

Supplemental Digital Content is Available in the Text.
In opioid-experienced patients (30-160 mg MSE/d), buccal buprenorphine was superior to placebo in 30%-50% pain reductions and PGIC, with similar constipation incidence.
Keywords: Chronic low back pain, Buccal buprenorphine, Opioid-experienced patients
Abstract
A buccal film of buprenorphine (BBUP) was evaluated for safety and efficacy in a multicenter, double-blind, placebo-controlled, enriched-enrollment, randomized-withdrawal study in opioid-experienced patients (30 to ≤160 mg/d morphine sulfate equivalent) with moderate to severe chronic low back pain taking around-the-clock opioid analgesics. Patients' opioid doses were tapered to ≤30 mg morphine sulfate equivalent before open-label titration with BBUP (range, 150-900 μg every 12 hours). Patients who responded (received adequate analgesia that was generally well tolerated for 14 days) were randomized to receive buprenorphine (n = 254) or placebo (n = 257) buccal film. The primary efficacy variable was the change from baseline to week 12 of double-blind treatment in mean average daily pain-intensity scores using a rating scale of 0 (no pain) to 10 (worst pain imaginable). In the intent-to-treat population, mean pain scores were 6.7 after opioid taper and declined to 2.8 after the BBUP titration period. After randomization, mean pain scores were lower in the BBUP group than in the placebo group; the difference between groups in the mean change from baseline to week 12 was −0.98 (95% CI, −1.32 to −0.64; P < 0.001). A significantly larger percentage of patients receiving BBUP than placebo had pain reductions ≥30% and ≥50% (P < 0.001 for both). In the double-blind portion of the study, the only adverse event reported more frequently with BBUP than placebo and in ≥5% of patients was vomiting (5.5% vs 2.3%). These findings demonstrate the efficacy and tolerability of BBUP in opioid-experienced patients taking around-the-clock opioid treatment for chronic low back pain.
1. Introduction
Buprenorphine is a Schedule III partial μ-opioid receptor agonist. Analgesic responses from buprenorphine at 0.3 to 0.4 mg intramuscular doses and 0.4 mg sublingual doses are reported to be more effective than a 10 mg intramuscular dose of morphine in postoperative pain patients.8,15,17,20,25,29,43,50 Relative to other opioids, buprenorphine has attributes that may provide an improved risk-benefit profile for treating chronic pain.36,43 The partial agonist activity at the μ-opioid receptor, combined with its high affinity for and slow dissociation from the receptor, differentiates buprenorphine from other μ-opioid agonists. Like other opioids, buprenorphine has been shown to depress respiration, but unlike other opioids, the effects are reported to have a ceiling.5,12,13,53 Buprenorphine seems to produce a lower level of “drug liking” than other opioids,15,30,53 leading to its approval for the treatment of opioid addiction. However, its poor oral bioavailability (∼10%) has limited its use for treating pain, and alternative formulations have been developed.
A transdermal formulation of buprenorphine is currently available in the United States for treating pain and is available in 5 dosage strengths: 5, 7.5, 10, 15, and 20 μg/h. Doses of 10 μg/h or higher have been shown to be effective in patients taking a morphine sulfate equivalent (MSE) dose <80 mg/d.51 However, the absolute bioavailability of transdermal relative to intravenous buprenorphine, following a 7-day application, is 15%.6 This low bioavailability and narrow range of effective doses (10-20 μg/h) may limit its use in some patients.6
A buccal film of buprenorphine (BBUP) has been developed and recently approved for management of chronic pain, severe enough to require around-the-clock, long-term opioid treatment (Belbuca; Endo Pharmaceuticals, Malvern, PA). Buccal buprenorphine uses a BioErodible MucoAdhesive (BEMA; BioDelivery Sciences International, Inc, Raleigh, NC) technology composed of flexible, water-soluble polymeric films that adhere to the moist buccal mucosa and erode over a period of minutes.3 In this delivery system, buprenorphine bioavailability is 46% to 65%, and its area under the curve increases in a dose proportional manner over approximately a 10-fold dose range.23,24 Buccal buprenorphine may therefore provide an additional treatment option for patients with chronic pain, including those taking >80 mg/d MSE for pain control.
The aim of this study was to determine the analgesic efficacy of BBUP every 12 hours (Q12h) in opioid-experienced patients (including those taking up to 160 mg/d MSE) with moderate to severe chronic low back pain (CLBP) using around-the-clock opioid analgesics for an extended period.
2. Methods
2.1. Participants
Adult (≥18 years) opioid-experienced patients (30-160 mg/d MSE) with moderate to severe CLBP were enrolled. Back pain was nonneuropathic (classes 1 and 2), neuropathic (classes 3-6), or symptomatic for >6 months after low-back surgery (class 9) at the time of screening based on the Quebec Task Force Classification for Spinal Disorders.44 Patients with cancer-related pain or any other chronic painful condition that in the investigator's opinion would interfere with the assessment of CLBP were excluded. In addition, patients with a history (<5 years) of alcohol abuse, substance abuse, or substance dependence according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition criteria, or with a positive urine toxicology screen for drugs of abuse (nonprescribed amphetamines, benzodiazepines, barbiturates, cannabinoids, or cocaine) were not eligible. The use of monoamine oxidase inhibitors, oral corticosteroids, chemotherapy, class IA and III antiarrhythmic medications, or any medication, nutraceutical, or herbal product with cytochrome P450 3A4 inhibiting or inducting properties was prohibited during the study.
2.2. Study design
This was a double-blind, placebo-controlled, enriched-enrollment, randomized-withdrawal (EERW) study comparing buprenorphine HCl buccal film (BEMA) to a placebo buccal film (ClinicalTrials.gov, Clinical Trial ID NCT01675167). The EERW study design is a well-accepted design for the evaluation of chronic pain in pivotal phase 3 studies.19,52 The EERW design is regarded as an appropriate and sensitive design to evaluate the efficacy of opioid analgesics, in part through a minimization of placebo response.
The study was conducted at 66 investigative sites throughout the United States. A total of 66 sites in the United States screened at least 1 patient (Addendum http://links.lww.com/PAIN/A328). The study design (Fig. 1) included a screening phase (2 weeks); an opioid taper phase (up to 4 weeks); an open-label BBUP titration phase (up to 8 weeks, including at least 2 weeks at a stable optimal dose); a double-blind, placebo-controlled, randomized, withdrawal treatment phase (12 weeks); and a follow-up phase (2 weeks). The buprenorphine buccal films were provided in dose strengths of 150, 300, 450, 600, 750, and 900 μg. All study medications were provided by the sponsor; investigators at each study center site enrolled patients and administered placebo or buprenorphine buccal films. All patients, investigators, and staff employed by the investigators or sponsor were blinded to treatment assignments.
Figure 1.

Enriched-enrollment randomized-withdrawal design.
Before the taper phase, additional as-needed (PRN) analgesic rescue medications were permitted on top of the stable daily maintenance dose of ≥30 mg MSE opioid analgesics, but they had to be included in the total daily MSE calculation and, in combination with the stable daily maintenance dose, were not allowed to exceed 160 mg/d MSE. Patients recorded their daily opioid and nonopioid analgesic medication use and completed the numerical rating scale (NRS) pain assessment (0 = no pain to 10 = pain as bad as you can imagine) daily throughout the study, using an interactive voice recognition/website system (IXRS). Opioid doses were tapered to ≤30 mg MSE per day, and before entering open-label titration with BBUP, patients had to report mean average daily pain-intensity scores ≥5 for 3 consecutive days during either screening or opioid taper. Patients were permitted to use hydrocodone/acetaminophen (HC/APAP) 5 mg/325 mg (PRN Q6h with a maximum of 4 tablets per day) as analgesic rescue throughout the rest of the opioid taper phase if required.
When patients received ≤30 mg MSE for at least 3 days and all other applicable inclusion criteria were met, they advanced to the open-label titration phase starting with a 150-μg or 300-μg dose of buprenorphine HCl buccal film Q12h, depending on their opioid dose at the end of screening. BBUP doses were increased progressively every 4 to 8 days until a 3-day mean pain score ≤4 or a dose of 900 μg Q12h was reached. Patient responders (NRS pain scores ≤4 for 14 days) to the dose range who found BBUP to be well tolerated (as judged by the investigator) and required no more than a single dose of HC/APAP (≤10/650 mg) as rescue during each of the previous 7 days were randomized to buprenorphine or placebo.
Patients were eligible for randomization if their mean pain-intensity score was ≤4 and at least 2 NRS points less than their pain score either at the end of the opioid taper or at screening. After titration to their optimal dose, patients were stratified by their buprenorphine buccal film dose and randomized 1:1 within the strata to receive buprenorphine HCl buccal film or placebo buccal film Q12h for 12 weeks. Patients were enrolled by investigators at each study center. Randomization codes were generated by the sponsor or designee to ensure that the appropriate number of patients were allocated to each treatment group at randomization. The randomization codes were stored in a secure electronic format within the IXRS system. To minimize the risk of opioid withdrawal in patients randomized to placebo, up to 2 doses per day of opioid rescue medication (1-2 tablets of 5/325 mg HC/APAP per dose) were permitted for the first 2 weeks in all patients; thereafter, 1 dose per day was permitted. Patients who required >1 dose per day of rescue medication on >2 occasions between visits were withdrawn from the study. In addition, patients who experienced moderate opioid withdrawal (Clinical Opiate Withdrawal Scale score ≥13) within 2 weeks of randomization were withdrawn from the study. At the completion of the 12-week study period, patients discontinued all study medication and either entered an open-label extension study or were treated as judged appropriate by their physician.
The trial was conducted in accordance with International Conference on Harmonisation Good Clinical Practice and the Declaration of Helsinki. A central institutional review board (Schulman Associates IRB, Cincinnati, OH) approved the study protocol and informed consent. All patients were informed of the risks and nature of participation and provided written informed consent prior to any study procedures being performed.
2.2.1. Efficacy evaluations
Patients reported their average daily pain-intensity scores, study medication use, and rescue medication use once daily using the IXRS. The Patient Global Impression of Change (PGIC; scale, 1 [no change since beginning treatment] to 7 [a great deal better])18 was completed at baseline (before randomization) and at the last visit of the double-blind treatment phase. The 24-item Roland Morris Disability Questionnaire (RMDQ; scale, 0 [best functionality] to 24 [worst functionality])2,47 was completed at the beginning of the open-label titration phase, at randomization, 28 days after randomization, and at the last study visit.
2.2.2. Safety evaluation
Adverse events (AEs), either reported by the patient or as noted by the investigator during physical examinations, were recorded.
2.3. Statistical analysis
The primary efficacy variable and secondary efficacy variables (except for patient-reported outcome measures) were analyzed using the intent-to-treat population, which comprised all patients who were randomized and received ≥1 dose of double-blind study medication. The primary endpoint was the change in mean average daily pain score from baseline (at randomization) to week 12 of the double-blind treatment period and was analyzed using an analysis of covariance (ANCOVA) with treatment and covariates of screen pain-intensity score (before the open-label titration phase) and baseline pain-intensity score (before randomization) as effects. Missing values due to study discontinuation were imputed before the analysis as follows: (1) AEs/tolerability: using the screening-observation-carried-forward imputation; (2) lack of efficacy: using the last-observation-carried-forward (LOCF) imputation; (3) opioid withdrawal: using the baseline-observation-carried-forward (BOCF) imputation; and (4) other: using multiple imputation procedures. The screening-observation-carried-forward, BOCF, and LOCF imputations were implemented before multiple imputations. The initial sample size calculation had 90% statistical power (2-sided test; alpha = 0.05) to detect a mean (SD) difference of 1.0 (2.6) between buprenorphine HCl buccal film and placebo and was determined to be 142 subjects per arm. Based on a prespecified sample size reestimation at the interim analysis, the study sample size was increased from 142/arm to 250/arm to give a conditional power of 83%. Thus, the protocol was updated (amendment #3) on November 6, 2013, to revise the number of patients to be enrolled as required by the prespecified sample size reestimation. The adjusted P value, estimate of treatment difference, and its corresponding 95% 2-sided CI were calculated for the primary analysis using Cui-Hung-Wang/Lawrence-Hung methods (with weighted Z-statistics) to preserve the overall type I error rate <0.05.11,39
Secondary efficacy endpoints included proportion of responders and change from baseline in PGIC and RMDQ scores. Patient responders were defined as the cumulative proportion of patients who completed the 12-week double-blind period and achieved percentage pain reductions (ie, ≥30% and ≥50%) from the start of the open-label titration phase to week 12 of the double-blind treatment phase. The patient responder analysis was conducted using the Cochran-Mantel-Haenszel χ2 test.
Multiple sensitivity analyses were also performed for the primary endpoint, including ANCOVA models analyzing the imputed data using a single BOCF or LOCF. A mixed-effects model repeated measures (MMRM) method was applied to the primary efficacy variable. The primary efficacy variable was also analyzed for the per-protocol subset by multiple imputation and the MMRM method.
Patient-reported outcomes (eg, PGIC and RMDQ) were analyzed using ANCOVA in a similar fashion as the primary endpoint. Safety variables included frequency of AEs and occurrence of opioid withdrawal. The safety analysis was based on all subjects who received at least 1 dose of study medication. Descriptive statistics (number and percentage) for patients reporting AEs in each treatment group were tabulated.
3. Results
3.1. Patient disposition
Patient disposition is presented in Figure 2. Most of the patients (62.7%; 511/815) successfully completed the open-label titration phase; the most frequent causes of discontinuation were AEs (9.9%), lack of efficacy (7.7%), and protocol violations (5.3%). The first patient enrolled on September 6, 2012; the last patient completed follow-up on June 6, 2014.
Figure 2.
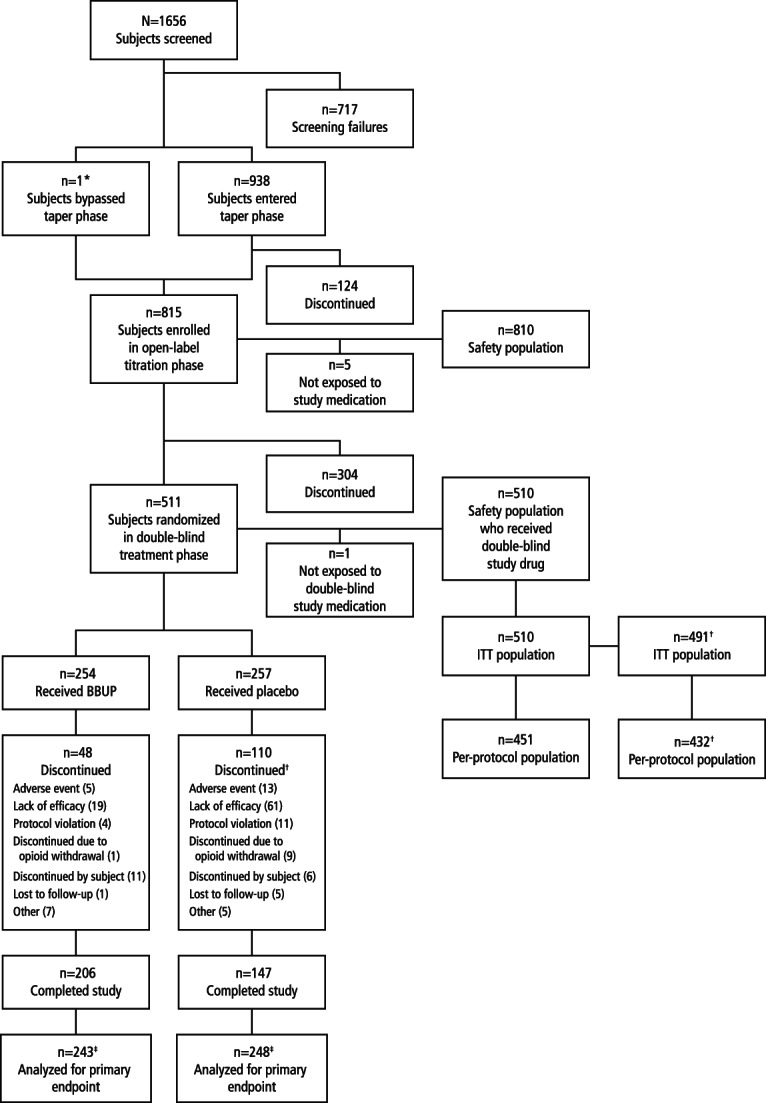
Patient disposition. BBUP, buccal buprenorphine; ITT, intent-to-treat. *One patient proceeded straight to titration from screening. †One site (19 patients) was excluded from the efficacy analysis because of inadequate control of data quality but was included in the safety analysis.
In the double-blind treatment phase, 254 patients were randomized to the BBUP group: 221 (87.0%) on <80 μg MSE, 26 (10.2%) on 80 to 120 μg MSE, and 7 (2.8%) on >120 μg MSE. In total, 257 patients were randomized to the placebo group, but 1 did not receive double-blind study medication. Of the 256 remaining placebo patients, 216 (84.4%) were on <80 μg MSE, 33 (12.9%) on 80 to 120 μg MSE, and 7 (2.7%) on >120 μg MSE. Among patients who were randomized to BBUP and received double-blind study drug, the distribution of BBUP doses at the end of titration was 150 µg (n = 10 [4%]), 300 μg (n = 30 [12%]), 450 μg (n = 36 [14%]), 600 μg (n = 43 [17%]), 750 μg (n = 42 [17%]), and 900 μg (n = 93 [36%]) twice daily (BID). Discontinuation rates were 18.9% in the BBUP group and 42.8% in the placebo group. Notably, discontinuations due to lack of efficacy were 7.5% in the BBUP group and 23.7% in the placebo group. During the double-blind treatment phase, 1 patient in the BBUP group (0.4%) and 9 in the placebo group (3.5%) discontinued the study because of opioid withdrawal. The 1 BBUP patient who withdrew because of opioid withdrawal received 450 μg BBUP BID. All 9 placebo patients who discontinued the study because of opioid withdrawal had been receiving doses of BBUP ≥600 μg BID at the end of the dose-titration phase.
3.2. Demographics
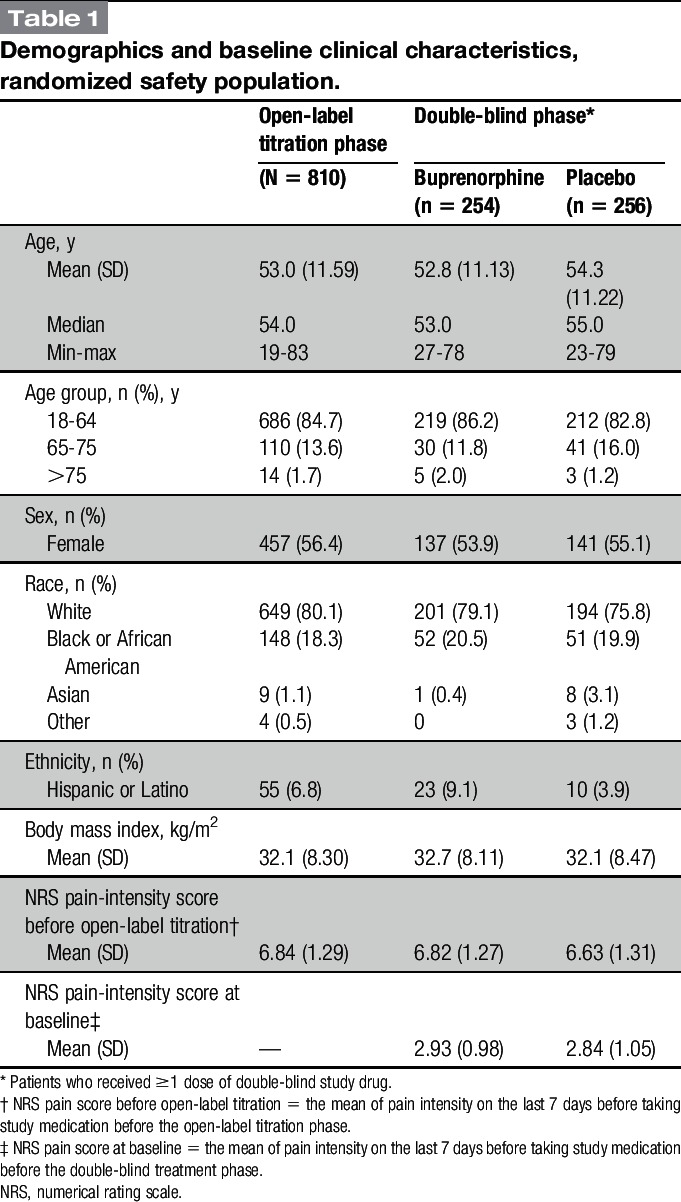
Demographic characteristics were similar for the 810 patients in the safety population who entered titration and the 510 who were randomized and treated (Table 1). Patient demographic data were representative of a CLBP population, with a mean age of 53 years, mean body mass index of 32.1 kg/m2, and the majority white (Table 1). The randomized treatment groups were well balanced with respect to age, body mass index, and sex, as well as baseline pain-intensity scores (Table 1).
Table 1.
Demographics and baseline clinical characteristics, randomized safety population.
3.3. Compliance
Compliance was based on return film counts and overall was high throughout the study. During the open-label phase, only 6.2% of patients failed to meet the 80% compliance requirement. During double-blind treatment, 0.4% of BBUP and 2.3% of placebo patients were less than 80% compliant.
3.4. Efficacy
3.4.1. Pain-intensity score
At the end of the opioid taper period and before buprenorphine titration, patients reported moderate to severe pain (mean [SD] NRS pain-intensity score, 6.84 [1.29]; Table 1). Pain decreased with buprenorphine such that in those patients randomized, the NRS score had decreased to a mean of <3 (mild). The randomized-withdrawal design examines efficacy by comparing the increase in pain experienced by those randomized to placebo with those who continue on active study treatment. From baseline to week 12 (primary endpoint), mean (SD) NRS pain scores increased significantly more in the placebo group (1.92 [1.87]) than in the BBUP group (0.88 [1.79]), with a between-group difference (favoring buprenorphine) of −0.98 (95% CI, −1.32 to −0.64; P < 0.001; Figs. 3 and 4). Compared with patients in the placebo group, patients in the BBUP group had significantly lower pain scores at week 1 and at all subsequent weekly time points through week 12 (Fig. 3).
Figure 3.
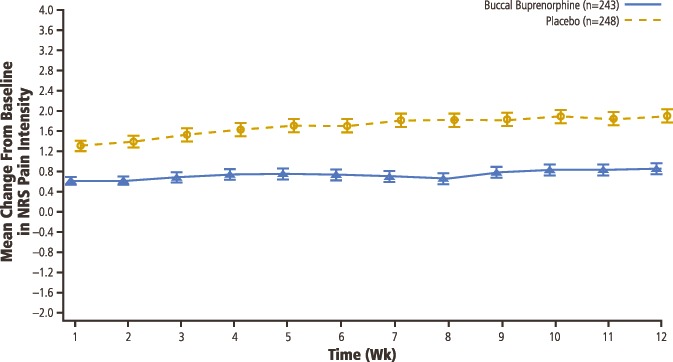
Mean (SE) change from baseline in NRS pain intensity in double-blind treatment period (with imputed values). NRS, numerical rating scale.
Figure 4.
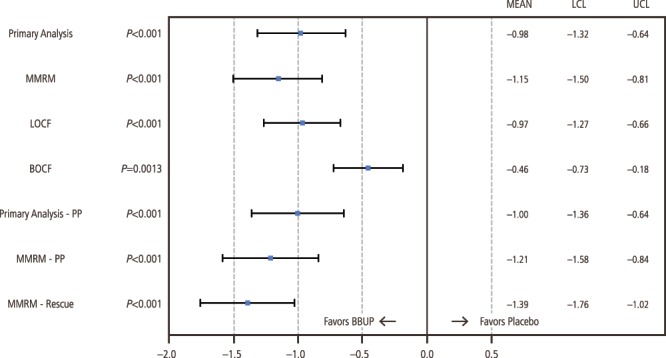
Primary, sensitivity, and supportive analyses of change from baseline to week 12 in numerical rating scale pain intensity in double-blind treatment phase. BBUP, buccal buprenorphine; BOCF, baseline-observation-carried-forward; LCL, lower confidence limit; LOCF, last-observation-carried-forward; MMRM, mixed-effects model repeated measures; PP, per protocol; UCL, upper confidence limit.
3.4.2. Sensitivity analyses
All sensitivity analyses confirmed that pain control was superior in the BBUP group (Fig. 4) compared with the placebo group, including the MMRM (−1.15, P < 0.001), LOCF (mean treatment difference, −0.97 [95% CI, −1.27 to −0.66]; P < 0.001), and BOCF (mean treatment difference, −0.46 [95% CI, −0.73 to −0.18]; P = 0.001) analyses. For the per-protocol population, a statistically significantly smaller mean change in NRS pain-intensity score from baseline to week 12 of the double-blind treatment phase was seen in the BBUP group compared with the placebo group, with a mean treatment difference of −1.00 (95% CI, −1.36 to −0.64; P < 0.001) on the primary efficacy analysis and −1.21 on the MMRM (P < 0.001).
3.4.3. Secondary endpoints
Patients who did not complete the 12-week study period were considered nonresponders. A significantly greater proportion of patients in the BBUP group compared with the placebo group were classified as responders based on achieving ≥30% pain reduction (BBUP group, 64.2%; placebo group, 30.6%; P < 0.001) or ≥50% pain reduction (BBUP group, 39.5%; placebo group, 16.9%; P < 0.001; Fig. 5). Consistent with this, the percentage of patients using rescue medication at week 12 was significantly lower in the buprenorphine group than in the placebo group (P < 0.001).
Figure 5.
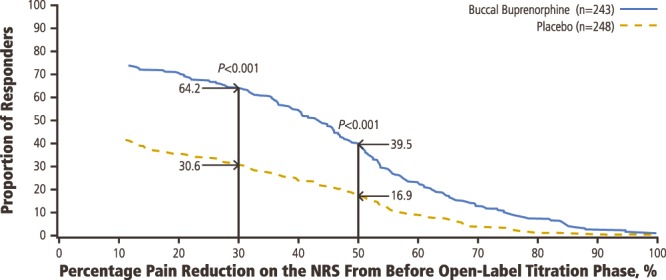
Proportion of responders with selected percentage pain reduction before open-label titration to week 12 in the double-blind treatment period.
Significant differences between groups were also observed for patient-reported outcomes. At baseline (ie, at randomization), the mean (SD) PGIC score was 5.4 (1.14) in the BBUP group and 5.3 (1.25) in the placebo group, indicating that patients perceived a benefit from treatment relative to study entry. Patient-reported impression of treatment benefit was significantly greater with BBUP: the mean (SD) PGIC score at week 12 was 4.5 (1.86) in the BBUP group vs 3.2 (1.98) in the placebo group (treatment difference, 1.3; 95% CI, 0.9-1.6; P < 0.001). Ninety-six (39.7%) patients in the BBUP group vs 49 (20.6%) in the placebo group showed a clinically meaningful improvement as indicated by a response of 6 or 7 on the PGIC. Patients reported a significantly greater increase in disability in the placebo group. The mean change from baseline to end of treatment in RMDQ disability score was 0.5 (5.03) in the BBUP group and 1.6 (5.63) in the placebo group (HR, −1.20; 95% CI, −2.08 to −0.31; P = 0.008).
3.5. Adverse events
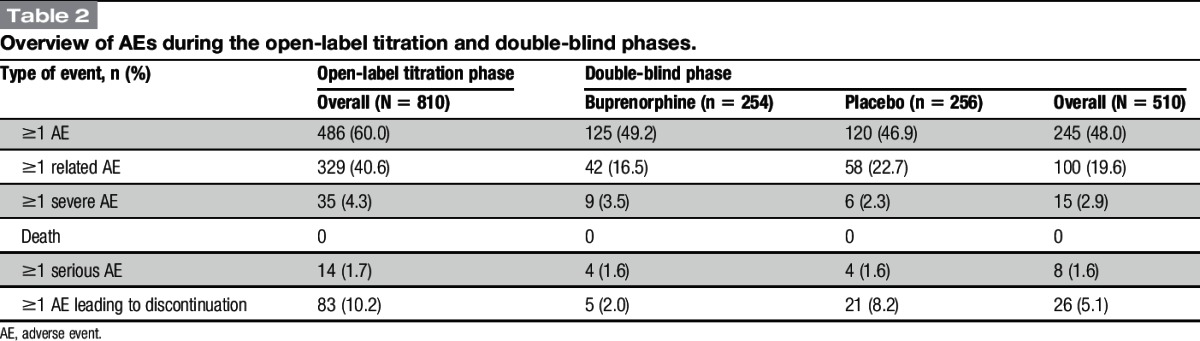
An overview of AEs is presented in Table 2. During the titration period (mean [SD] duration of exposure, 38.7 [16.53] days), 60% of patients experienced 1 or more AEs, and 10.2% of patients discontinued because of AEs. Serious AEs were reported by 1.7% of patients during titration, and there were no deaths. During the double-blind period, AEs were reported by 48% of patients, and 5.1% discontinued because of AEs: 5 (2.0%) randomized to BBUP and 21 (8.2%) randomized to placebo. Serious AEs were reported by 1.6% of patients, and there were no deaths.
Table 2.
Overview of AEs during the open-label titration and double-blind phases.
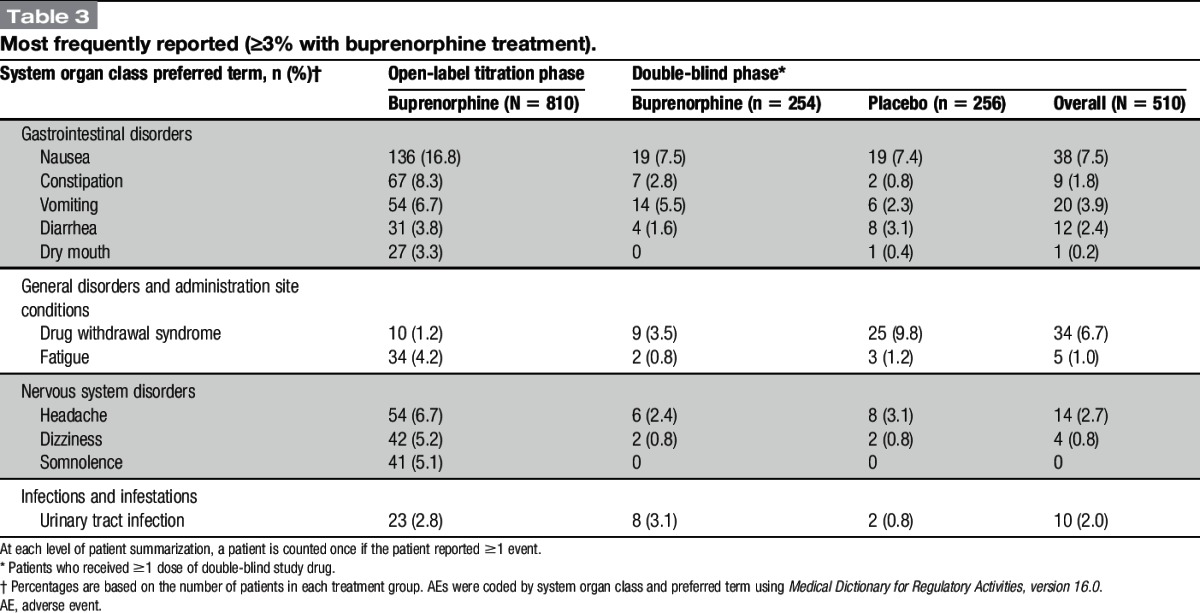
The most frequent AEs are presented in Table 3. The most common AEs during titration phase were those typically associated with opioids (nausea, constipation, vomiting, headache, dizziness, and somnolence). The incidence of drug withdrawal during this period was low at 1.2%. During the double-blind period, constipation and vomiting were reported more frequently on buprenorphine, whereas diarrhea, drug withdrawal syndrome, fatigue, and headache were reported more frequently with placebo. Notably, nausea was reported by a similarly low percentage of patients on buprenorphine (7.5%) and placebo (7.4%). There were no cases of accidental overdose and no AEs associated with misuse or abuse of the study medication. One patient in the BBUP group experienced an AE of dyspnea; no cases of respiratory depression were observed.
Table 3.
Most frequently reported (≥3% with buprenorphine treatment).
4. Discussion
A high percentage of patients with chronic pain receive opioids, with estimates ranging from 60% to 90%.4,16,40 The opioid-associated risks for misuse, abuse, addiction, respiratory depression, diversion, and other adverse effects limit the use of these drugs for the management of chronic pain. Although buprenorphine, like other opioids, is positively reinforcing, and therefore possesses some abuse liability, in opioid-experienced individuals, the risk of buprenorphine abuse is believed to be lower compared with other opioids.9,55 Respiratory depression is an opioid titration–limiting factor.15 In addition, the activation of μ-opioid receptors in the gastrointestinal (GI) tract results in lowered gut motility, and constipation often becomes problematic with long-term use.4,37
The pharmacologic properties of buprenorphine differentiate it from other μ-opioid receptor agonists. In particular, the partial agonist activity of buprenorphine results in a ceiling effect on opioid receptors associated with respiratory depression10,14,53 and may contribute to a lesser effect on GI motility.10 It has been suggested in previous studies that there may be an improved risk-benefit ratio with buprenorphine relative to other opioids.5,25 However, the utility of buprenorphine as an analgesic has been limited by a perceived weakness in analgesic activity, particularly in patients requiring >80 mg/d MSE.
The present double-blind, placebo-controlled, randomized-withdrawal study evaluated BBUP in patients with CLBP being treated with 30 to 160 mg MSE per day. The study used an enriched population that met the criteria for response to and tolerance of BBUP to determine the analgesic efficacy of buprenorphine buccal film relative to placebo. Patients entered the open-label titration phase with moderate to severe pain as indicated by their NRS pain assessments, and with a significant amount of self-perceived disability due to their CLBP (mean scores of approximately 15/24 on the RMDQ).7,48,49 Most of the patients were successfully titrated to an effective and tolerated dose that reduced pain to a mild level and reduced self-perceived disability. Randomized patients were first titrated to effect with buprenorphine and then either randomized to continue BBUP or switched to placebo. The fact that most of the placebo-treated patients completed 12 weeks of double-blind treatment would seem to indicate that most patients did not subjectively detect the switch from BBUP. Our study design is generally regarded as the most appropriate to minimize bias in patients who have experience with the drug class under study.26,41 After randomization, mean pain scores increased significantly in the placebo group vs patients randomized to continue their stable dose of BBUP. The difference in analgesic effect was evident early and was sustained for the entire observation period. The treatment difference was significant at week 12 vs baseline (primary endpoint); all 6 sensitivity analyses were significant vs placebo, even with BOCF analysis (considered the most conservative approach; P = 0.002). On randomization, pain in the placebo group increased by approximately 2 points on the NRS scale, which is a clinically meaningful increase,22 whereas pain in those who continued double-blind BBUP increased by approximately 1 point, which is below the threshold for clinical significance. This between-group difference in change was highly statistically significant (P < 0.001), indicating that BBUP-treated patients experienced a clinically significant improvement compared with their pain before treatment and, on randomization, maintained statistically significantly better pain control compared with placebo-treated patients. Moreover, twice as many patients on BBUP had an improvement of ≥30% compared with placebo-treated patients, and this result was also statistically significant (P < 0.001) and a clinically meaningful response. In addition, this difference was statistically significant (P < 0.001) whether a criterion of 30% or 50% pain reduction was used.22 The patient-reported measure of PGIC was also significantly different between the groups and clinically meaningful with 40% of BBUP patients reporting a PGIC of 6 or 7 (highest 2 response categories that are considered indicative of clinical improvement) vs only 21% of placebo patients.31
In a previous study, the buprenorphine transdermal patch demonstrated efficacy in opioid-experienced patients, but only in those previously treated with ≤80 mg/d MSE.51 In contrast, BBUP used in this study was effective in a population that included patients taking up to 160 mg/d MSE. One factor that may account for the efficacy observed in this study in patients requiring up to 160 mg/d MSE is the pharmacokinetics of buprenorphine based on different formulations, particularly the improved bioavailability (of approximately 50%) of BBUP. As a result, there is greater exposure (area under the curve) with BBUP,23,24 and the dose range provides more flexibility to titrate to an optimal dose.
The efficacy data presented here are generally comparable with results observed with full μ-agonists in randomized, double-blind, placebo-controlled trials, including OROS hydromorphone extended release (ER; 12-64 mg once daily),27 oxymorphone ER (up to 130 mg BID),28 and single-entity hydrocodone (20-100 mg BID),45 in opioid-experienced patients with moderate to severe CLBP. Changes in pain-intensity scores more than 12 weeks relative to placebo were similar in magnitude to those observed with BBUP. Across the studies, the percentage of patients achieving ≥30% reduction in pain intensity at 12 weeks was comparable (61% with OROS hydromorphone ER, 68% with single-entity hydrocodone, 60% with oxymorphone ER, and 64% with BBUP in this study), as was the percentage of patients with ≥50% reduction in pain intensity (42% with OROS hydromorphone ER, 48% with single-entity hydrocodone, 48% with oxymorphone ER, and 40% with BBUP in this study).27,42,45 The efficacy data reported here with BBUP in opioid-experienced patients are also similar to results of a similarly designed trial assessing BBUP in opioid-naive patients with moderate to severe CLBP.46
In this study, discontinuations due to AEs were low in patients receiving BBUP (10.2% during the open-label titration period and 2.0% during the double-blind treatment period). Only 2 serious AEs (ileus and abdominal pain) considered related to BBUP occurred during the study, and no cases of respiratory depression were observed. The rate of opioid withdrawal syndrome was 1.2% in the open-label phase and 3.5% (n = 9) in the buprenorphine group during the double-blind phase (Table 3), but only 1 patient discontinued double-blind buprenorphine treatment because of opioid withdrawal syndrome.
In this study, the frequency of constipation as an AE with buprenorphine was 2.8% during 12 weeks of double-blind treatment. By comparison, in the randomized-withdrawal phase 3 study that examined BBUP in opioid-naive patients with CLBP, 4% of patients had constipation.46 For other opioids, the frequency of constipation as an AE has been reported as 8% to ∼70%.1,27,32,34,45 Although rates as low as 6% have been reported, they were achieved using an active bowel regimen to reduce constipation. The low incidence of constipation with BBUP was achieved without a bowel regimen, although patients were certainly permitted treatment for constipation.28,35 There may be a pharmacoeconomic value with buprenorphine because of reduced costs for treating constipation, which is a problem with all opioids.1,33,37,38,54
However, it is important to note that this study with BBUP was not a head-to-head trial; the potential clinical and pharmacoeconomic advantages of BBUP over other available opioid formulations with respect to reduced AEs (eg, constipation or respiratory depression) would need to be directly demonstrated in well-controlled studies. Another limitation, and one inherent to clinical trials, is the generalizability of the data. Clinical studies in opioid-experienced patients do not require taper of the patients' opioid before conversion to a full μ-agonist. Although the population enrolled in this study was opioid experienced, only patients willing and able to taper their opioid treatment to ≤30 mg/d MSE were recruited and entered.
In summary, treatment with BBUP at doses up to 900 μg twice daily was found to be efficacious in controlling moderate to severe CLBP in an opioid-experienced population (30-160 mg/d MSE). BBUP was generally well tolerated. There was no evidence of respiratory depression in this large patient population. In addition, rates of opioid-associated GI AEs (nausea, constipation, and vomiting) were low, ranging from 3% to 8%. During open-label treatment, when opioid-experienced patients were switched from their current around-the-clock opioid to BBUP, the rate of drug withdrawal syndrome was low (∼1%). Furthermore, there was a high level of compliance with the buccal film. Patient satisfaction was indicated by the high percentage of completers and those that continued in the subsequent long-term safety study (NCT01755546).21
5. Conclusion
In opioid-experienced patients (requiring 30-160 mg/d MSE), BBUP (with approximately 50% bioavailability) at doses up to 900 μg twice daily produced analgesia superior to placebo over the 12-week, double-blind period with significant differences in PGIC, as well as percentages of patients with ≥30% or ≥50% reductions in pain. There were few differences between BBUP and placebo in the incidence of AEs including constipation, and there were no reports of respiratory depression.
Conflict of interest statement
J. Gimbel has received investigator payments from Endo Pharmaceuticals and serves as a consultant for Endo. E. L.H. Spierings has received investigator payments from Endo Pharmaceuticals and was the recipient of an ISR grant from Endo. N. Katz has received payments for consulting for BioDelivery Sciences International. Q. Xiang is an employee and shareholder of Endo Pharmaceuticals. E. Tzanis is a former employee of Endo Pharmaceuticals. A. Finn is a former employee and a current shareholder of BioDelivery Sciences International.
Funding for research and editorial support was provided by Endo Pharmaceuticals Inc, Malvern, PA.
E. L.H. Spierings participated as an investigator in the study and presented the results as a poster entitled “BEMA Buprenorphine Efficacy and Tolerability in Opioid-Naive Patients With Moderate-to-Severe Chronic Low Back Pain” at the International Conference on Opioids, Boston, MA, June 7 to 9, 2015.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Annemans L. Pharmacoeconomic impact of adverse events of long-term opioid treatment for the management of persistent pain. Clin Drug Investig 2011;31:73–86. [DOI] [PubMed] [Google Scholar]
- [2].Ashworth J, Green DJ, Dunn KM, Jordan KP. Opioid use among low back pain patients in primary care: is opioid prescription associated with disability at 6-month follow-up? PAIN 2013;154:1038–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].BELBUCA™ [package insert]. Malvern, PA: Endo Pharmaceuticals; 2015. [Google Scholar]
- [4].Benyamin R, Trescot AM, Datta S, Buenaventura R, Adlaka R, Sehgal N, Glaser SE, Vallejo R. Opioid complications and side effects. Pain Physician 2008;11:S105–S120. [PubMed] [Google Scholar]
- [5].Budd K. High dose buprenorphine for postoperative analgesia. Anaesthesia 1981;36:900–903. [DOI] [PubMed] [Google Scholar]
- [6].Butrans® [package insert]. Stamford, CT: Purdue Pharma L.P; 2014. [Google Scholar]
- [7].Chou R, Shekelle P. Will this patient develop persistent disabling low back pain? JAMA 2010;303:1295–1302. [DOI] [PubMed] [Google Scholar]
- [8].Christoph T, Kogel B, Schiene K, Meen M, De Vry J, Friderichs E. Broad analgesic profile of buprenorphine in rodent models of acute and chronic pain. Eur J Pharmacol 2005;507:87–98. [DOI] [PubMed] [Google Scholar]
- [9].Comer SD, Collins ED. Self-administration of intravenous buprenorphine and the buprenorphine/naloxone combination by recently detoxified heroin abusers. J Pharmacol Exp Ther 2002;303:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cowan A, Lewis JW, Macfarlane IR. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol 1977;60:537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cui L, Hung HM, Wang SJ. Modification of sample size in group sequential clinical trials. Biometrics 1999;55:853–857. [DOI] [PubMed] [Google Scholar]
- [12].Dahan A. Opioid-induced respiratory effects: new data on buprenorphine. Palliat Med 2006;20(suppl 1):s3–8. [PubMed] [Google Scholar]
- [13].Dahan A, Yassen A, Bijl H, Romberg R, Sarton E, Teppema L, Olofsen E, Danhof M. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth 2005;94:825–834. [DOI] [PubMed] [Google Scholar]
- [14].Dahan A, Yassen A, Romberg R, Sarton E, Teppema L, Olofsen E, Danhof M. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth 2006;96:627–632. [DOI] [PubMed] [Google Scholar]
- [15].Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol 2012;10:209–219. [DOI] [PubMed] [Google Scholar]
- [16].Deyo RA, Smith DH, Johnson ES, Donovan M, Tillotson CJ, Yang X, Petrik AF, Dobscha SK. Opioids for back pain patients: primary care prescribing patterns and use of services. J Am Board Fam Med 2011;24:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dobkin AB, Esposito B, Philbin C. Double-blind evaluation of buprenorphine hydrochloride for post-operative pain. Can Anaesth Soc J 1977;24:195–202. [DOI] [PubMed] [Google Scholar]
- [18].Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, Kerns RD, Stucki G, Allen RR, Bellamy N, Carr DB, Chandler J, Cowan P, Dionne R, Galer BS, Hertz S, Jadad AR, Kramer LD, Manning DC, Martin S, McCormick CG, McDermott MP, McGrath P, Quessy S, Rappaport BA, Robbins W, Robinson JP, Rothman M, Royal MA, Simon L, Stauffer JW, Stein W, Tollett J, Wernicke J, Witter J, IMMPACT. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. PAIN 2005;113:9–19. [DOI] [PubMed] [Google Scholar]
- [19].Dworkin RH, Turk DC, Peirce-Sandner S, Baron R, Bellamy N, Burke LB, Chappell A, Chartier K, Cleeland CS, Costello A, Cowan P, Dimitrova R, Ellenberg S, Farrar JT, French JA, Gilron I, Hertz S, Jadad AR, Jay GW, Kalliomaki J, Katz NP, Kerns RD, Manning DC, McDermott MP, McGrath PJ, Narayana A, Porter L, Quessy S, Rappaport BA, Rauschkolb C, Reeve BB, Rhodes T, Sampaio C, Simpson DM, Stauffer JW, Stucki G, Tobias J, White RE, Witter J. Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. PAIN 2010;149:177–193. [DOI] [PubMed] [Google Scholar]
- [20].Edge WG, Cooper GM, Morgan M. Analgesic effects of sublingual buprenorphine. Anaesthesia 1979;34:463–467. [DOI] [PubMed] [Google Scholar]
- [21].Endo Pharmaceuticals. Data on file. A 52-week, open-label, long-term treatment evaluation of the safety and efficacy of BEMA® buprenorphine in subjects with moderate to severe chronic pain. Malvern, PA: 2015. [Google Scholar]
- [22].Farrar JT, Young JP, Jr, LaMoreaux L, Werth JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. PAIN 2001;94:149–158. [DOI] [PubMed] [Google Scholar]
- [23].Finn A, Bai SA, Xiang Q. An evaluation of the bioavailability and dose linearity of BEMA® buprenorphine buccal film in healthy subjects. Paper presented at: PAINWeek; September 2-6, 2014; Las Vegas, NV.
- [24].Finn A, Bai SA, Xiang Q. An evaluation of the pharmacokinetics, safety, and tolerability of BEMA® buprenorphine following multiple-dose administration to healthy subjects. Paper presented at: PAINWeek; September 2-6, 2014; Las Vegas, NV.
- [25].Freye E, Anderson-Hillemacher A, Ritzdorf I, Levy JV. Opioid rotation from high-dose morphine to transdermal buprenorphine (Transtec) in chronic pain patients. Pain Pract 2007;7:123–129. [DOI] [PubMed] [Google Scholar]
- [26].Furlan A, Chaparro LE, Irvin E, Mailis-Gagnon A. A comparison between enriched and nonenriched enrollment randomized withdrawal trials of opioids for chronic noncancer pain. Pain Res Manag 2011;16:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hale M, Khan A, Kutch M, Li S. Once-daily OROS hydromorphone ER compared with placebo in opioid-tolerant patients with chronic low back pain. Curr Med Res Opin 2010;26:1505–1518. [DOI] [PubMed] [Google Scholar]
- [28].Hale ME, Ahdieh H, Ma T, Rauck R, Oxymorphone ER Study Group 1. Efficacy and safety of OPANA ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: a 12-week, randomized, double-blind, placebo-controlled study. J Pain 2007;8:175–184. [DOI] [PubMed] [Google Scholar]
- [29].Hovell BC, Ward AE. Pain relief in the post-operative period: a comparative trial of morphine and a new analgesic buprenorphine. J Int Med Res 1977;5:417–421. [DOI] [PubMed] [Google Scholar]
- [30].Huang P, Kehner GB, Cowan A, Liu-Chen LY. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther 2001;297:688–695. [PubMed] [Google Scholar]
- [31].Hurst H, Bolton J. Assessing the clinical significance of change scores recorded on subjective outcome measures. J Manipulative Physiol Ther 2004;27:26–35. [DOI] [PubMed] [Google Scholar]
- [32].Hyup Lee J, Lee CS, Ultracet ER Study Group. A randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy and safety of the extended-release tramadol hydrochloride/acetaminophen fixed-dose combination tablet for the treatment of chronic low back pain. Clin Ther 2013;35:1830–1840. [DOI] [PubMed] [Google Scholar]
- [33].Iyer S, Davis KL, Candrilli S. Opioid use patterns and health care resource utilization in patients prescribed opioid therapy with and without constipation. Manag Care 2010;19:44–51. [PubMed] [Google Scholar]
- [34].Kalso E, Edwards JE, Moore RA, McQuay HJ. Opioids in chronic non-cancer pain: systematic review of efficacy and safety. PAIN 2004;112:372–380. [DOI] [PubMed] [Google Scholar]
- [35].Katz N, Rauck R, Ahdieh H, Ma T, Gerritsen van der Hoop R, Kerwin R, Podolsky G. A 12-week, randomized, placebo-controlled trial assessing the safety and efficacy of oxymorphone extended release for opioid-naive patients with chronic low back pain. Curr Med Res Opin 2007;23:117–128. [DOI] [PubMed] [Google Scholar]
- [36].Koppert W, Ihmsen H, Korber N, Wehrfritz A, Sittl R, Schmelz M, Schuttler J. Different profiles of buprenorphine-induced analgesia and antihyperalgesia in a human pain model. PAIN 2005;118:15–22. [DOI] [PubMed] [Google Scholar]
- [37].Kurz A, Sessler DI. Opioid-induced bowel dysfunction: pathophysiology and potential new therapies. Drugs 2003;63:649–671. [DOI] [PubMed] [Google Scholar]
- [38].Kwong WJ, Diels J, Kavanagh S. Costs of gastrointestinal events after outpatient opioid treatment for non-cancer pain. Ann Pharmacother 2010;44:630–640. [DOI] [PubMed] [Google Scholar]
- [39].Lawrence J, Hung HMJ. Estimation and confidence intervals after adjusting the maximum information. Biom J 2003;45:143–152. [Google Scholar]
- [40].Manchikanti L, Damron KS, McManus CD, Barnhill RC. Patterns of illicit drug use and opioid abuse in patients with chronic pain at initial evaluation: a prospective, observational study. Pain Physician 2004;7:431–437. [PubMed] [Google Scholar]
- [41].Moore RA, Wiffen PJ, Eccleston C, Derry S, Baron R, Bell RF, Furlan AD, Gilron I, Haroutounian S, Katz NP, Lipman AG, Morley S, Peloso PM, Quessy SN, Seers K, Strassels SA, Straube S. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. PAIN 2015;156:1382–1395. [DOI] [PubMed] [Google Scholar]
- [42].OPANA ER® [package insert]. Malvern, PA: Endo Pharmaceuticals Inc; 2014. [Google Scholar]
- [43].Pergolizzi J, Aloisi AM, Dahan A, Filitz J, Langford R, Likar R, Mercadante S, Morlion B, Raffa RB, Sabatowski R, Sacerdote P, Torres LM, Weinbroum AA. Current knowledge of buprenorphine and its unique pharmacological profile. Pain Pract 2010;10:428–450. [DOI] [PubMed] [Google Scholar]
- [44].Quebec Task Force on Spinal Disorders. Scientific approach to the assessment and management of activity-related spinal disorders. A monograph for clinicians. Report of the Quebec Task Force on Spinal Disorders. Spine (Phila Pa 1976) 1987;12:S1–59. [PubMed] [Google Scholar]
- [45].Rauck RL, Nalamachu S, Wild JE, Walker GS, Robinson CY, Davis CS, Farr SJ. Single-entity hydrocodone extended-release capsules in opioid-tolerant subjects with moderate-to-severe chronic low back pain: a randomized double-blind, placebo-controlled study. Pain Med 2014;15:975–985. [DOI] [PubMed] [Google Scholar]
- [46].Rauck RL, Potts J, Xiang Q, Tzanis E, Finn A. Efficacy and tolerability of buccal buprenorphine in opioid-naive patients with moderate to severe chronic low back pain. Postgrad Med 2016;128:1–11. [DOI] [PubMed] [Google Scholar]
- [47].Roland M, Fairbank J. The Roland-Morris Disability Questionnaire and the Oswestry Disability Questionnaire. Spine (Phila Pa 1976) 2000;25:3115–3124. [DOI] [PubMed] [Google Scholar]
- [48].Roland M, Morris R. A study of the natural history of back pain. Part I: development of a reliable and sensitive measure of disability in low-back pain. Spine (Phila Pa 1976) 1983;8:141–144. [DOI] [PubMed] [Google Scholar]
- [49].Roland M, Morris R. A study of the natural history of low-back pain. Part II: development of guidelines for trials of treatment in primary care. Spine (Phila Pa 1976) 1983;8:145–150. [DOI] [PubMed] [Google Scholar]
- [50].Sittl R, Likar R, Nautrup BP. Equipotent doses of transdermal fentanyl and transdermal buprenorphine in patients with cancer and noncancer pain: results of a retrospective cohort study. Clin Ther 2005;27:225–237. [DOI] [PubMed] [Google Scholar]
- [51].Steiner D, Munera C, Hale M, Ripa S, Landau C. Efficacy and safety of buprenorphine transdermal system (BTDS) for chronic moderate to severe low back pain: a randomized, double-blind study. J Pain 2011;12:1163–1173. [DOI] [PubMed] [Google Scholar]
- [52].US Department of Health and Human Services. Guidance for Industry: Enrichment Strategies for Clinical Trials to Support Approval of Human Drugs and Biological Products. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm332181.pdf. Accessed April 26, 2016.
- [53].Walsh SL, Preston KL, Bigelow GE, Stitzer ML. Acute administration of buprenorphine in humans: partial agonist and blockade effects. J Pharmacol Exp Ther 1995;274:361–372. [PubMed] [Google Scholar]
- [54].Wan L, Corman S, Gao X, Liu S, Patel H, Mody R. Economic burden of opioid-induced constipation among long-term opioid users with noncancer pain. Am Health Drug Benefits 2015;8:93–102. [PMC free article] [PubMed] [Google Scholar]
- [55].Yokell MA, Zaller ND, Green TC, Rich JD. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011;4:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]