

Abstract
Patients under 5 years were not evaluated in the phase-3 study for enzyme replacement therapy (ERT) in MPS IV A. Here we describe the evolution of a severe Morquio A pediatric patient who was diagnosed at 19 months old and treated by ERT at 21 months old for the next 30 months.
Applying the standard ERT protocol on this very young patient appeared to reduce his urinary excretion of glycosaminoglycans (GAGs); the improvements in both the 6 minute-walk test (6MWT) and the stair climb test, however, were no different than those reported in the nature history study. Additionally, this young patient experienced many ERT-associated side effects, and as a result a specific corticosteroid protocol (1 mg/kg of betamethasone the day before and 1 h before the ERT infusion) was given to avoid adverse events. Under these treatments, the height of this patient increased during the first year of the ERT although no more height gain was observed thereafter for 18 months. However, despite of ERT, his bone deformities (including severe pectus carinatum) actually worsened and his medullar cervical spine compression showed no improvement (thus needed decompression surgery).
Conclusion: early ERT treatment did not improve the bone outcome in this severe MPS IV A patient after the 30 months-long treatment. A longer term follow up is required to further assess the efficacy of ERT on both the motor and the respiratory function of the patient.
1. Introduction
Morquio A syndrome (mucopolysaccharidosis IVA (MPS IVA); OMIM#253000) is a rare autosomal recessive lysosomal storage disorder caused by N-acetylglactosamine-6-sulfatase (GALNS) deficiency due to genetic mutations [1]. In severe cases, the patients can suffer from serious skeletal dysplasia characterized by vertebral platyspondyly, hip dysplasia and genu valgum. Until recently, apart from possible hematopoietic stem cells transplantation [2], [3], the mainstay of the treatment remained symptomatic [4].
In 2014, the results of a phase-3 placebo-controlled trial indicated positive outcomes associated with using elosulfase alfa as an enzyme replacement therapy (ERT) for MPS IVA in both urinary excretion of glycosaminoglycans (GAGs) and 6 minute-walk test [5]. However, patients under 5 years old were not included in this study. Recently, to address younger patients' need, Jones et al. examined the effects of a 52 weeks-long ERT in 15 patients aged < 5 years old; they found positive effects of ERT on patients' height compared to the natural history study (MorCAP) [6].
We report here the results of a 30 months-long ERT treatment in a severe case of Morquio A of 21 months old.
2. Clinical report
The patient is the second child of a consanguineous Moroccan family. The pregnancy and birth were uneventful (birth parameters: weight 3.2 kg; height 49 cm; head circumference 36 cm). He was seen at 19 months of age for thoraco-lumbar kyphosis and pectus carinatum. Initially, the child weighted 12.2 kg (75th percentile), heighted 81 cm (25th percentile) and had a head circumference at 53 cm (98th percentile).
The clinical examination showed typical bone deformities, no hepatosplenomegaly and neurological examination was normal. There was no corneal clouding on ophthalmologic examination. Initial X-rays showed costal widening, roster-shaped vertebra, widening and verticalization of the acetabulum, coxa valga and ovoid-shaped proximal femoral epiphysis. The patient was diagnosed with MPS IV A based on the results of (1) an increased urinary GAGs excretion (increased keratan sulfate), (2) a null GALNS activity in leucocytes, (3) the identification of homozygous c.871G > A mutation in GALNS gene — already described as responsible for a severe MPS IVA phenotype [7].
Elosulfase alfa treatment (2 mg/kg/week) was initiated for this patient at 21 months of life. During the 30 months-long ERT treatments, 3 administrations were missed due to fevers caused by the intercurrent diseases or surgery.
2.1. Biological efficacy
As we could not evaluate the blood levels of keratan sulfate (KS) and chondroitin-6-sulfate (C6S), and the urine level of C6S, we examined only the urinary keratan sulfate (uKS) during the course of the disease. Initially, uKS excretion was at 13 mg per mmol of creatinine (normal values: < 4 mg/mmol creatinine). After 3 months of ERT, we observed a 72% decrease in uKS excretion; the level of uKs decrease reached 82% after 18 months and stabilized around 60% after 30 months of treatment (at 5 mg/mmol creatinine). We further noticed that the decreases in uKs were entirely dependent on the ERT since when the treatment was interrupted during one week for the vertebral surgery, uKS immediately increased; its level returned to pre-surgery values only after two weeks of ERT.
2.2. Clinical efficacy
2.2.1. Height (Fig. 1)
Fig. 1.
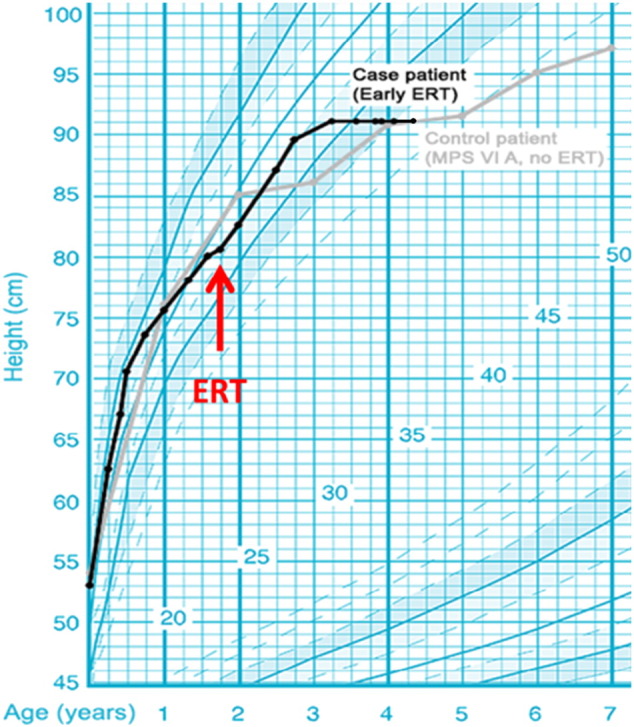
Patient height (black line) compared to a MPS IV patient without ERT (gray line). Enzyme replacement therapy initiation (red arrow). French auxology reference curves [23].
After 30 months of ERT, we observed an initial improvement of height (+ 10 cm in 12 months), but this evolution stopped and the height stabilized at 92 cm without any height gain in the last 12 months (Fig. 1).
2.2.2. Bone and spine evolutions
The pectus carinatum and the kyphosis actually worsened during the 30 months of ERT although no limbs morphological change was detectable during the same period. Cervical cord compression (diagnosed on systematic medullar MRI) occurred despite of the treatment, and as a result, the child required surgical decompression after 13 months of ERT. The MRI control at 4 years old (after 27 months of ERT) showed continuation of slight medullary hypersignal, suggesting persistent medullar involvement due to the cervicothoracic junction kyphosis. The diameter of the cervical canal, however, remains stable.
2.2.3. Functional parameters (motor and respiratory functions)
We performed the 6 minute-walk and the stairs climbing tests every year once the child acquired the ability to walk. We observed improvements in walking distance and in stairs climbing of the patient every year (Table 1). Respiratory functions test were not done because of the young age of the child
Table 1.
6 minute-walk test and stair climbing test at 6, 13 and 30 months of ERT.
| Nov 2013 | July 2014 | Feb 2015 | July 2016 | |
|---|---|---|---|---|
| Age (months) | 21 | 27 | 34 | 46 |
| ERT (months) | 0 | 6 | 13 | 30 |
| 6-Min walk test | ||||
| Distance (m) | Walking not acquired | 209 | 217 | 257 |
| Heart rate (/min) | 172 | 165 | 135 | |
| Saturation (%) | 98 | 96 | 97 | |
| Stairs climbing test | ||||
| Number of stairs (min) | Walking not acquired | 10 (1′22) | 22 (2′) | 44 (2′23) |
| Heart rate (/min) | 168 | 157 | 130 | |
| Saturation (%) | 100 | 96 | 100 |
2.2.4. Liver, spleen and cardiac follow-up
Abdominal and cardiac ultrasounds were normal at diagnosis and remained normal throughout the treatment period.
2.2.5. Neurocognitive development
Neuropsychological follow-up (WPPSI IV) showed normal cognitive development.
2.3. Safety
The ERT in this young patient induced frequent adverse events such as vomiting, chills, tremors and fever despite medications, including dexchlorpheniramine (anti-histamine), acetaminophen, and corticosteroids given 1 h prior to ERT infusion. However, the child presented a severe adverse events after 16 months of ERT (shock necessitating emergency management by fluid infusion and corticosteroid administration). We then modified the pre-treatment to betamethasone at 1 mg/kg po the day before and 1 mg/kg iv 1 h before ERT. Since this time, the patient never presented any further adverse events. The posology of betamethasone administered the day before was progressively decreased after 8 months without adverse events, and was stopped 16 months after the beginning of this protocol.
3. Discussion
The management of Morquio A syndrome has until recently been limited to supportive and symptom-based care [4]. Some experiences [2], [3] in HSCT have been published without clear improvement in height outcome; for example, in the study of Yabe et al. [3], 4 cases of Morquio A patients had undergone HSCT, the youngest patient, who suffered a severe form of the disease and had the HSCT at 4 years old, showed no clear improvement on growth as his final height at 18 years old was only 112 cm [3]. At present, the first line treatment is the enzyme replacement therapy (ERT) with elosulfase alfa [5] because of the morbidity and mortality related to HSCT.
We report here a 30 months follow-up of ERT treatment in a patient with severe Morquio A disease whose treatment began at 21 months of age. Our patient was diagnosed with a severe MPS IVA disease based on his clinical presentation and his homozygous mutation c. 871G > A of the GALNS gene, already described as responsible for a severe form of MPS IVA [7]. Our report here is valuable as it fills in the gap of knowledge previously established by two main studies on Elosulfase alfa ERT: first included only patients older than 5 years old [5], and second included younger MPS IV A patients (below 5 years of age) for only 52 weeks of treatment [6].
In our case study, the ERT rapidly reduced the GAGs urinary excretion level (72% reduction in 3 months) of the patient, although this level was not normalized after 30 months of ERT. The maximum decrease observed in our case study was 82% after 18 months of ERT although this reduced later to 60%. These reductions in uKs are larger than those reported previously by Jones et al.; these authors found an average decrease of 43.5% in uKS excretion for the 15 patients studied after 52 weeks of ERT [6]. Interestingly, 100% of patients produced antibodies to the replacement enzyme in the study of Jones et al. (15/15) [6]. As we had no access to antibodies measurements, we wondered if our patient also synthesized antibody against Elosulfase alfa. This is important as our patient presented severe adverse events (SAE) which obliged us to administrate emergency treatment. However, our observation that there were persistent reductions in uKS excretion during the entire 30 months of ERT made us believe that there may be no neutralizing antibody produced in our patient. Another question we had concerns the posology of Elosulfase alfa. As uKS excretion level was not normalized but rather stabilized at much reduced but above normal level, we wondered whether higher dose of Elosulfase alfa may improve further the uKS excretion and thus ameliorate the clinical outcome [8].
During the course of ERT, our patient experienced many adverse events. This is consistent with the general experiences that more frequent adverse events are associated with ERT in the more severe forms of lysosomal storage diseases in which no important residue enzyme activity is present due to null mutations. For example, while SAEs indeed are most frequently observed in severe maltase acid deficiency or Hurler disease during ERT, in the cases where residual enzyme activities remain significantly such as in Gaucher type I, less SAEs are observed [9].
Although we observed an improvement on the growth of our patient during the first year of ERT, his growth stopped when the child reached 3 years old (after 12 month ERT). Based on the natural history data of the severe Morquio A patients, normal growth is expected in patient's first two years of life; this is then followed by a rather small growth between 2 and 4 years of age, and thereafter a much impaired growth [10]. Our patient stopped growing soon after reaching 3 years old, even though he was still under ERT. It is possible that the high dose of corticosteroid administered right before ERT (to avoid SAE) can affect the height evolution [11]. Presently, at 4 years of age, our patient's height reaches only 92 cm, a height corresponding to the third centile of specific Morquio A patients growth curves [10]. As we are slowly decreasing the dose of corticosteroid administered to him, it is possible that his growth may progress further. Nevertheless, we remain doubtful for his future improvement on growth since there is an absolute absence of improvement in his height during the last 18 months under ERT. Based on the study of Jones et al., growth improvement can be expected in the first six months of ERT; these authors found that the effect on growth does not last after this time as the curve of standing height/length (Z-score) decrease after 39 weeks of treatment [6].
In MPS I, the medical treatment (ERT as well as bone marrow transplant [BMT]) does not modifies the natural history of the skeletal deformities [12], [13]. In MPS I, II or VI, apart when the treatment is administered in the very first weeks of life, ERT or BMT can improve the joint mobility through decreasing the infiltration of GAGs into ligaments, tendons, joint capsules, and other soft tissues; these treatments, however, never influence the outcome of bone morphology [14], [15], [16]. This poor evolution is due to the accumulation of GAGs which not only abrogates skeletal remodeling, but also causes disordered endochondral and intramembranous ossification which begins even during fetal development [13]. These are the reasons why the orthopedic complications in these patients such as the cervical spine narrowing, for example, (one of the most severe complications of this disease [17]) cannot be corrected by medical interventions.
Our patient showed statistically significant improvements in endurance tests, including 6 minute-walk test and 3 min stairs climbing test, during ERT. Harmatz et al. published the natural evolution of the results of these tests in untreated patients. Our patient shared the same evolutions as the similar aged patients in this study, who managed to walk 250 m in 6 MWT at 4 years of age [18]. Despite the fact that our patient stopped to climb during the 3 min climbing test before the end of time, he managed to climb 44 stairs in 2 min and 23 s at 4 years of age, a steady improvement since 3 years. As there is no age specific data for this test in the paper of Harmatz et al. [18], it is not possible to compare our case results to the published data. To summary, we suggest that it is difficult to determine whether the improvements made by our patient are related to the ERT treatment or they reflect the natural improvement in motor functions normally occurs to these diseased children at this age.
The French guidelines for MPS IV [19] recommend the use of ERT in very young MPS IVA patients and when they are still capable of walking. For patients presenting mild form of the disease in which the benefit of ERT is not proven, such indication must be discussed in front of the French MPS expert group. Based on the French guidelines, ERT can only be terminated in cases of severe adverse events or after discussions with patient's family when the efficacy of ERT appears doubtful. This contrasts the situation in UK where clear criteria are needed to be met to stop ERT [20]. In UK, to stop ERT, a patient must meet 4 of the 5 criteria (6MWT, respiratory function, cardiac evaluation, urinary KS excretion and quality of life, depression and pain evaluations) under naïve responder or long term trial to continue ERT [20]. In the Netherlands, the reimbursement of ERT in MPS IV was stopped in 2016 because of insufficient cost/effectiveness ratio [21]. Long term studies are needed to determinate clear criteria for prescription and for stopping this treatment which can induce more adverse events than clinical benefits for these patients.
Very early ERT has been described in other lysosomal storage disorders. For example, Leroux et al. described the case of a patient with MPS type I (Hurler) with early Laronidase ERT (began 5.5 months of age) and bone marrow transplantation at 11 months, with good evolution, although follow-up was described only for 4 months [14]. Lampe et al. reported the benefits of early ERT in a Hunter patient under 1 year of age, including somatic improvements, markedly decreased urinary GAG excretion, stabilized dysostosis multiplex, and minor joint stiffness [15]. In MPS type VI, McGill et al. started ERT at 8-weeks of age in a sibling control study which provided valuable evidence that initiating ERT improves overall morphology, clinical outcome and quality of life in this very early treated patient compared to his older sibling [16]. These observations allude to the need to treat MPS IVA patients by ERT as early as possible to obtain the best results of this high cost treatment. This goal could only be achieved by a systematic neonatal screening which is now possible for lysosomal storage disorders [22]. Although Morquio A disease is very rare condition, the absence of neurological disease in this MPS would make its neonatal diagnosis and treatment very beneficial while the treatment at 21 months of age is still very far from satisfactory as shows our observation.
References
- 1.Tomatsu S., Fukuda S., Masue M., Sukegawa K., Fukao T., Yamagishi A., Hori T., Iwata H., Ogawa T., Nakashima Y. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem. Biophys. Res. Commun. 1991;181:677–683. doi: 10.1016/0006-291x(91)91244-7. [DOI] [PubMed] [Google Scholar]
- 2.Wang J., Luan Z., Jiang H., Fang J., Qin M., Lee V., Chen J. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis-a ten-year report from the China children transplant group biology of blood and marrow transplantation. Journal of the American Society for Blood and Marrow Transplantation. 2016 doi: 10.1016/j.bbmt.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Yabe H., Tanaka A., Chinen Y., Kato T., Sawamoto K., Yasuda E., Shintaku H., Suzuki H., Orii T., Tomatsu S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016;117:84–94. doi: 10.1016/j.ymgme.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomatsu S., Yasuda E., Patel P., Ruhnke K., Shimada T., Mackenzie W.G., Mason R., Thacker M.M., Theroux M., Montano A.M., Almeciga-Diaz C.J., Barrera L.A., Chinen Y., Sly W.S., Rowan D., Suzuki Y., Orii T. Morquio A syndrome: diagnosis and current and future therapies. Pediatr. Endocrinol. Rev. 2014;PER 12(Suppl. 1):141–151. [PMC free article] [PubMed] [Google Scholar]
- 5.Hendriksz C.J., Burton B., Fleming T.R., Harmatz P., Hughes D., Jones S.A., Lin S.P., Mengel E., Scarpa M., Valayannopoulos V., Giugliani R., Investigators S., Slasor P., Lounsbury D., Dummer W. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014;37:979–990. doi: 10.1007/s10545-014-9715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S.A., Bialer M., Parini R., Martin K., Wang H., Yang K., Shaywitz A.J., Harmatz P. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) < 5 y. Pediatr. Res. 2015;78:717–722. doi: 10.1038/pr.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomatsu S., Montano A.M., Nishioka T., Gutierrez M.A., Pena O.M., Tranda Firescu G.G., Lopez P., Yamaguchi S., Noguchi A., Orii T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A) Hum. Mutat. 2005;26:500–512. doi: 10.1002/humu.20257. [DOI] [PubMed] [Google Scholar]
- 8.Tomatsu S., Sawamoto K., Shimada T., Bober M.B., Kubaski F., Yasuda E., Mason R.W., Khan S., Almeciga-Diaz C.J., Barrera L.A., Mackenzie W.G., Orii T. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opinion on Orphan Drugs. 2015;3:1279–1290. doi: 10.1517/21678707.2015.1086640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J., Lozier J., Johnson G., Kirshner S., Verthelyi D., Pariser A., Shores E., Rosenberg A. Neutralizing antibodies to therapeutic enzymes: considerations for testing, prevention and treatment. Nat. Biotechnol. 2008;26:901–908. doi: 10.1038/nbt.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montano A.M., Tomatsu S., Brusius A., Smith M., Orii T. Growth charts for patients affected with Morquio A disease. Am. J. Med. Genet. A. 2008;146A:1286–1295. doi: 10.1002/ajmg.a.32281. [DOI] [PubMed] [Google Scholar]
- 11.Loke Y.K., Blanco P., Thavarajah M., Wilson A.M. Impact of inhaled corticosteroids on growth in children with asthma: systematic review and meta-analysis. PLoS One. 2015;10 doi: 10.1371/journal.pone.0133428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langereis E.J., den Os M.M., Breen C., Jones S.A., Knaven O.C., Mercer J., Miller W.P., Kelly P.M., Kennedy J., Ketterl T.G., O'Meara A., Orchard P.J., Lund T.C., van Rijn R.R., Sakkers R.J., White K.K., Wijburg F.A. Progression of hip dysplasia in mucopolysaccharidosis type I Hurler after successful hematopoietic stem cell transplantation. J. Bone Joint Surg. 2016;98:386–395. doi: 10.2106/JBJS.O.00601. (American volume) [DOI] [PubMed] [Google Scholar]
- 13.Weisstein J.S., Delgado E., Steinbach L.S., Hart K., Packman S. Musculoskeletal manifestations of Hurler syndrome: long-term follow-up after bone marrow transplantation. J. Pediatr. Orthop. 2004;24:97–101. doi: 10.1097/00004694-200401000-00019. [DOI] [PubMed] [Google Scholar]
- 14.Leroux S., Muller J.B., Boutaric E., Busnel A., Lemouel F., Andro-Garcon M., Neven B., Valayannopoulos V., Vinceslas C. Hurler syndrome: early diagnosis and treatment. Archives de pediatrie: organe officiel de la Societe francaise de pediatrie. 2014;21:501–506. doi: 10.1016/j.arcped.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Lampe C., Atherton A., Burton B.K., Descartes M., Giugliani R., Horovitz D.D., Kyosen S.O., Magalhaes T.S., Martins A.M., Mendelsohn N.J., Muenzer J., Smith L.D. Enzyme replacement therapy in mucopolysaccharidosis II patients under 1 year of age. JIMD Reports. 2014;14:99–113. doi: 10.1007/8904_2013_289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGill J.J., Inwood A.C., Coman D.J., Lipke M.L., de Lore D., Swiedler S.J., Hopwood J.J. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age—a sibling control study. Clin. Genet. 2010;77:492–498. doi: 10.1111/j.1399-0004.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- 17.Mollmann C., Lampe C.G., Muller-Forell W., Scarpa M., Harmatz P., Schwarz M., Beck M., Lampe C. Development of a scoring system to evaluate the severity of craniocervical spinal cord compression in patients with mucopolysaccharidosis IVA (Morquio A syndrome) JIMD Reports. 2013;11:65–72. doi: 10.1007/8904_2013_223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harmatz P.R., Mengel K.E., Giugliani R., Valayannopoulos V., Lin S.P., Parini R., Guffon N., Burton B.K., Hendriksz C.J., Mitchell J.J., Martins A.M., Jones S.A., Guelbert N., Vellodi A., Wijburg F.A., Yang K., Slasor P., Decker C. Longitudinal analysis of endurance and respiratory function from a natural history study of Morquio A syndrome. Mol. Genet. Metab. 2015;114:186–194. doi: 10.1016/j.ymgme.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 19.F.d.S.M.R. G2M, Protocole National de Diagnostic et de Soins (PNDS) Mucopolysaccharidoses. http://www.sfce.sfpediatrie.com/sites/default/files/recommandations/pnds_-_mucopolysaccharidoses.pdf (2016).
- 20.NIH, Managed Access Agreement, Elosulfase alfa for treating mucopolysaccharidosis type IVa https://www.nice.org.uk/guidance/hst2/resources/managed-access-agreement-december-2015-2238935869 (2015).
- 21.M.v. Volksgezondheid, https://www.zorginstituutnederland.nl/binaries/content/documents/zinl-www/actueel/nieuws/2016/medicijn-vimizim-onvoldoende-effectief-bij-zeldzame-stofwisselingsziekte/medicijn-vimizim-onvoldoende-effectief-bij-zeldzame-stofwisselingsziekte/zinl%3ADocument/1603-medicijn-vimizim-onvoldoende-effectief-bij-zeldzame-stofwisselingsziekte/Medicijn + Vimizim + onvoldoende + effectief + bij + zeldzame + stofwisselingsziekte.pdf (2016).
- 22.Spacil Z., Tatipaka H., Barcenas M., Scott C.R., Turecek F., Gelb M.H. High-throughput assay of 9 lysosomal enzymes for newborn screening. Clin. Chem. 2013;59:502–511. doi: 10.1373/clinchem.2012.189936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sempe M., Pedron G., Roy-Pernot M.P. 1979. Auxologie méthode et séquences. [Google Scholar]