

Coherent neutron scattering determines the forms, time scales, and amplitudes of protein functional collective modes.
Keywords: Protein dynamics, coherent neutron scattering, molecular dynamics simulation, normal mode analysis, collective motion
Abstract
Protein function often depends on global, collective internal motions. However, the simultaneous quantitative experimental determination of the forms, amplitudes, and time scales of these motions has remained elusive. We demonstrate that a complete description of these large-scale dynamic modes can be obtained using coherent neutron-scattering experiments on perdeuterated samples. With this approach, a microscopic relationship between the structure, dynamics, and function in a protein, cytochrome P450cam, is established. The approach developed here should be of general applicability to protein systems.
INTRODUCTION
Large-amplitude, collective atomic motions in proteins play a crucial role in many functional processes, including the entry of substrates into catalytic sites (1), allosteric conformational change (2), and enzymatic reactions (3). These motions have been extensively investigated using molecular dynamics (MD) simulations (4, 5). However, the corresponding quantitative experimental characterization of their amplitudes, forms, and time dependences remains challenging.
Complete, atomic-detail information on collective internal protein motions requires the simultaneous direct determination of time and space correlations in the interatomic displacements. Generally speaking, diffraction techniques provide no information on time correlations, whereas techniques deriving motional spectra lack the spatial component (6). However, in principle, both can be derived from inelastic coherent neutron scattering (7). Although inelastic coherent neutron scattering has been widely applied to characterize collective atomic motions in soft matter systems, including small-molecule liquids (8), polymers (9), and glasses (10), its application to protein dynamics has been rare (7, 11, 12).
Recently, neutron spin echo (NSE) spectroscopy has been used to measure inelastic coherent neutron-scattering signals from protein motions (11, 12). Although this technique has been shown to successfully detect collective internal protein motion in aqueous solution, the global rotation and translation of the macromolecule dominate the neutron signal measured and render the quantitative characterization of the protein internal dynamics difficult (11, 13). To overcome this problem, we performed high-flux backscattering experiments on perdeuterated protein powders, where the global motion of the protein molecules is shown to contribute negligibly to the neutron signals measured. The experiments reveal that in-phase collective dynamic modes, that is, protein residues that move synchronously in time with similar amplitudes, extend through several nanometers across the protein molecule on a time scale of ~100 ps, and the findings are consistent with corresponding MD simulations. Moreover, it is found that the wave vector dependence of the fluctuation rate of interatomic motion measured experimentally can be used to derive the most probable forms of the large-scale protein motions at atomic resolution while also furnishing the explicit amplitudes of the motions sampled on the resolution time of the instrument. Hence, coherent neutron-scattering signals can be used to characterize large-scale functional modes in a protein.
RESULTS
Neutron scattering
The model protein studied is cytochrome P450cam (CYP101). P450s are an important enzyme family that catalyzes a variety of biochemical reactions involved in carcinogenesis, drug metabolism, lipid and steroid biosynthesis, and degradation of pollutants in higher organisms (14). CYP101 from Pseudomonas putida has long served as a model system for studying the dynamic characteristics of substrate binding in P450s (15). Here, neutron-scattering experiments were conducted on both camphor-bound CYP101 and its perdeuterated counterpart using the high-flux backscattering spectrometer (HFBS) at the National Institute of Standards and Technology (NIST). For simplicity, these two samples are referred to as H-CYP101 and D-CYP101. Details of the experimental conditions and MD protocols are provided in Materials and Methods. The experimentally measured quantity is the dynamic structure factor (eqs. S4 and S5), S(q,ΔE), which is the time Fourier transform of the intermediate scattering function (eqs. S1 and S2), I(q,t), and furnishes the distribution of the dynamic modes in the sample over frequency at a given scattering wave vector, q. Typical spectra of S(q,ΔE) measured at 285 K for D-CYP101 and H-CYP101 are presented in Fig. 1 (A and B). S(q,ΔE) contains incoherent and coherent components: Sinc(q,ΔE) and Scoh(q,ΔE). Sinc(q,ΔE) characterizes self-correlations in atomic motions, whereas Scoh(q,ΔE) probes mostly cross-correlations, that is, interatomic fluctuations. As we performed neutron-scattering experiments on both D-CYP101 and H-CYP101, the contributions of coherent and incoherent scattering to the neutron signals measured on these two samples can be explicitly separated; the procedures followed are presented in the Supplementary Materials.
Fig. 1. Quasi-elastic neutron spectra of CYP101.

Dynamic structure factor, S(q,ΔE), averaged over the q windows from 0.35 to 1.75 Å−1, measured on (A) D-CYP101 and (B) H-CYP101 at 285 K. The red curves are the resolution function measured on vanadium. The raw data have been smoothed by averaging five neighboring data points. Comparison of the q dependence of γ(q) with that of I(q) obtained (C) experimentally and (D) from simulation. γ(q) is the ratio of the coherent to incoherent S(q,ΔE) intensity with both integrated over the energy window from 5 to 10 μeV at a given q at 285 K. Error bars throughout the text represent 1 standard deviation (SD). arb. units, arbitrary units; Const., constant.
A system undergoing in-phase collective motions, that is, constituting units moving synchronously in time with similar amplitudes, will exhibit Scoh(q, ΔE) ∝ I(q)Sinc(q, ΔE), whereas conversely Scoh(q, ΔE) ∝ Sinc(q,ΔE) is found if the motion is completely random and uncorrelated (7, 9, 16). Here, I(q) is the overall static structure factor, characterizing the atomic structure of the system (eq. S3).
Figure 1C presents γ(q), the ratio between the integral of Scoh(q,ΔE) in the energy window from 5 to 10 μeV, which corresponds to ~100 to 200 ps, and the corresponding integral for Sinc(q,ΔE) at 285 K. As seen in Fig. 1C, the q dependence of γ(q) resembles that of I(q).
To verify the above result, all-atom MD simulation was performed on CYP101 in the same conditions as the experiment, and the same analysis as above was performed using the MD-derived scattering functions (Fig. 1D). Again, the MD-derived γ(q) resembles the q dependence of I(q). Hence, as shown by both experiment and simulation (Fig. 1, C and D), γ(q) exhibits a q dependence similar to I(q) down to 0.35 Å−1 (~2 nm). This demonstrates the existence of collective in-phase atomic motions across several nanometers in the protein on the corresponding time scale, ~100 to 200 ps. A closer examination of the experimental data (Fig. 1C) reveals that γ(q) resembles I(q) more closely in the low-q region (<0.8 Å−1) than at high q. This arises from the fact that large-scale collective modes spanning a large portion of the protein molecule dominate the low-q neutron signal, whereas local modes, which are less correlated, smear the behavior at high q (13).
Earlier inelastic coherent neutron-scattering work on glasses, fully deuterated green fluorescent protein, and polymers using time-of-flight neutron spectroscopy demonstrated the collective, in-phase nature of vibrational modes on subpicosecond time scales in these systems (7, 9, 10). The present study reveals that the relaxation processes in protein molecules on much longer time scales, that is, 100 to 200 ps, can also be collective, in-phase and can span nanometer length scales.
To quantitatively characterize the spatial and temporal features of the interatomic motion in CYP101, the decay of the coherent intermediate scattering function at instrument resolution was derived as
| (1) |
The experimental ζcoh(q,Δt) was calculated using the intensity of the elastic peak of the coherent dynamic structure factor, Scoh(q,Δt), which is derived from the elastic intensity measured on both D-CYP101 and H-CYP101 (see eq. S8 and Fig. 2, A and B) at 285 K. The simulation-derived ζcoh(q,Δt) was estimated using the coherent intermediate scattering function, Icoh(q,Δt), computed directly from the MD trajectories, and Δt is the instrument resolution ~1 ns. ζcoh(q,Δt) thus defined furnishes a quantitative measure of how much the intermediate scattering function decays at Δt and has the dimensions of a diffusion coefficient. ζcoh(q,Δt) characterizes the fluctuation rate of the interatomic motion at a given distance (~2π/q) on the experimental time scale and thus contains the characteristic spatiotemporal information on the prevalent collective dynamic modes in the protein (17). Experimentally, ζcoh(q,Δt) exhibits a prominent peak at ~0.7 Å−1 (~9 Å) (Fig. 2C). The corresponding MD-derived ζcoh(q,Δt) qualitatively resembles the experiment, with a peak at ~0.8 Å−1 and a minimum at ~1.3 Å−1 (Fig. 2D), indicating that the interatomic motion reaches maximum and minimum fluctuation rates at corresponding distances (~8 and ~5 Å) (17).
Fig. 2. Intensity of the elastic peak of the dynamic structure factor and the derived decay of the intermediate scattering function.

Experimental temperature dependence of S(q,Δt) for (A) H-CYP101 and (B) D-CYP101 at three different q values: 0.56, 1.1, and 1.6 Å−1, normalized by the values at 4 K. The data points have been averaged in every 10-K window. The decay of the coherent intermediate scattering function, ζcoh(q,Δt), derived (C) experimentally and (D) from simulation at 285 K.
When comparing ζcoh(q,Δt) (Fig. 2, C and D) with I(q) (Fig. 1, C and D), it can be seen that the maxima in ζcoh(q,Δt) occurs roughly at the minima in I(q). This observation can be explained by the theory of “de Gennes narrowing” such that the system concerned exhibits the fastest fluctuations at the least-favored configurations (17–19).
A close examination of Fig. 2 (C and D) shows that ζcoh_exp(q,Δt) differs quantitatively from ζcoh_MD(q,Δt). This is largely due to the experimental data having been collected on protein powders, which involve interprotein scattering, whereas the simulation-derived values are averages over the results calculated from each of the eight protein molecules in the simulation box. The interprotein scattering effect can be evaluated using the interprotein structure factor, S(q), defined as the ratio between the overall static scattering intensity I(q) measured on the powder sample and the form factor F(q) of a single protein molecule (Fig. 3A) (11, 18). As seen in Fig. 3B, after correction for S(q), the simulation-derived ζcoh(q,Δt) agrees quantitatively with the experimental values. This correction procedure is further supported by the MD results presented in Fig. 3C. Moreover, unlike the earlier NSE experiments performed on protein solutions, where the neutron signals measured are dominated by the global rotation and translation of the biomacromolecules (11, 13), rendering the quantitative characterization of the protein internal dynamics difficult, we used powder samples here, where the dynamic neutron signals come almost entirely from protein internal motions (see Fig. 3D).
Fig. 3. Interprotein structure factor and its correction to obtain single-protein dynamics.
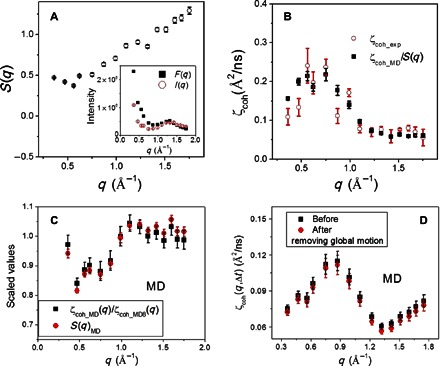
(A) Experimental interprotein structure factor, S(q) = I(q)/F(q). To obtain S(q) on an absolute scale, I(q) was multiplied by a constant to match the values of F(q) at q > 1 Å−1 (inset), because the interprotein scattering at high q is much smaller than at low q (32). (B) Comparison of ζcoh_exp(q,Δt) with ζcoh_MD(q,Δt) corrected by experimental S(q). (C) Comparison of MD-derived S(q)MD with the ratio between the single-protein ζcoh_MD(q) and the overall value, ζcoh_MD8(q). ζcoh_MD(q) is the average over the values calculated from each of the eight protein molecules in the simulation box, whereas ζcoh_MD8(q) is estimated by taking the eight protein molecules as a whole solution. S(q)MD arises from the interprotein packing in the MD box. (D) Comparison of ζcoh_MD(q,Δt) before and after removing the global motion of the protein molecule. Removal of the global motion was performed by fitting the MD trajectory of each protein molecule to its structure in the first frame.
Normal mode analysis
To determine the forms of the large-scale collective in-phase protein motions probed experimentally, we performed a normal mode analysis using a commonly used elastic network model (20), which assumes that all protein residues move synchronously along the eigenvectors of the normal modes. Because low-frequency normal modes are those most likely to be global, only the 10 lowest-frequency normal modes are considered. ζcoh(q,Δt) derived individually from the first three lowest-frequency normal modes is presented in Fig. 4A. The q dependence of ζcoh(q,Δt) differs significantly for these modes, especially in the low-q region, <1 Å−1. To determine the normal mode that matches best the experimental ζcoh(q,Δt), a dimensionless scoring function is defined as below
| (2) |
where < x2(Δt) > NM is the mean-squared atomic displacement of mode α (eq. S10), quantitatively characterizing the amplitude of the mode, and serves as a q-independent fitting parameter adjusted to minimize φα. S(q) is the experimental interprotein structure factor (Fig. 3A), and is the decay of the coherent intermediate scattering function resulting from mode α when its amplitude is normalized, that is, < x2(Δt) > NM = 1 Å2 (Fig. 4A and eq. S11). The so-obtained φα characterizes how far the dynamic form of mode α differs from that measured experimentally. The results (Fig. 4B) identify mode 1 as best matching the experiment. A schematic picture of mode 1 is presented in Fig. 5A, showing a relative rotational motion of three domains in the protein.
Fig. 4. Normal mode analysis.

(A) ζαcoh_norm(q,Δt) derived from the first three lowest-frequency normal modes (see eq. S11 and the corresponding text in the Supplementary Materials). (B) Scoring function φα for different normal modes (Eq. 2). (C) Comparison of ζcoh(q,Δt) measured experimentally and that derived from the first normal mode, the latter being corrected by the interprotein structure factor (Fig. 3A) and multiplied by the amplitude (< x2 > NM = 0.41 Å2) required to minimize φα. (D) Overlap parameter, Oα, between the eigenvector of a given normal mode and the direction of the conformational change between the open, substrate-free and the closed, substrate-bound forms of CYP101 (Eq. 4).
Fig. 5. Schematic pictures of the first normal mode.

(A) Three parts (marked as green, yellow, and pink) in the protein rotate against each other (shown by the colored arrows). The region marked red is the catalytic site. The entrance of the substrate access channel formed by the B′ helices and F/G loops is marked brown. (B) Cross-sectional view schematically illustrating the opening of the substrate access channel step by step via mode 1. The black arrow points to the entrance of the channel.
ζcoh(q,Δt) derived from mode 1 with the minimized value of φα is shown in Fig. 4C, and this explains the experimental results well. The minimization of φα is achieved when the amplitude of the normal mode < x2(Δt) > NM = 0.41 Å2 (Eq. 2). This value is only slightly smaller than that of the average mean-squared atomic displacement, 0.57 Å2, estimated experimentally from the q dependence of the intensity of the elastic peak in the incoherent dynamic structure factor, Sinc(q,Δt) (see fig. S4). The small difference is consistent with the fact that the coherent scattering is primarily sensitive to correlated interatomic motions, whereas the mean-squared atomic displacement estimated from Sinc(q,Δt) includes both correlated and uncorrelated dynamics.
The catalytic site of CYP101 is deeply buried, and the substrate enters the site via a channel (Fig. 5), the entrance of which is formed by the B′ helix and the F/G loop (Fig. 5A). Setting the amplitude of mode 1 as < x2 > NM = 0.41 Å2, the gate of the substrate access channel, approximated here as the distance between Gly189 in the F/G loop and Arg89 in the B′ helix, varies by ~0.5 Å on an experimental time scale of ~1 ns (eq. S10).
To test the above method, we also calculated the scoring function for different normal modes with respect to the MD results using Eq. 3
| (3) |
Unlike Eq. 2, Eq. 3 does not contain potential contamination from the interprotein structure factor, S(q), because the MD-derived result is just an average over the values calculated from the individual protein molecules in the MD box. φα thus obtained for different normal modes is presented in Fig. 6A. Again, mode 1 matches best the MD results, with the amplitude of the mode at < x2 > NM = 0.44 Å2 (see Fig. 6B). This amplitude leads to the fluctuation of the gate for the substrate access channel again being ~0.5 Å, in good agreement with the value of 0.4 Å calculated directly from the MD trajectory, quantitatively validating our method.
Fig. 6. Testing the method using MD-derived results.
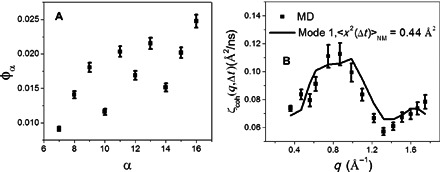
(A) Scoring function φα (Eq. 3) for different normal modes with reference to the MD-derived ζcoh(q,Δt). (B) Comparison of ζcoh(q,Δt) derived from MD with that calculated from the first normal mode with < x2 > NM = 0.44 Å2 required to minimize φα.
DISCUSSION
The opening and closing of the substrate access channel in CYP101 (Fig. 5) regulate the access of the substrate into the catalytic site (21) and are thus crucial for the protein function. To quantify the relevance of the dynamical pathway in each normal mode for the opening and closing of the channel, we calculated the overlap parameter, Oα, between the direction vector of a given normal mode, α, and that for the conformational change between the open, substrate-free and the closed, substrate-bound forms of CYP101. Oα is a dot product defined as
| (4) |
where is the displacement of atom i in normal mode α and Δri is the displacement of that atom from the open, substrate-free structure [Protein Data Bank (PDB): 3L62] to the closed, substrate-bound form (PDB: 3L63) of CYP101 (21). Oα for mode 1 is about one order of magnitude larger than that for other modes (Fig. 4D). Therefore, mode 1, identified through the coherent neutron-scattering experiment in the present work, is also the mode most likely to determine the opening and closing of the substrate access channel in CYP101. Figure 5B schematically illustrates the opening-closing process of the substrate access channel via mode 1 viewed from a cross section of the protein molecule. The present work demonstrates that a complete characterization of large-scale functional protein dynamics, that is, atomic-detail information on the forms, time scales, and amplitudes of modes, can be obtained from backscattering spectroscopic experiments that measure the inelastic coherent neutron-scattering signal from a perdeuterated protein powder.
We found that collective, in-phase dynamic modes, that is, residues moving synchronously in time with similar amplitudes, span several nanometer length scales across the P450 molecule on a ~100-ps time scale. Moreover, the q dependence of the decay of the coherent intermediate scattering function obtained experimentally and from simulation is shown to be able to determine the most probable pathway for the collective motion in the protein molecule at atomic resolution and, furthermore, to derive the explicit amplitude of the motion on the instrumentally accessible time scale. Both the experimental and simulation results presented here indicate that CYP101 uses a specific collective mode in which three domains rotate against each other to create access to the catalytic site for the substrate, a mechanism crucial for the functionality of the enzyme. The experimental method developed here for the quantitative characterization of functional modes is generally applicable to protein systems.
MATERIALS AND METHODS
Backscattering
Dynamic neutron-scattering experiments were performed on camphor-bound CYP101 (H-CYP101) and its perdeuterated counterpart (D-CYP101) using the NG2 HFBS at the NIST Center for Neutron Research. To suppress the contributions of water and of global rotational and translational motion of the protein molecule to the scattering signal, lyophilized powder samples were used (22–24). The dynamic neutron signal thus obtained arose almost entirely from protein internal motions (Fig. 3D). However, the collective motion observed in the present work may exhibit a dependence on the hydration level of the protein, and this deserves further experimental exploration. Details of the sample preparation can be found in the study of Miao et al. (25). The quasi-elastic dynamic structure factor, S(q,ΔE), was measured at 285 K. Higher temperatures than this can lead the protein to denature over the lengthy experimental time (~2 days). Typical experimental spectra are presented in Fig. 1 (A and B).
In addition to quasi-elastic scattering, the intensity of the elastic peak in the dynamic structure factor, S(q,Δt), at an instrumental resolution of HFBS ~ 0.8 μeV, which corresponds to Δt ~ 1 ns, was also measured over the q range of 0.35 to 1.75 Å−1 in the temperature window from 4 to 298 K. The temperature dependence of S(q,Δt) at three different q values, normalized by the values at 4 K, is presented in Fig. 2 (A and B, for H-CYP101 and D-CYP101, respectively).
Neutron spin echo
The overall static structure factor I(q) (Fig. 1C) was measured on fully deuterated CYP101 powder using an NSE spectrometer at the NIST Center for Neutron Research (Gaithersburg, MD). A standard annular sample can was used in which the sample was loaded into a thin aluminum pouch under helium to avoid any water intake. The wavelength of the incoming neutron beam was set to 5 Å with a Δλ/λ ~ 0.2. With this setup, measurements were made over the q range of 0.20 to 1.70 Å−1 with a q resolution of 0.05 Å−1. To obtain good statistics, each point at any given q was measured for about 5 min. Background measurements were made in an empty annular sample can and were subtracted.
Simulations
All-atom MD simulations of camphor-bound CYP101 were carried out using NAMD22 (26) with the 3L63 crystal structure [1.5 Å resolution (21)]. The simulation box contained eight proteins, obtained by replicating the unit cell (four proteins) along the Z direction, and 943 water molecules, corresponding to the experimental hydration level, 0.05 g of protein per gram of water. MD simulation at this low hydration level is often used to mimic the neutron-scattering experiments performed on dry protein powders (23, 24). Seventy-two K+ ions were added to neutralize the system. The CHARM22 force field (27) was used for the protein, and TIP3P (28) was used for water molecules. The particle mesh Ewald method (29) was applied for the electrostatics with a real space cutoff of 12 Å and a Fourier grid spacing of 1 Å. The systems were energy-minimized with 2000 steepest-descent steps. Constant pressure and temperature (NPT) equilibration using velocity-rescaling temperature (30) and Parrinello-Rahman pressure coupling (31) was carried out for 200 ns at T = 285 K and P = 1 atm with coupling time constants of τ = 0.1 ps and τ = 0.4 ps, respectively. The last 100 ns were used for the analysis, and the coordinates were written out every 1 ps. More details are presented in the Supplementary Materials of Miao et al. (25). The coherent neutron-scattering profiles were calculated using the same MD trajectories used for deriving the incoherent scattering results but by replacing the scattering length of hydrogen atoms by that of deuterium atoms to mimic the experimental conditions. All atoms in the protein were taken into account to derive both the coherent and incoherent scattering profiles. The incoherent signal derived from H-CYP101 was dominated by the signal of hydrogen atoms (>95%), whereas the coherent signal derived from D-CYP101 arose about equally from deuterium atoms and from other elements (carbon, nitrogen, and oxygen).
Supplementary Material
Acknowledgments
We thank Y. Miao for providing simulation trajectories and A. Faraone for the NSE measurement. Funding: L.H. acknowledges support from the High-Performance Computer Facility at Shanghai Jiao Tong University and NSF China (grant 11504231). The Oak Ridge National Laboratory (ORNL) group acknowledges support from the ORNL Adaptive Biosystems Imaging project funded by the Biological and Environmental Research Directorate of the Office of Science of the U.S. Department of Energy. This work used facilities supported in part by the NSF (agreement no. DM-1508249). Author contributions: L.H. conducted the experiment and simulation, designed the work, and wrote the paper. X.C. and J.C.S. contributed to the writing and the design of the work. M.T. participated in the neutron experiment. A.B. and N.J. participated in the experiment and made the samples. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1600886/DC1
Supplementary Text
fig. S1. Coherent and incoherent contribution into the neutron-scattering signal arising from the hydrogenated and deuterated protein samples.
fig. S2. Comparison of and .
fig. S3. Comparison of Scoh(q,Δt) with SD(q,Δt).
fig. S4. q2 dependence of the intensity of the elastic peak in the incoherent dynamic structure factor, Sinc(q,Δt) (eq. S9), at 285 K.
REFERENCES AND NOTES
- 1.Johs A., Harwood I. M., Parks J. M., Nauss R. E., Smith J. C., Liang L., Miller S. M., Structural characterization of intramolecular Hg2+ transfer between flexibly linked domains of mercuric ion reductase. J. Mol. Biol. 413, 639– 656 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Martin G. S., The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2, 467–475 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Banks R. D., Blake C. C. F., Evans P. R., Haser R., Rice D. W., Hardy G. W., Merrett M., Phillips A. W., Sequence, structure and activity of phosphoglycerate kinase: A possible hinge-bending enzyme. Nature 279, 773–777 (1979). [DOI] [PubMed] [Google Scholar]
- 4.Henzler-Wildman K. A., Thai V., Lei M., Ott M., Wolf-Watz M., Fenn T., Pozharski E., Wilson M. A., Petsko G. A., Karplus M., Hübner C. G., Kern D., Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838–844 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Noskov S. Y., Bernèche S., Roux B., Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 431, 830–834 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Henzler-Wildman K., Kern D., Dynamic personalities of proteins. Nature 450, 964–972 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Nickels J. D., Perticaroli S., O’Neill H., Zhang Q., Ehlers G., Sokolov A. P., Coherent neutron scattering and collective dynamics in the protein, GFP. Biophys. J. 105, 2182–2187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venkataraman G., Dasannacharya B. A., Rao K. R., Cold neutron scattering from liquid Cd4. Phys. Lett. 23, 226–227 (1966). [Google Scholar]
- 9.Buchenau U., Wischnewski A., Richter D., Frick B., Is the fast process at the glass transition mainly due to long wavelength excitations?. Phys. Rev. Lett. 77, 4035 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Sokolov A. P., Vibrations at the boson peak: Random- and coherent-phase contributions. J. Phys. Condens. Matter 11, A213 (1999). [Google Scholar]
- 11.Biehl R., Hoffman B., Monkenbusch M., Falus P., Préost S., Merkel R., Richter D., Direct observation of correlated interdomain motion in alcohol dehydrogenase. Phys. Rev. Lett. 101, 138102 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Bu Z., Biehl R., Monkenbusch M., Richter D., Callaway D. J. E., Coupled protein domain motion in Taq polymerase revealed by neutron spin-echo spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 102, 17646 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong L., Sharp M. A., Poblete S., Biehl R., Zamponi M., Szekely N., Appavou M.-S., Winkler R. G., Nauss R. E., Johs A., Parks J. M., Yi Z., Cheng X., Liang L., Ohl M., Miller S. M., Richter D., Gompper G., Smith J. C., Structure and dynamics of a compact state of a multidomain protein, the mercuric ion reductase. Biophys. J. 107, 393–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sligar S. G., Glimpsing the critical intermediate in cytochrome P450 oxidations. Science 330, 924–925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G., The catalytic pathway of cytochrome p450cam at atomic resolution. Science 287, 1615–1622 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Carpenter J. M., Pelizzari C. A., Inelastic neutron scattering from amorphous solids. I. Calculation of the scattering law for model structures. Phys. Rev. B 12, 2391 (1975). [Google Scholar]
- 17.Hong L., Smolin N., Smith J. C., de Gennes narrowing describes the relative motion of protein domains. Phys. Rev. Lett. 112, 158102 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Ackerson B. J., Pusey P. N., Tough R. J. A., Interpretation of the intermediate scattering function at short times. J. Chem. Phys. 76, 1279 (1982). [Google Scholar]
- 19.De Gennes P. G., Liquid dynamics and inelastic scattering of neutrons. Phys. Chem. Chem. Phys. 25, 825–839 (1959). [Google Scholar]
- 20.Suhre K., Sanejouand Y.-H., ElNémo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee Y.-T., Wilson R. F., Rupniewski I., Goodin D. B., P450cam visits an open conformation in the absence of substrate. Biochemistry 49, 3412–3419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rupley J. A., Careri G., Protein hydration and function. Adv. Protein Chem. 41, 37–172 (1991). [DOI] [PubMed] [Google Scholar]
- 23.Hong L., Cheng X., Glass D. C., Smith J. C., Surface hydration amplifies single-well protein atom diffusion propagating into the macromolecular core. Phys. Rev. Lett. 108, 238102 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Miao Y. L., Yi Z., Glass D. C., Hong L., Tyagi M., Baudry J., Jain N., Smith J. C., Temperature-dependent dynamical transitions of different classes of amino acid residue in a globular protein. J. Am. Chem. Soc. 134, 19576–19579 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Miao Y. L., Yi Z., Cantrell C., Glass D. C., Baudry J., Jain N., Smith J. C., Coupled flexibility change in cytochrome P450cam substrate binding determined by neutron scattering, NMR, and molecular dynamics simulation. Biophys. J. 103, 2167–2176 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K., Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L. Jr, Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M., All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 29.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G., A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 30.Bussi G., Donadio D., Parrinello M., Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Parrinello M., Rahman A., Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 32.Inoue R., Biehl R., Rosenkranz T., Fitter J., Monkenbusch M., Radulescu A., Farago B., Richter D., Large domain fluctuations on 50-ns timescale enable catalytic activity in phosphoglycerate kinase. Biophys. J. 99, 2309–2317 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaspar A. M., Busch S., Appavoua M.-S., Haeussler W., Georgiib R., Su Y., Dostera W., Using polarization analysis to separate the coherent and incoherent scattering from protein samples. Biochim. Biophys. Acta 1804, 76–82 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Lehnert U., Réat V., Weik M., Zaccaï G., Pfister C., Thermal motions in bacteriorhodopsin at different hydration levels studied by neutron scattering: Correlation with kinetics and light-induced conformational changes. Biophys. J. 75, 1945–1952 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tirion M. M., Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77, 1905–1908 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Tama F., Gadea F. X., Marques O., Sanejouand Y.-H., Building-block approach for determining low-frequency normal modes of macromolecules. Proteins 41, 1–7 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1600886/DC1
Supplementary Text
fig. S1. Coherent and incoherent contribution into the neutron-scattering signal arising from the hydrogenated and deuterated protein samples.
fig. S2. Comparison of and .
fig. S3. Comparison of Scoh(q,Δt) with SD(q,Δt).
fig. S4. q2 dependence of the intensity of the elastic peak in the incoherent dynamic structure factor, Sinc(q,Δt) (eq. S9), at 285 K.