

Abstract
Interferon-gamma (IFNγ), a pleiotropic cytokine, is expressed in diverse neurodegenerative and neuroinflammatory conditions. Its protective mechanisms are well documented during viral infections in the brain, where IFNγ mediates non-cytolytic viral control in infected neurons. However, IFNγ also plays both protective and pathological roles in other central nervous system (CNS) diseases. Of the many neural cells that respond to IFNγ, neural stem/progenitor cells (NSPCs), the only pluripotent cells in the developing and adult brain, are often altered during CNS insults. Recent studies highlight the complex effects of IFNγ on NSPC activity in neurodegenerative diseases. However, the mechanisms that mediate these effects, and the eventual outcomes for the host, are still being explored. Here, we review the effects of IFNγ on NSPC activity during different pathological insults. An improved understanding of the role of IFNγ would provide insight into the impact of immune responses on the progression and resolution of neurodegenerative diseases.
Keywords: neural stem/progenitor cells, interferon-gamma, STAT1, neuroinflammation, neural stem cells, virus
Introduction
Historically, the brain was considered as an immune-privileged organ due to the absence of a lymphatic system and the maintenance of transplanted tissue grafts. However, we now understand that the immune system readily interacts with cells in the brain parenchyma during stress, disease, and infections.1–3 The interactions between resident and infiltrating immune cells and the brain tissue contribute to both neuroprotection and neurotoxicity, depending on the type of insult, the type of injured neural cell, and even the age of the host.4 Within the brain, innate immune cells, which respond nonspecifically to damaged or infected cells, and adaptive immune cells, which recognize specific antigens, are active during inflammatory responses. Recent discoveries describe mechanisms that allow exchange of antigenic information through channels present on astrocytes that encircle brain vasculature.4,5 This allows antigenic exchange between the central nervous system (CNS) and the periphery and results in specific, targeted immune responses within the CNS. The generation of effective innate and adaptive immune responses is critical, as many neurons in the brain are nonrenewable and cannot be readily replaced.
The CNS can be exposed to diverse insults and injuries, including stroke, viral infections, neurodegenerative diseases, and autoimmune disorders. During these insults, inflammation in the brain is characterized by the activation of resident microglial cells as well as infiltration of peripheral macrophages and lymphocytes. These cells release pro- and anti-inflammatory cytokines and chemokines, which cause further immune activation and infiltration of other immune cell subsets. Ideally, the infiltration and activation of immune cells leads to resolution of the insult. However, excessive or chronic immune activation during insults such as stroke, Alzheimer’s disease (AD), and multiple sclerosis (MS) can cause neurotoxicity and damage to the CNS.6 Often, the mediators of neuronal loss are the cytokines and/or chemokines released by immune cells. The challenge for the body is to strike a balance that resolves the adverse event, limits damage to CNS cells, and avoids excessive immune activation. In conditions where the body fails to rescue or preserve neurons, pharmacological interventions must be devised to promote neuroprotection or neuroregeneration. In order to do so, it is important to understand the effects of inflammatory and anti-inflammatory mediators on neural cells.
One of the cytokines released as part of the inflammatory milieu is interferon-gamma (IFNγ). It is the only member of the Type II family of interferons and is secreted predominately by activated immune cells such as T cells and natural killer (NK) cells.7 In contrast, members of the type I family (IFNα and β) are secreted by almost all cells when the cell is infected or damaged. As part of the host response, IFNγ activates other immune cells and increases the expression of major histocompatibility complex class I and II on its target cells, helping to mount a robust immune response.8,9 In addition to these functions, IFNγ also acts directly on neural cells.10–15 For example, IFNγ induces non-cytolytic clearance of several neurotropic viruses, such as Sindbis virus and measles virus, from CNS neurons.10,11,16 On the other hand, IFNγ also plays a role in neurodegeneration in many CNS diseases.17–19 Therefore, the precise role of IFNγ during CNS inflammation is still unclear but likely involves a complex set of responses from different cell types.
Neural stem/progenitor cells (NSPCs) are the only multipotent population of cells in the CNS. These cells are capable of self-renewal, thereby maintaining the NSPC pool, and of differentiation into neurons, astrocytes, and oligodendrocytes (Fig. 1). During CNS development, NSPCs populate the CNS broadly, while their anatomical localization becomes restricted as the brain matures.20,21 A small population of NSPCs persists in specific niches in the adult brain, namely within the subventricular zone (SVZ) and the subgranular zone (SGZ) of the dentate gyrus (DG).20,22,23 The SVZ is located along the lateral wall of the lateral ventricle. NSPCs in the SVZ differentiate into neuroblasts that migrate along the rostral migratory stream into the olfactory bulb. In the olfactory bulb, these neuroblasts terminally differentiate into granule and periglomerular neurons.24 NSPCs in the SGZ of the DG give rise to granule neurons, which are important in learning and memory. The role of NSPCs under physiological conditions and in neurodegenerative diseases is an active area of study. In addition to normal NSPC functions such as CNS development, learning and memory, and maintenance of olfaction, NSPCs are affected differentially in CNS diseases such as epilepsy, depression, and even natural aging.25–27 A number of studies have looked at the role of IFNγ in mediating changes in NSPC activity. However, the outcomes of these experiments differ depending on the source of NSPCs (fetal or adult), the models used (cell lines or primary cells), and the species of the host.28–32 In this review, we consolidate the current literature on the role of IFNγ in modifying NSPC activity in CNS diseases. We also try to understand the implications of IFNγ-mediated changes in NSPC activity, and how they contribute to neurological sequelae. We also discuss future directions for understanding the interactions of NSPCs with IFNγ and other inflammatory cytokines during neuroinflammation.
Figure 1.
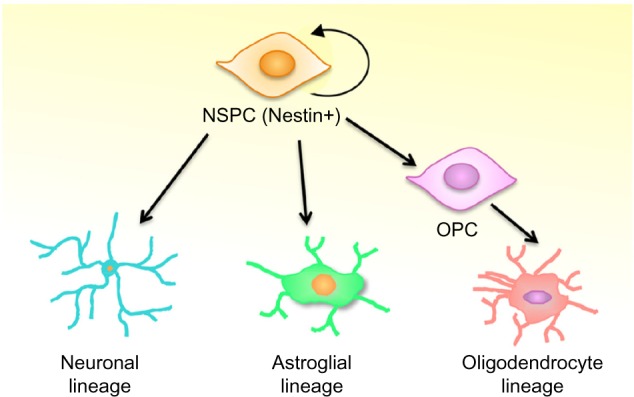
Neural stem/precursor cells (NSPCs) differentiate into the neural cells of the central nervous system. NSPCs are capable of self-renewal and differentiation into distinct neural cell lineages. Depending on external cues, NSPCs can differentiate into neurons (neuronal lineage) or astrocytes (glial lineage). NSPCs can also differentiate in oligodendrocyte precursor cells (OPCs), which are more restricted stem cells that can give rise to oligodendrocytes.
IFNγ Signaling: Canonical and Non-Canonical Pathways
In order to understand the effects of IFNγ, we need to acknowledge the diversity of signaling pathways that it initiates. IFNγ binds to the IFNγ receptor (IFNGR), which consists of two IFNGR1 subunits and two IFNGR2 subunits. Binding of IFNγ to IFNGR1 causes heterotetramerization of the receptor, which then leads to the activation of downstream kinases.33,34 IFNγ predominantly activates the Janus associated kinase/signal transducer and activator of transcription-1 (JAK/STAT) signaling pathway (Fig. 2). Activation of JAKs results in the recruitment and activation (phosphorylation) of STATs at the receptor. Out of the seven STAT family members, STAT1 is the main downstream effector of IFNγ. Upon phosphorylation by JAKs, STAT1 homodimerizes and translocates to the nucleus, where it initiates the transcription of IFNγ-stimulated genes (ISGs). There are approximately 500 ISGs that can be stimulated by IFNγ, including genes involved in viral clearance, cell cycle control, and inflammatory signaling.33 For example, IFNγ increases major histocompatibility complex (MHC) expression in a STAT1-dependent manner, leading to the recognition of tumor cells by the immune system.35 IFNγ also inhibits the proliferation of fibroblasts by reducing cyclin and cyclin-dependent kinase (CDK) expression, particularly that of cyclin D/CDK4.36 The profile of ISGs is dependent both on cell type and on other inflammatory signals that are received by the target cell (reviewed by van Boxel-Dezaire and Stark37). Thus, the phenotypic response to IFNγ also varies depending upon the cell type, which is reflected in the conflicting reports of neuroprotection and toxicity with IFNγ treatment.15,38,39
Figure 2.
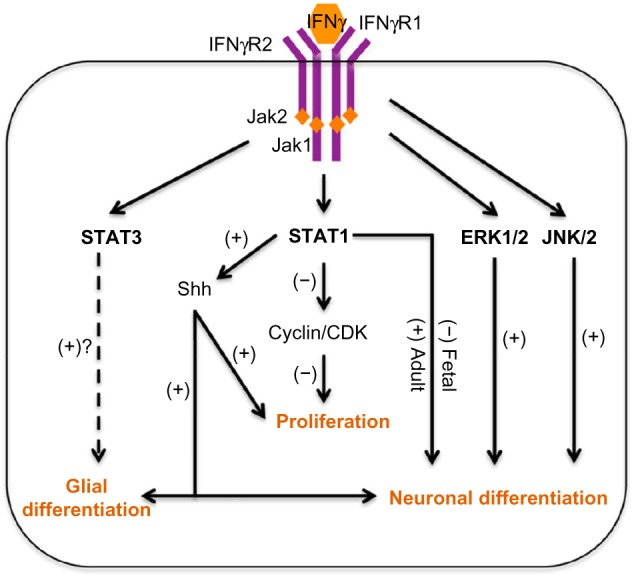
Current hypotheses for IFNγ-mediated effect on NSPC activity.
Notes: IFNγ binds to the IFNγ receptor (IFNGR1 and 2) and activates Janus-activated kinases 1 and 2 and signal transducers and activators of transcription-1 (JAK-STAT1) pathway. In neural stem/progenitor cells (NSPCs), STAT1 activation blocks NSPC proliferation through decreased expression of cyclin/cyclin-dependent kinase (CDK) complexes. In fetal NSPCs, IFNγ-mediated STAT1 activation decreases NSPC differentiation in the neuronal lineage (A. Kulkarni, unpublished data). However, studies with adult NSPCs report that IFNγ-mediated STAT1 activation results in increased neuronal differentiation. STAT1 activation leads to increased expression and secretion of the sonic hedgehog (Shh) protein, which in turn causes anomalous and concurrent expression of both glial and neuronal markers in the same cell.57 Moreover, IFNγ-mediated STAT1 activation and Shh expression in granule precursor cells leads to increased NSPC proliferation. In Paju cells, IFNγ-induced activation of the extracellular-signal regulated kinase-1/2 (ERK-1/2) increases neuronal differentiation, which may be independent of STAT1 activation.61 In C17.2 cells, IFNγ-mediated activation of c-jun N-terminal kinase (JNK) pathway causes neuronal differentiation, without the activation of ERK1/2 pathway.62 Developmental cytokines such as the leukemia inhibitory factor and ciliary neurotropic factor mediate glial differentiation through STAT3. IFNγ-mediated activation of STAT3 is observed in fetal NSPCs; however, the role of this pathway in NSPC differentiation is yet unclear.
To evade clearance from the body, many viruses inhibit STAT1 function and expression, thereby abrogating the antiviral response of the cell.40,41 However, IFNγ also signals through STAT1-independent mechanisms, which may be activated alone or in parallel with STAT1-dependent pathways.42 These pathways result in protective as well as pathological outcomes. Primary hippocampal neurons utilize STAT1-independent pathways for viral control, possibly because endogenous STAT1 expression is inherently low in these cells.11 Neurons also activate extracellular regulated kinase-1/2 (ERK-1/2) signaling in response to IFNγ, which confers neuroprotection against apoptotic insults. In contrast, primary astrocytes activate STAT3 upon IFNγ treatment, which leads to the production of neurotoxic factors.43 IFNγ stimulation may also lead to recruitment of adaptor molecules such as the c-Cbl proto-oncogene and the GTPases Ras and Rap1.44 Like STAT1-dependent signals, activation of Rap1 signaling inhibits cell proliferation in human embryonic kidney cells.45 Therefore, IFNγ may activate multiple pathways that limit cell growth, which could be advantageous for controlling viral replication in a rapidly dividing cell. An interesting observation is seen in lung epithelial cells, where STAT1 is activated through the activation of the phospholipase C-gamma2/protein kinase C/Src (PLCγ2-PKCα-src) pathway in a JAK-dependent manner. This pathway causes increased expression of intercellular adhesion molecule (ICAM)-146 and facilitates binding and transmigration of immune cells into tissues. Therefore, in addition to pathways that are STAT1-independent, other signaling proteins and adaptor proteins may link JAK and STAT1 indirectly in certain cell types.
IFNγ-Mediated Signaling in NSPCs
The diversity of IFNγ-mediated signaling pathways raises the question of how IFNγ may affect NSPCs. The role of IFNγ is particularly important because the JAK-STAT family of proteins has been implicated in NSPC proliferation and differentiation.32,47,48 A number of reports show that IFNγ inhibits the proliferation of murine NSPCs derived from the adult SVZ in vitro.30,49–52 Moreover, NSPCs derived from mice lacking IFNγ show enhanced neurogenesis and proliferation.53 There is ample evidence that IFNγ can alter NSPC function through activation of the STAT family of proteins. Lum et al showed that when adult SVZ-derived NSPCs were treated with IFNγ, there was activation of both STAT1 and STAT3.50 Conversely, Pereira et al observed that IFNγ-treated NSPCs derived from postnatal brains (P7–P9) showed increased STAT1 activation but no change in STAT3 activation.51 One reason for this discrepancy could be the time point at which STAT3 activation was measured; the former study assessed STAT3 activation 4 days post treatment, whereas the latter measured activation at 15 minutes post treatment. Studies from our lab also report robust STAT1 activation and transient STAT3 activation in NSPCs derived from fetal mouse cortex (A. Kulkarni, unpublished data). Consistent with an inhibition of growth, IFNγ decreased NSPC proliferation in a STAT1-dependent manner, with restriction in the late G1 phase of the cell cycle. Studies in many cell types indicate that STAT1 and STAT3 play opposing roles in cell proliferation; STAT1 is generally anti-proliferative and STAT3 is pro-proliferative.54 This explains the role of STAT1 in mediating anti-proliferative effects of IFNγ on NSPCs, but the role of STAT3 is yet unclear. In NSPCs from STAT1 knockout mice, we observed substantial STAT3 activation post IFNγ treatment, suggesting potential crosstalk between the STAT1 and STAT3 pathways. It is interesting to note that the temporal response of NSPCs to IFNγ is distinct from other neural cells. Neurons show a delayed but sustained upregulation of STAT1 activation and expression, whereas astrocytes show spontaneous but transient STAT1 activation.15 NSPCs demonstrate rapid and sustained activation of STAT1 (over 72 hours), suggesting that neural cells possess unique mechanisms for regulating STAT1 expression and dephosphorylation.
The sonic hedgehog (Shh) protein also plays an important role in NSPC proliferation and fate specification.55 In NSPCs, IFNγ induces Shh expression. These studies show that IFNγ-mediated Shh expression and signaling results in increased proliferation of cerebellar neural precursor cells.56 Other studies demonstrate that induction of Shh by IFNγ results in a dysregulated cell fate characterized by expression of glial and neuronal markers in the same cell.57 IFNγ also increases Shh expression in adipocyte precursors, suggesting that IFNγ may act broadly on undifferentiated cells to induce differentiation or growth.58
One outstanding question is how NSPC differentiation is affected by IFNγ during pathological insults. In vitro studies on adult murine NSPCs indicate that IFNγ induces neuronal differentiation.30,49,50 Pereira et al showed that infusion of IFNγ into the mouse SVZ increased neuronal differentiation in a STAT1-dependent manner.51 However, Ben-Hur et al did not observe any changes in differentiation in IFNγ-treated NSPCs derived from neonatal rat striatum.52 In contrast to many studies on adult NSPCs, embryonic NSPCs exhibit decreased neuronal differentiation in response to IFNγ.59 Together, these studies suggest that NSPCs may differ in their responsiveness to IFNγ depending upon the anatomical location and the age of the host.
A number of other cytokines, including leukemia inhibitory factor (LIF) and ciliary neurotropic factor (CNTF), activate JAK-STAT signaling and regulate NSPC cell fate. During CNS development, these cytokines trigger glial differentiation in NSPCs through activation of STAT1 and STAT3. However, they cause glial differentiation only during late gestational periods (embryonic day 16 and later). STAT-dependent gliogenic activity is repressed during the neurogenic period (embryonic days 10–14) through epigenetic inhibition of glial gene expression.48,60 Whether IFNγ synergistically affects LIF and CNTF signaling during development is unknown. However, one could conjecture that IFNγ may augment glial differentiation at later stages in development through the activation of JAK-STAT signaling.
In addition to STAT1, mitogen-activated protein kinases (MAPKs) have also been implicated in mediating neuronal differentiation in cell lines. IFNγ induces neuronal differentiation through the ERK-1/2 pathway in the human neuroblastoma Paju cell line.61 In a murine cerebellar cell line, IFNγ causes neuronal differentiation through the activation of c-jun N-terminal kinase (JNK) pathway.62 Moreover, inhibition of the JNK pathway but not the ERK-1/2 pathway reversed the effects of IFNγ. Other MAPKs, such as p38, have also been implicated in mediating neuronal differentiation.28 Admittedly, these studies were conducted in transformed cell lines and not in primary NSPCs.61 However, they highlight the importance of MAPK signaling as an alternative pathway for influencing cell-fate decisions downstream of IFNγ.
The fact that IFNγ affects NSPC proliferation and cell-fate specification is well established, although the necessary signaling pathways are still being defined. Variables such as the age of the host, brain region, and species may impact on the NSPC response to IFNγ. Regardless of the differences in model systems, STATs play a major role in mediating the effects of IFNγ on NSPC activity. Activation of non-canonical pathways, such as MAPKs, and crosstalk with other STAT signaling pathways may also be involved. It will be important to account for the mutable responses of NSPCs to IFNγ when considering how these cells react in in vivo disease models.
A central role for inflammation has been acknowledged in many CNS diseases.63–65 However, the role of inflammation, and specifically of IFNγ, in modulating NSPC functions is under active study. IFNγ is one variable that affects how NSPCs respond in inflammatory environments. Because IFNγ is a pleiotropic cytokine, alterations in IFNγ expression often affect multiple neural and immune cells, which can further impact on NSPC function. Taking into account the diversity of signaling pathways activated by IFNγ, and variability of its effects on NSPCs in different systems, IFNγ may exert subtle alterations in pathological outcomes in neuroinflammatory conditions. The discovery of multipotent NSPCs in the adult brain has also generated interest in how these NSPCs are affected by inflammation in the mature brain, particularly during neurodegenerative disease. Here, we discuss current studies that focus on the role of IFNγ and its effects in altering NSPC activity in models of Alzheimer’s disease, multiple sclerosis, and viral neurotropic infections.
Alzheimer’s Disease
AD is the leading cause of dementia in the United States, with estimates of more than 5 million AD cases in that country alone. AD patients experience progressive memory loss, cognitive decline, and functional and behavioral impairments that are irreversible with current therapies.66 Pathologically, the AD brain is characterized by widespread neuronal loss and by the accumulation of misfolded protein aggregates, which include amyloid plaques and neurofibrillary tangles. The amyloid plaques contain oligomers of the β-amyloid (Aβ) peptide, a cleavage product derived from the amyloid precursor protein (APP) after processing by β- and γ-secretases. Neurofibrillary tangles are comprised of hyperphosphorylated tau protein, which is normally associated with the microtubules. The accumulation of Aβ oligomers is thought to be responsible for synaptic dysfunction, neuronal death, and the activation of neighboring glial cells including astrocytes and oligodendrocytes.67–69 Despite the recognition that Aβ and tau are the main components of AD plaques and tangles, effective treatments to prevent aggregation and subsequent neurodegeneration are unavailable.
Although the initiating factors that lead to AD pathology are not completely understood, it is clear that inflammation plays a role in the progression of the disease. It has been hypothesized that neurodegeneration triggers expression of pro-inflammatory cytokines that lead to hyperphosphorylation of tau, thus contributing to the formation of neurofibrillary tangles.70 Elevated levels of inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNFα) are found in proximity to amyloid plaques and in the plasma and CSF of AD patients.71–74 Evidence for both protective and pathogenic effects of IFNγ have been noted in AD. Astrocytes, the major source of Aβ in the brain, are stimulated to produce Aβ peptides when co-stimulated with IFNγ and TNFα or IL-1β.75 IFNγ alone can stimulate β-secretase expression in human astrocytes, suggesting that IFNγ might enhance processing of Aβ.76 Mononuclear cells from moderately severe AD patients produce elevated levels of IFNγ in comparison to cells from healthy controls or mild AD patients.77,78 Moreover, IFNγ increases the death of primary neurons treated with Aβ peptides.39 Although human studies are inconclusive as to whether IFNγ is elevated in the AD brain, polymorphisms in the IFNγ promoter that lead to high IFNγ expression are associated with slower progression of AD.79 These findings suggest that the neuroinflammatory response, including IFNγ production, may have differential effects on neural cells in AD pathogenesis.
To better elucidate the role of IFNγ, transgenic mouse models of AD have examined disease progression in relation to IFNγ expression and signaling, with varying effects on AD pathology. In Tg2576 mice, which harbor the Swedish mutation of human APP, deletion of the IFNγ receptor is associated with reduced glial activation and Aβ deposition due to a decrease in APP processing.80 In contrast, overexpression of IFNγ in APP transgenic mice leads to activation of microglia and astrocytes and expression of MHC-II and complement cascade proteins. In this case, the enhanced inflammation confers neuroprotection by promoting Aβ phagocytosis and reducing plaque formation in the forebrain and hippocampus.81 IFNγ also leads to increased phosphorylation of tau in mouse models of tauopathy, although the phosphorylated tau does not aggregate into tangles or contribute to neuropathology.82 These contradictory findings not only highlight the challenge of modeling distinct aspects of AD pathology in mice, but also show that IFNγ may have both protective and toxic effects on disease progression and the inflammatory response.
Accumulation of Aβ begins in the hippocampus in early stages of disease, which correlates with the loss of short-term memory as an early indicator of AD. In severely affected Alzheimer’s brains, expression of neurogenic markers (doublecortin, NeuroD) is enhanced in the dentate gyrus and CA1 region of the hippocampus,83 indicative of enhanced neurogenesis in AD. In presenile patients, there is little evidence of changes in neurogenesis, suggesting that the course of disease as well as differences in methodology may complicate interpretations of NSPC function in human tissues.84 Further evaluation of human AD brains is needed to fully understand changes in NSPC activity as a function of disease severity. In mouse models of AD, accumulation of Aβ through transgene expression or intraventricular injection reduces NSPCs and newly born neurons in the hippocampus.85 Aβ treatment of primary NSPCs in vitro also inhibits NSPC proliferation and production of new neurons. These investigations show that NSPCs are responsive to the types of insults that occur in AD, although it is unclear how such changes in NSPC activity impact on disease progression in humans.
Several studies directly address the role of IFNγ in neurogenesis and NSPC proliferation in AD models. Using triple-transgenic mice (3 × Tg-AD), which harbor mutations in presenilin, APP, and tau, prolonged expression of IFNγ reveals opposing effects on AD pathology.86 IFNγ increases microglial activation and intracellular accumulation of Aβ, which is an early marker of AD. However, IFNγ also promotes neurogenesis in the hippocampus and reduces tau pathology in the 3 × Tg-AD model. Baron and colleagues observed that low levels of IFNγ expression increase neurogenesis and synaptic activity in the dentate gyrus of aged wild-type mice and transgenic mice with a mutation in APP.87 Moreover, IFNγ-induced neurogenesis correlates with improved spatial learning and memory, suggesting that changes in neurogenesis may improve neurological outcomes. Although IFNγ may play a neuroprotective role in AD models, injection of Aβ peptides into wild-type mice impairs IFNγ expression, neurogenesis, and NSPC proliferation in the hippocampus.85 Thus, whether IFNγ is able to influence NSPC function may depend largely on the stage of the disease and the inflammatory milieu that is expressed at each stage. Regardless, the sensitivity of NSPCs to IFNγ and the multiple factors that impact on IFNγ expression in AD suggest that modulation of IFNγ could slow the course of AD through encouraging neuroprotection and repair by NSPCs.
Multiple Sclerosis and Experimental Autoimmune Encephalitis
MS is a chronic inflammatory disease of the brain and spinal cord characterized by demyelination of axons and eventual neuronal death.88 Patients with MS exhibit motor impairment, sensory and visual disturbances, pain, fatigue, and cognitive deficits.89 The brain and spinal cord of MS patients have demyelinated areas called plaques or lesions that indicate a loss of the myelin sheath and death of oligodendrocytes in the white matter. The infiltration of immune cells causes the formation of lesions, with associated activation of glial cells and disturbances in neuronal signaling due to axonal degeneration90 (reviewed by Dendrou et al.91). Experimental autoimmune encephalomyelitis (EAE) is a rodent model of MS that is widely used to study the mechanism of the disease and to test the efficacy of therapies.92 This model recapitulates several clinical, pathological, and immunological features of MS by immunizing animals with myelin proteins, such as myelin basic protein or proteolipid protein, in adjuvants.93,94 Studies suggest that the immune response in MS and EAE causes apoptosis of oligodendrocytes, which contributes to demyelination and ultimately neurodegeneration.95,96
IFNγ is part of the inflammatory milieu in MS patients and can be found within MS lesions.97,98 Early clinical trials using recombinant IFNγ therapy exacerbated MS symptoms and elevated multiple inflammatory markers during intravenous administration.99 Serum levels of IFNγ also increase prior to clinical attacks, whereas IFNα expression increases during periods of remission.100 A number of single nucleotide polymorphisms (SNPs) within different inflammatory and immune-related genes have been associated with susceptibility to MS.101 Sex-based differences are also observed in susceptibility to MS, which is more common in women than in men.102 Interestingly, population-based studies suggest that polymorphisms in the 3′ untranslated region of the IFNγ gene are associated with the development of MS in men.103 Although there is evidence for an association of IFNγ with MS, many questions remain about its role in the pathology of the disease. Most in vivo studies have focused on the effects of IFNγ on peripheral immune cells and mature oligodendrocytes, while its action on NSPCs is less defined in the pathogenesis and progression of MS and EAE.
Several studies demonstrate that IFNγ contributes to the death of oligodendrocytes by macrophage/microglia activation, upregulation of MHC molecules, and induction of inflammatory mediators (reviewed by Goverman104). Studies in transgenic mice with temporally regulated expression of IFNγ show that the duration of IFNγ expression dictates its beneficial or detrimental effects on the development of EAE.105,106 IFNγ expression in the CNS before the onset of EAE improves the course of disease and prevents loss of oligodendrocytes, demyelination, and axon degeneration. This protective effect of IFNγ is mediated by pancreatic endoplasmic stress kinase (PERK) activation in oligodendrocytes.106 Further studies suggest that enhanced PERK signaling inhibits apoptosis of oligodendrocytes before the start of clinical disease and blocks EAE-induced demyelination and axonal degeneration at the peak of the disease.107 These protective effects on axons are not due to a decrease in the inflammatory response but may be due to maintenance of myelin integrity. PERK signaling activates an antiapoptotic transcription factor, nuclear factor kappa-B, which may be a possible protective mechanism in oligodendrocytes.107 In contrast, IFNγ expression at the recovery stage of EAE suppresses oligodendrocyte regeneration and remyelination in lesions.105 However, how IFNγ impacts NSPC function, and the potential differentiation into oligodendrocyte precursors, remains largely unexplored in the chronic inflammatory environment of MS.
In MS, there is the dual challenge of replacing or protecting both degenerating neurons and damaged/dying oligodendrocytes. NSPCs are capable of giving rise to new neurons and to oligodendrocyte precursor cells (OPCs), which are more restricted stem cells that can ultimately produce new oligodendrocytes (Fig. 1). Both NSPCs and OPCs are of potential therapeutic importance in MS.108 Acute inflammation may contribute to remyelination by inducing development of OPCs.109 Yet, in MS brains, OPCs do not fully differentiate into mature oligodendrocytes or participate in remyelination, despite the localization of OPCs in the white matter lesions.110 Because MS is a chronic inflammatory disease, the response of NSPCs and OPCs must be considered in the context of prolonged exposure to inflammatory mediators.
Studies suggest that EAE induces the proliferation of NSPCs and OPCs in the SVZ. The subsequent migration of mitotically active SVZ cells to the olfactory bulb and regions of demyelinated white matter is also enhanced.111 In the brains of postmortem MS patients, there is increased proliferation of NSPCs in the SVZ, which also express the oligodendroglial markers Sox10 and Olig2,112 although the eventual migration and maturation of the cells in lesions is limited.110 These studies suggest that NSPCs respond to inflammatory changes in MS, but may not fully participate in remyelination of lesioned areas. One possible explanation for the disrupted activity of NSPCs and OPCs is the reactivation of Shh by IFNγ. Shh is a member of the hedgehog family of morphogens that are critical in the regulation of stem cell niches and proliferation of NSPCs in postnatal telencephalon,113 adult hippocampus,114 and SVZ.115 Typically, Shh activates the downstream transcription factor Gli1 in order to mediate differentiation of neurons and oligodendrocytes.116,117 However, in MS lesions and EAE mice, Shh expression is highly upregulated while Gli1 expression is decreased.116 The authors found that IFNγ treatment of embryonic and adult NSPCs recapitulated their in vivo observations, with increased Shh expression and inhibition of Shh-induced Gli1 expression.116 Thus, IFNγ inhibits the differentiation of NSPCs by downregulation of Shh-induced Gli1 expression. This paradoxical effect of IFNγ on Shh-Gli1 signaling may contribute to the lack of differentiation and maturation of NSPCs and OPCs in MS.116 These findings further imply that prolonged IFNγ exposure may have a negative impact on maturation and remyelination by new oligodendrocytes. In support of this idea, IFNγ limits remyelination and OPC recruitment in a model of chronic toxin-induced demyelination, suggesting that IFNγ may have long-term impacts on remyelination.118
Due to the dysregulation of the endogenous stem cell pool, many studies have attempted to transplant NSPCs or other stem cell lineages into mouse models of EAE. The administration of NSPCs derived from mesenchymal stem cells correlates with reduced T-cell infiltration, less demyelination, and an increase in the number of nestin-positive cells.119 Importantly, this study and others demonstrate that transplanted stem cells can be immunosuppressive and highlights the crosstalk that can occur between NSPCs and immune cells.119,120 Furthermore, transplant of NSPCs into a cuprizone-induced demyelination model demonstrated remyelination by endogenous OPCs, without migration or differentiation by the transplanted cells.121 The authors found that the transplanted NSPCs encouraged differentiation of resident OPCs through the release of growth factors. These studies suggests that the protective mechanisms of NSPCs may be due to a trophic effect, allowing transplanted cells to communicate with resident neural cells in the brain.119 As there is a need for therapies that not only modulate the immune response but also repair CNS injury in MS, NSPCs that retain their multipotential capacity are good candidates for repair of MS lesions. Thus, further studies are warranted to consider how inflammatory cytokines such as IFNγ may influence the differentiation or proliferation of endogenous and transplanted NSPCs.
Viral CNS Diseases
Neurotropic viruses damage the CNS by directly killing infected neurons or as a result of the immune response to the virally infected cells.122 In addition to neurons, NSPCs, a mitotically active population of cells, are also altered during viral infections. NSPCs are permissible to several viruses including murine and human cytomegalovirus, herpes simplex virus, Japanese encephalitic virus, and Zika virus.123–126 These infections result in reduced NSPC proliferation and increased apoptosis, which could impair neuronal repair and neurogenesis.125,126 In addition to direct viral infection, NSPC activity may be affected through a bystander effect from antiviral cytokines.108,127 IFNγ is critical in controlling the spread of many neurotropic viruses including measles virus, Theiler's virus, herpes simplex virus, and Sindbis virus.10,128–130 Even though the antiviral and immunomodulatory roles of IFNγ are well documented, its role in affecting NSPC activity in the context of viral infections is less clear. Recent research indicates that IFNγ may affect NSPC survival, proliferation, and neurogenic potential during infections, depending on the model system and on the cellular tropism of the virus. Here, we review the role of IFNγ on NSPCs in different models of neurotropic viral infections.
Herpes simplex virus-1 (HSV-1)
HSV-1 is a DNA virus that infects approximately 54% of adults in the United States.131 In most cases, the virus resides latently in sensory neurons of the trigeminal ganglion with intermittent bouts of reactivation. During reactivation, new infectious viral particles are produced that travel down the axon and infect epithelial cells at the site of neuronal innervation, leading to the typical “cold sore” lesions associated with HSV-1. However, in some cases, HSV-1 may spread from the trigeminal ganglia to the temporal and inferior frontal lobes to establish a more severe, widespread CNS infection. The resultant herpes simplex encephalitis (HSE) can be fatal or cause long-term cognitive deficits. The factors that lead to HSE and unrestricted HSV-1 spread in the brain are unknown, although genetic factors, including deficits in the type I interferons, have been implicated.132
NSPCs, astrocytes, and young and mature neurons are all permissible to HSV-1 infection.133 In vivo studies involving nasal HSV-1 inoculation in mice implicate two mechanisms of damage to the CNS. First is the infection and lysis of neurons that control critical physiological functions.134 In addition, Lundberg et al showed that the antiviral immune response against HSV-1 played a major role in CNS pathology.135 In their study, mice susceptible to HSV-1 developed fatal focal lesions in the brain that consisted on infiltrating macrophages and neutrophils. When the macrophages and neutrophils were depleted, the mice showed delayed mortality as compared to nondepleted animals. Moreover, treatment with acyclovir decreased viral load to undetectable levels, but did not reduce mortality. Therefore, these studies show that the inflammatory response in the brain contributes to the pathology and death in HSV-1-infected mice.
Studies using intranasal delivery of HSV to the brain demonstrate an initial increase in the number of NSPCs during the acute phase (6 days post infection; dpi) of HSV infection. However, NSPC numbers decline during the chronic phase (10–30 dpi), wherein the adaptive immune response is active.136 Interestingly, the NSPCs are not infected by HSV-1 in the intranasal model, suggesting that changes in NSPC function are due to the inflammatory environment. During the chronic phase of HSV-1 infection, activated CD8+ T cells are the major source of IFNγ in the CNS. Coculture of virus-activated CD8+ T cells and NSPCs showed reduced NSPC proliferation and differentiation into glial cells.31 When antibodies blocking IFNγ binding to its receptor were used, the decrease in proliferation was abrogated. Together, these findings demonstrate that IFNγ may be a key factor in dictating how NSPCs respond to cytotoxic T cells. These studies further suggest that the IFNγ-mediated effects on NSPCs may affect functional recovery post-HSV-1 infection.
Cytomegalovirus (CMV)
CMV, like HSV-1, is also a DNA virus belonging to the herpesvirus family. Examination of serum samples from 1999 to 2004showed that 50.4% of the US population was infected with CMV.137 In adults, CMV mostly acts as an opportunistic pathogen affecting immunocompromised adults including transplant recipients and HIV-infected patients. Clinical manifestations in immunocompromised adults include retinitis, encephalitis, and subcortical dementia among others. Congenital CMV infections are a major cause of birth defects in the United States, with approximately 2% of newborns infected by transplacental transfer from the mother. Ten to fifteen percent of these infections develop neurological sequelae such as hearing loss, mental impairments, and microcephaly.138 Thus, the developing brain is especially susceptible to the pathogenic effects of CMV infection.
CMV preferentially targets NSPCs in brain tissue, although it is capable of infecting other neural cells. CMV infection of NSPCs inhibits proliferation and neurogenesis and induces apoptosis.139 These observations have been made both in human NSPCs and in mouse models of murine CMV (mCMV) infection.125,140 Similarly, NSPCs infected with human CMV (hCMV) display reduced proliferation and decreased neuronal differentiation.140 Studies with hCMV also show that, when infected, human NSPCs undergo apoptosis due to improper folding of proteins in the endoplasmic reticulum (ER), triggering the ER stress response.139 These observations indicate that CMV not only inhibits NSPC function but also results in cell death.
mCMV models have been used extensively to study the effect of viral infection on NSPCs in vivo. mCMV demonstrates a strong cellular tropism for NSPCs. Thus, the majority of the mCMV models involve direct infection of NSPCs by the virus. mCMV infection of newborn mice leads to extensive infection of NSPCs and a loss of NSPCs and newly born neurons.125 The immune response against the virus begins with infiltration of macrophages and NK cells and the activation of CNS resident microglia.141 CD8+ T cells, which are critical in controlling mCMV infection, follow the activation of innate immune cells.142 CD8+ T cells mediate clearance of CMV through cytolytic and non-cytolytic mechanisms, including IFNγ production.143 IFNγ increases the expression of MHC class I and II on NSPCs, which would allow recognition by infiltrating T cells.144 mCMV, however, is able to counteract the IFNγ-mediated induction of MHC class I in NSPCs.145 These mechanisms may allow the virus to evade immune clearance and establish latency in the NSPCs. Moreover, MHC expression is important for neuronal development and synaptic refinement.146 Therefore, a CMV-mediated decrease in MHC expression could hamper the development of neuronal networks, particularly during the formative stages of brain development.
Measles virus (MV)
MV is a negative-strand RNA virus and a member of the Morbilivirus genus and Paramyxoviridae family. Upon exposure, the virus enters the upper respiratory tract and is taken up by dendritic cells, B cells, and T cells expressing the CD150 receptor.147 The route of MV entry into the brain is unknown, but may involve transmigration of infected leukocytes across the blood–brain barrier, infection of endothelial cells of the brain microvasculature, infection of the choroid plexus and the nasal epithelium, and/or antegrade transport via the olfactory nerves.148 MV infection in the CNS can cause severe neurological disorders: primary measles encephalitis, measles inclusion body encephalitis, and subacute sclerosing panencephalitis (SSPE).149
Mice are not infectable with MV, but transgenic mice expressing the human isoforms of the MV receptors are available. These mouse models include global or tissue-specific expression of the human receptors used by circulating MV strains (CD150/SLAM) or by vaccine strains (CD46). Our laboratory uses a mouse model that expresses human CD46 under the control of the neuron-specific enolase promoter (CD46+ mice), thereby restricting MV infection to mature CNS neurons.150 In CD46+ adult mice, IFNγ is required for non-cytolytic clearance of MV from infected neurons.128 In neonatal CD46+ mice, we explored how the antiviral immune in the brain affected NSPCs, which were spared from MV infection. IFNγ preserved the NSPC pool during MV infection, but could not prevent a decline in neurogenesis. Moreover, newly differentiated neurons (doublecortin+) were lost regardless of IFNγ expression.151 These data indicate that IFNγ may be critical in protecting uninfected NSPCs during an antiviral immune response, but cannot protect new neurons from the effects of neuroinflammation.
There are considerable functional differences in the immune response of adults and neonates, with the neonatal immune response often failing to produce adequate levels of IFNγ during infections (reviewed by Adkins et al.152). In the CD46+ model of MV infection, neonatal mice succumb to MV despite immune cell infiltration in the brain, whereas adults survive the infection with effective viral control.153 This provides an opportunity to examine how age-dependent differences in the immune response affect NSPC activity as well as how the antiviral response impacts on brain development. As NSPC growth and differentiation is altered during infection by many neurotropic viruses,123 the role of IFNγ will be central to understanding how antiviral immunity disrupts brain development and repair.
Conclusion
Neurogenesis occurs under both physiological and pathological conditions with unique stimuli and neurological outcomes. An understanding of the factors that dictate the proliferation and differentiation of NSPCs will inform the development of pharmaceutical strategies to manipulate neurogenesis. In order to design treatments that provide a therapeutic benefit, we must better define how new neurons are integrated into existing neural networks, and even determine whether production of new neural cells is beneficial in pathological settings. Neuroinflammation is apparent in many degenerative disorders and infectious diseases. IFNγ is only one factor that influences the inflammatory milieu and the response of neural cells to damage. Thus, the role of IFNγ must be considered in the context of a complete immune response, with a variable accompaniment of cytokines and chemokines, when considering the ultimate impact of IFNγ on NSPC function.
Acknowledgments
We are very grateful to Debbie Willson, Mary Caruso, and Jackie Farrer for administrative support.
Footnotes
ACADEMIC EDITOR: Dama Laxminarayana, Editor in Chief
PEER REVIEW: Six peer reviewers contributed to the peer review report. Reviewers’ reports totaled 644 words, excluding any confidential comments to the academic editor.
FUNDING: This work was funded by the Samuel and Emma Winters Foundation (LOD), the Duquesne University Mylan School of Pharmacy (AK, PG, LOD), and R15-NS087606-01A1 (LOD). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Wrote the first draft of the manuscript: AK, PG, LOD. Contributed to the writing of the manuscript: AK, PG, LOD. Agree with manuscript results and conclusions: AK, PG, LOD. Jointly developed the structure and arguments for the paper: AK, PG, LOD. Made critical revisions and approved final version: AK, PG, LOD. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Konstantinos AP, Sheridan JF. Stress and influenza viral infection: modulation of proinflammatory cytokine responses in the lung. Respir Physiol. 2001;128(1):71–7. doi: 10.1016/s0034-5687(01)00266-3. [DOI] [PubMed] [Google Scholar]
- 2.Binder GK, Griffin DE. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293(5528):303–6. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- 3.Rempel JD, Quina LA, Blakely-Gonzales PK, Buchmeier MJ, Gruol DL. Viral induction of central nervous system innate immune responses. J Virol. 2005;79(7):4369–81. doi: 10.1128/JVI.79.7.4369-4381.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solomos AC, Rall GF. Get it through your thick head: emerging principles in neuroimmunology and neurovirology redefine central nervous system “immune privilege”. ACS Chem Neurosci. 2016;7(4):435–41. doi: 10.1021/acschemneuro.5b00336. [DOI] [PubMed] [Google Scholar]
- 5.Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The glymphatic system: a beginner’s guide. Neurochem Res. 2015;40(12):2583–99. doi: 10.1007/s11064-015-1581-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(suppl 1):S232–40. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 8.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265(5168):106–9. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 9.Fruh K, Yang Y. Antigen presentation by MHC class I and its regulation by interferon gamma. Curr Opin Immunol. 1999;11(1):76–81. doi: 10.1016/s0952-7915(99)80014-4. [DOI] [PubMed] [Google Scholar]
- 10.Burdeinick-Kerr R, Griffin DE. Gamma interferon-dependent, non-cytolytic clearance of sindbis virus infection from neurons in vitro. J Virol. 2005;79(9):5374–85. doi: 10.1128/JVI.79.9.5374-5385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Donnell LA, Conway S, Rose RW, et al. STAT1-independent control of a neurotropic measles virus challenge in primary neurons and infected mice. J Immunol. 2012;188(4):1915–23. doi: 10.4049/jimmunol.1101356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podolsky MA, Solomos AC, Durso LC, Evans SM, Rall GF, Rose RW. Extended JAK activation and delayed STAT1 dephosphorylation contribute to the distinct signaling profile of CNS neurons exposed to interferon-gamma. J Neuroimmunol. 2012;251(1–2):33–8. doi: 10.1016/j.jneuroim.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee M, McGeer E, McGeer PL. Neurotoxins released from interferon-gamma-stimulated human astrocytes. Neuroscience. 2013;229:164–75. doi: 10.1016/j.neuroscience.2012.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW. Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. MBio. 2016;7(1):e2145–e2115. doi: 10.1128/mBio.02145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Donnell LA, Henkins KM, Kulkarni A, et al. Interferon gamma induces protective non-canonical signaling pathways in primary neurons. J Neurochem. 2015;135(2):309–22. doi: 10.1111/jnc.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burdeinick-Kerr R, Govindarajan D, Griffin DE. Noncytolytic clearance of sindbis virus infection from neurons by gamma interferon is dependent on Jak/STAT signaling. J Virol. 2009;83(8):3429–35. doi: 10.1128/JVI.02381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seifert HA, Collier LA, Chapman CB, Benkovic SA, Willing AE, Pennypacker KR. Pro-inflammatory interferon gamma signaling is directly associated with stroke induced neurodegeneration. J Neuroimmune Pharmacol. 2014;9(5):679–89. doi: 10.1007/s11481-014-9560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seifert HA, Leonardo CC, Hall AA, et al. The spleen contributes to stroke induced neurodegeneration through interferon gamma signaling. Metab Brain Dis. 2012;27(2):131–41. doi: 10.1007/s11011-012-9283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bombeiro AL, D’Imperio Lima MR, Chadi G, Alvarez JM. Neurodegeneration and increased production of nitrotyrosine, nitric oxide synthase, IFN-gamma and S100beta protein in the spinal cord of IL-12p40-deficient mice infected with Trypanosoma cruzi. Neuroimmunomodulation. 2010;17(2):67–78. doi: 10.1159/000258689. [DOI] [PubMed] [Google Scholar]
- 20.Temple S. The development of neural stem cells. Nature. 2001;414(6859):112–7. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- 21.Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24(7):1726–33. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kempermann G, Song H, Gage FH. Neurogenesis in the adult hippocampus. Cold Spring Harb Perspect Biol. 2015;7(9):a018812. doi: 10.1101/cshperspect.a018812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707–10. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22(3):629–34. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jessberger S, Zhao C, Toni N, Clemenson GD, Jr, Li Y, Gage FH. Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J Neurosci. 2007;27(35):9400–7. doi: 10.1523/JNEUROSCI.2002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anacker C. Adult hippocampal neurogenesis in depression: behavioral implications and regulation by the stress system. Curr Top Behav Neurosci. 2014;18:25–43. doi: 10.1007/7854_2014_275. [DOI] [PubMed] [Google Scholar]
- 27.Lugert S, Basak O, Knuckles P, et al. Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 2010;6(5):445–56. doi: 10.1016/j.stem.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 28.Morooka T, Nishida E. Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J Biol Chem. 1998;273(38):24285–8. doi: 10.1074/jbc.273.38.24285. [DOI] [PubMed] [Google Scholar]
- 29.Guzhova I, Hultquist A, Cetinkaya C, Nilsson K, Pahlman S, Larsson LG. Interferon-gamma cooperates with retinoic acid and phorbol ester to induce differentiation and growth inhibition of human neuroblastoma cells. Int J Cancer. 2001;94(1):97–108. doi: 10.1002/ijc.1443. [DOI] [PubMed] [Google Scholar]
- 30.Wong G, Goldshmit Y, Turnley AM. Interferon-gamma but not TNF alpha promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Exp Neurol. 2004;187(1):171–7. doi: 10.1016/j.expneurol.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Hu S, Rotschafer JH, Lokensgard JR, Cheeran MC. Activated CD8+ T lymphocytes inhibit neural stem/progenitor cell proliferation: role of interferon-gamma. PLoS One. 2014;9(8):e105219. doi: 10.1371/journal.pone.0105219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter J, Dihne M. Species-dependent differences of embryonic stem cell-derived neural stem cells after Interferon gamma treatment. Front Cell Neurosci. 2012;6:52. doi: 10.3389/fncel.2012.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23(2):96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 34.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–86. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95(13):7556–61. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimco G, Knight RA, Latchman DS, Stephanou A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle. 2010;9(23):4638–49. doi: 10.4161/cc.9.23.13955. [DOI] [PubMed] [Google Scholar]
- 37.van Boxel-Dezaire AH, Stark GR. Cell type-specific signaling in response to interferon-gamma. Curr Top Microbiol Immunol. 2007;316:119–54. doi: 10.1007/978-3-540-71329-6_7. [DOI] [PubMed] [Google Scholar]
- 38.Mizuno T, Zhang G, Takeuchi H, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J. 2008;22(6):1797–806. doi: 10.1096/fj.07-099499. [DOI] [PubMed] [Google Scholar]
- 39.Bate C, Kempster S, Last V, Williams A. Interferon-gamma increases neuronal death in response to amyloid-beta1-42. J Neuroinflammation. 2006;3:7. doi: 10.1186/1742-2094-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. 2003;77(13):7635–44. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons JD, White LJ, Morrison TE, et al. Venezuelan equine encephalitis virus disrupts STAT1 signaling by distinct mechanisms independent of host shutoff. J Virol. 2009;83(20):10571–81. doi: 10.1128/JVI.01041-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gil MP, Bohn E, O’Guin AK, et al. Biologic consequences of Stat1-independent IFN signaling. Proc Natl Acad Sci U S A. 2001;98(12):6680–5. doi: 10.1073/pnas.111163898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hashioka S, Klegeris A, Qing H, McGeer PL. STAT3 inhibitors attenuate interferon-gamma-induced neurotoxicity and inflammatory molecule production by human astrocytes. Neurobiol Dis. 2011;41(2):299–307. doi: 10.1016/j.nbd.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 44.Alsayed Y, Uddin S, Ahmad S, et al. IFN-gamma activates the C3G/Rap1 signaling pathway. J Immunol. 2000;164(4):1800–6. doi: 10.4049/jimmunol.164.4.1800. [DOI] [PubMed] [Google Scholar]
- 45.Schmitt JM, Stork PJ. Cyclic AMP-mediated inhibition of cell growth requires the small G protein Rap1. Mol Cell Biol. 2001;21(11):3671–83. doi: 10.1128/MCB.21.11.3671-3683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang YJ, Holtzman MJ, Chen CC. Differential role of Janus family kinases (JAKs) in interferon-gamma-induced lung epithelial ICAM-1 expression: involving protein interactions between JAKs, phospholipase Cgamma, c-Src, and STAT1. Mol Pharmacol. 2004;65(3):589–98. doi: 10.1124/mol.65.3.589. [DOI] [PubMed] [Google Scholar]
- 47.Fan G, Martinowich K, Chin MH, et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 2005;132(15):3345–56. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- 48.He F, Ge W, Martinowich K, et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8(5):616–25. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turbic A, Leong SY, Turnley AM. Chemokines and inflammatory mediators interact to regulate adult murine neural precursor cell proliferation, survival and differentiation. PLoS One. 2011;6(9):e25406. doi: 10.1371/journal.pone.0025406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lum M, Croze E, Wagner C, McLenachan S, Mitrovic B, Turnley AM. Inhibition of neurosphere proliferation by IFNgamma but not IFNbeta is coupled to neuronal differentiation. J Neuroimmunol. 2009;206(1–2):32–8. doi: 10.1016/j.jneuroim.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 51.Pereira L, Medina R, Baena M, Planas AM, Pozas E. IFN gamma regulates proliferation and neuronal differentiation by STAT1 in adult SVZ niche. Front Cell Neurosci. 2015;9:270. doi: 10.3389/fncel.2015.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ben-Hur T, Ben-Menachem O, Furer V, Einstein O, Mizrachi-Kol R, Grigoriadis N. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Mol Cell Neurosci. 2003;24(3):623–31. doi: 10.1016/s1044-7431(03)00218-5. [DOI] [PubMed] [Google Scholar]
- 53.Li L, Walker TL, Zhang Y, Mackay EW, Bartlett PF. Endogenous interferon gamma directly regulates neural precursors in the non-inflammatory brain. J Neurosci. 2010;30(27):9038–50. doi: 10.1523/JNEUROSCI.5691-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: a matter of balance. JAKSTAT. 2012;1(2):65–72. doi: 10.4161/jkst.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komada M. Sonic hedgehog signaling coordinates the proliferation and differentiation of neural stem/progenitor cells by regulating cell cycle kinetics during development of the neocortex. Congenit Anom (Kyoto) 2012;52(2):72–7. doi: 10.1111/j.1741-4520.2012.00368.x. [DOI] [PubMed] [Google Scholar]
- 56.Sun L, Tian Z, Wang J. A direct cross-talk between interferon-gamma and sonic hedgehog signaling that leads to the proliferation of neuronal precursor cells. Brain Behav Immun. 2010;24(2):220–8. doi: 10.1016/j.bbi.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter J, Hartung HP, Dihne M. Interferon gamma and sonic hedgehog signaling are required to dysregulate murine neural stem/precursor cells. PLoS One. 2012;7(8):e43338. doi: 10.1371/journal.pone.0043338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Todoric J, Strobl B, Jais A, et al. Cross-talk between interferon-gamma and hedgehog signaling regulates adipogenesis. Diabetes. 2011;60(6):1668–76. doi: 10.2337/db10-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahn J, Lee J, Kim S. Interferon-gamma inhibits the neuronal differentiation of neural progenitor cells by inhibiting the expression of Neurogenin2 via the JAK/STAT1 pathway. Biochem Biophys Res Commun. 2015;466(1):52–9. doi: 10.1016/j.bbrc.2015.08.104. [DOI] [PubMed] [Google Scholar]
- 60.Bonni A, Sun Y, Nadal-Vicens M, et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278(5337):477–83. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 61.Song JH, Wang CX, Song DK, Wang P, Shuaib A, Hao C. Interferon gamma induces neurite outgrowth by up-regulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation of ERK1/2 pathway. J Biol Chem. 2005;280(13):12896–901. doi: 10.1074/jbc.M412139200. [DOI] [PubMed] [Google Scholar]
- 62.Kim SJ, Son TG, Kim K, Park HR, Mattson MP, Lee J. Interferon-gamma promotes differentiation of neural progenitor cells via the JNK pathway. Neurochem Res. 2007;32(8):1399–406. doi: 10.1007/s11064-007-9323-z. [DOI] [PubMed] [Google Scholar]
- 63.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2(1):a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(suppl 1):S210–2. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- 65.Walker L, Sills GJ. Inflammation and epilepsy: the foundations for a new therapeutic approach in epilepsy? Epilepsy Curr. 2012;12(1):8–12. doi: 10.5698/1535-7511-12.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014;76(pt A):27–50. doi: 10.1016/j.neuropharm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 67.Roth AD, Ramirez G, Alarcon R, Von Bernhardi R. Oligodendrocytes damage in Alzheimer’s disease: beta amyloid toxicity and inflammation. Biol Res. 2005;38(4):381–7. doi: 10.4067/s0716-97602005000400011. [DOI] [PubMed] [Google Scholar]
- 68.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1(2):120–9. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 69.Kurz A, Perneczky R. Novel insights for the treatment of Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(2):373–9. doi: 10.1016/j.pnpbp.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 70.Rojo LE, Fernandez JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39(1):1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 71.Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol. 2014;50(2):534–44. doi: 10.1007/s12035-014-8657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Griffin WS, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86(19):7611–5. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hull M, Berger M, Volk B, Bauer J. Occurrence of interleukin-6 in cortical plaques of Alzheimer’s disease patients may precede transformation of diffuse into neuritic plaques. Ann N Y Acad Sci. 1996;777:205–12. doi: 10.1111/j.1749-6632.1996.tb34420.x. [DOI] [PubMed] [Google Scholar]
- 74.van der Wal EA, Gomez-Pinilla F, Cotman CW. Transforming growth factor-beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport. 1993;4(1):69–72. doi: 10.1097/00001756-199301000-00018. [DOI] [PubMed] [Google Scholar]
- 75.Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol Dis. 2000;7(6 pt B):682–9. doi: 10.1006/nbdi.2000.0321. [DOI] [PubMed] [Google Scholar]
- 76.Hong HS, Hwang EM, Sim HJ, et al. Interferon gamma stimulates beta-secretase expression and sAPPbeta production in astrocytes. Biochem Biophys Res Commun. 2003;307(4):922–7. doi: 10.1016/s0006-291x(03)01270-1. [DOI] [PubMed] [Google Scholar]
- 77.Huberman M, Shalit F, Roth-Deri I, et al. Correlation of cytokine secretion by mononuclear cells of Alzheimer patients and their disease stage. J Neuroimmunol. 1994;52(2):147–52. doi: 10.1016/0165-5728(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 78.Reale M, Iarlori C, Feliciani C, Gambi D. Peripheral chemokine receptors, their ligands, cytokines and Alzheimer’s disease. J Alzheimers Dis. 2008;14(2):147–59. doi: 10.3233/jad-2008-14203. [DOI] [PubMed] [Google Scholar]
- 79.Asselineau D, Benlhassan K, Arosio B, et al. Interleukin-10 production in response to amyloid-beta differs between slow and fast decliners in patients with Alzheimer’s disease. J Alzheimers Dis. 2015;46(4):837–42. doi: 10.3233/JAD-142832. [DOI] [PubMed] [Google Scholar]
- 80.Yamamoto M, Kiyota T, Horiba M, et al. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170(2):680–92. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chakrabarty P, Ceballos-Diaz C, Beccard A, et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184(9):5333–43. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li A, Ceballos-Diaz C, DiNunno N, et al. IFN-gamma promotes tau phosphorylation without affecting mature tangles. FASEB J. 2015;29(10):4384–98. doi: 10.1096/fj.15-275834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101(1):343–7. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boekhoorn K, Joels M, Lucassen PJ. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis. 2006;24(1):1–14. doi: 10.1016/j.nbd.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 85.Zheng M, Liu J, Ruan Z, et al. Intrahippocampal injection of Abeta1-42 inhibits neurogenesis and down-regulates IFN-gamma and NF-kappaB expression in hippocampus of adult mouse brain. Amyloid. 2013;20(1):13–20. doi: 10.3109/13506129.2012.755122. [DOI] [PubMed] [Google Scholar]
- 86.Mastrangelo MA, Sudol KL, Narrow WC, Bowers WJ. Interferon-{gamma} differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am J Pathol. 2009;175(5):2076–88. doi: 10.2353/ajpath.2009.090059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baron R, Nemirovsky A, Harpaz I, Cohen H, Owens T, Monsonego A. IFN-gamma enhances neurogenesis in wild-type mice and in a mouse model of Alzheimer’s disease. FASEB J. 2008;22(8):2843–52. doi: 10.1096/fj.08-105866. [DOI] [PubMed] [Google Scholar]
- 88.Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354(9):942–55. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 89.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 90.Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(pt 5):1175–89. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–58. doi: 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- 92.Croxford AL, Kurschus FC, Waisman A. Mouse models for multiple sclerosis: historical facts and future implications. Biochim Biophys Acta. 2011;1812(2):177–83. doi: 10.1016/j.bbadis.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 93.Steinman L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron. 1999;24(3):511–4. doi: 10.1016/s0896-6273(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 94.Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129(pt 8):1953–71. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- 95.Matute C, Perez-Cerda F. Multiple sclerosis: novel perspectives on newly forming lesions. Trends Neurosci. 2005;28(4):173–5. doi: 10.1016/j.tins.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 96.Mc Guire C, Volckaert T, Wolke U, et al. Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J Immunol. 2010;185(12):7646–53. doi: 10.4049/jimmunol.1000930. [DOI] [PubMed] [Google Scholar]
- 97.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 98.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16(1):34–8. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 99.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37(7):1097–102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 100.Dettke M, Scheidt P, Prange H, Kirchner H. Correlation between interferon production and clinical disease activity in patients with multiple sclerosis. J Clin Immunol. 1997;17(4):293–300. doi: 10.1023/a:1027374615106. [DOI] [PubMed] [Google Scholar]
- 101.Pravica V, Popadic D, Savic E, Markovic M, Drulovic J, Mostarica-Stojkovic M. Single nucleotide polymorphisms in multiple sclerosis: disease susceptibility and treatment response biomarkers. Immunol Res. 2012;52(1–2):42–52. doi: 10.1007/s12026-012-8273-y. [DOI] [PubMed] [Google Scholar]
- 102.Ahlgren C, Oden A, Lycke J. High nationwide prevalence of multiple sclerosis in Sweden. Mult Scler. 2011;17(8):901–8. doi: 10.1177/1352458511403794. [DOI] [PubMed] [Google Scholar]
- 103.Kantarci OH, Hebrink DD, Schaefer-Klein J, et al. Interferon gamma allelic variants: sex-biased multiple sclerosis susceptibility and gene expression. Arch Neurol. 2008;65(3):349–57. doi: 10.1001/archneurol.2007.66. [DOI] [PubMed] [Google Scholar]
- 104.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9(6):393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129(pt 5):1306–18. doi: 10.1093/brain/awl044. [DOI] [PubMed] [Google Scholar]
- 106.Lin W, Bailey SL, Ho H, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117(2):448–56. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin W, Lin Y, Li J, et al. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci. 2013;33(14):5980–91. doi: 10.1523/JNEUROSCI.1636-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carpentier PA, Palmer TD. Immune influence on adult neural stem cell regulation and function. Neuron. 2009;64(1):79–92. doi: 10.1016/j.neuron.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Foote AK, Blakemore WF. Inflammation stimulates remyelination in areas of chronic demyelination. Brain. 2005;128(pt 3):528–39. doi: 10.1093/brain/awh417. [DOI] [PubMed] [Google Scholar]
- 110.Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131(pt 7):1749–58. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- 111.Picard-Riera N, Decker L, Delarasse C, et al. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci U S A. 2002;99(20):13211–6. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nait-Oumesmar B, Picard-Riera N, Kerninon C, et al. Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci U S A. 2007;104(11):4694–9. doi: 10.1073/pnas.0606835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tekki-Kessaris N, Woodruff R, Hall AC, et al. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development. 2001;128(13):2545–54. doi: 10.1242/dev.128.13.2545. [DOI] [PubMed] [Google Scholar]
- 114.Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6(1):21–7. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 115.Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437(7060):894–7. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- 116.Wang Y, Imitola J, Rasmussen S, O’Connor KC, Khoury SJ. Paradoxical dysregulation of the neural stem cell pathway sonic hedgehog-Gli1 in autoimmune encephalomyelitis and multiple sclerosis. Ann Neurol. 2008;64(4):417–27. doi: 10.1002/ana.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jacob J, Briscoe J. Gli proteins and the control of spinal-cord patterning. EMBO Rep. 2003;4(8):761–5. doi: 10.1038/sj.embor.embor896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mana P, Linares D, Fordham S, Staykova M, Willenborg D. Deleterious role of IFNgamma in a toxic model of central nervous system demyelination. Am J Pathol. 2006;168(5):1464–73. doi: 10.2353/ajpath.2006.050799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Harris VK, Yan QJ, Vyshkina T, Sahabi S, Liu X, Sadiq SA. Clinical and pathological effects of intrathecal injection of mesenchymal stem cell-derived neural progenitors in an experimental model of multiple sclerosis. J Neurol Sci. 2012;313(1–2):167–77. doi: 10.1016/j.jns.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 120.Einstein O, Fainstein N, Vaknin I, et al. Neural precursors attenuate autoimmune encephalomyelitis by peripheral immunosuppression. Ann Neurol. 2007;61(3):209–18. doi: 10.1002/ana.21033. [DOI] [PubMed] [Google Scholar]
- 121.Einstein O, Friedman-Levi Y, Grigoriadis N, Ben-Hur T. Transplanted neural precursors enhance host brain-derived myelin regeneration. J Neurosci. 2009;29(50):15694–702. doi: 10.1523/JNEUROSCI.3364-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ludlow M, Kortekaas J, Herden C, et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016;131(2):159–84. doi: 10.1007/s00401-015-1511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Das S, Basu A. Viral infection and neural stem/progenitor cell’s fate: implications in brain development and neurological disorders. Neurochem Int. 2011;59(3):357–66. doi: 10.1016/j.neuint.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 124.Ariff IM, Thounaojam MC, Das S, Basu A. Japanese encephalitis virus infection alters both neuronal and astrocytic differentiation of neural stem/progenitor cells. J Neuroimmune Pharmacol. 2013;8(3):664–76. doi: 10.1007/s11481-013-9455-7. [DOI] [PubMed] [Google Scholar]
- 125.Mutnal MB, Cheeran MC, Hu S, Lokensgard JR. Murine cytomegalovirus infection of neural stem cells alters neurogenesis in the developing brain. PLoS One. 2011;6(1):e16211. doi: 10.1371/journal.pone.0016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tang H, Hammack C, Ogden SC, et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. 2016;18(5):587–90. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kokaia Z, Martino G, Schwartz M, Lindvall O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15(8):1078–87. doi: 10.1038/nn.3163. [DOI] [PubMed] [Google Scholar]
- 128.Patterson CE, Lawrence DM, Echols LA, Rall GF. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J Virol. 2002;76(9):4497–506. doi: 10.1128/JVI.76.9.4497-4506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rodriguez M, Zoecklein LJ, Howe CL, et al. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler’s murine encephalomyelitis virus infection. J Virol. 2003;77(22):12252–65. doi: 10.1128/JVI.77.22.12252-12265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smith PM, Wolcott RM, Chervenak R, Jennings SR. Control of acute cutaneous herpes simplex virus infection: T cell-mediated viral clearance is dependent upon interferon-gamma (IFN-gamma) Virology. 1994;202(1):76–88. doi: 10.1006/viro.1994.1324. [DOI] [PubMed] [Google Scholar]
- 131.Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2 – United States, 1999–2010. J Infect Dis. 2014;209(3):325–33. doi: 10.1093/infdis/jit458. [DOI] [PubMed] [Google Scholar]
- 132.Rozenberg F. Genetic susceptibility to herpes simplex encephalitis. Pathol Biol (Paris) 2013;61(1):21–7. doi: 10.1016/j.patbio.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 133.Braun E, Zimmerman T, Hur TB, et al. Neurotropism of herpes simplex virus type 1 in brain organ cultures. J Gen Virol. 2006;87(pt 10):2827–37. doi: 10.1099/vir.0.81850-0. [DOI] [PubMed] [Google Scholar]
- 134.Armien AG, Hu S, Little MR, et al. Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol. 2010;20(4):738–50. doi: 10.1111/j.1750-3639.2009.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lundberg P, Ramakrishna C, Brown J, et al. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol. 2008;82(14):7078–88. doi: 10.1128/JVI.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rotschafer JH, Hu S, Little M, Erickson M, Low WC, Cheeran MC. Modulation of neural stem/progenitor cell proliferation during experimental Herpes Simplex encephalitis is mediated by differential FGF-2 expression in the adult brain. Neurobiol Dis. 2013;58:144–55. doi: 10.1016/j.nbd.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988–2004. Clin Infect Dis. 2010;50(11):1439–47. doi: 10.1086/652438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17(5):355–63. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 139.Nakamura H, Liao H, Minami K, et al. Human cytomegalovirus induces apoptosis in neural stem/progenitor cells derived from induced pluripotent stem cells by generating mitochondrial dysfunction and endoplasmic reticulum stress. Herpesviridae. 2013;4(1):2. doi: 10.1186/2042-4280-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Soderberg-Naucler C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol. 2006;80(18):8929–39. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kosugi I, Kawasaki H, Arai Y, Tsutsui Y. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am J Pathol. 2002;161(3):919–28. doi: 10.1016/S0002-9440(10)64252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bantug GR, Cekinovic D, Bradford R, Koontz T, Jonjic S, Britt WJ. CD8+ T lymphocytes control murine cytomegalovirus replication in the central nervous system of newborn animals. J Immunol. 2008;181(3):2111–23. doi: 10.4049/jimmunol.181.3.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cheeran MC, Gekker G, Hu S, Min X, Cox D, Lokensgard JR. Intracerebral infection with murine cytomegalovirus induces CXCL10 and is restricted by adoptive transfer of splenocytes. J Neurovirol. 2004;10(3):152–62. doi: 10.1080/13550280490441130. [DOI] [PubMed] [Google Scholar]
- 144.Johansson S, Price J, Modo M. Effect of inflammatory cytokines on major histocompatibility complex expression and differentiation of human neural stem/progenitor cells. Stem Cells. 2008;26(9):2444–54. doi: 10.1634/stemcells.2008-0116. [DOI] [PubMed] [Google Scholar]
- 145.Cheeran MC, Jiang Z, Hu S, Ni HT, Palmquist JM, Lokensgard JR. Cytomegalovirus infection and interferon-gamma modulate major histocompatibility complex class I expression on neural stem cells. J Neurovirol. 2008;14(5):437–47. doi: 10.1080/13550280802356845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290(5499):2155–9. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.de Swart RL, Ludlow M, de Witte L, et al. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 2007;3(11):e178. doi: 10.1371/journal.ppat.0030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Oglesbee M, Niewiesk S. Measles virus neurovirulence and host immunity. Future Virol. 2011;6(1):85–99. doi: 10.2217/fvl.10.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Buchanan R, Bonthius DJ. Measles virus and associated central nervous system sequelae. Semin Pediatr Neurol. 2012;19(3):107–14. doi: 10.1016/j.spen.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 150.Rall GF, Manchester M, Daniels LR, Callahan EM, Belman AR, Oldstone MB. A transgenic mouse model for measles virus infection of the brain. Proc Natl Acad Sci U S A. 1997;94(9):4659–63. doi: 10.1073/pnas.94.9.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fantetti KN, Gray EL, Ganesan P, Kulkarni A, O’Donnell LA. Interferon gamma protects neonatal neural stem/progenitor cells during measles virus infection of the brain. J Neuroinflammation. 2016;13(1):107. doi: 10.1186/s12974-016-0571-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4(7):553–64. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 153.Lawrence DM, Vaughn MM, Belman AR, Cole JS, Rall GF. Immune response-mediated protection of adult but not neonatal mice from neuron-restricted measles virus infection and central nervous system disease. J Virol. 1999;73(3):1795–801. doi: 10.1128/jvi.73.3.1795-1801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]