

Abstract
Background
Activation of oncogenes downstream the EGFR gene contributes to colorectal tumorigenesis and determines the sensitivity to anti-EGFR treatments. The aim of this study was to evaluate the prognostic value of KRAS, BRAF, NRAS and PIK3CA mutations in a large collection of CRC patients from genetically-homogeneous Sardinian population.
Methods
A total of 1284 Sardinian patients with histologically-proven diagnosis of colorectal carcinoma (CRC) and presenting with metastatic disease were included into the study. Genomic DNA was isolated from formalin-fixed, paraffin-embedded primary tumour tissue samples of CRC patients and screened for mutations in RAS and BRAF genes, using pyrosequencing assays, and in PIK3CA gene, using automated DNA sequencing assays.
Results
Overall, mutation rates were 35.6 % for KRAS, 4.1 % for NRAS, and 2.1 % for BRAF. Among available DNA samples, 114/796 (14.3 %) primary CRCs were found to carry a mutation in the PIK3CA gene. In this subset of patients analysed in all four genes, a pathogenetic mutation of at least one gene was discovered in about half (378/796; 47.5 %) of CRC cases. A mutated BRAF gene was found to steadily act as a negative prognostic factor for either time to progression as metastatic disease (from detection of primary CRC to diagnosis of first distant metastasis; p = 0.009) or partial survival (from diagnosis of advanced disease to the time of death or last control; p = 0.006) or overall survival (p < 0.001). No significant impact on prognosis was observed for mutated KRAS, NRAS, and PIK3CA genes or combined RAS mutations (all RAS).
Conclusions
Our study defines both prevalence and prognostic role of main activated oncogenes in a population-based large collection of CRC patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-016-1053-z) contains supplementary material, which is available to authorized users.
Keywords: Colorectal cancer, KRAS, NRAS, BRAF, PIC3CA
Background
Colorectal cancer (CRC) is the third most incident malignancy in both sexes, after lung and prostate cancer in men and breast and cervix cancer in women; it also represents the fourth most frequent neoplastic cause of death after lung, stomach, and liver cancer [1]. The disease is more common in men than women. In 2015, 132,700 new cases of colorectal cancer and 49,700 disease-related deaths were estimated in USA [2]. The percent of colon and rectum cancer deaths is highest among people aged 75-84 [2].
From the pathogenetic point of view, CRC is a complex and heterogeneous neoplastic disease, exhibiting multiple genetic and epigenetic alterations. The accumulation of acquired molecular aberrations is involved in colorectal tumorigenesis, able to transform normal colonic epithelieum into adenocarcinomas. Starting from the model proposed by Fearon and Vogelstein in 1990, not only tubular and tubulovillous adenoma may progress to invasive adenocarcinoma, but it is now recognized that the serrated adenomas (SSA) and traditional serrated adenomas (TSA), originally excluded, may also undergo malignant transformation [3, 4].
At least four kinds of different pathogenic mechanisms have been proposed to be relevant in CRC classification: chromosomal instability (CIN), microsatellite instability (MSI), CpG island methylator phenotype (CIMP) and global DNA hypomethylation. The CIN pathway is the most common feature, occurring in 85 % of colorectal cancers and showing numerical chromosome changes and/or multiple structural aberrations. Different studies suggest that CIN promotes cancer progression by increasing clonal diversity [5]. It involves mutations in the APC gene and/or the loss of chromosome 5q where it is located, mutations in the KRAS oncogene, the loss of chromosomal arm 18q, and deletion of chromosome 17p where is mapped the TP53 tumour suppressor gene [6]. The MSI phenotype is present in the remaining 15 % of CRCs. It is usually mutually exclusive with CIN aberrations and involves the impairment of the DNA mismatch repair system. This damage causes replication errors at genomic level—detectable as numerical alterations in the repetitive units of DNA microsatellites—and inactivating mutations in target tumour suppressor genes [7]. The CIMP feature is characterized by hypermethylation of genomic DNA located in CpG islands, specific regulatory sites enriched in CpG motifs and mapped in the promoter regions of tumour suppressor genes. Although the mechanism is still under investigation, different studies suggest a pathogenetic correlation between CIMP and occurrence of BRAF mutations in CRC [7, 8]. Finally, a reduction of DNA methylation rates has also been reported in majority of CIN-positive CRCs [9].
Altogether, the above-mentioned genetic and epigenetic alterations have a powerful impact on different cellular functions: cell proliferation, apoptosis, differentiation, angiogenesis, invasion and immortalization [10, 11]. Among others, the EGFR-RAS-RAF-MEK-ERK and the PI3 K-AKT-mTOR signalling cascades play the main roles in CRC development and progression. In particular, activated RAS (mainly, KRAS and NRAS) proteins promote cell proliferation through constitutive stimulation of the downstream RAF-MEK-ERK effectors in the so-called mitogen-activated protein-kinase (MAPK) pathway [12].
In recent past years, the EGFR-depending pathway has been largely exploited for personalized therapies and, in particular, EGFR has become a key target of specific inhibitors to treat metastatic CRCs [13, 14]. Activating mutations in K-/N-RAS are recognized as a strong predictor of resistance to EGFR-targeted agents (monoclonal antibodies, mAbs), since they cause a constitutive phosphorylation of the RAS proteins—independent on activation status of the upstream EGFR protein—which in turn permanently promote cell proliferation and drastically reduce the effects of the EGFR inhibition [15–18].
The role of RAS mutations as prognostic factors in CRC patients remains uncertain and may be somehow dependent on populations’ origin [19–22]. Conversely, oncogenic BRAF mutations seem not to be predictive of insensitivity to the anti-EGFR therapy, but are recognized as predictors of poor survival [21, 23–27].
In Sardinia, whose population is genetically homogeneous due to its historical isolation and, thus, presents a high rate of inbreeding, colorectal cancer represents the second principal death-causing malignancy, with an incidence quite comparable with that observed in Western countries (standardized rate, 117.4 per 100.000 inhabitants per year; Sardinian population includes about one million and half inhabitants) [28, 29]. The contribution of KRAS, BRAF, and PIK3CA mutations to CRC pathogenesis was previously investigated by our group on a limited subset of patients from Sardinia [30]. The aim of this study was to evaluate the prognostic role of the somatic mutations in all candidate genes (KRAS, BRAF, NRAS, and PIK3CA) using a very large population-based collection of Sardinian patients with metastatic CRC.
Methods
Samples
One thousand two hundred and eighty-four patients with histologically-proven diagnosis of colorectal carcinoma (CRC) were included into the study. At the time of enrolment, only patients presenting with metastatic disease (stage IV, according to the American Joint Committee on Cancer (AJCC) guidelines [31]) entered the study. For all included patients, medical records and pathology reports were used to assess the histological classification and disease stage at the time of the diagnosis of the primary CRC. To avoid any bias, CRC patients were consecutively collected from September 2009 to December 2015; they were included regardless of age at diagnosis and disease characteristics of the primary tumour. No CRC case from our series was associated with clinically relevant colorectal polyposis. Sardinian origin was ascertained in all cases through verification of the place of birth for all patients and their parents.
Formalin-fixed, paraffin-embedded tissue samples from CRC patients were obtained from the archives of the Institutes and Services of Pathology participating to the study. Tissue sections were estimated to contain at least 80 % neoplastic cells by light microscopy.
All patients were informed about the aims of this study and, before the tissue sample was collected, gave a written informed consent. The study was reviewed and approved by the ethical review board of the University of Sassari.
Mutation analysis
Genomic DNA was isolated from tissue sections using standard protocols, as previously described [28]. Briefly, somatic DNA was purified using the QIAamp DNA FFPE Tissue kit (Qiagen Inc., Valencia, CA, USA) and the DNA quality assessed for each specimen.
Mutation analysis was conducted in the coding sequence of the following genes: KRAS (exons 2, 3, and 4), NRAS (exons 2, 3, and 4), BRAF (exon 15, where almost all of the oncogenic mutations are located), and PIK3CA (exons 9 and 20, which are mostly implicated in the protein kinase activity). For KRAS, NRAS, and BRAF genes, quantitative measurements of mutations were based on pyrosequencing assays, which were performed on a PyroMark Q24 system (Qiagen Inc., USA), following the manufacturer’s instructions. For PIK3CA gene, exons 9 and 20 were investigated for mutations by direct sequencing, using an automated fluorescence-based cycle sequencer (ABIPRISM 3130, Life Technologies-ThermoFisher Scientific, Waltham, MA, USA), as previously described by our group [30]. Protocols for PCR-based assays will be available upon request. For PIK3CA gene, mutation analysis was missing in about two-fifths of cases (488/1284; 38 %) due to the low amount of available tumour tissue samples.
Statistical analysis
The Cox regression model was performed using raw mortality and tumor-specific mortality. The time of overall survival was expressed in months, and the independent variables (KRAS, NRAS, all-RAS, BRAF, RAS-BRAF, PIK3CA, all genes, pT, pN, M) were stratified in age groups: 32–45 (N = 60), 46–64 (N = 440), and 65–98 (N = 491). Evaluation of the impact of all independent variables on prognosis was calculated regardless the type of treatment (EGFR inhibition or others) administered to patients. Kaplan–Meier estimates were executed through stratification by mutation data. The above-mentioned variables (mutational status, survivals, pT, pN, M) were included in a logistic regression for multivariate analysis by Pearson’s Chi Square test. The exact coefficient for sample proportion analysis was performed to determine whether there was any significant difference (below 0.05 level) between considering a mutated gene alone and considering mutations in multiple genes. All analyses were performed using the statistical package SPSS/7.5 per Windows.
Results
Genomic DNA from primary tumour tissues of 1284 consecutively-collected patients with metastatic colorectal carcinoma (mCRC), originating from different geographical areas within Sardinia island, was screened for somatic mutations in KRAS, NRAS, BRAF, and PIK3CA genes. As reported in Table 1, the most frequent anatomical site and degree of differentiation of the primary tumour were the left colon (516; 40 %) and the moderately differentiated grade (1041; 81 %), respectively. The median age was 64 years (range, 32-88 years), with a preponderance of males (772; 60 %) (Table 1). At the time of diagnosis, minority of patients presented with localized disease (AJCC stage II: 286; 22 %) (Table 1).
Table 1.
Distribution of RAS-BRAF mutations according to the characteristics of CRC patients
| Characteristic | No. | % | KRAS mut | NRAS mut | BRAF mut | All mut |
|---|---|---|---|---|---|---|
| Sex | ||||||
| Male | 772 | 60.1 | 253 (32.8 %) | 31 (4.0 %) | 13 (1.7 %) | 297 (39.8 %) |
| Female | 512 | 39.9 | 204 (39.8 %) | 22 (4.3 %) | 14 (2.7 %) | 240 (46.9 %) |
| Anatomical tumour site | ||||||
| Right-transverse colon | 441 | 34.3 | 148 (35.6 %) | 18 (4.1 %) | 9 (2.0 %) | 175 (39.7 %) |
| Left colon | 516 | 40.2 | 185 (35.9 %) | 21 (4.1 %) | 10 (1.9 %) | 216 (41.9 %) |
| Rectum | 327 | 25.5 | 124 (37.9 %) | 14 (4.3 %) | 8 (2.4 %) | 146 (44.6 %) |
| Disease stage at diagnosis | ||||||
| Stage II (T3–4N0M0) | 286 | 22.3 | 108 (37.8 %) | 11 (3.8 %) | 6 (2.1 %) | 125 (43.7 %) |
| Stage III (TXN1–3M0) | 567 | 44.1 | 191 (33.7 %) | 24 (4.2 %) | 9 (1.6 %) | 224 (39.5 %) |
| Stage IV (TXNXM1) | 431 | 33.6 | 158 (36.7 %) | 18 (4.2 %) | 12 (2.8 %) | 188 (43.6 %) |
| Tumour grading | ||||||
| Well differentiated | 138 | 10.7 | 49 (35.5 %) | 7 (5.1 %) | 3 (2.2 %) | 59 (42.8 %) |
| Moderately differentiated | 1041 | 81.1 | 370 (35.5 %) | 41 (3.9 %) | 21 (2.0 %) | 432 (41.5 %) |
| Poorly differentiated | 105 | 8.2 | 38 (36.2 %) | 5 (4.8 %) | 3 (2.9 %) | 46 (43.8 %) |
| Age, years | ||||||
| ≤50 | 126 | 9.8 | 47 (37.3 %) | 10 (7.9 %) | 3 (2.4 %) | 60 (47.6 %) |
| 51–60 | 325 | 25.3 | 118 (36.3 %) | 12 (3.7 %) | 6 (1.8 %) | 136 (41.8 %) |
| 61–70 | 492 | 38.3 | 173 (35.2 %) | 19 (3.9 %) | 9 (1.8 %) | 201 (40.9 %) |
| >70 | 341 | 26.6 | 119 (34.9 %) | 12 (3.5 %) | 9 (2.6 %) | 140 (41.1 %) |
Pyrosequencing assays were conducted for identification of mutations in the exons 2, 3 and 4 of both the KRAS and NRAS oncogenes as well as in the exon 15 of the BRAF oncogene. Somatic mutations in these genes were detected in 537 of 1284 (41.8 %) primary tumours: 457 (35.6 %) KRAS, 53 (4.1 %) NRAS, and 27 (2.1 %) BRAF mutations. Overall, no concurrent mutations of KRAS, NRAS, and BRAF genes were detected. Six patients had two mutations in KRAS gene: G12C and Q61H, G12D and Q61L, G12D and Q61H, G12 V and Q61H, G12 V and Q61R, G13 V and Q61H. In Fig. 1, spectrum and distribution of the 543 mutations identified is shown. The codon 12 of KRAS (345/463; 74.5 %) and the codon 61 of NRAS (25/53; 47 %) were the most affected in our series (Additional file 1: Table S1). For BRAF, all 27 patients carrying a mutation in this gene presented the substitution of valine by a glutamic acid at position 600 (V600E), which has been widely demonstrated to account for vast majority of the BRAF mutations reported in literature (Fig. 1; Additional file 1: Table S1) [32, 33].
Fig. 1.
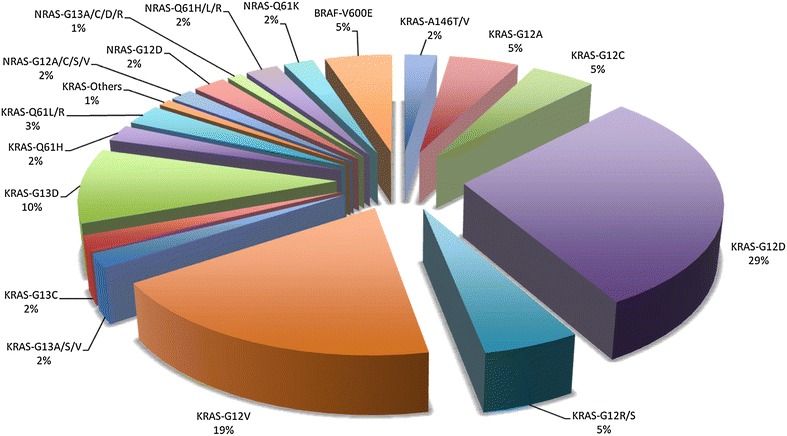
Somatic mutations in candidate genes among Sardinian CRC patients
All mutations identified in the present study are reported in both the Human Gene Mutation Database (HGMD) at http://www.hgmd.cf.ac.uk/ac/index.php and the catalogue of somatic mutations in cancer (COSMIC) at http://www.sanger.ac.uk/genetics/CGP/cosmic/.
Among available DNA samples, 796 primary tumours were also evaluated for the occurrence of pathogenetic mutations in exons 9 and 20 of the PIK3CA gene (see “Methods” section). Overall, PIK3CA mutations were detected in 114 (14.3 %) patients; one primary CRC tissue presented two mutations (E545G and Q1033L). Screening revealed the occurrence of mutations in three codons of exon 9 (95/115; 82.6 %) and four codons of exon 20 (20/115; 17.4 %) in the PIK3CA gene (Additional file 1: Table S2).
Again, all identified PIK3CA mutations have been reported in gene mutation databases (HGMD and COSMIC; see above) as commonly associated with CRC, with a recognized functional role of the corresponding mutated proteins.
Table 2 summarizes the distribution and relationship of the somatic mutations identified in the series of 796 CRC patients analysed in all four genes. Altogether, a mutation of at least one gene was discovered in about half (378/796; 47.5 %) of CRC cases; in other words, one out of two primary tumours displayed an extended wild-type genetic status, with lack of any pathogenic sequence variation in these four main candidate genes.
Table 2.
Frequencies of gene mutations in the series of 796 patients screened for all four genes
| Mutated genes | KRAS | KRAS + PIK3CA | NRAS | NRAS + PIK3CA | BRAF | BRAF + PIK3CA | PIK3CA | Wild-type |
|---|---|---|---|---|---|---|---|---|
| Cases | 227 | 36 | 21 | 8 | 16 | 6 | 64 | 418 |
| % | 28.5 | 4.5 | 2.7 | 1.0 | 2.0 | 0.8 | 8.0 | 52.5 |
Considering the patients’ origin, distribution of RAS mutations was confirmed to be heterogeneous in Sardinia, as previously reported by our group [30]: 247/537 (46.0 %) mutated cases in North Sardinia versus 263/747 (35.2 %) in Middle-South Sardinia (Fig. 2). No difference in BRAF mutation distribution was instead observed within the island: 12/537 (2.2 %) in North Sardinia versus 15/747 (2.0 %) in Middle-South Sardinia (Fig. 2). It should be kept in mind, that all somatic samples from the entire series of Sardinian CRC patients were analysed for mutations with the same methodological procedures (see “Methods” section).
Fig. 2.
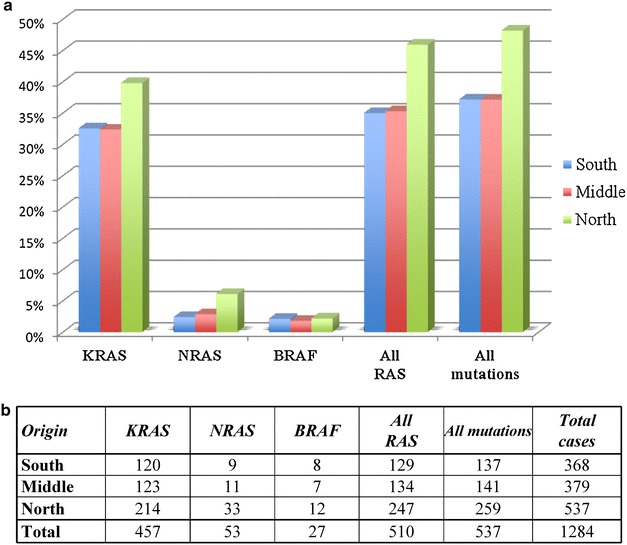
Geographical distribution of RAS and BRAF mutation carriers in Sardinia. a Frequencies in percentage; b number of cases
Mutations in KRAS, NRAS, BRAF, and PIK3CA genes were evaluated for association with several pathological parameters: sex, age at diagnosis, anatomical location of primary CRC, tumour grading, AJCC stage of the disease. No significant correlation was found between the occurrence of mutations in any of the four genes and all analysed parameters (see Table 1).
Logistic regression multivariate analysis was performed on the totality of tested patients to estimate relative risk of survival variations and to adjust potential confounding effects, as well as to assess possible multiplicative interactions. Using the Cox model adjusted according to disease stage and diagnosis age, the occurrence of mutations in BRAF gene was found to have a highly-significant negative impact on prognosis, on either partial survival (from the diagnosis of advanced disease to the time of death or last control, PS) [p = 0.006; HR: 3.21; 95 % CI: 1.38−7.44] or overall survival (from the disease onset to the time of death or last control, OS) [p < 0.001; HR: 4.12; 95 % CI: 2.49–6.81] (Table 3). No prognostic values on both survivals of either KRAS or NRAS or all RAS (KRAS + NRAS) or PIK3CA mutated status were instead observed in our series (Table 3).
Table 3.
Statistical correlation of gene mutations with prognostic parameters
| Mutated gene | Risk ratio | 95 % CI | p |
|---|---|---|---|
| Partial survival (from metastatic disease onset to death or last control) | |||
| KRAS | 1.257 | 0.628–1.739 | 0.4906 |
| NRAS | 1.634 | 1.018–2.712 | 0.0763 |
| All RAS | 1.345 | 0.968–2.158 | 0.0924 |
| BRAF | 3.214 | 1.387–7.445 | 0.0064 |
| All RAS + BRAF | 1.733 | 0.997–2.994 | 0.0511 |
| PIK3CA | 1.403 | 0.877–1.479 | 0.1365 |
| Overall survival (from disease diagnosis to death or last control) | |||
| KRAS | 1.219 | 0.983–1.512 | 0.0703 |
| NRAS | 1.052 | 0.612–1.810 | 0.8522 |
| All RAS | 1.108 | 0.932–1.316 | 0.2432 |
| BRAF | 4.120 | 2.491–6.812 | <0.001 |
| All RAS + BRAF | 1.878 | 1.238–3.996 | 0.0394 |
| PIK3CA | 0.934 | 0.711–1.226 | 0.6246 |
| Time to progression as metastatic disease (from disease diagnosis to first metastasis) | |||
| KRAS | 1.077 | 0.843–1.677 | 0.1508 |
| NRAS | 1.154 | 0.853–1.851 | 0.1006 |
| All RAS | 1.277 | 1.025–2.371 | 0.0943 |
| BRAF | 2.972 | 1.261–6.332 | 0.0091 |
| All RAS + BRAF | 2.217 | 1.125–4.371 | 0.0214 |
| PIK3CA | 0.964 | 0.691–1.346 | 0.8337 |
Considering the time to progression as metastatic disease (from the detection of primary CRC to the diagnosis of first distant metastasis, TTPM), the occurrence of BRAF mutations again remained a statistically-independent negative prognostic factor [p = 0.009; HR: 2.97; 95 % CI: 1.26–6.33] (Table 3). Noteworthy, the impact on OS and TTPM of the BRAF mutations was so highly significant to negatively affect such survivals in the combined group of mutated patients (considering all mutations in RAS and BRAF genes) (Table 3).
Using the Kaplan–Meier method, survival curves indicated that patients carrying a BRAF mutation presented strongly significant poorer partial [p < 0.001] and overall [p < 0.001] survivals in comparison with those carrying a BRAF wild-type gene (Fig. 3). With the exception of the NRAS mutations on partial survival [p = 0.047], no significant association with both types of survival was instead observed for mutations in the other genes (a prognostic value close to be significant was found for all-RAS mutation-positive status [p = 0.059] on overall survival) (Fig. 3).
Fig. 3.
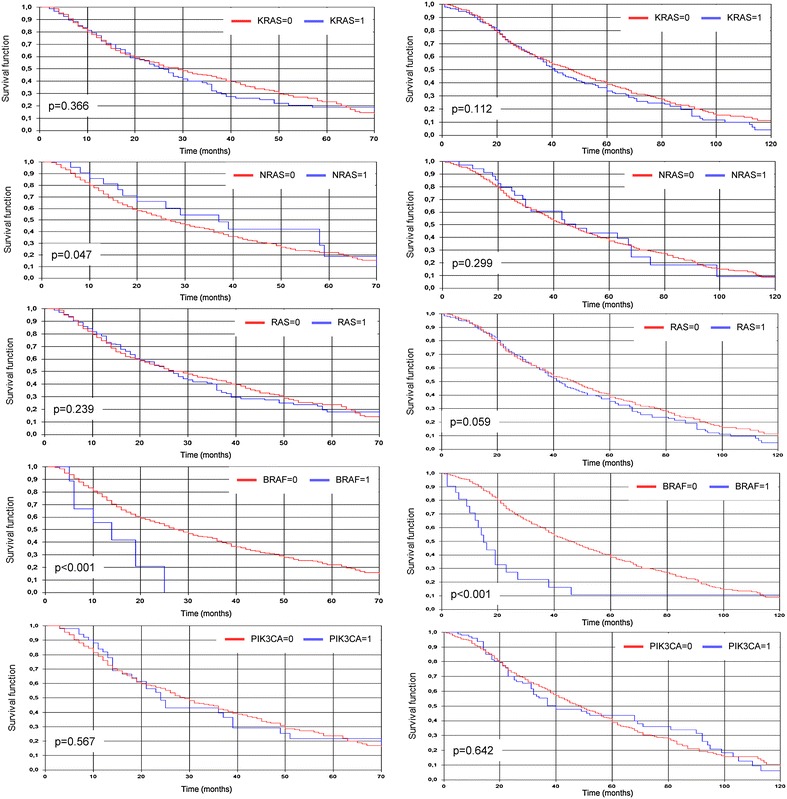
Kaplan-Meier cumulative survival analyses according to gene mutation status: partial survival in the left column, overall survival on the right; 0 wild-type, 1 mutated
No correlation between occurrence of gene mutations and response to therapies (EGFR inhibitors or others) has been inferred into the present study (such clinical data are being collected for a successive study in next future).
Discussion
Colorectal cancer is one of the most prevalent malignancies worldwide [1, 2, 28]. Great efforts have been made in the last decades for the comprehension of the pathophysiological and molecular mechanisms of the disease, and improvements in its clinical management have been made. Despite this, a lot more has to be done to improve the outcomes in the treatment of advanced stage patients, in whom the prognosis remains relatively poor [34]. On this purpose, the most relevant advances in recent years regard the targeted therapies inhibiting the EGFR-RAS cascade. The EGFR is a transmembrane protein for the epidermal growth (EGF) that explicates its functions through the activation of the RAS protein family (HRAS, KRAS, and NRAS). Activated RAS proteins promote cell proliferation and tumour progression and invasiveness through several mechanisms, including constitutive stimulation of the kinases into the MAPK pathway [15]. EGFR-targeted agents, like cetuximab and panitumumab that compete with EGF for binding to the receptor, have been employed in clinical practice, in order to reduce cell proliferation, migration, invasion, and metastasis formation [18].
KRAS mutations have been demonstrated to reduce the effectiveness of the anti-neoplastic agents mentioned, and they are currently a validated predictive marker of negative pharmacological response to anti-EGFR therapies. Mutations on codons 12 and 13 in exon 2 have been initially established as biomarkers of resistance to anti-EGFR antibodies; soon after, mutations on codons 61 and 146, as well as mutations on exons 3 and 4, have been discovered to produce the same effect [35–38]. As we mentioned before, these mutations cause a constitutive phosphorylation of the RAS proteins, which permanently promote cell proliferation and drastically reduce the effects of the EGFR inhibition [15–18]. Mutations in the entire coding region of the NRAS oncogene as well as in the exon 20 of the PIK3CA gene, which are rarer in CRC patients, have also been demonstrated to be predictors of reduced response to anti-EGFR therapies [36–38].
In our series, KRAS, NRAS, BRAF, and PIK3CA mutations were observed in 47.5 % of the CRC cases examined, mostly confirming the mutation prevalence observed in our previous study (a mutation of at least one among the KRAS, BRAF, and PIK3CA genes was discovered in about 45 % of Sardinian CRC cases) [30]. In Italian population, higher rates were recently reported in a considerably smaller collection of CRC patients from North Italy [39]. Using the same mutation screening tests of our present study (pyrosequencing assays; see “Methods” section), Foltran and colleagues reported that the 39.2 % of their cases was wild-type, defined as lack of mutations in KRAS exons 2-4, NRAS exons 2 and 3, BRAF exon 15, and PIK3CA exons 9 and 20 [39]. Conversely, the mutation rates were 47.4, 3.6, 5.2, and 16.5 % for KRAS, NRAS, BRAF, and PIK3CA, respectively (about 12 % of cases carried both KRAS and PIK3CA mutations) [39]. In the Sardinian population, mutation rates were much lower for KRAS (35.6 %) and BRAF (2.1 %), slightly lower for PIK3CA (14.3 %) and slightly higher for NRAS (4.1 %). Focusing on KRAS mutations only, Sardinian CRC patients were found to carry a mutated KRAS gene in 30 to 36 % of the cases [30, 40].
The distribution of the mutations within the territory of the island was confirmed to be heterogeneous, with the northern populations presenting higher rates of mutations, especially in the RAS genes [30]. This pattern confirms older evidences, regarding not only CRC but also other malignancies, and suggests that the genetic background may influence the occurrence of cancer gene mutations, even within relatively homogeneous populations such as that from Sardinia [30, 41–43]. Globally, the mutation rates found in our cohort are however within the ranges published in other series throughout the world [30, 44].
In our study, we did not evidence any relation between the mutation rates and the clinical and/or pathological parameters examined (age, sex, anatomical location, stage of the disease at diagnosis, grading). Most of the lesions were located in the left colon, but most of the mutations involved the rectum, even if the differences were not statistically significant. Some authors advocate that the anatomical location, histology and grading of the primary lesions has a relevant prognostic role, especially because BRAF mutations have been more frequently observed in right-sided, poorly differentiated mucinous tumors [26, 45–47].
Somatic mutations in the exon 15 of BRAF have been widely demonstrated, indeed, to negatively impact the prognosis of patients with CRC, independently of any clinical parameter studied [26, 45–47]. This was observed also in the present study; patients with substitution of a valine by a glutamic acid at position 600 (V600E), which was the only mutation found in our cases and the most commonly reported in the scientific literature, had a worst PS, OS, and TTPM in comparison to those without this alteration [32, 33]. Furthermore, the group of mutated patients—combining carriers of all mutually exclusive mutations in RAS and BRAF genes—presented a worst OS. This finding is in line with those of other authors who found that the all-wild type malignancies (RAS, BRAF, and in some cases PIK3CA included) had globally a better prognosis in comparison to the mutated ones [36, 39]. This is particularly relevant considering that a significant portion of the patients (approximately a half of them in our series) with CRC do not present any mutation of the genes mentioned above. The prognostic role of PIK3CA is less clear; it is known that it often coexists with KRAS mutations and predicts resistance to anti-EGFR therapies, but its exact prognostic role is poorly understood [39]. Ogino et al. evidenced that PIK3CA is associated with poor prognosis among patients with curatively resected colon cancer, but such an association was not confirmed neither in our series, nor in other studies in early or advanced stage patients [39, 48, 49].
The prognostic role of KRAS mutations as a global prognostic factor of disease progression and survival in CRC patients is controversial. Several studies have reported a statistically significant reduction in disease-free survival (DFS) and OS in the presence of KRAS mutations [50–52]. Ogino and colleagues performed an evaluation of the independent effect of the CIMP and MSI alterations as well as of the KRAS and BRAF mutations on the prognosis of 649 patients with IV stage CRC [53]. The study did not identify a relevant role for the KRAS mutations on prognosis, but the authors provided evidence that CIMP high appears to be an independent predictor of a low CRC specific mortality, while BRAF mutations are associated with a high CRC specific mortality [53]. By contrast, in their combined analysis of specific KRAS gene alterations, BRAFV600E status, and MSI, Zlobec et al. found that specific KRAS mutations act as informative prognostic factors in both sporadic and hereditary CRC [54]. In another recent series of 342 cases, Winder et al. were the first to report an improved OS in patients with KRAS codon 13 mutations, compared to those with wild-type genes, further suggesting that different types of KRAS mutations may be differently associated with OS in patients with CRC [55]. Finally, a multivariate analysis by Bazan et al. in a series of 160 cases found that KRAS codon 13 mutations were associated with poorer OS but not DFS [56]. Finally, Ren et al. in a recent well-designed meta-analysis performed a careful selection of 23 articles published from 1992, including 25 sets of data with 4687 patients and 1364 (29 %) KRAS-mutated cases [57]. Among them, 9 papers indicated that KRAS mutations were associated with worse prognosis, 15 failed to demonstrate any statistically significant association between such mutations and prognosis, and, finally, one data set identified an improved survival rate for patents with CRC and KRAS mutations [57]. Globally, the authors found that KRAS mutations were not associated with CRC prognosis, either before or after adjustment for the effect of publication bias [57].
In a previous study on Sardinian patients, we observed an improved TTPM in male patients with CRC harbouring KRAS mutations; we hypothesized that such a finding may be associated with the specific types of KRAS mutations observed in males and females as consequence of exposure to different lifestyle factors [57]. Nevertheless, such a finding was not confirmed in the present study, performed in a considerably greater number of patients. On the other hand, we found in the present series that a mutated NRAS negatively affects partial survival, even if it did not show any impact on OS or TTPM. Global cases with RAS mutations had a prognostic value on PS and TTPM close to be significant, and this further enhances the above-mentioned observation that colorectal malignancies with wild-type status in all these candidate genes generally present a better prognosis.
The discrepancy of results among the mentioned studies on the prognostic role of RAS and PIK3CA mutations in CRC may be caused by some confounding factors. The most frequent are: (1) heterogeneity of the study populations; (2) discrepancies in study designs and methodologies; (3) strategies for mutational detection; (4) presence of underlying predisposing conditions (i.e. ulcerative colitis); (5) variability of the staging systems (Dukes, AJCC, different editions); and (6) differences in clinical management and treatment. The advantage of our study is to avoid several bias through a prospective collection of data on a consistent high number of patients from a homogeneous population, processed and analysed in a single institution. To date, we can consider as validated the prognostic role of BRAF mutations as well as that of the wild-type status in all main candidate genes. Further studies are probably necessary to better assess the prognostic role of the single RAS and PIK3CA mutations.
Conclusions
Our findings evidence that approximately a half of the Sardinian patients with CRC present one or more mutations in one or more of the KRAS, NRAS, BRAF, and PIK3CA genes. The KRAS mutations are the most frequent, followed by those in NRAS, and BRAF, as observed in other studies, and they are strongly confirmed to be mutually exclusive. On the other hand, PIK3CA mutations often coexist with the RAS mutations. With the exclusion of mutations in any of the other genes, harbouring a mutated BRAF was demonstrated to negatively impact the prognosis—regardless the type of survival taken into consideration—in CRC patients from Sardinian population. This makes the role of the latter mutations clear; as for the others, further investigations are necessary to better comprehend their prognostic impact.
Authors’ contributions
GrP, performed vast majority of mutation analyses and participated in design of the study; VD, AC, and GP made substantial contributions to the conception and design of the study, as well as in all the phases of the collection of data and preparation of the manuscript; AM, MiC, and MC contributed to part of the mutation screening and data acquisition; AL, ED, AP, GS, CP, SO, MS, MTI, GB, GiuS, FC, TS, LV, MiB, and GG collected patients’ samples and contributed to the analysis of all the clinical data included into the study; MB, performed statistical analysis of biomolecular data; GrP, VD, and PP wrote the initial manuscript; AC, PP, and FT provided critical revisions related to the intellectual content of the manuscript, GP performed the final revision and approved the manuscript to be submitted. All authors read and approved the final manuscript.
Acknowledgements
Authors are grateful to patients for their important contribution to this study.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets during and/or analysed during the current study are available from the corresponding author on request.
Consent for publication
All the participants into the study were informed about the aims of this study and, before the tissue sample was collected, gave a written informed consent.
Ethics approval and consent to participate
Ethical review board of the University of Sassari.
Funding
Work was supported by the Sardinian Regional Government/Regione Autonoma della Sardegna (project no. CRP-59690) and the Italian Minister of Health “Progetto Ricerca Finalizzata” (project no. RF-2010-2315581). Funders had no role in the design of the study, samples’ collection and analysis, data interpretation, and manuscript writing.
Abbreviations
- CIMP
CpG islant methylator phenotype
- CIN
chromosomal instability
- CRC
colorectal cancer
- DFS
disease free survival
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- mAbs
monoclonal antibodies
- MAPK
mitogen-activated protein-kinase
- mCRC
metastatic colorectal cancer
- MSI
microsatellite instability
- OS
overall survival
- PS
partial survival
- SSA
serrated adenomas
- TSA
traditional serrated adenomas
- TTPM
time to metastatic progression
Additional file
10.1186/s12967-016-1053-z Additional tables S1 and S2. the file contains two supplementary tables depicting the exact distribution of the 543 mutations identified in RAS and BRAF genes (Table S1), as well as the distribution of the 115 PIK3CA mutations (Table S2) in our cohort.
Footnotes
Grazia Palomba, Valentina Doneddu and Antonio Cossu contributed equally to this work
Contributor Information
Grazia Palomba, Email: graziap68@yahoo.it.
Valentina Doneddu, Email: valentinadoneddu@gmail.com.
Antonio Cossu, Email: cossu@uniss.it.
Panagiotis Paliogiannis, Phone: +393405931590, Email: panospaliogiannis@gmail.com.
Antonella Manca, Email: antonella.manca@icb.cnr.it.
Milena Casula, Email: casulam@yahoo.it.
Maria Colombino, Email: colombinom@yahoo.it.
Annamaria Lanzillo, Email: am.lanzillo@tiscali.it.
Efisio Defraia, Email: defraiae@gmail.com.
Antonio Pazzola, Email: antonio.pazzola@tiscali.it.
Giovanni Sanna, Email: gisanna@uniss.it.
Carlo Putzu, Email: cputzu@gmail.com.
Salvatore Ortu, Email: ortu.salv@tiscali.it.
Mario Scartozzi, Email: marioscartozzi@gmail.com.
Maria Teresa Ionta, Email: mtionta@yahoo.it.
Giovanni Baldino, Email: giovannibaldino@tiscali.it.
Giuseppina Sarobba, Email: gi.sarobba@tiscali.it.
Francesca Capelli, Email: francapelli@tiscali.it.
Tito Sedda, Email: tito.sedda@alice.it.
Luciano Virdis, Email: virdis@tiscali.it.
Michela Barca, Email: kikkyboat@yahoo.it.
Giulia Gramignano, Email: g.gramignano@libero.it.
Mario Budroni, Email: marbudroni@gmail.com.
Francesco Tanda, Email: tandaf@uniss.it.
Giuseppe Palmieri, Email: gpalmieri@yahoo.com.
References
- 1.Jemal A, Center MM, DeSantis C, et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 2.SEER cancer statistics factsheets: colon and rectum cancer. Bethesda: National Cancer Institute; http://seer.cancer.gov/statfacts/html/colorect.html. Accessed 20 May 2016.
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 4.Sakai E, Fukuyo M, Ohata K, et al. Genetic and epigenetic aberrations occurring in colorectal tumors associated with serrated pathway. Int J Cancer. 2016;138:1634–1644. doi: 10.1002/ijc.29903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens JB, Liu G, Abdallah BY, et al. Unstable genomes elevate transcriptome dynamics. Int J Cancer. 2014;134:2074–2087. doi: 10.1002/ijc.28531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/A:1025861527711. [DOI] [PubMed] [Google Scholar]
- 7.Boland CR, Thibodeau SN, Hamilton SR, et al. National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 8.Barault L, Charon-Barra C, Jooste V, et al. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res. 2008;68:8541–8546. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 9.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 10.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 11.Peltomäki P. Mutations and epimutations in the origin of cancer. Exp Cell Res. 2012;318:299–310. doi: 10.1016/j.yexcr.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Burotto M, Chiou VL, Lee JM, et al. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120:3446–3456. doi: 10.1002/cncr.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bronte G, Silvestris N, Castiglia M, et al. New findings on primary and acquired resistance to anti-EGFR therapy in metastatic colorectal cancer: do all roads lead to RAS? Oncotarget. 2015;6:24780–24796. doi: 10.18632/oncotarget.4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Hu J, Cheng L, et al. Biomarkers predicting resistance to epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer with wild-type KRAS. Onco Targets Ther. 2016;9:557–565. doi: 10.2147/OTT.S86966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorissen RN, Walker F, Pouliot N, et al. Epidermal growth factor receptor: mechanisms of activation and signaling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/S0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 16.Markman B, Javier Ramos F, Capdevila J, et al. EGFR and KRAS in colorectal cancer. Adv Clin Chem. 2010;51:71–119. doi: 10.1016/S0065-2423(10)51004-7. [DOI] [PubMed] [Google Scholar]
- 17.Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- 18.Yuan ZX, Wang XY, Qin QY, et al. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: a meta-analysis. PLoS ONE. 2013;8:e65995. doi: 10.1371/journal.pone.0065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu LX, Mao C, Zhang J, et al. Predictive and prognostic value of KRAS mutations in metastatic colorectal cancer patients treated with cetuximab: a meta-analysis of 22 studies. Eur J Cancer. 2010;46:2781–2787. doi: 10.1016/j.ejca.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Guo F, Shi X, et al. BRAF V600E mutation and KRAS codon 13 mutations predict poor survival in Chinese colorectal cancer patients. BMC Cancer. 2014;14:802. doi: 10.1186/1471-2407-14-802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadowaki S, Kakuta M, Takahashi S, et al. Prognostic value of KRAS and BRAF mutations in curatively resected colorectal cancer. World J Gastroenterol. 2015;21:1275–1283. doi: 10.3748/wjg.v21.i4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tol J, Nategaal ID, Punt CJA. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–99. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 24.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 25.Yokota T, Ura T, Shibata N, et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014;53:852–864. doi: 10.3109/0284186X.2014.895036. [DOI] [PubMed] [Google Scholar]
- 27.Zhu L, Dong C, Cao Y, et al. Prognostic role of BRAF mutation in stage II/III colorectal cancer receiving curative resection and adjuvant chemotherapy: a meta-analysis based on randomized clinical trials. PLoS ONE. 2016;1:e0154795. doi: 10.1371/journal.pone.0154795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmieri G, Paliogiannis P, Scognamillo F, et al. Colorectal cancer epidemiology in an area with a spontaneous screening program. Acta Medica Mediterr. 2013;29:231–234. [Google Scholar]
- 29.Budroni M, Cesaraccio R, Pirino D, et al. Cancer incidence in Sassari Province (2003–2007) In: Forman D, Bray F, Brewster DH, Gombe Mbalawa G, Kohler B, Pineros M, Steiliarova-Foucher E, Swaminatham R, Ferlay J, et al., editors. Cancer incidence in five continents. Lyon: International Agency for Research on Cancer; 2014. [Google Scholar]
- 30.Palomba G, Colombino M, Contu A, et al. Prevalence of KRAS, BRAF, and PIK3CA somatic mutations in patients with colorectal carcinoma may vary in the same population: clues from Sardinia. J Transl Med. 2012;10:178. doi: 10.1186/1479-5876-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobin LH, Gospodarowicz MK, Wittekind C, editors. TNM classification of malignant tumors. 7. Chichester: Wiley; 2010. [Google Scholar]
- 32.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 33.Casula M, Colombino M, Satta MP, et al. BRAF gene is somatically mutated but does not make a major contribution to malignant melanoma susceptibility. J Clin Oncol. 2004;22:286–292. doi: 10.1200/JCO.2004.07.112. [DOI] [PubMed] [Google Scholar]
- 34.Paliogiannis P, Cossu A, Tanda F, et al. KRAS mutational concordance between primary and metastatic colorectal adenocarcinoma. Oncol Lett. 2014;8:1422–1426. doi: 10.3892/ol.2014.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–721. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 37.Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, Phase 3 trial. Lancet Oncol. 2014;15:1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 38.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 39.Foltran L, De Maglio G, Pella N, et al. Prognostic role of KRAS, NRAS, BRAF and PIK3CA mutations in advanced colorectal cancer. Future Oncol. 2015;11:629–640. doi: 10.2217/fon.14.279. [DOI] [PubMed] [Google Scholar]
- 40.Palomba G, Cossu A, Paliogiannis P, et al. Prognostic role of KRAS mutations in Sardinian patients with colorectal carcinoma. Oncology letters. 2016;12:1415–1421. doi: 10.3892/ol.2016.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palomba G, Pisano M, Cossu A, et al. Spectrum and prevalence of BRCA1 and BRCA2 germline mutations in Sardinian breast cancer patients through a hospital-based screening. Cancer. 2005;104:1172–1179. doi: 10.1002/cncr.21298. [DOI] [PubMed] [Google Scholar]
- 42.Palomba G, Loi A, Uras A, et al. A role of BRCA1 and BRCA2 germline mutations in breast cancer susceptibility within Sardinian population. BMC Cancer. 2009;9:245. doi: 10.1186/1471-2407-9-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casula C, Muggiano A, Cossu A, et al. Role of key-regulator genes in melanoma susceptibility and pathogenesis among patients from South Italy. BMC Cancer. 2009;9:352. doi: 10.1186/1471-2407-9-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldus SE, Schaefer KL, Engers R, et al. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16:790–799. doi: 10.1158/1078-0432.CCR-09-2446. [DOI] [PubMed] [Google Scholar]
- 45.Richman SD, Seymour MT, Chambers P, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–5937. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- 46.Souglakos J, Philips J, Wang R, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer. 2009;101:465–472. doi: 10.1038/sj.bjc.6605164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tol J, Dijkstra JR, Klomp M, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer. 2010;46:1997–2009. doi: 10.1016/j.ejca.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 48.Ogino S, Nosho K, Kirkner GJ, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009;27:1477–1484. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eklöf V, Wikberg ML, Edin S, et al. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer. 2013;108:2153–2163. doi: 10.1038/bjc.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imamura Y, Morikawa T, Liao X, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type. Clin Cancer Res. 2012;18:4753–4763. doi: 10.1158/1078-0432.CCR-11-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pentheroudakis G, Kotoula V, De Roock W, et al. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer. 2013;13:49. doi: 10.1186/1471-2407-13-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zlobec I, Kovac M, Erzberger P, et al. Combined analysis of specific KRAS mutation, BRAF and microsatellite instability identifies prognostic subgroups of sporadic and hereditary colorectal cancer. Int J Cancer. 2010;127:2569–2575. doi: 10.1002/ijc.25265. [DOI] [PubMed] [Google Scholar]
- 55.Winder T, Mündlein A, Rhomberg S, et al. Different types of K-Ras mutations are conversely associated with overall survival in patients with colorectal cancer. Oncol Rep. 2009;21:1283–1287. doi: 10.3892/or_00000352. [DOI] [PubMed] [Google Scholar]
- 56.Bazan V, Agnese V, Corsale S, et al. Specific TP53 and/or Ki-ras mutations as independent predictors of clinical outcome in sporadic colorectal adenocarcinomas: results of a 5-year Gruppo Oncologico dell’Italia Meridionale (GOIM) prospective study. Ann Oncol. 2005;16:50–55. doi: 10.1093/annonc/mdi908. [DOI] [PubMed] [Google Scholar]
- 57.Ren J, Li G, Ge J, et al. Is K-ras gene mutation a prognostic factor for colorectal cancer: a systematic review and meta-analysis. Dis Colon Rectum. 2012;55:913–923. doi: 10.1097/DCR.0b013e318251d8d9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets during and/or analysed during the current study are available from the corresponding author on request.