

Supplemental Digital Content is Available in the Text.
Tumor-derived glutamate may significantly contribute to cancer-induced bone pain. The glutamate transporter system xc− is a promising therapeutic target in this pain state.
Keywords: Pain, Cancer, Glutamate, Peroxynitrite, Transporter, Cystine/glutamate antiporter, Oxidative stress, Nitrosative stress, Reactive oxygen species, Reactive nitrogen species, Superoxide
Abstract
Bone is one of the leading sites of metastasis for frequently diagnosed malignancies, including those arising in the breast, prostate and lung. Although these cancers develop unnoticed and are painless in their primary sites, bone metastases result in debilitating pain. Deeper investigation of this pain may reveal etiology and lead to early cancer detection. Cancer-induced bone pain (CIBP) is inadequately managed with current standard-of-care analgesics and dramatically diminishes patient quality of life. While CIBP etiology is multifaceted, elevated levels of glutamate, an excitatory neurotransmitter, in the bone-tumor microenvironment may drive maladaptive nociceptive signaling. Here, we establish a relationship between the reactive nitrogen species peroxynitrite, tumor-derived glutamate, and CIBP. In vitro and in a syngeneic in vivo model of breast CIBP, murine mammary adenocarcinoma cells significantly elevated glutamate via the cystine/glutamate antiporter system xc−. The well-known system xc− inhibitor sulfasalazine significantly reduced levels of glutamate and attenuated CIBP-associated flinching and guarding behaviors. Peroxynitrite, a highly reactive species produced in tumors, significantly increased system xc− functional expression and tumor cell glutamate release. Scavenging peroxynitrite with the iron and mangano-based porphyrins, FeTMPyP and SRI10, significantly diminished tumor cell system xc− functional expression, reduced femur glutamate levels and mitigated CIBP. In sum, we demonstrate how breast cancer bone metastases upregulate a cystine/glutamate co-transporter to elevate extracellular glutamate. Pharmacological manipulation of peroxynitrite or system xc− attenuates CIBP, supporting a role for tumor-derived glutamate in CIBP and validating the targeting of system xc− as a novel therapeutic strategy for the management of metastatic bone pain.
1. Introduction
Cancer pain is reportedly experienced by 75% to 90% of patients with late-stage cancer27 with metastatic cancer–induced bone pain (CIBP) being the most common complaint.21 Although reports of pain with primary breast, lung and prostate cancers are rare, bone metastases produce excruciating pain, the etiology of which is not fully understood. Patients with metastatic breast cancer survive on average 1.5 to 3 years, during which time they have a number of comorbidities, including intractable pain.5 Current management of CIBP involves a treatment progression from radiation to strong opioids with adjuvant supplementation to treat worsening pain.44 In many cases, however, pain relief may be incomplete, unsatisfactory,40 or plagued with dose-limiting side effects further compromising patient quality of life.37 Additionally, narcotics for cancer pain are associated with the substantial rise in rates of diversion and substance abuse, contributing to the “opioid epidemic.”2 Novel analgesics are desperately needed for the >42% of patients with cancer pain who do not achieve acceptable pain relief.40
Cancer-induced bone pain elicits neurochemical changes unique from other chronic pain states.17 Although the bone itself is densely innervated by sympathetic and nociceptive fibers,20 the majority of metastatic tumors arising in bone lack detectable nerve fibers within the tumor mass acutely24 but can begin to gain access to the tumor during late stages and often after bone degradation.3 Rather, it is suggested that the acidic and enzymatic tumor environment and the substances released from tumor and tumor-associated cells (ie, growth factors, cytokines/chemokines) provoke nociceptive fibers in the bone.17,22,43 New analgesic drug targets for CIBP are being identified as more is learned about this distinctly complex pain state.
Recently, Singh and colleagues have demonstrated that several breast tumor cell lines release glutamate in vitro through the cystine/glutamate antiporter system xc−.31,32 System xc− plays a critical role in maintaining intracellular and extracellular antioxidant (eg, glutathione, cysteine) levels by exchanging extracellular cystine for intracellular glutamate. Glutamate released from cancer cells may stimulate nociceptors by activating N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and metabotropic-type glutamate receptors on peripheral endings17 leading to the persistent nociceptive state found in CIBP. The validity of system xc− as an analgesic target in CIBP is bolstered by evidence that repeated administration of sulfasalazine (SSZ), a system xc− inhibitor, attenuates pain-related behaviors in a nonsyngeneic model of CIBP.38
Although SSZ can block tumor cell glutamate release in vitro and assuage CIBP,31,33,38 neither the contribution of system xc− to tumor cell glutamate release in vivo nor the functional regulation of this transporter in the bone–tumor microenvironment are known. We explored the effect of direct and indirect pharmacological inhibition of system xc−-mediated glutamate transport in the bone–tumor microenvironment on pain behaviors in a syngeneic murine model of breast CIBP. For the first time, we demonstrate that increased levels of glutamate in the bone–tumor environment is, in part, responsible for CIBP and identify the reactive species peroxynitrite as a driver of tumor cell system xc− expression and glutamate release. Thus, the system xc− antagonist SSZ, a clinically available drug, and an inhibitor of peroxynitrite may have utility as adjunct therapies for CIBP.
2. Materials and methods
2.1. In vitro
2.1.1. Cell culture
Murine mammary tumor line 66.1 was a kind gift from Dr. Amy M. Fulton.42 66.1 cells were cultured in Eagle minimum essential medium (MEM) with 10% fetal bovine serum, 100 IU−1 penicillin, and 100 μg/mL streptomycin.16 For all assays, cells were counted using a gridded hemacytometer (Hausser Scientific, Horsham, PA).
2.1.2. Immunofluorescence microscopy
66.1 cells were seeded on gelatin-coated glass coverslips, fixed at 48 hours with ice-cold 70% ethanol and permeabilized with 0.25% Triton. Coverslips were incubated with anti-XCT polyclonal antibody (NB300-318; Novus Biologicals, Littleton, CO; 1:50 dilution in 5% BSA). AlexaFluor 488 goat anti-rabbit secondary antibody (2.5 μg/mL; Life Technologies, Carlsbad, CA) was prepared in 5% BSA with 1% normal donkey serum. Coverslips were mounted on glass slides with ProLong Gold antifade reagent with DAPI (Molecular Probes, Eugene, OR). Slides were imaged on a Zeiss Axioskop 40 using a 63x/0.08 numerical aperture Achroplan objective. Images were captured with a Zeiss AxioCam-Cm 1.
2.1.3. Western blot analysis
Whole-cell lysates were analyzed for expression of the functional subunit of system xc− XCT. Cell protein samples (10 μg) were resolved on 10% SDS–polyacrylamide gels (TGX Criterion XT; Bio-Rad, Hercules, CA) and transferred to a polyvinylidene difluoride membrane. Protein transfer was verified by Ponceau S staining, and the polyvinylidene difluoride membrane was incubated in 5% nonfat dry milk in Tris-buffered saline containing 0.05% Tween-20 (vol/vol; TBST). Membranes were incubated with rabbit polyclonal anti-XCT (Novus Biologicals NB300-318) or mouse monoclonal anti-actin AC40 (Abcam ab8226), appropriate secondary antibodies, and developed using Clarity ECL Substrate (Bio-Rad). Bands were quantitated and corrected for background using ImageJ software (Wayne Rasband, Research Services Branch, NIMH, Bethesda, MD). All data were normalized to β-actin as loading control and reported as fold change over untreated control.
2.1.4. Glutamate release
66.1 cells were seeded on 12-well plates and pretreated for 2 hours with Fe(III) meso-tetrakis (N-methylpyridinium-4-yl)porphyrin chloride (FeTMPyP; Cayman Chemical, Ann Arbor, MI), SSZ (Sigma-Aldrich, St. Louis, MO), Mn(III)-5,15-di-phenyl-tetracyclohexenylporphyin chloride (SRI10), or vehicle-containing (0.01 or 0.02% DMSO) MEM and then 6 hours with 3-morpholinosydnonimine (SIN-1) with and without FeTMPyP, SSZ, or SRI10. Cells were then incubated in a drug-containing, l-glutamine-, phenol red-, and serum-free Opti-MEM (Life Technologies, Carlsbad, CA) for 18 hours. Media was then harvested for glutamate analysis while cells were trypsinized and counted. Glutamate release was quantified with the AMPLEX Red glutamic acid assay kit (Invitrogen/Molecular Probes, Eugene, OR) that was optimized for measurement of glutamate concentrations above 0.5 μM by omitting l-alanine and l-glutamate pyruvate transaminase from the reaction, thus eliminating signal amplification through repeated cycling of glutamate through α-ketoglutarate31 and analyzed on a Synergy Bio-Tek Plate Reader.
2.2. In vivo
2.2.1. Animals
All procedures were approved by the University of Arizona Animal Care and Use Committee and conform to the Guidelines by the National Institutes of Health and the International Association for the Study of Pain. Adult female BALBc/cAnNHsd mice (15-18 g; Harlan, IN) were maintained in a climate-controlled room on a 12-hour light–dark cycle and allowed food and water ad libitum.
2.2.2. Intramedullary implantation of 66.1 cells
66.1 cells were implanted into the femur intramedullary space as described.16,18 The condyles of the right distal femoris were exposed and a hole was drilled to create a space for injection of 4 × 104 66.1 cells in 5 μL complete MEM or 5 μL complete MEM alone as a control into the intramedullary space.
2.2.3. Drug treatment
Animals received SSZ, FeTMPyP, or SRI10 dissolved in vehicle solutions of dimethyl sulfoxide, Tween-80, and 0.9% saline (1:1:8, SSZ and SRI10) or 0.9% saline (FeTMPyP) for injection (10 mL/kg, i.p.). Repeated dosing studies consisted of once-daily SSZ (30 mg/kg, i.p., days 7–10), FeTMPyP (10 mg/kg, i.p., days 7–14), SRI10 (3 mg/kg, i.p., day 7–day 14) or vehicle (10 mL/kg, i.p.) after femoral inoculation.
2.2.4. Analysis of chronic pain
Animals were assessed for spontaneous pain presurgery and on postsurgery days 7, 10 and/or 14 before and 60 minutes after treatment. All testing was performed by a blinded observer during the animals' light cycle.
2.2.5. Spontaneous pain
Spontaneous pain-related behaviors were recorded as previously described.16,18 Flinching and guarding were observed for 2 minutes during a resting state after a 30-minute acclimation period. Flinching was defined as the lifting and rapid flexing of the ipsilateral hind paw not associated with walking or movement. The number of flinches was recorded within the 2-minute observation period. Guarding was characterized by fully retracting the ipsilateral hind limb under the torso. The time the hindpaw was retracted during the 2-minute period was recorded. Studies were performed in a blinded manner (the observer was blinded to all treatments).
2.3. Ex vivo
2.3.1. Glutamate in femur aspirate
Animals were killed on day 10 or day 14, and both femurs were removed. Proximal and distal ends of the femur were clipped and the intramedullary contents rinsed into a vial with 0.1M PBS containing protease inhibitor cocktail and EDTA (Pierce, Rockford, IL). Femur contents from 2 to 4 animals per treatment were pooled per sample. AMPLEX Red glutamic acid assay kit (Invitrogen/Molecular Probes) was used to determine glutamate concentration from pooled samples.
2.3.2. Bone marrow extrudate immunoblotting
Animals were killed on day 14, and ipsilateral and contralateral femurs were removed. Proximal and distal ends of the femur were clipped, and the intramedullary content was collected and pooled as described above. Extrudates were sonicated and spun at 10,000g for 10 minutes to remove insoluble debris. Femur marrow protein samples (10 μg) were analyzed for expression of XCT by Western blot analysis, 2.1.3.
2.3.3. Bone histology
Animal femurs were inoculated with breast cancer cells (66.1), and FeTMPyP (10 mg/kg, i.p., q.d.) or vehicle (saline, 10 mL/kg, i.p., q.d.) was administered on postsurgery days 7 to 14. After behavioral testing on postsurgery day 14, animals were anesthetized (ketamine 80 mg/kg:xylazine 12 mg/kg, i.p.) and perfused transcardially with 0.1 M PBS followed by 4% neutral-buffered formalin and 12.5% picric acid (Sigma). Femurs were collected, postfixed overnight at 4°C and decalcified in 10% EDTA (RDO-Apex, Aurora, IL) for 14 days, and then paraffin embedded. Femurs were cut in the frontal plane into 5-μm sections and stained with hematoxylin and eosin (H&E) to visualize normal marrow elements and cancer cells under bright field microscopy on a Nikon E800 at ×4 magnification. Tumor or marrow areas within the femur (6 bones per treatment) were measured between the epiphyseal plates using ImageJ software (National Institutes of Health) by a blinded observer.
2.3.4. Radiography
Animal femurs were inoculated with breast cancer cells (66.1) or cell-free media (Sham), and FeTMPyP (10 mg/kg, i.p., q.d.) or vehicle (saline, 10 mL/kg, i.p., q.d.) was administered on postsurgery days 7 to 14. A digital Faxitron machine was used to acquire live radiographs of mice anesthetized with ketamine/xylazine on day 14. Bone loss was rated by 3 blinded observers trained in scoring animal radiographs according to the following scale: 0 = normal bone, 1 = 1 to 3 radiographic lesions indicating bone loss, 2 = 4 to 6 radiographic lesions indicating bone loss, 3 = full-thickness unicortical bone loss indicating unicortical bone fracture, and 4 = full-thickness bicortical bone loss indicating bicortical bone fracture. Observer scores (3) for each bone on day 14 were averaged.
2.3.5. Statistical analysis
All data are presented as mean ± SEM. N values are as follows: Western blot analysis, 3 to 4 independent experiments; extracellular glutamate, 4 to 6 from ≥2 independent experiments; femur glutamate, 3 to 4 pooled samples; behavior, 6 to 12 animals/treatment; histology, 6 animals/treatment; radiography, 6 to 12 animals/treatment. Statistical significance between treatment groups was determined by Western blot, glutamate release and radiography studies, 1-way analysis of variance (ANOVA) followed by the Bonferroni post hoc multiple comparisons test; femur glutamate studies, 1-way ANOVA followed by the Bonferroni post hoc multiple comparisons test or Kruskal–Wallis test followed by the Dunn multiple comparison test; behavioral studies, 2-way ANOVA with the Bonferroni post hoc multiple comparisons test; histology, Student t test. A value of P < 0.05 was accepted as statistically significant. Statistical analyses were run and plots were generated in GraphPad Prism 5.0 (Graph Pad Inc, San Diego, CA).
3. Results
3.1. 66.1. Cells release glutamate through system xc−
Several tumor cell lines, including those of mouse, rat, and human origin, release glutamate in vitro through the cysteine/glutamate antiporter system xc−.32 We investigated the expression and function of system xc− in the murine mammary adenocarcinoma cell line 66.1; this cell line is highly tumorigenic in vitro and in vivo.11,16,18,42 To visualize system xc− in 66.1 cells, we used immunocytochemistry. System xc− comprises a heavy subunit (4f2hc) and a light subunit (XCT). XCT is specific to system xc− and is essential for amino acid exchange. Positive immunoreactivity for XCT was evident in untreated 66.1 cells (Fig. 1A).
Figure 1.
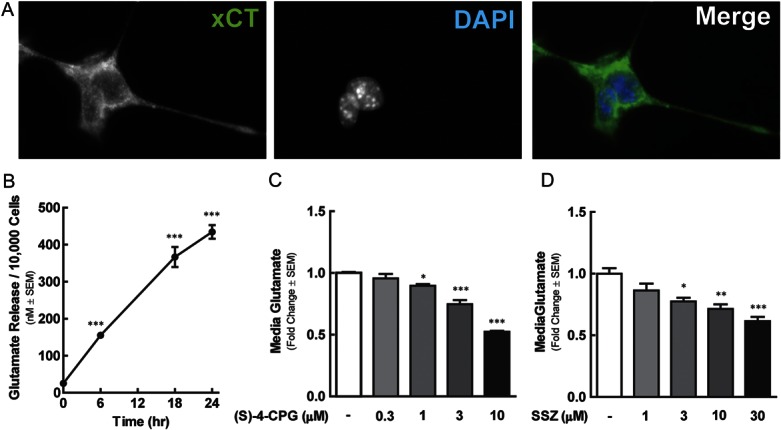
The murine mammary adenocarcinoma cell line 66.1 expresses the cystine/glutamate antiporter system xc−. (A) Representative images of 66.1 XCT and DAPI staining, as indicated. 66.1 cell glutamate release was quantified over a 24-hour period in untreated cells (B), and at 18 hours in those exposed to vehicle, sulfasalazine (SSZ) (C) or (S)-4-carboxyphenylglycine (CPG-4) (D). Asterisks denote values statistically different from time 0 or vehicle control group, *P < 0.05, **P < 0.01, ***P < 0.001.
To assess whether 66.1 system xc− is functional, 66.1 glutamate released into l-glutamine-free media was measured over time (Fig. 1B). Media glutamate concentration increased significantly from 10 nM/10,000 (t = 0 hours) to 400 nM/10,000 cells over 24 hours (1-way ANOVA F(3) = 130.2, P < 0.0001). System xc− is the only glutamate transporter implicated in breast tumor cell glutamate release to date32; we verified its role in 66.1 glutamate release using 2 established system xc− inhibitors: (S)-4-carboxyphenylglycine (CPG-4, Fig. 1C) and SSZ33 (Fig. 1D). Both CPG-4 (1, 3, and 10 μM) and SSZ (3, 10, and 30 μM) concentration-dependently reduced 66.1-derived glutamate release after an 18-hour incubation period (1-way ANOVA F(3) = 10.11, P = 0.0001). Media glutamate concentration, normalized to the cell number, was reduced 0.5 fold and 0.6 fold with 10 μM CPG-4 and 30 μM SSZ, respectively. Together, these data suggest that 66.1 cells extrude glutamate through system xc−.
3.2. Blockade of system xc− with SSZ attenuates CIBP-related behaviors and reduces femur glutamate
We next determined whether systemic SSZ treatment reduced pain in a murine model of CIBP by modulating tumor-derived glutamate release. Acute antihyperalgesic studies with SSZ in CIBP (Fig. S1, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A322) informed our dose selection of SSZ for the repeated exposure study. On day 7 after 66.1 intrafemoral inoculation, mice displayed significant bone cancer–induced flinching (Fig. 2A) and guarding (Fig. 2B). Repeated SSZ administration (30 mg/kg, i.p., q.d., from day 7 to day 10) in cancer-inoculated mice significantly decreased flinching (2-way ANOVA F(3,2) = 3.015, P = 0.0309) and guarding (2-way ANOVA F(3,2) = 10.51, P < 0.0001) on day 10, as compared with vehicle-treated animals (Fig. 2A, B). Media-inoculated sham mice had minor, surgery-associated spontaneous pain on postsurgery day 7 that was significantly blunted relative to cancer animals, not affected by SSZ treatment, and did not increase over the experimental time course.
Figure 2.
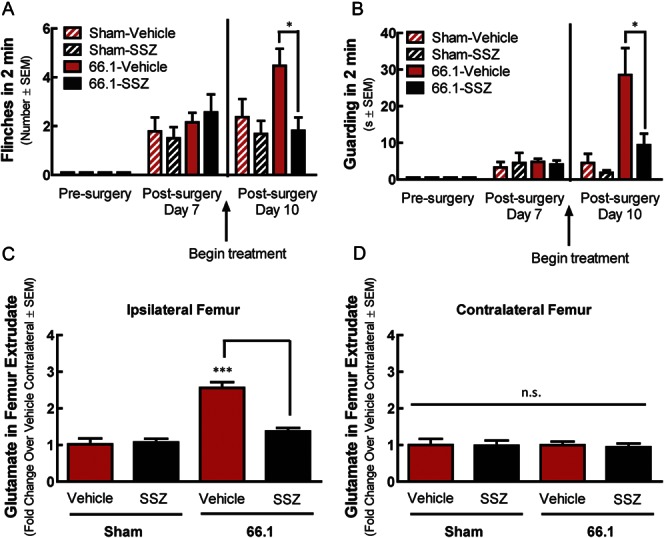
Blockade of system xc− with sulfasalazine (SSZ) attenuates cancer-induced bone pain behaviors and reduces femur glutamate. Animal femurs were injected with breast cancer cells (66.1) or cell-free media (sham) after baseline (presurgery) behavioral measurements. SSZ (30 mg/kg, i.p.) or vehicle (10 mL/kg, i.p.) was administered after behavioral measurements on postsurgery day 7 and continued for 3 days (q.d.). Cancer-induced spontaneous flinching (A) and guarding (B) were significantly reduced 60 minutes after SSZ treatment on day 10, as compared with vehicle-treated animals. Femur contents from inoculated femur (ipsilateral) (C) and contralateral femur (D) were collected on day 10 for glutamate analysis; *P < 0.05, **P < 0.01, ***P < 0.001.
Assessment of glutamate concentration in femur extrudates from these mice revealed a significant increase in glutamate in the ipsilateral femur of 66.1 animals treated with vehicle, as compared with glutamate concentration in both the contralateral femur of tumor-bearing animals and the ipsilateral and contralateral femurs of non–tumor-bearing sham animals (Fig. 2C, D; 1-way ANOVA F(3) = 17.93, P = 0.0002). This finding is consistent with the idea that tumor cells release glutamate in vivo. In contrast to tumor-bearing, vehicle-treated animals, glutamate concentration was not elevated, relative to contralateral bone or sham animals, in femur extrudates from cancer-inoculated, SSZ-treated animals (1-way ANOVA F(3) = 0.06, 104, P = 0.9792). These data demonstrate, for the first time, that the antihyperalgesic effect of SSZ in CIBP corresponds with a decrease in glutamate in the bone–tumor microenvironment.
3.3. Peroxynitrite regulates tumor cell system xc− functional expression
As an alternative to direct inhibition of system xc− with SSZ, we next asked whether we could reduce tumor cell glutamate release by modulating system xc− functional expression. System xc− is involved in mobilizing a cell's antioxidant defenses; upregulation occurs in response to oxidative challenge.15 66.1 system xc−-mediated glutamate transport may be driven by peroxynitrite, a potent oxidant formed by the spontaneous, diffusion-limited reaction of superoxide and nitric oxide that is significantly elevated in invasive breast carcinomas39 and our murine CIBP model (Fig. S2, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A322). To test this hypothesis, we applied the superoxide- and nitric oxide-donating compound SIN-1, to 66.1 cells in vitro.
66.1 cells were treated with concentrations of SIN-1 ranging from 50 nM to 5 µM for 18 hours; concentrations and a time point were selected from those known to increase system xc−-mediated transport in other cell types.4 SIN-1 significantly increased XCT levels in whole-cell lysates at both 5 and 50 µM, as compared with vehicle (0.01% DMSO)-treated lysates (Fig. 3A, B; 1-way ANOVA F(4) = 4.119, P = 0.0316).
Figure 3.

Peroxynitrite regulates system xc− functional expression. 66.1 cells were treated SIN-1 (50 nm–5 μM) or vehicle. Eighteen hours after treatment, cells were harvested and analyzed by Western blot analysis for XCT (A and B). SIN-1 stimulated 66.1 glutamate release in the absence (C) or presence (D) of sulfasalazine was quantified. 66.1 cells were exposed to SIN-1 in the presence of FeTMPyP or vehicle. Eighteen hours after treatment, cells were harvested and analyzed through Western blot analysis for expression of XCT (E and F). 66.1 glutamate release in the presence of SIN-1 and FeTMPyP, SRI10, or vehicle was quantified (G and H). Asterisks represent data points that are significantly different from those of vehicle control, unless otherwise indicated; *P < 0.05, **P < 0.01, ***P < 0.001.
The functional relevance of peroxynitrite-driven increases in XCT protein expression in 66.1 cells was investigated as a function of glutamate export in the presence of SIN-1. SIN-1 concentration-dependently increased tumor cell glutamate release as compared with vehicle-treated cells (Fig. 3C). At concentrations of 500 nM and 5 μM, SIN-1 significantly increased 66.1-mediated cell glutamate release up to 1.5 fold over an 18-hour period (1-way ANOVA F(3) = 15.60, P < 0.0001). Inclusion of the established system xc− inhibitor SSZ (3, 10, and 30 μM) significantly reduced SIN-1-induced glutamate release (Fig. 3D; 1-way ANOVA F(4) = 26.52, P < 0.0001). Together, these data suggest that peroxynitrite increases system xc−-mediated glutamate expulsion from 66.1 cells.
Peroxynitrite can be targeted pharmacologically with oxidoreductant compounds that detoxify peroxynitrite by catalyzing its isomerization and/or reduction to innocuous nitrate or nitrite. The iron-containing cationic alkylpyridyl porphyrin FeTMPyP catalyzes the isomerization of peroxynitrite to more innocuous NO3− (log kred (ONOO−) = 6.34) and the dismutation of superoxide (log kcat(O2•−) = 7.2).29 In 66.1 cells, FeTMPyP treatment concentration-dependently blocked SIN-1-induced increases in XCT protein expression (Fig. 3E, F). SIN-1 upregulation of XCT protein was blocked by 10 μM FeTMPyP (1-way ANOVA F(4) = 49.54, P < 0.0001).
We then determined whether FeTMPyP treatment reduced SIN-1-induced increase in 66.1 glutamate release. FeTMPyP (1 and 10 μM), significantly blocked SIN-1-induced increase in 66.1 glutamate release (Fig. 3G; 1-way ANOVA F(4) = 14.50, P < 0.0001). To verify the role of peroxynitrite in system xc− regulation, we used the more selective peroxynitrite decomposition catalyst SRI10. SRI10 removes peroxynitrite (log kred (ONOO−) = 5 to 6), but lacks superoxide dismutase activity.26 SRI10 treatment at 100 nM, 1 μM, and 10 μM significantly blocked SIN-1-induced increase in 66.1 glutamate release (Fig. 3H; 1-way ANOVA F(5) = 18.64, P < 0.0001). Together, these data show that elimination of peroxynitrite blocks the induction of system xc− functional expression in tumor cells.
3.4. The redox-active porphyrin FeTMPyP attenuates CIBP-related behaviors and reduces femur glutamate
To determine whether FeTMPyP was antihyperalgesic in CIBP, cancer-associated flinching and guarding behaviors were assessed in tumor-bearing mice before and after surgery and before and after FeTMPyP treatment. Acute, systemic (i.p.) FeTMPyP treatment resulted in a profound and long-lasting dose-dependent reduction in pain behaviors (Fig. S3A, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A322), B; 2-way ANOVA Fflinching(4, 9) = 60.95, Fguarding(4, 9) = 68.59, P < 0.0001. The peak FeTMPyP antinociceptive effect occurred 60 minutes after administration. At higher doses, cancer-induced hypersensitivity did not return to baseline levels until 6 to 8 hours after drug treatment. Vehicle treatment did not alter CIBP behaviors (1-way ANOVA F(8) = 0.1833, P = 0.9924).
Dose–response curves for flinching and guarding behavior were constructed by fitting 4-parameter logistic curves to percent maximum possible effect (% MPE) data; the FeTMPyP A90 was calculated to be 10.72 and 2.79 mg/kg for flinching and guarding, respectively. Based on acute studies, a dose between the flinching and guarding A90 (10 mg/kg) was selected for repeated administration studies. This dose did not impair motor coordination or alter weight gain over the experimental timeline (Fig. S4A, B, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A322).
Mice with CIBP were repeatedly treated with either vehicle (10 mL/kg 0.9% saline, i.p., q.d.) or FeTMPyP (10 mg/kg, i.p., q.d.) from day 7 to day 14. Vehicle-treated cancer-inoculated mice showed increasing flinching and guarding behavior day 7 through day 14, mirroring disease progression (Fig. 4A, B). Daily administration of FeTMPyP to cancer-inoculated mice decreased ipsilateral flinching and guarding 60 minutes after treatment on postsurgery day 10 (Fig. 4A, B; 2-way ANOVA followed by the Bonferroni post hoc test tflinching = 4.057, tguarding = 3.331, P < 0.01). Mice were again evaluated for CIBP behaviors on day 14; unexpectedly, before treatment, FeTMPyP-treated animals displayed significantly more spontaneous flinching and guarding behaviors than did their vehicle-treated counterparts (2-way ANOVA followed by Bonferroni posttests tflinching = 4.621, tguarding = 3.713, P < 0.01). Despite this enhanced manifestation of CIBP, FeTMPyP treatment on day 14 to cancer-inoculated mice significantly decreased guarding and flinching 60 minutes after injection (Fig. 4A, B) (2-way ANOVA followed by Bonferroni posttests, tflinching = 8.903, tguarding = 8.027, P < 0.001). Sham mice showed minor, surgery-associated spontaneous flinching and guarding on postsurgery day 7 to day 14 that were significantly blunted relative to cancer animals (2-way ANOVA followed by Bonferroni posttests tflinching = 3.233, tguarding = 2.727, P < 0.05, on day 7) and was unaffected by FeTMPyP treatment (2-way ANOVA followed by Bonferroni posttests tflinching = 0.0, tguarding = 0.08, 214, P > 0.05, on day 7). Repeated FeTMPyP treatment did not alter tumor burden as determined by hematoxylin and eosin staining of tumor-bearing femur (Fig. 5; Student t test t = 0.5439, P = 0.6034) or bone integrity (Fig. 6; Student t test t = 0.9830, P = 0.3310) on day 14.
Figure 4.
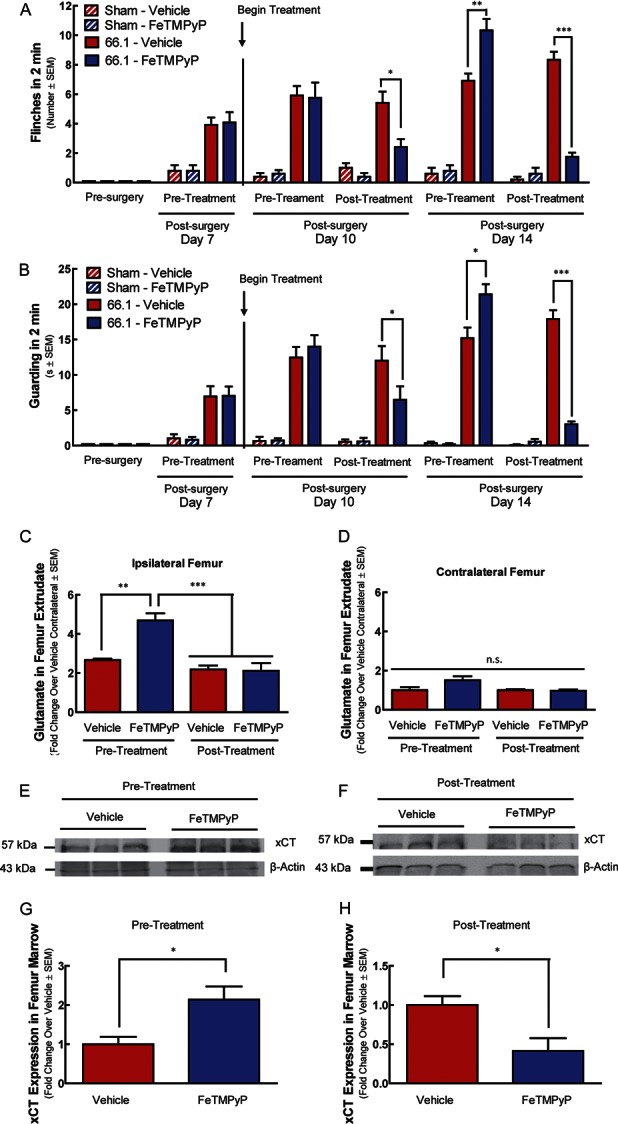
Repeated FeTMPyP treatment modulates spontaneous cancer-induced bone pain behaviors and system xc− functional expression in vivo. Animal femurs were injected with breast cancer cells (66.1) or cell-free media (sham) after baseline (presurgery) behavioral measurements. On day 7 after femoral inoculation, 66.1 animals demonstrated bone cancer–induced flinching (A) and guarding (B). FeTMPyP (10 mg/kg, i.p.) or vehicle (saline, 10 mL/kg, i.p.) was administered after behavioral measurements day 7 and continued to day 14 (q.d.). Spontaneous flinching and guarding were assessed before and after treatment on days 10 and 14 (A and B). Femur marrow from inoculated femur (ipsilateral) (C) and contralateral femur (D) was collected from tumor-bearing animals before or after the final treatment on day 14 for glutamate analysis. Day 14 ipsilateral marrow samples from tumor-bearing animals were analyzed for XCT expression using Western blot analysis (E–H); *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 5.

Repeated FeTMPyP does not alter tumor burden. Animal femurs were inoculated with cell-free media (sham) or breast cancer cells (66.1). FeTMPyP (10 mg/kg, i.p., q.d.) or vehicle (saline, 10 mL/kg, i.p., q.d.) was administered on postsurgery days 7 to 14. On day 14, femurs were collected, paraffin-embedded, and stained with hematoxylin and eosin (H&E) to visualize normal marrow elements and cancer cells under bright field microscopy. Representative images (A) and quantification of cancer cell marrow occupancy (B) are shown.
Figure 6.

Repeated FeTMPyP does not alter bone integrity. Animal femurs were inoculated with cell-free media (sham) or breast cancer cells (66.1). FeTMPyP (10 mg/kg, i.p., q.d.) or vehicle (saline, 10 mL/kg, i.p., q.d.) was administered on postsurgery days 7 to 14. On day 14, live radiographs were taken. Bone loss was rated by 3 blinded observers according to an established scale (A). Elevated bone loss scores were found in the tumor-bearing animals as compared with sham controls, but no treatment differences were evident (B); ***P < 0.001.
Femur extrudates were collected from 2 distinct experimental groups to determine glutamate levels relative to CIBP behaviors: (1) “pretreatment” mice that were killed before treatment on day 14 and (2) “posttreatment” mice that received drug or vehicle treatment on day 14. 66.1-inoculated, FeTMPyP-treated mice in the pretreatment group displayed increased CIBP-related behaviors, whereas those in the FeTMPyP posttreatment group displayed reduced CIBP-related behaviors, relative to their respective vehicle controls (Fig. 4A, B). Likewise, glutamate concentration in ipsilateral femur extrudates was significantly increased in 66.1-inoculated, FeTMPyP-treated mice in the pretreatment group, as compared with the vehicle control (Fig. 4C; 1-way ANOVA followed by Bonferroni posttests t = 4.468, P < 0.01), and significantly reduced in the 66.1-inoculated, FeTMPyP-treated posttreatment group, as compared with pretreatment levels (1-way ANOVA followed by Bonferroni posttests t = 6.075, P < 0.001), illustrating reduced glutamate in the bone-tumor microenvironment with FeTMPyP intervention. Glutamate in the contralateral bone was not significantly affected by 66.1 inoculation or drug treatment (Fig. 4D; 1-way ANOVA followed by Bonferroni posttests t = 0.0-3.265, all P > 0.05). Femur extrudate XCT protein expression was significantly elevated in the FeTMPyP-treated pretreatment group (Fig. 4E, G; Student t test t = 3.509, P = 0.0247) and reduced in the FeTMPyP-treated posttreatment group (Fig. 4F, H; Student t test t = 5.153, P = 0.0067), corresponding to both glutamate levels and pain behaviors. Thus, system xc− expression and tumor-derived glutamate levels may drive pain behaviors in vivo.
3.5. The peroxynitrite decomposition catalyst SRI10 attenuates CIBP-related behaviors and reduces femur glutamate
To determine whether peroxynitrite elimination, superoxide dismutation, or both actions contribute to FeTMPyP's efficacy in CIBP, we used the superoxide-sparing peroxynitrite decomposition catalyst SRI10. Mice with CIBP were treated with either vehicle (10 mL/kg 0.9% saline, i.p., q.d.) or SRI10 (3 mg/kg, i.p., q.d.) from day 7 to day 14. Vehicle-treated, cancer-inoculated mice displayed increasing spontaneous flinching and guarding behavior over the experimental time course (Fig. 7A, B). Daily administration of SRI10 to cancer-inoculated mice decreased ipsilateral guarding and flinching 60 minutes after treatment on postsurgery day 7 (Fig. S5, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A322) and day 10 (2-way ANOVA followed by Bonferroni posttests, tflinching = 4.329, tguarding = 3.187 P < 0.05) and day 14 (Fig. 7A, B; 2-way ANOVA followed by Bonferroni posttests, tflinching = 3.849, tguarding = 3.763 P < 0.01). The decrease in pain behaviors pre- to post-SRI10 treatment day 14 was paralleled by a reduction in ipsilateral femur marrow glutamate (Fig. 7C, D; Kruskal–Wallis test followed by the Dunn multiple comparison test, difference in rank sum = 6.667, P < 0.05). These data implicate peroxynitrite in the antihyperalgesic effects and glutamate reduction seen with FeTMPyP and suggest that elimination of peroxynitrite is sufficient to reduce tumor-derived glutamate and CIBP.
Figure 7.

Repeated SRI10 treatment attenuates spontaneous cancer-induced bone pain behaviors and reduces femur glutamate. Animal femurs were injected with breast cancer cells (66.1) or cell-free media (sham) after baseline (presurgery) behavioral measurements. SRI10 (3 mg/kg, i.p.) or vehicle (10 mL/kg, i.p.) was administered after behavioral measurements day 7 and continued to day 14 (q.d.). Spontaneous flinching (A) and guarding (B) were assessed before and after treatment on days 10 and 14. Femur marrow from inoculated femur (ipsilateral) (C) and contralateral femur (D) was collected from tumor-bearing animals before or after the final treatment on day 14 for glutamate analysis; *P < 0.05, **P < 0.01, ***P < 0.001.
4. Discussion
Many solid tumors, including breast, do not result in pain in their native tissue but cause excruciating pain once they metastasize to bone; our laboratory focuses on the contribution of the bone microenvironment to this pain. Nearly 200,000 women are diagnosed with breast cancer each year in the United States alone.1 Notably, breast cancer is the most frequent malignant tumor in women worldwide and accounts for the greatest number of bone metastases of all cancer types.6,7 Bone metastases are detected in up to 70% of patients with advanced breast cancer, making bone the most common site for breast cancer metastasis and distal breast cancer relapse.
Determining the mechanisms underlying breast CIBP is critical for the identification of new analgesic targets and improving quality of life for patients with cancer. Recently, oxidative stress, an established hallmark of tumor burden and known contributor to chronic pain,28 was implicated in CIBP.34,37 The pronociceptive action of oxidative stress in CIBP has been hypothesized to be a function of tumor-derived nitrating species that alters the response of primary afferent neurons to glutamate and the release of glutamate by cancer cells, but the mechanism is unknown.17,31,37,38 Our current findings build on the previous findings of the Singh laboratory in which they demonstrated in vitro that system xc− in cancer cell lines result in increased levels of glutamate and result in pain behaviors that are attenuated by SSZ.38 In this study, we provide evidence that (1) tumor cell glutamate release through system xc− in vivo is a critical contributor to CIBP, (2) reactive oxygen/nitrogen species regulate the cystine/glutamate antiporter system xc− in tumor cells, and (3) upregulation of system xc− increases glutamate in the bone–tumor microenvironment, resulting in behavioral signs of CIBP.
We found that the murine mammary adenocarcinoma cell line 66.1 releases glutamate through system xc−. Although our studies were limited to only a single cell line, this finding places 66.1 on a growing list of tumor cell lines that release glutamate through system xc−, including C3L5 (mouse breast cancer), B16F1 (mouse melanoma), MAT-LyLu (rat prostate cancer), CNS-1 (rat astrocytoma), MDA-MB-231, and MCF-7 (human breast cancers).32 Thus, glutamate release from cancer cells is a general phenomenon. We elected to use the 66.1 cell line because it is highly tumorigenic, yet less aggressive than the 4T1 line, allowing for colonization of bone with reduced risk of extraskeletal metastasis with disease progression.
The hypothesis that the system xc− inhibitor SSZ attenuates CIBP-related behaviors by reducing system xc−-mediated glutamate release in vivo has been previously proposed38 but with the need for further experimental verification. Here, blockade of tumor cell glutamate transport with SSZ both attenuated cancer-associated flinching and guarding behaviors and decreased marrow glutamate in a murine model of CIBP. That is, direct system xc− inhibition with SSZ reduced CIBP-related behaviors and glutamate in the bone–tumor microenvironment. The lack of SSZ effect on glutamate levels in sham animals or the contralateral femur of cancer-bearing animals affirms that SSZ reduces glutamate release from tumor cells but not native bone cells. The measurements of cancer-induced flinching and guarding are believed to be more clinically relevant than evoked behaviors because patients are not routinely evoked/poked with von Frey filaments with translational relevance.8,9 These data provide a crucial link between tumor-derived glutamate through system xc− and CIBP.
The major physiological role of system xc− is to support pathways that mediate cellular protection from damaging ROS/RNS: (1) the synthesis of the antioxidant glutathione and (2) the cystine/cysteine redox cycle.37 The XCT gene and, thus system xc−-mediated transport, is upregulated in response to oxidative/nitrosative challenge.30 Rapid proliferation and tumor burden make cancer just such an oxidative/nitrosative challenge.36 In the tumor–bone microenvironment, sources of oxidative and nitrosative stress include both tumor and associated immune cells (eg, macrophages, neutrophils, T-lymphocytes).17 Relevant to both cancer and pain is the reactive species peroxynitrite.
Production of peroxynitrite is significantly increased under pathological conditions (eg, cancer).14,25,41 Although the short half-life of peroxynitrite makes direct detection of this RNS difficult,10 nitration of tyrosine residues is used as an in vivo marker of peroxynitrite generation. Invasive breast carcinomas with associated CIBP have high levels of nitrotyrosine.39 Clinically, high nitrotyrosine levels are associated with reduced disease-free and overall survival for patients with breast cancer and are a significant, independent predictor for overall survival.19,23
Given elevated peroxynitrite generation in breast cancer and the ability of this oxidant species to increase system xc− transport, we hypothesized that peroxynitrite may drive tumor cell system xc− expression, glutamate release, and CIBP. In line with literature reports, we demonstrate elevated system xc− protein expression and glutamate transport in 66.1 tumor cells after application of SIN-1, a peroxynitrite generator. The role of peroxynitrite was confirmed by sensitivity to the redox-active porphyrin FeTMPyP and superoxide-sparing peroxynitrite decomposition catalyst SRI10. The role of system xc− in SIN-1-induced glutamate transport was confirmed with the established system xc− inhibitor SSZ. Thus, system xc− could be targeted directly using an inhibitor or indirectly by pharmacologically modulating its functional expression through the elimination of peroxynitrite.
In vivo, chronic FeTMPyP or SRI10 treatment reduced behavioral evidence of CIBP. Our data support the idea that the antihyperalgesic effect of FeTMPyP and SRI10 in CIBP is attributable, in part, to a reduction of tumor-derived glutamate. After a 7-day treatment course, cancer-bearing, FeTMPyP- or SRI10-treated mice showed less pain compared with vehicle-treated controls. Both femur marrow glutamate and system xc− protein expression were reduced in these animals, as compared with pretreatment levels. These data suggest an association between reductions in pain behaviors with decreased glutamate in the bone–tumor microenvironment. Not all pain was abolished, which suggests that CIBP is complex and multifaceted, and although glutamate is an important part of the algogenic factors that may play a role as an activator of nociception due to its interaction with NMDA and AMPA channels, it is only one component of the several algogenic substances that are present in the bone–tumor microenvironment. The importance of glutamate may be linked to initiating changes in the electrical potential of nociceptive fibers that may be further sensitized by the many cytokines and chemokines in CIBP.13,30 Critically, FeTMPyP did not alter tumor burden or bone integrity, which suggests that the reduction in pain behaviors and femur marrow glutamate was not secondary to a reduction in tumor size or osteolytic lesions. Interestingly, cancer-bearing, FeTMPyP-treated mice exhibited more pain behaviors with elevated femur marrow glutamate and system xc− protein expression 24 hours after injection. This pain and femur marrow glutamate “overshoot” on experimental day 14 may be the result of modulating a dynamic already challenged system and model what is referred to in patients with metastatic cancer as “breakthrough” pain. Maintaining redox balance and the redoxome is particularly challenging for rapidly dividing, malignant cells. Perturbing this balance with a redox modulator, particularly one in the iron–porphyrin class known to undergo Fenton chemistry,35 may have led to further dysregulation. In the future, this glutamate and pain rebound may be prevented by addressing the pharmacokinetic characteristics of FeTMPyP and maintaining constant drug serum levels. Compounds that remove peroxynitrite or prevent its formation show great therapeutic promise in animal models of disease and several are currently in development for use as anticancer single agents, chemosensitizers, and radioprotectants.35
Despite marked advances in chemotherapeutics, very little progress has been made in understanding the pain associated with metastatic bone disease. Recent reports have emphasized the need for treatments that not only promote survival in patients with terminally illness but also manage pain and preserve functional status.12,13 Such interventions will improve patient, family, and caregiver quality of life.12
In sum, we demonstrate that both direct and indirect pharmacological inhibition of system xc− significantly reduces advanced-stage bone cancer pain by reducing extracellular glutamate (Fig. 8). We also show for the first time that peroxynitrite, generated in the cancer microenvironment, significantly upregulates system xc− and is itself a novel analgesic target in CIBP. These studies provide a critical, previously missing, mechanistic link to explain the efficacy of SSZ in CIBP. Further research is warranted to explore the full therapeutic potential of redox modulators in metastatic bone disease and evaluate the repurposing of SSZ as an adjunct therapy for patients with CIBP.
Figure 8.

System xc− can be targeted directly or indirectly to reduce glutamate in the bone-tumor microenvironment and assuage cancer-induced bone pain (CIBP). System xc− can be targeted pharmacologically through direct blockage with sulfasalazine or through elimination of peroxynitrite with the decomposition catalysts FeTMPyP and SRI10. Both therapeutic strategies reduced glutamate in the bone–tumor microenvironment and CIBP-related behaviors.
Conflict of interest statement
The authors have no conflicts of interest to declare.
This work was supported by NIH/NCI RO1 CA142115.
Supplementary Material
Appendix A. Supplemental Digital Content
Supplemental Digital Content associated with this article can be found online at http://links.lww.com/PAIN/A322.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].American Cancer Society. Global Cancer Facts & Figures 2nd Edition Atlanta: American Cancer Society; 2011. [Google Scholar]
- [2].Barclay JS, Owens JE, Blackhall LJ. Screening for substance abuse risk in cancer patients using the Opioid Risk Tool and urine drug screen. Support Care Cancer 2014;22:1883–8. [DOI] [PubMed] [Google Scholar]
- [3].Bloom AP, Jimenez-Andrade JM, Taylor RN, Castaneda-Corral G, Kaczmarska MJ, Freeman KT, Coughlin KA, Ghilardi JR, Kuskowski MA, Mantyh PW. Breast cancer-induced bone remodeling, skeletal pain, and sprouting of sensory nerve fibers. J Pain 2011;12:698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Buckley BJ, Whorton AR. Adaptive responses to peroxynitrite: increased glutathione levels and cystine uptake in vascular cells. Am J Physiol Cell Physiol 2000;279:C1168–76. [DOI] [PubMed] [Google Scholar]
- [5].Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev 2001;27:165–76. [DOI] [PubMed] [Google Scholar]
- [6].Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res 2006;12(20 pt 2):6243s-9s. [DOI] [PubMed] [Google Scholar]
- [7].Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer 1987;55:61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Currie GL, Delaney A, Bennett MI, Dickenson AH, Egan KJ, Vesterinen HM, Sena ES, Macleod MR, Colvin LA, Fallon MT. Animal models of bone cancer pain: systematic review and meta-analyses. PAIN 2013;154:917–26. [DOI] [PubMed] [Google Scholar]
- [9].Delaney A, Fleetwood-Walker SM, Colvin LA, Fallon M. Translational medicine: cancer pain mechanisms and management. Br J Anaesth 2008;101:87–94. [DOI] [PubMed] [Google Scholar]
- [10].Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proc Natl Acad Sci U S A 1998;95:3566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dexter DL, Kowalski HM, Blazar BA, Fligiel Z, Vogel R, Heppner GH. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res 1978;38:3174–81. [PubMed] [Google Scholar]
- [12].Fisch MJ, Lee JW, Weiss M, Wagner LI, Chang VT, Cella D, Manola JB, Minasian LM, McCaskill-Stevens W, Mendoza TR, Cleeland CS. Prospective, observational study of pain and analgesic prescribing in medical oncology outpatients with breast, colorectal, lung, or prostate cancer. J Clin Oncol 2012;30:1980–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys 1998;356:1–11. [DOI] [PubMed] [Google Scholar]
- [15].Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 2013;18:522–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lozano-Ondoua AN, Hanlon KE, Symons-Liguori AM, Largent-Milnes TM, Havelin JJ, Ferland HL, III, Chandramouli A, Owusu-Ankomah M, Nikolich-Zugich T, Bloom AP, Jimenez-Andrade JM, King T, Porreca F, Nelson MA, Mantyh PW, Vanderah TW. Disease modification of breast cancer-induced bone remodeling by cannabinoid 2 receptor agonists. J Bone Miner Res 2013;28:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lozano-Ondoua AN, Symons-Liguori AM, Vanderah TW. Cancer-induced bone pain: mechanisms and models. Neurosci Lett 2013;(557 pt A):52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lozano-Ondoua AN, Wright C, Vardanyan A, King T, Largent-Milnes TM, Nelson M, Jimenez-Andrade JM, Mantyh PW, Vanderah TW. A cannabinoid 2 receptor agonist attenuates bone cancer-induced pain and bone loss. Life Sci 2010;86:646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest 2011;121:4015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O'Leary P, Mantyh PW. Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience 2002;113:155–66. [DOI] [PubMed] [Google Scholar]
- [21].Mercadante S. Malignant bone pain: pathophysiology and treatment. PAIN 1997;69:1–18. [DOI] [PubMed] [Google Scholar]
- [22].Nagae M, Hiraga T, Yoneda T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J Bone Miner Metab 2007;25:99–104. [DOI] [PubMed] [Google Scholar]
- [23].Nakamura Y, Yasuoka H, Tsujimoto M, Yoshidome K, Nakahara M, Nakao K, Nakamura M, Kakudo K. Nitric oxide in breast cancer: induction of vascular endothelial growth factor-C and correlation with metastasis and poor prognosis. Clin Cancer Res 2006;12:1201–7. [DOI] [PubMed] [Google Scholar]
- [24].O'Connell JX, Nanthakumar SS, Nielsen GP, Rosenberg AE. Osteoid osteoma: the uniquely innervated bone tumor. Mod Pathol 1998;11:175–80. [PubMed] [Google Scholar]
- [25].Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007;87:315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rausaria S, Ghaffari MM, Kamadulski A, Rodgers K, Bryant L, Chen Z, Doyle T, Shaw MJ, Salvemini D, Neumann WL. Retooling manganese(III) porphyrin-based peroxynitrite decomposition catalysts for selectivity and oral activity: a potential new strategy for treating chronic pain. J Med Chem 2011;54:8658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sabino MA, Mantyh PW. Pathophysiology of bone cancer pain. J Support Oncol 2005;3:15–24. [PubMed] [Google Scholar]
- [28].Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med 2011;51:951–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Salvemini D, Wang ZQ, Stern MK, Currie MG, Misko TP. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci U S A 1998;95:2659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 2002;277:44765–71. [DOI] [PubMed] [Google Scholar]
- [31].Seidlitz EP, Sharma MK, Saikali Z, Ghert M, Singh G. Cancer cell lines release glutamate into the extracellular environment. Clin Exp Metastasis 2009;26:781–7. [DOI] [PubMed] [Google Scholar]
- [32].Sharma MK, Seidlitz EP, Singh G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun 2010;391:91–5. [DOI] [PubMed] [Google Scholar]
- [33].Shukla K, Thomas AG, Ferraris DV, Hin N, Sattler R, Alt J, Rojas C, Slusher BS, Tsukamoto T. Inhibition of xc(-) transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg Med Chem Lett 2011;21:6184–7. [DOI] [PubMed] [Google Scholar]
- [34].Slosky LM, Largent-Milnes TM, Vanderah TW. Use of animal models in understanding cancer-induced bone pain. Cancer Growth Metastasis 2015;8(suppl 1):47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Slosky LM, Vanderah TW. Therapeutic potential of peroxynitrite decomposition catalysts: a patent review. Expert Opin Ther Pat 2015;25:443–66. [DOI] [PubMed] [Google Scholar]
- [36].Tas F, Hansel H, Belce A, Ilvan S, Argon A, Camlica H, Topuz E. Oxidative stress in breast cancer. Med Oncology 2005;22:11–15. [DOI] [PubMed] [Google Scholar]
- [37].Ungard RG, Seidlitz EP, Singh G. Oxidative stress and cancer pain. Can J Physiol Pharmacol 2013;91:31–7. [DOI] [PubMed] [Google Scholar]
- [38].Ungard RG, Seidlitz EP, Singh G. Inhibition of breast cancer-cell glutamate release with sulfasalazine limits cancer-induced bone pain. PAIN 2014;155:28–36. [DOI] [PubMed] [Google Scholar]
- [39].Vakkala M, Kahlos K, Lakari E, Paakko P, Kinnula V, Soini Y. Inducible nitric oxide synthase expression, apoptosis, and angiogenesis in in situ and invasive breast carcinomas. Clin Cancer Research 2000;6:2408–16. [PubMed] [Google Scholar]
- [40].van den Beuken-van Everdingen MH, de Rijke JM, Kessels AG, Schouten HC, van Kleef M, Patijn J. Prevalence of pain in patients with cancer: a systematic review of the past 40 years. Ann Oncol 2007;18:1437–49. [DOI] [PubMed] [Google Scholar]
- [41].Virag L, Szabo E, Gergely P, Szabo C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett 2003;140:113–24. [DOI] [PubMed] [Google Scholar]
- [42].Walser TC, Rifat S, Ma X, Kundu N, Ward C, Goloubeva O, Johnson MG, Medina JC, Collins TL, Fulton AM. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res 2006;66:7701–7. [DOI] [PubMed] [Google Scholar]
- [43].Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain Res 1994;654:15–26. [DOI] [PubMed] [Google Scholar]
- [44].WHO. Cancer pain relief. Geneva, World Health Organziation, 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.