | Function unknown |
| - | 135 | 3.63 | Not in COGs |
Abbreviation: n.a. not assigned
The total is based on the total number of protein coding genes in the genome
Insights from the genome sequence
Several pathways in the aerobic hydrocarbon degradation by ring modifications and alkane hydroxylases are known and used by members of the Roseobacter group [9]. Yet, analysis of genomic homology could be difficult due to the low amount of gene synteny among genomes of strains even on species level, and the high distribution of functionally related genes across multiple loci [69]. In general, strain O3.65 is not able to hydroxylate an aromatic ring via specific ring hydroxylating dioxygenases, such as benzoate 1,2-dioxygenase or naphthalene 1,2-dioxygenase; genes of the protein families 00355, 00848 and 00866 were not found [9]. The draft genome of strain O3.65 is carrying none or only a low number of genes (given in parentheses) encoding for enzymes involved in the cleavage of gentisate (gdo; 0), the benzoyl-CoA pathway (box; 0) and the meta cleavage of homoprotocatechuate (hgd; 2 of 7). In contrast, strain O3.65 does contain several putative ring-cleaving dioxygenases: Two aromatic ring-opening dioxygenases, catalytic subunit, LigB family (TRIHO_09370; TRIHO_18120; pfam02900), hydroquinol and 1,2-catechol dioxygenases (TRIHO_05060; TRIHO_09430; pfam04444/pfam 00775), protocatechuate 3,4-dioxygenase alpha and beta subunit (TRIHO_21670/60; pfam00755) and at least four catechol 2,3-dioxygenases (TRIHO_03150; TRIHO_07560; TRIHO_29300; TRIHO_43160 pfam00903, TRIHO_09100; TRIHO_20770 pfam12681). All those ring-cleaving enzymes are essential for degrading substances like protocatechuate, vanillin, 4-hydroxybenzoate, ferulic acid or p-coumarin, which is consistent with our growth experiments (see discussion of morphology and physiology above).
However, genes for degradation of hydroxylated aromatic compounds like p-hydroxybenzoate via protocatechuate (pca, β-ketoadipate pathway) are present in the genome of O3.65. For example, the genes pobA and pcaDCHGB (TRIHO_21630-80) are homologues to genes found in Silicibacter sp. TM1040 and Ruegeria mobilis 45A6. The genes pcaIJ (TRIHO_43620/30) of strain O3.65 coding for the 3-oxoadipate:succinyl CoA transferase are arranged in the same way as in Citreicella sp. SE45, but the entire neighboring gene arrangement of both strains differs completely from those of other Roseobacter representatives. Comparative analysis shows that all Phaeobacter, Pseudophaeobacter, Leisingera and Ruegeria spp. do not have the genes pcaIJ for an 3-oxoadipate:succinyl CoA transferase (EC 2.8.3.6); instead, it seems to be replaced by an 3-oxoacid CoA-transferase (EC 2.8.3.5) with an AA-composition similarity of 32 %. Also missing for the above mentioned genomes, but present for strain O3.65 and located next to the subunit pcaIJ, is a regulatory protein (coded by pcaR; 2609025149, TRIHO_43610) needed for functionality of the enzyme 3-oxiadipate CoA transferase. PcaR, characterized for Pseudomonas putida [70] was blasted against the Phaeobacter-Leisingera-group finding genes with ~30 % similarity, but in distinctly different neighborhoods than in O3.65, which could imply other functions of the IclR family (transcriptional regulator, Pfam01614) to which pcaR belongs. Moreover, no similar pcaR-genes were found in any genomes of Ruegeria spp., underlining its distinctiveness from these two groups. We assume that strain O3.65 is able to metabolize phenylacetic acids via the phenylacetyl-CoA pathway (paa) having all the necessary genes (paaABCDE), except the catalytic subunit. However, strain O3.65 is able to grow on phenylalanine, which is degraded via the paa-pathway, like in P. inhibens DSM 17395 [71]. Besides, strain O3.65 is able to carry out the degradation of the aromatic intermediate homogentisate by a specific homogentisate 1,2-dioxygenase (TRIHO_32660; pfam04209).
Even though strain O3.65 is carrying the gene for an alkane 1-monooxygenase (pAH1; coded by alkB locus tag TRIHO_03510) and all genes for the following pathway steps for metabolizing an alkane into a fatty acid, it did not exhibit any growth in experiments on nonane, decane, hexadecane or paraffin. In contrast, Pseudophaeobacter arcticus DSM 23566T was able to grow on all those alkanes. Maybe this is caused by the missing gene coding for rubredoxin reductase (EC 1.18.1.1/4) in strain O3.65, required for the reducing step of rubredoxin. Rubredoxin and rubredoxin reductase are essential electron transfer proteins and present in known alkane degraders like Alcanivorax dieselolei B5 [72]. Notably, this gene is also missing in strain DSM 23566T, leading to the conclusion that there might be other ways of alkane degradation, as already stated by Buchan and Gonzalez (2010) [9]. Perhaps EPS [73] or unknown substances from other oil degrading bacteria in contaminated seawater could help solubilizing oil substances, what has to be shown for strain O3.65. If this can be confirmed, strain O3.65 is involved in the microbial degradation of n-alkanes, which were found in enhanced concentrations in the oil-slick as well as polycyclic aromatic hydrocarbons of high-molecular weight [18, 74, 75].
In summary, we observed that strain O3.65 is able to degrade several oil-derived compounds via different pathways for hydrocarbon degradation. However, the missing pathways, especially the missing RHD, indicate that strain O3.65 does not belong to the group of specialized primary oil-degrading microbes within the hydrocarbon-degrading metabolic network. Instead, strain O3.65 belongs to the group of secondary hydrocarbon consumers feeding on special oil-derived components, i.e. “predigested” hydrocarbon fragments or on non-oil exudates from primary oil degraders. Matching to this was the found of an Alcanivorax affiliated isolate in our sample (unpublished data), which are well-known primary petroleum degraders, commonly rising in numbers during oil spills [76].
Using a whole genome comparison approach by multilocus sequence analysis, based on 684 orthologous single-copy genes and by gene content analysis of the same strains considered as in the 16S rRNA gene analysis above, separate clustering of strain O3.65 is supported (Fig. 2). By MLSA and gene content analysis, the closest related genus of strain O3.65 is not Phaeobacter (Fig. 1) but Ruegeria, supported by bootstrap values of 100 and 98 %, respectively. Four Ruegeria strains (R. sp. 39RL_GOM-46 m, R. mobilis 45A6, R. sp. TrichCH4B and R. sp. TM1040), separated from other Ruegeria spp., form the sequence cluster adjacent to strain O3.65. While Ruegeria sp. 39RL_GOM-46 m was obtained from the same oil-polluted environment and has an identical 16S rRNA gene sequence (Fig. 1), the MLSA or the gene content approach separate this strain from strain O3.65, and indicate a different genetic potential and evolution of both strains. The other three closely related Ruegeria strains have a 16S rRNA gene dissimilarity of 3.5 % and 4.6 %, respectively. Strains affiliated to Phaeobacter, Pseudophaeobacter and Leisingera clustered separately within the single genera in distinct groups, at which their clustering pattern is nearly identical by both calculation methods, emphasizing a high stability of the phylogenetic analyses.
Furthermore, we compared all available genome-sequenced Phaeobacter, Pseudophaeobacter, Leisingera and Ruegeria strains covering most of the type strains as well as type species of those genera and the draft genome of strain O3.65 by in silico DNA-DNA hybridization using the online tool genome to genome distance calculator (GGDC 2.0; [77–79]). The DDH similarities of strain O3.65 to the above mentioned reference strains are listed in Additional file 1: Table S1. The highest similarity was found for the genome of strain Ruegeria sp. 39RL_GOM-46 m with a maximal value of 100 ± 0.1 % implying that strain 39RL_GOM-46 m is another strain of a new proposed species represented by Rhodobacteraeae strain O3.65. This high similarity is in agreement with the 16S rRNA gene sequence similarity. Further, the GGDC analysis revealed a distinctly low mean similarity of the O3.65 genome (20.5 ± 4.8 %) compared to all other genomes considered in our study, including the other three closely related Ruegeria strains clustering together with strain 39RL_GOM-46 m and all available genome sequenced types species/strains of the genera Phaeobacter, Pseudophaeobacter, Leisingera and Ruegeria. This low similarity on genome level indicates a significant different genomic repertoire of strain O3.65 compared to its most closely-related neighbors, supporting that strain O3.65 represents a new species of a new genus within the Rhodobacteraceae, not distinguishable by 16S rRNA gene phylogeny only.
While aerobic anoxygenic photosynthesis is a widespread but phylogenetically dispersed feature among the Roseobacter group [8] strain O3.65 is not able to use light via aerobic anoxygenic photosynthesis or rhodopsins. However, both types of the coxL gene for the carbon monoxide dehydrogenase are present, implying a role within the marine carbon monoxide cycling, because only strains with both coxL forms (I and II; TRIHO_01790-60 and TRIHO_28700-40) are able to oxidize carbon monoxide [80, 81]. This could provide an additional energy source for strain O3.65 not available for other non-chemolithotrophic microbes [82].
Some Roseobacter species are able to synthesize the essential cofactor biotin, e.g. P. gallaeciensis BS107 and Ruegeria sp. R11 [83]. No genes for biotin synthesis were found in the genome of strain O3.65, as shown previously for Ruegeria sp. TM1040 and R. pomeroyi DSS-3 [83]. Therefore, bacteria missing the synthesis apparatus of biotin are equipped with a highly affine (or high-affinity) biotin uptake system present in strain O3.65, and homologous to those in Leisingera caerulea DSM 24564T and Leisingera methylohalidivorans MB2T/DSM 14336T.
An in silico analysis for secondary metabolites via the online tool antiSMASH 3.0 [84] revealed secondary metabolite clusters for bacteriocin, lassopeptide, ectoine and a type 1 polyketide synthase (PKS). PKSs mediate the biosynthesis of bioactive natural substances and are known for the genus Phaeobacter [85]. Genes encoding for iron-chelating siderophore biosynthesis and transport, commonly found in Phaeobacter and Leisingera species [29, 32, 33], are also present in genome of strain O3.65. The operon for biosynthesis (TRIHO_27280) is homologous to those in P. inhibens T5T and the Ruegeria sp. strains TrichCH4B and TM1040. The operon coding for the uptake of siderophores (TRIHO_36570) is homolog to those in R. mobilis 45A6 and Ruegeria sp. TrichCH4B. Strain O3.65 is lacking genes coding for AHL synthetase proteins, described for P. inhibens T5T [29] and P. gallaeciensis DSM 26640T [34]. Moreover, the AHL synthetase protein was found in all genomes of the type strains of the Leisingera, Pseudophaeobacter and Ruegeria group listed in this study (Additional file 1: Table S1) with the exception of R. mobilis NBRC101030T. Several Phaeobacter strains [35, 85–89], including the P. inhibens strains DSM 17395 and T5T as next described species to strain O3.65, are able to produce the antibiotic TDA and a brownish pigment [85]. These Phaeobacter-typical characteristics were not found to be encoded in the genome of strain O3.65 and could not be observed phenotypically.
Strain O3.65 is carrying at least three operons for the secretion system type IV (virB), which are versatile and involved in conjugation, DNA uptake or in effector translocation [90]. (TRIHO_37480, homolog to Roseovarius sp. 217, TRIHO_40140, TRIHO_41580 homolog to Oceanibulbus indolifex HEL-45T). Furthermore, genes for the flp pilus type IV are present in genome of strain O3.65, known to play important roles in surface adhesion, biofilm formation, motility, conjugation, and DNA transfer and uptake, with significant effects for pathogenicity [91] (TRIHO_20800 homolog to R. mobilis 45A6 and Ruegeria sp. TrichCH4BTRIHO_30860 homolog to R. mobilis 45A6 and Ruegeria sp. TM1040).
Our data of the draft genome revealed a diverse composition of several genes and functional operons of strain O3.65 originated from different phylogenetic groups, which was derived by their homologies. Having both opportunities to exchange or uptake DNA by pilus and secretion systems could be an explanation for carrying such a brought mixture of Ruegeria-, Phaeobacter- and Leisingera-like genes. Besides, this could elucidate the discrepancy of the phylogenetic classification based on 16S rRNA gene sequences and the genome based approaches (Figs. 1 and 2, see above).
Conclusion
The differences detected based on the genomic and physiological data of strain O3.65 compared to previously described organisms within the Rhodobacteraceae , especially to the genus Phaeobacter, suggests that strain O3.65 represents a member of a new species within a new genus. The multitude of recent reclassifications of several strains within the Rhodobacteraceae, especially within the genera Phaeobacter and Leisingera [29–31, 59] shows the difficulty to accurately classify (new) species related to these phylogenetic clades based only on 16S rRNA gene level, and supports our suggestion of a new genus to avoid a misleading phylogenetic classification a priori. Strain O3.65 is lacking several features typical for the genus Phaeobacter, e.g. production of the antibiotic TDA and AHLs, pigmentation, the hgd-pathway and biotin synthesis. Even though based on 16S rRNA gene comparison the closest described strain is Phaeobacter inhibens DSM 17395, high genetic exchange of strain O3.65 with members of the genus Ruegeria is indicated by the MLSA and gene content analysis based on whole genome information. Strain O3.65 is able to degrade hydroxylated aromatic compounds by several pathways, but is lacking genes to utilize alkanes. However, strain O3.65 represents a new, abundant and ecologically relevant microbial species within the hydrocarbon degrading microbial community of the DWH oil spill. We assume that strain O3.65 belongs to the group of secondary hydrocarbon consumers feeding on special oil-components, on “predigested” hydrocarbon fragments, or on non-oil exudates from primary oil degraders.
Acknowledgement
We thank Lisa Nigro and Tingting Yang for sampling and the crew of the RV Walton Smith for their support. We thank Marco Dogs and Mathias Wolterink for assistance in cultivation. We thank Frauke-Dorothee Meyer and Kathleen Gollnow for technical support. The work was supported by the German Research Foundation (DFG) Transregio-SFB 51 (Roseobacter). This research was made possible in part by a grant from The Gulf of Mexico Research Initiative to Andreas Teske. Data are publicly available through the Gulf of Mexico Research Initiative Information & Data Cooperative (GRIIDC) at https://data.gulfresearchinitiative.org.
Authors’ contribution
HAG, TB and AT designed the study. HAG and TB coordinated the study. HAG drafted the manuscript with help from all co-authors, performed genomic, phylogenetic and physiological analyses and interpretation of the data. FK carried out isolation of bacteria, molecular biological and physiological studies, phylogenetic and genomic analysis. KG isolated bacteria and performed molecular biological screenings and phylogenetic analysis. SV and AP carried out sequencing, assembly and annotation of the genome, and performed phylogenomic analysis. TB and AT interpreted the data and revised the ms critically for intellectual content. All authors reviewed and improved the final manuscript and declare no financial interest or any conflict of interest. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Abbreviations
- AHL
N-acyl-L-homoserine lactone
- DDH
DNA-DNA hybridization
- DWH
Deepwater Horizon
- EPS
Exopolymeric substances
- MLSA
Multilocus sequence analysis
- RHD
Ring hydroxylating dioxygenase
- TDA
Tropodithietic acid
Additional file
Enrichment culture of surface seawater contaminated with weathered oil (slicks) from the Deepwater Horizon (DWH) oil spill with oil and oil–water phase. From the latter, indicated by an arrow, Rhodobacteraceae strain O3.65 was isolated. Table S1. Dissimilarity (%) based on 16S rRNA gene sequence comparison 29 and in silico DNA-DNA hybridization (DDH) of strain O3.65 using 16S rRNA gene sequences and genomes of (typeT31) strains of the genera Phaeobacter, Pseudophaeobacter, Leisingera, Ruegeria and other relevant strains. The neighbor-joining distance matrix tool of the ARB software was used for calculation of 16S rRNA gene similarity. DDH was done using the genome to genome distance calculator (GGDC 2.0, DSMZ, http://ggdc.dsmz.de/distcalc2.php, [77, 78]) and represents values of the recommended formula 2 [79]. Table sorted by increasing dissimilarity. (PDF 262 kb)
References
- 1.Buchan A, LeCleir GR, Gulvik CA, Gonzalez JM. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol. 2014;12(10):686–698. doi: 10.1038/nrmicro3326. [DOI] [PubMed] [Google Scholar]
- 2.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang SX, Mann AJ, Waldmann J, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336(6081):608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 3.Wemheuer B, Gullert S, Billerbeck S, Giebel HA, Voget S, Simon M, Daniel R. Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. Fems Microbiol Ecol. 2014;87(2):378–389. doi: 10.1111/1574-6941.12230. [DOI] [PubMed] [Google Scholar]
- 4.Buchan A, Gonzalez JM, Moran MA. Overview of the marine Roseobacter lineage. Appl Environ Microbiol. 2005;71(10):5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinkhoff T, Giebel HA, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 2008;189(6):531–539. doi: 10.1007/s00203-008-0353-y. [DOI] [PubMed] [Google Scholar]
- 6.Giebel HA, Brinkhoff T, Zwisler W, Selje N, Simon M. Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ Microbiol. 2009;11(8):2164–2178. doi: 10.1111/j.1462-2920.2009.01942.x. [DOI] [PubMed] [Google Scholar]
- 7.Pujalte MJ, Lucena T, Ruvira MA, Arahal DR, Macián MC. The family Rhodobacteraceae. In: Rosenberg E, DeLong EF, Stackebrandt E, Lory S, Thompson F, editors. The prokaryotes-alphaproteobacteria and betaproteobacteria. 4. Berlin: Springer; 2014. pp. 439–512. [Google Scholar]
- 8.Wagner-Döbler I, Biebl H. Environmental biology of the marine Roseobacter lineage. Annu Rev Microbiol. 2006;60:255–280. doi: 10.1146/annurev.micro.60.080805.142115. [DOI] [PubMed] [Google Scholar]
- 9.Buchan A, González JM. Roseobacter. In: Timmis K, editor. Handbook of Hydrocarbon and Lipid Microbiology. Berlin Heidelberg: Springer; 2010. pp. 1335–1343. [Google Scholar]
- 10.Brakstad OG, Lodeng AGG. Microbial diversity during biodegradation of crude oil in seawater from the North Sea. Microb Ecol. 2005;49(1):94–103. doi: 10.1007/s00248-003-0225-6. [DOI] [PubMed] [Google Scholar]
- 11.McKew BA, Coulon F, Osborn AM, Timmis KN, McGenity TJ. Determining the identity and roles of oil-metabolizing marine bacteria from the Thames estuary, UK. Environ Microbiol. 2007;9(1):165–176. doi: 10.1111/j.1462-2920.2006.01125.x. [DOI] [PubMed] [Google Scholar]
- 12.Arnosti C, Ziervogel K, Yang T, Teske A. Oil-derived marine aggregates – hot spots of polysaccharide degradation by specialized bacterial communities. Deep-Sea Res II. 2016;129:179–186. doi: 10.1016/j.dsr2.2014.12.008. [DOI] [Google Scholar]
- 13.Sauret C, Böttjer D, Talarmin A, Guigue C, Conan P, Pujo-Pay M, Ghiglione J-F. Top-Down Control of Diesel-Degrading Prokaryotic Communities. Microb Ecol. 2015;70(2):445–458. doi: 10.1007/s00248-015-0596-5. [DOI] [PubMed] [Google Scholar]
- 14.Størdal IF, Olsen AJ, Jenssen BM, Netzer R, Hansen BH, Altin D, Brakstad OG. Concentrations of viable oil-degrading microorganisms are increased in feces from Calanus finmarchicus feeding in petroleum oil dispersions. Mar Pollut Bull. 2015;98(1–2):69–77. doi: 10.1016/j.marpolbul.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Brito EM, Guyoneaud R, Goni-Urriza M, Ranchou-Peyruse A, Verbaere A, Crapez MAC, Wasserman JCA, Duran R. Characterization of hydrocarbonoclastic bacterial communities from mangrove sediments in Guanabara Bay, Brazil. Res Microbiol. 2006;157(8):752–762. doi: 10.1016/j.resmic.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Harwati TU, Kasai Y, Kodama Y, Susilaningsih D, Watanabe K. Characterization of diverse hydrocarbon-degrading bacteria isolated from Indonesian seawater. Microbes Environ. 2007;22(4):412–415. doi: 10.1264/jsme2.22.412. [DOI] [Google Scholar]
- 17.McNutt MK, Camilli R, Crone TJ, Guthrie GD, Hsieh PA, Ryerson TB, Savas O, Shaffer F. Review of flow rate estimates of the Deepwater Horizon oil spill. Proc Natl Acad Sci U S A. 2012;109(50):20260–20267. doi: 10.1073/pnas.1112139108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, Jansson JK, Probst A, Borglin SE, Fortney JL, et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science. 2010;330(6001):204–208. doi: 10.1126/science.1195979. [DOI] [PubMed] [Google Scholar]
- 19.Redmond MC, Valentine DL. Natural gas and temperature structured a microbial community response to the Deepwater Horizon oil spill. Proc Natl Acad Sci. 2012;109(50):20292–20297. doi: 10.1073/pnas.1108756108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang T, Nigro LM, Gutierrez T, D’Ambrosio L, Joye SB, Highsmith R, Teske A. Pulsed blooms and persistent oil-degrading bacterial populations in the water column during and after the Deepwater Horizon blowout. Deep-Sea Res II. 2016;129:282–291. doi: 10.1016/j.dsr2.2014.01.014. [DOI] [Google Scholar]
- 21.Kleindienst S, Paul JH, Joye SB. Using dispersants after oil spills: impacts on the composition and activity of microbial communities. Nat Rev Microbiol. 2015;13(6):388–396. doi: 10.1038/nrmicro3452. [DOI] [PubMed] [Google Scholar]
- 22.Passow U, Ziervogel K, Asper V, Diercks A. Marine snow formation in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ Res Lett. 2012;7(3):035301. doi: 10.1088/1748-9326/7/3/035301. [DOI] [Google Scholar]
- 23.Ziervogel K, McKay L, Rhodes B, Osburn CL, Dickson-Brown J, Arnosti C, Teske A. Microbial Activities and Dissolved Organic Matter Dynamics in Oil-Contaminated Surface Seawater from the Deepwater Horizon Oil Spill Site. Plos One. 2012;7(4):e34816. doi: 10.1371/journal.pone.0034816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joye SB, Teske AP, Kostka JE. Microbial Dynamics Following the Macondo Oil Well Blowout across Gulf of Mexico Environments. Bioscience. 2014;64(9):766–777. doi: 10.1093/biosci/biu121. [DOI] [Google Scholar]
- 25.Zech H, Thole S, Schreiber K, Kalhofer D, Voget S, Brinkhoff T, Simon M, Schomburg D, Rabus R. Growth phase-dependent global protein and metabolite profiles of Phaeobacter gallaeciensis strain DSM 17395, a member of the marine Roseobacter-clade. Proteomics. 2009;9(14):3677–3697. doi: 10.1002/pmic.200900120. [DOI] [PubMed] [Google Scholar]
- 26.Giebel HA, Kalhoefer D, Gahl-Janssen R, Choo YJ, Lee K, Cho JC, Tindall BJ, Rhiel E, Beardsley C, Aydogmus OO, et al. Planktomarina temperata gen. nov., sp nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int J Syst Evol Microbiol. 2013;63:4207–4217. doi: 10.1099/ijs.0.053249-0. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1–2):203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 28.Ludwig W. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32(4):1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dogs M, Voget S, Teshima H, Petersen J, Davenport K, Dalingault H, Chen A, Pati A, Ivanova N, Goodwin LA, et al. Genome sequence of Phaeobacter inhibens type strain (T5(T)), a secondary metabolite producing representative of the marine Roseobacter clade, and emendation of the species description of Phaeobacter inhibens. Stand Genomic Sci. 2013;9(2):334–350. doi: 10.4056/sigs.4448212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beyersmann PG, Chertkov O, Petersen J, Fiebig A, Chen A, Pati A, Ivanova N, Lapidus A, Goodwin LA, Chain P, et al. Genome sequence of Phaeobacter caeruleus type strain (DSM 24564(T)), a surface-associated member of the marine Roseobacter clade. Stand Genomic Sci. 2013;8(3):403–419. doi: 10.4056/sigs.3927626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breider S, Scheuner C, Schumann P, Fiebig A, Petersen J, Pradella S, Klenk HP, Brinkhoff T, Göker M. Genome-scale data suggest reclassifications in the Leisingera-Phaeobacter cluster including proposals for Sedimentitalea gen. nov and Pseudophaeobacter gen. nov. Frontiers in. Microbiology. 2014;5:416. doi: 10.3389/fmicb.2014.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riedel T, Teshima H, Petersen J, Fiebig A, Davenport K, Daligault H, Erkkila T, Gu W, Munk C, Xu Y, et al. Genome sequence of the Leisingera aquimarina type strain (DSM 24565(T)), a member of the marine Roseobacter clade rich in extrachromosomal elements. Stand Genomic Sci. 2013;8(3):389–402. doi: 10.4056/sigs.3858183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buddruhs N, Chertkov O, Petersen J, Fiebig A, Chen A, Pati A, Ivanova N, Lapidus A, Goodwin LA, Chain P, et al. Complete genome sequence of the marine methyl-halide oxidizing Leisingera methylohalidivorans type strain (DSM 14336 T), a representative of the Roseobacter clade. Stand Genomic Sci. 2013;9(1):128–141. doi: 10.4056/sigs.4297965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frank O, Pradella S, Rohde M, Scheuner C, Klenk HP, Goker M, Petersen J. Complete genome sequence of the Phaeobacter gallaeciensis type strain CIP 105210(T) (= DSM 26640(T) = BS107(T) Stand Genomic Sci. 2014;9(3):914–932. doi: 10.4056/sigs.5179110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thole S, Kalhoefer D, Voget S, Berger M, Engelhardt T, Liesegang H, Wollherr A, Kjelleberg S, Daniel R, Simon M, et al. Phaeobacter gallaeciensis genomes from globally opposite locations reveal high similarity of adaptation to surface life. Isme J. 2012;6(12):2229–2244. doi: 10.1038/ismej.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buddruhs N, Pradella S, Goker M, Pauker O, Pukall R, Sproer C, Schumann P, Petersen J, Brinkhoff T. Molecular and phenotypic analyses reveal the non-identity of the Phaeobacter gallaeciensis type strain deposits CIP 105210(T) and DSM 17395. Int J Syst Evol Microbiol. 2013;63:4340–4349. doi: 10.1099/ijs.0.053900-0. [DOI] [PubMed] [Google Scholar]
- 37.Vandecandelaere I, Nercessian O, Segaert E, Achouak W, Faimali M, Vandamme P. Ruegeria scottomollicae sp. nov., isolated from a marine electroactive biofilm. Int J Syst Evol Microbiol. 2008;58:2726–2733. doi: 10.1099/ijs.0.65843-0. [DOI] [PubMed] [Google Scholar]
- 38.Fernandes N, Case RJ, Longford SR, Seyedsayamdost MR, Steinberg PD, Kjelleberg S, Thomas T. Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. Plos One. 2011;6(12):e27387. doi: 10.1371/journal.pone.0027387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park S, Park D-S, Bae KS, Yoon J-H. Phaeobacter aquaemixtae sp. nov., isolated from the junction between the ocean and a freshwater spring. Int J Syst Evol Microbiol. 2014;64(4):1378–1383. doi: 10.1099/ijs.0.057646-0. [DOI] [PubMed] [Google Scholar]
- 40.Oh KH, Jung YT, Oh TK, Yoon JH. Ruegeria faecimaris sp nov., isolated from a tidal flat sediment. Int J Syst Evol Microbiol. 2011;61:1182–1188. doi: 10.1099/ijs.0.025999-0. [DOI] [PubMed] [Google Scholar]
- 41.Collins AJ, Fullmer MS, Gogarten JP, Nyholm SV. Comparative genomics of Roseobacter clade bacteria isolated from the accessory nidamental gland of Euprymna scolopes. Front Microbiol. 2015;6:123. doi: 10.3389/fmicb.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YG, Hwang CY, Cho BC. Pelagicola litoralis gen.nov., sp nov., isolated from coastal water in Korea. Int J Syst Evol Microbiol. 2008;58:2102–2106. doi: 10.1099/ijs.0.65820-0. [DOI] [PubMed] [Google Scholar]
- 43.Freese HM, Dalingault H, Petersen J, Pradella S, Davenport K, Teshima H, Chen A, Pati A, Ivanova N, Goodwin LA, et al. Genome sequence of the phage-gene rich marine Phaeobacter arcticus type strain DSM 23566(T) Stand Genomic Sci. 2013;8(3):450–464. doi: 10.4056/sigs.383362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon JH, Kang SJ, Lee JS, Oh TK. Lutimaribacter saemankumensis gen. nov., sp nov., isolated from a tidal flat of the Yellow Sea. Int J Syst Evol Microbiol. 2009;59:48–52. doi: 10.1099/ijs.0.000109-0. [DOI] [PubMed] [Google Scholar]
- 45.Uchino Y, Hirata A, Yokota A, Sugiyama J. Reclassification of marine Agrobacterium species: Proposals of Stappia stellulata gen. nov., comb. nov., Stappia aggregata sp. nov., nom. rev., Ruegeria atlantica gen. nov., comb. nov., Ruegeria gelatinovora comb. nov., Ruegeria algicola comb. nov., and Ahrensia kieliense gen. nov., sp. nov., nom. rev. J Gen Appl Microbiol. 1998;44(3):201–210. doi: 10.2323/jgam.44.201. [DOI] [PubMed] [Google Scholar]
- 46.Lee K. Ruegeria pelagia sp. nov., isolated from the Sargasso Sea, Atlantic ocean. Int J Syst Evol Microbiol. 2007;57:1815–1818. doi: 10.1099/ijs.0.65032-0. [DOI] [PubMed] [Google Scholar]
- 47.Lee J, Roh SW, Whon TW, Shin NR, Kim YO, Bae JW. Genome sequence of strain TW15, a novel member of the genus Ruegeria, belonging to the marine Roseobacter clade. J Bacteriol. 2011;193(13):3401–3402. doi: 10.1128/JB.05067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee M-H, Song E-J, Seo M-J, Hyun D-W, Bae J-W, Lee S-Y, Roh S, Nam Y-D. Phaeobacter marinintestinus sp. nov., isolated from the intestine of a sea cucumber (Apostichopus japonicus) Antonie Van Leeuwenhoek. 2015;107(1):209–216. doi: 10.1007/s10482-014-0318-x. [DOI] [PubMed] [Google Scholar]
- 49.Rivers A, Smith C, Moran M. An Updated genome annotation for the model marine bacterium Ruegeria pomeroyi DSS-3. Stand Genomic Sci. 2014;9(1):1–9. doi: 10.1186/1944-3277-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park S, Yoon JH. Ruegeria arenilitoris sp. nov., isolated from the seashore sand around a seaweed farm. Anton Leeuw Int J Gen Mol Microbiol. 2012;102(4):581–589. doi: 10.1007/s10482-012-9753-8. [DOI] [PubMed] [Google Scholar]
- 51.Kim YO, Park S, Nam BH, Kang SJ, Hur YB, Lee SJ, Oh TK, Yoon JH. Ruegeria halocynthiae sp. nov., isolated from the sea squirt Halocynthia roretzi. Int J Syst Evol Microbiol. 2012;62:925–930. doi: 10.1099/ijs.0.031609-0. [DOI] [PubMed] [Google Scholar]
- 52.Huo YY, Xu XW, Li X, Liu C, Cui HL, Wang CS, Wu M. Ruegeria marina sp. nov., isolated from Marine Sediment. Int J Syst Evol Microbiol. 2011;61:347–350. doi: 10.1099/ijs.0.022400-0. [DOI] [PubMed] [Google Scholar]
- 53.Jin HM, Lee HJ, Kim JM, Park MS, Lee K, Jeon CO. Litorimicrobium taeanense gen. nov., sp nov., isolated from a sandy beach. Int J Syst Evol Microbiol. 2011;61:1392–1396. doi: 10.1099/ijs.0.025007-0. [DOI] [PubMed] [Google Scholar]
- 54.Moran MA, Belas R, Schell MA, Gonzalez JM, Sun F, Sun S, Binder BJ, Edmonds J, Ye W, Orcutt B, et al. Ecological genomics of marine roseobacters. Appl Environ Microbiol. 2007;73(14):4559–4569. doi: 10.1128/AEM.02580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doberva M, Sanchez-Ferandin S, Ferandin Y, Intertaglia L, Croué J, Suzuki M, Lebaron P, Lami R. Genome Sequence of the Sponge-Associated Ruegeria halocynthiae strain MOLA R1/13b, a marine roseobacter with two quorum-sensing-based communication systems. Genome Announce. 2014;2(5):e00998–14. doi: 10.1128/genomeA.00998-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim YO, Park S, Nam BH, Jung YT, Kim DG, Yoon JH. Ruegeria meonggei sp. nov., an alphaproteobacterium isolated from ascidian Halocynthia roretzi. Anton Leeuw Int J Gen Mol Microbiol. 2014;105(3):551–558. doi: 10.1007/s10482-013-0107-y. [DOI] [PubMed] [Google Scholar]
- 57.Gaboyer F, Tindall BJ, Ciobanu MC, Duthoit F, Le Romancer M, Alain K. Phaeobacter leonis sp. nov., an alphaproteobacterium from Mediterranean Sea sediments. Int J Syst Evol Microbiol. 2013;63:3301–3306. doi: 10.1099/ijs.0.046128-0. [DOI] [PubMed] [Google Scholar]
- 58.Kampfer P, Arun AB, Rekha PD, Busse HJ, Young CC, Glaeser SP. Ruegeria intermedia sp. nov., a moderately thermophilic bacterium isolated from a coastal hot spring. Int J Syst Evol Microbiol. 2013;63:2538–2544. doi: 10.1099/ijs.0.047910-0. [DOI] [PubMed] [Google Scholar]
- 59.Martens T, Heidorn T, Pukall R, Simon M, Tindall BJ, Brinkhoff T. Reclassification of Roseobacter gallaeciensis Ruiz-Ponte et al. 1998 as Phaeobacter gallaeciensis gen. nov., comb. nov., description of Phaeobacter inhibens sp nov., reclassification of Ruegeria algicola (Lafay et al. 1995) Uchino et al. 1999 as Marinovum algicola gen. nov., comb. nov., and emended descriptions of the genera Roseobacter, Ruegeria and Leisingera. Int J Syst Evol Microbiol. 2006;56:1293–1304. doi: 10.1099/ijs.0.63724-0. [DOI] [PubMed] [Google Scholar]
- 60.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43(D1):D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 66.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340(4):783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 67.Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang JH, Woyke T, Huntemann M, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42(D1):D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petersen J, Frank O, Göker M, Pradella S. Extrachromosomal, extraordinary and essential—the plasmids of the Roseobacter clade. Appl Microbiol Biotechnol. 2013;97(7):2805–2815. doi: 10.1007/s00253-013-4746-8. [DOI] [PubMed] [Google Scholar]
- 69.Buchan A, Neidle EL, Moran MA. Diverse organization of genes of the beta-ketoadipate pathway in members of the marine Roseobacter lineage. Appl Environ Microbiol. 2004;70(3):1658–1668. doi: 10.1128/AEM.70.3.1658-1668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Romero-Steiner S, Parales RE, Harwood CS, Houghton JE. Characterization of the pcaR regulatory gene from Pseudomonas putida, which is required for the complete degradation of p-hydroxybenzoate. J Bacteriol. 1994;176(18):5771–5779. doi: 10.1128/jb.176.18.5771-5779.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berger M, Brock NL, Liesegang H, Dogs M, Preuth I, Simon M, Dickschat JS, Brinkhoff T. Genetic analysis of the upper phenylacetate catabolic pathway in the production of tropodithietic acid by Phaeobacter gallaeciensis. Appl Environ Microbiol. 2012;78(10):3539–3551. doi: 10.1128/AEM.07657-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu C, Shao Z. Alcanivorax dieselolei sp. nov., a novel alkane-degrading bacterium isolated from sea water and deep-sea sediment. Int J Syst Evol Microbiol. 2005;55(3):1181–1186. doi: 10.1099/ijs.0.63443-0. [DOI] [PubMed] [Google Scholar]
- 73.Gutierrez T, Berry D, Yang TT, Mishamandani S, McKay L, Teske A, Aitken MD. Role of bacterial exopolysaccharides (EPS) in the fate of the oil released during the Deepwater Horizon oil spill. Plos One. 2013;8(6):e67717. doi: 10.1371/journal.pone.0067717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Camilli R, Reddy CM, Yoerger DR, Van Mooy BAS, Jakuba MV, Kinsey JC, McIntyre CP, Sylva SP, Maloney JV. Tracking hydrocarbon plume transport and biodegradation at Deepwater Horizon. Science. 2010;330(6001):201–204. doi: 10.1126/science.1195223. [DOI] [PubMed] [Google Scholar]
- 75.Diercks AR, Highsmith RC, Asper VL, Joung D, Zhou Z, Guo L, Shiller AM, Joye SB, Teske AP, Guinasso N. Characterization of subsurface polycyclic aromatic hydrocarbons at the Deepwater Horizon site. Geophys Res Lett. 2010;37(20):160–164. doi: 10.1029/2010GL045046. [DOI] [Google Scholar]
- 76.Harayama S, Kishira H, Kasai Y, Shutsubo K. Petroleum biodegradation in marine environments. J Mol Microbiol Biotechnol. 1999;1(1):63–70. [PubMed] [Google Scholar]
- 77.Auch AF, Klenk HP, Goker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci. 2010;2(1):142–148. doi: 10.4056/sigs.541628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Auch AF, von Jan M, Klenk HP, Goker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2(1):117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. Bmc Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cunliffe M. Correlating carbon monoxide oxidation with cox genes in the abundant Marine Roseobacter Clade. Isme J. 2011;5(4):685–691. doi: 10.1038/ismej.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cunliffe M. Physiological and metabolic effects of carbon monoxide oxidation in the model marine bacterioplankton ruegeria pomeroyi DSS-3. Appl Environ Microbiol. 2013;79(2):738–740. doi: 10.1128/AEM.02466-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moran MA, Miller WL. Resourceful heterotrophs make the most of light in the coastal ocean. Nat Rev Microbiol. 2007;5(10):792–800. doi: 10.1038/nrmicro1746. [DOI] [PubMed] [Google Scholar]
- 83.Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, Howard EC, King E, Oakley CA, Reisch CR, et al. Genome characteristics of a generalist marine bacterial lineage. Isme J. 2010;4(6):784–798. doi: 10.1038/ismej.2009.150. [DOI] [PubMed] [Google Scholar]
- 84.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Muller R, Wohlleben W, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martens T, Gram L, Grossart HP, Kessler D, Muller R, Simon M, Wenzel SC, Brinkhoff T. Bacteria of the Roseobacter clade show potential for secondary metabolite production. Microb Ecol. 2007;54(1):31–42. doi: 10.1007/s00248-006-9165-2. [DOI] [PubMed] [Google Scholar]
- 86.Bruhn JB, Nielsen KF, Hjelm M, Hansen M, Bresciani J, Schulz S, Gram L. Ecology, inhibitory activity, and morphogenesis of a marine antagonistic bacterium belonging to the Roseobacter clade. Appl Environ Microbiol. 2005;71(11):7263–7270. doi: 10.1128/AEM.71.11.7263-7270.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bruhn JB, Gram L, Belas R. Production of antibacterial compounds and biofilm formation by Roseobacter species are influenced by culture conditions. Appl Environ Microbiol. 2007;73(2):442–450. doi: 10.1128/AEM.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prado S, Montes J, Romalde JL, Barja JL. Inhibitory activity of Phaeobacter strains against aquaculture pathogenic bacteria. Int Microbiol. 2009;12:107–114. [PubMed] [Google Scholar]
- 89.Brinkhoff T. Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl Environ Microbiol. 2004;70(4):2560–2565. doi: 10.1128/AEM.70.4.2560-2565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Micro. 2003;1(2):137–149. doi: 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Craig L, Pique ME, Tainer JA. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol. 2004;2(5):363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- 92.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Woese CR, Kandler O, Wheelis ML. Towards a Natural System of Organisms - Proposal for the Domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology. Second ed. Volume 2 (The Proteobacteria), Part B (The Gammaproteobacteria). New York: Springer; 2005. p 1.
- 95.Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov..In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second ed. Volume 2, Part C. New York: Springer; 2005. p 1.
- 96.107 VLN List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56(1):1–6. doi: 10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- 97.Garrity GM, Bell JA, Lilburn T. Family I. Rhodobacteraceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s Manual of Systematic Bacteriology. Second ed. Volume 2, Part C. New York: Springer; 2005. p 1.
- 98.BAuA Classification of Bacteria and Archaea in risk groups. TRBA. 2010;466:93. [Google Scholar]
- 99.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. Bmc Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]