

Abstract
We used a single-molecule localization technique called direct stochastic optical reconstruction microscopy (dSTORM) to quantify both colocalization and spatial distribution on a cellular level for two conceptually different N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer conjugates. Microscopy images were acquired of entire cells with resolutions as high as 25 nm revealing the nanoscale distribution of the fluorescently labeled therapeutic components. Drug-free macromolecular therapeutics consisting of two self-assembling nanoconjugates showed slight increase in nanoclusters on the cell surface with time. Additionally, dSTORM provided high resolution images of the nanoscale organization of the self-assembling conjugates at the interface between two cells. A conjugate designed for treating ovarian cancer showed that the model drug (Cy3) and polymer bound to Cy5 were colocalized at an early time point before the model drug was enzymatically cleaved from the polymer. Using spatial descriptive statistics it was found that the drug was randomly distributed after 24 h while the polymer bound dye remained in clusters. Four different fluorescent dyes were used and two different therapeutic systems were tested to demonstrate the versatility and possible general applicability of dSTORM for use in studying drug delivery systems.
Keywords: Drug delivery, N-(2-hydroxypropyl)methacrylamide (HPMA), dSTORM, Super resolution
1. Introduction
Nano drug delivery systems often involve complex assemblies of various materials including drugs, synthetic polymers [1,2], lipids [3], peptides [4], carbohydrates [5] or oligonucleotides [6]. The system complexity is required to overcome barriers between the point of injection and the target site [7]. Therapeutics must evade immunogenic triggers, cross cellular membranes, cross nuclear pore complex, etc. to reach their target. Often nano drug delivery systems need to undergo chemical reactions or assembly steps to properly cross a barrier or exert their therapeutic effect [8]. To properly study the mechanisms of nano drug delivery systems, new tools are needed to visualize and quantify their effects on individual cells and track their distribution at a cellular level [9]. We used a single-molecule point localization technique called direct stochastic optical reconstruction microscopy (dSTORM) to visualize and quantify cellular distribution of two conceptually different HPMA copolymer therapeutic systems to demonstrate the tool’s general applicability in drug delivery.
Discoveries related to controlling fluorescence of proteins and synthetic dyes fueled advances in optical imaging where individual fluorescent molecules can be localized [10–13]. Some single molecule localization approaches include photoactivated localization microscopy (PALM) [10], STORM [13], dSTORM [12], fluorescence PALM [14], bleaching/blinking-assisted localization microscopy (BaLM) [15], ground-state depletion microscopy followed by individual molecule return (GDSDIM) [16], and generalized single-molecule high-resolution imaging with photobleaching (gSHRiMP) [17]. In dSTORM imaging, a single fluorescent dye is used along with an appropriate imaging buffer that has thiol-containing compounds (such as mercaptoethylamine) in solution [12]. The thiol groups react with the dye upon laser irradiation to put the dye into a metastable dark state thereby controlling its fluorescence [18]. Controlling dye fluorescence enables the activation of sparse subsets of fluorophores that can be individually localized because the fluorescence signal from each molecule is spaced far enough apart that signals do not overlap. Since the fluorescent signals do not overlap the centroid of the fluorescent spot can be precisely localized by fitting the signal to a Gaussian point spread function. Each localized molecule is fit to the point spread function and a centroid of the signal is easily obtained. The uncertainty of the centroid depends upon the number of photons collected and how well the signal fits the point spread function. The process of activating and localizating sparse subsets of molecules in individual frames is repeated thousands of times after which a super-resolution image is constructed from the fitted localizations of all the molecules in every frame. A good fit and a sufficient number of photons can result in images with resolutions as high as 10 nm. Image resolution is enhanced by an order of magnitude over traditional optical microscopy methods such as confocal microscopy and total internal reflection fluorescence microscopy (TIRFM). These discoveries and advances have resulted in elucidation of cellular structures such as Tar clustering in Escherichia coli [19], MreB helical organization, an actin analog [20], and the hemispherical clathrin coat [21]. Many commercially available fluorescent probes can be used in dSTORM imaging [22]. Because dSTORM localizes individual molecules, the resulting image contains coordinate data, photon count, and single molecule resolution precision.
The coordinate data can then be used to quantitatively study drug delivery mechanisms using spatial descriptive statistical techniques such as pair-correlation analysis [23,24]. Spatial descriptive statistical techniques allow researchers to quantify the distribution of drugs, delivery vehicles, or proteins (i.e. cluster size, packing density) [23,25]. For example pair-correlation functions can be determined from an image and give rise to correlation lengths, which are related to the size of structures in the image [26]. Correlation functions can also be used to determine other important biophysical parameters in complex systems [26]. Recently, we used pair-correlation analysis to calculate CD20 protein cluster size and estimate the density of CD20 in the clusters from 2D dSTORM images [27]. Traditional optical imaging techniques (confocal, TIRFM) cannot produce the resolution needed to observe and calculate the size of nanoscale clusters, nor can they provide individual coordinate data for each molecule, thereby precluding use of statistical methods to extract valuable biophysical data.
The two N-(2-hydroxypropyl)methacrylamide (HPMA) therapeutic systems we imaged differ in the function of the HPMA copolymer and in the cellular target. The drug-free macromolecular therapeutic system relies on the biorecognition at the cell surface of oligonucleotides or coiled-coil peptides conjugated to a HPMA polymer and an anti-CD20 Fab′ fragment to physically crosslink CD20 and induce apoptosis [6,28,29]. In the drug-free system, the HPMA copolymer acts as a physical crosslinker between hybridized pairs of complementary oligonucleotides or peptides and CD20 on the cell surface rather than a carrier of a cytotoxic drug. The biorecognition of the oligonucleotides or coiled-coil peptides resulted in crosslinked CD20 on the surface of the cell, thereby initiating apoptosis. One of the nanoconjugates is an anti-CD20 Fab′ fragment from the monoclonal antibody 1F5 covalently attached to a 25 base pair oligonucleotide (Fig. 1). Multiple copies of the complementary morpholino are covalently attached to a HPMA copolymer, forming a hybrid graft copolymer (Fig. 1). This conjugate system has shown efficacy in Burkitt’s lymphoma mouse models and has recently shown promising results in vitro against patient cells of mantle cell lymphoma [30] and chronic lymphocytic leukemia (CLL) [31] patient samples. Currently, the oligonucleotides (MORF1/MORF2) are used in our lab due to their superior binding affinity and apoptosis inducting in vitro and in vivo compared to coiled-coil peptides (CCE/CCK) [6,32].
Fig. 1.
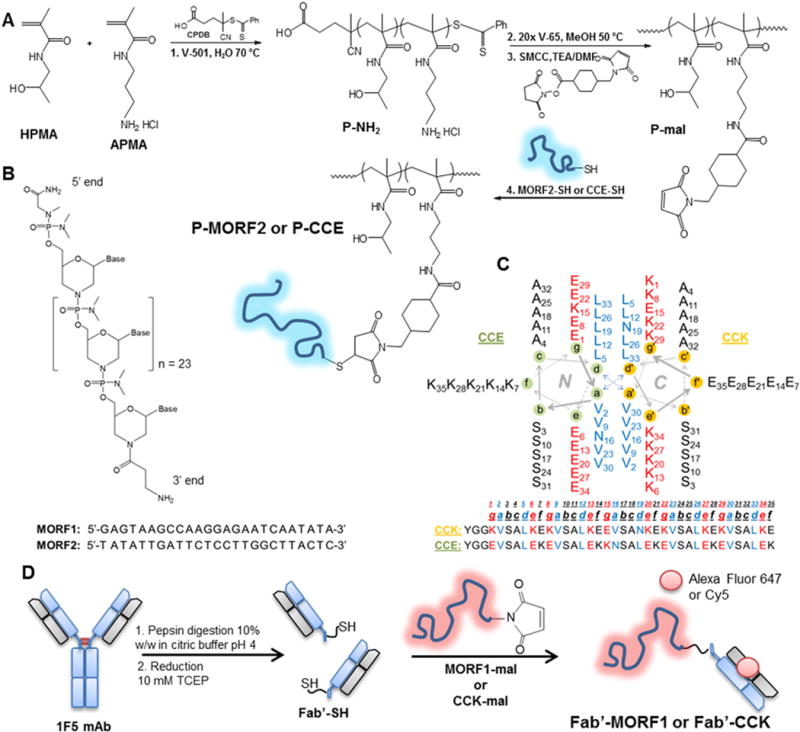
Schematic illustration of the polymer and Fab′ conjugate synthesis. A) RAFT polymerization of HPMA and APMA followed by end group modification and finally bioconjugation to either MORF2-SH or CCE-SH; B) morpholino structure and base sequence for MORF1 and MORF2; C) helical wheel diagram of the coiled-coil peptides CCE and CCK; D) 1F5 mAb digestion, reduction and conjugation to MORF1-mal or CCK-mal.
The HPMA conjugate designed to treat ovarian cancer is a diblock copolymer with a GFLG peptide sequence linking the two blocks, and the model drug (Cy3) was tethered to the polymer via an enzyme degradable peptide sequence, GFLG (Fig. 2) [33]. The GFLG peptide sequence was introduced into the copolymer backbone so that higher molecular weight polymer conjugates (~80–100 kDa) could be synthesized [34,35], otherwise only lower molecular weight polymers (~40–50 kDa), which have lower circulation times and therefore lower accumulation in solid tumors, could be used to ensure excretion by the kidneys. For the conjugate to deliver the model drug the cell must internalize it, and then the “drug” (Cy3) is cleaved enzymatically from the polymer. The polymer functions as a carrier for the cytotoxic drug or agent to prevent its release while in circulation to prevent adverse effects from the drug cytotoxicity and to increase circulation time of the polymer drug conjugate to enhance accumulation inside the tumor.
Fig. 2.
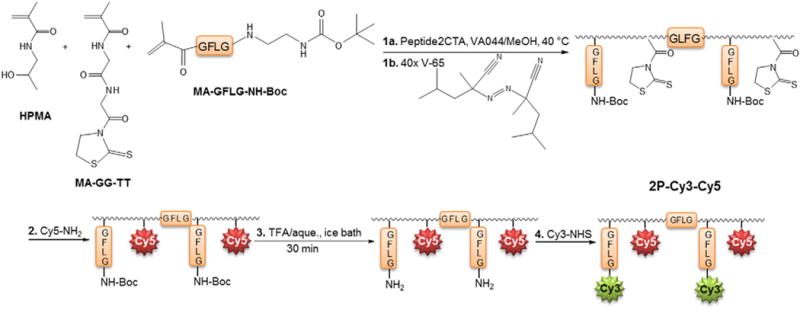
Schematic illustration of synthetic steps to construct the backbone degradable conjugate 2P-Cy3–Cy5.
The principal aims of this paper were to quantify nanoconjugate localization and to show that dSTORM is versatile in that it can be applied to functionally different therapeutic systems. We synthesized drug-free nanoconjugates representing both biorecognition strategies of hybridization and coiled-coil formation, and we also synthesized a conjugate for ovarian cancer with a cleavable diblock copolymer backbone. Components of the conjugates were labeled with one of five different fluorophores (Cy5, Alexa Fluor 647, Cy3B, Cy3, and FITC) four of which are dSTORM compatible. We used 3D dSTORM to track the labeled drug-free nanoconjugates and diblock conjugate in human Burkitt’s lymphoma cells and human ovarian cancer cells respectively. The localization of the drug-free conjugates was imaged at 2 h and 6 h while the distribution of the diblock components in ovarian cancer cells was imaged at 4 h and 24 h.
2. Materials and methods
2.1. Materials
The solvents dichloromethane (DCM), methanol, diethyl ether, acetone and dimethylformamide (DMF) were purchased from Fisher Scientific (Pittsburgh, PA). Cysteamine, glucose oxidase type seven from Aspergillus niger, catalase from bovine liver, piperidine, trifluoroacetic acid (TFA), triisopropylsilane (TIS), and diisopropylethylamine (DIPEA) were purchased from Sigma-Aldrich (St. Louis, MO). Amino acids and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) were purchased from AAPPTEC (Louisville, KY). The fluorescent probes Cy3-NHS ester, Cy5-amine, and Cy5-NHS ester were purchased from Lumiprobe (Hallandale Beach, FL). Alexa Fluor 647 NHS ester was purchased from Life Technologies (Carlsbad, CA). The Cy3B-NHS ester was purchased from GE Healthcare Life Sciences (Pittsburgh, PA). The heterobifunctional linker succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) was purchased from Highfine Biotech (Suzhou, China). The initiators 2,2′-azobis[2-(2-imidazolin-2-yl)-propane]dihydrochloride (VA-044), 2,2-azobis(2,4-dimethyl valeronitrile) (V-65), and 4,4′-azobis(4-cyanopentanoic acid) (V-501) were purchased from Wako Chemicals (Richmond, VA). The monomer N-(3-aminopropyl)methacrylamide hydrochloride (APMA) was purchased from Polysciences, Inc. (Warrington, PA). Other monomers and chain transfer agents were synthesized as described previously: HPMA [36], 3-(N-methacryloylglycylglycyl) thiazolidine-2-thione (MA-GG-TT) [37], 2-(N-methacryloylglycylphenyl alanylleucylglycine)-N′-Boc-ethylenediamine (MA-GFLG-NH-Boc) [38], methacryloylated fluorescein (MA-FITC), and RAFT chain transfer agents, 4-cyanopentanoic acid dithiobenzoate (CPDB) [39], and peptide2CTA (Nα,Nε-bis(4-cyano-4-(phenylcarbonothioylthio)pentanoylglycylphenyl alanylleucylglycyl)lysine) [35]. Tris/HCL, 1 M solution, pH 8.0 ultrapure was purchased from USB Corporation (Cleveland, OH). Glass bottom microwell dishes used for dSTORM imaging were purchased from MatTek (Ashland, MA). The reducing agent tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was purchased from Gold Biotechnology (St. Louis, MO). Oligonucleotides (MORF1-NH2 and MORF2-SH) were purchased from Gene Tools (Philomath, OR).
2.2. Coiled-coil peptide synthesis
The peptides, CCE and CCK, [40] were synthesized using solid-phase peptide synthesis with an Fmoc/tBu strategy on 2-chlorotrityl chloride resin. A spacer consisting of tyrosine and two glycines was added to the N terminus, and then the peptide was finally modified with either Cys (for conjugation to HPMA) or SMCC. The peptides were cleaved from the resin using 95% TFA, 2.5% TIS, and 2.5% H2O (EDT was added for the peptide modified with Cys). The beads were removed and the solution condensed before precipitating the peptides in cold diethyl ether. Peptide purification was performed using reverse-phase high pressure liquid chromatography (RP-HPLC) on a semi-preparative Zorbax 300SB-C18 column (Agilent, Santa Clara, CA). The peptide purity was confirmed using analytical HPLC and MALDI-TOF mass spectrometry (Voyager-DE STR Biospectrometry Workstation, Perseptive Biosystems, Framingham, MA). The amino acid sequences of the peptides are shown in Fig. 1.
2.3. HPMA copolymer conjugates synthesis and characterization
2.3.1. Synthesis and characterization of FITC-labeled graft copolymer P-CCE-FITC
RAFT polymerization was used to synthesize polyHPMA copolymers as described previously [41,42]. The molar ratios in the polymerization of HPMA, APMA, and MA-FITC were 94.5%, 5% and 0.5% respectively. The initiator V-501 was used, and the polymerization was carried out at 70 °C in a mixture of water and methanol (~3:1 by volume). The polymers were precipitated in acetone/ether. The copolymer was analyzed on an ÄKTA FPLC system (Amersham Pharmacia Biotech) equipped with miniDAWN and OptilabREX detectors with a Superose 6 HR10/30 column. The mobile phase was sodium acetate buffer and 30% acetonitrile (v/v) (pH = 6.5). Amine content was analyzed using a ninhydrin assay. The amino groups were converted to maleimide groups using SMCC, the polymer (P-NH2) and SMCC were dissolved in DMF and allowed to react for 24 h. The final maleimide content on the polymer was analyzed using a modified Ellman’s assay. The polymer weight average molecular weight (Mw), number average molecular weight (Mn), and polydispersity index (PDI) were determined using the RI signal of the elution from the Superose 6 column. The peptide CCE-SH was conjugated to P-FITC-mal in 0.1 M PBS, pH 7.2, with 10 mM TCEP for 24 h. Unreacted CCE-SH was removed using dialysis. Conjugation of the CCE-SH peptide to the polymer was confirmed using RP-HPLC. Peptide content in the purified P-CCE-FITC conjugate was determined using UV absorbance of tyrosine.
2.3.2. Synthesis of Cy3B-labeled HPMA copolymer conjugate P-MORF2-Cy3B
Conjugates for dSTORM imaging were prepared similarly as described above. Instead of copolymerizing a Cy3B monomer, we conjugated a small amount of Cy3B-NHS with P-NH2 prior to converting the amino groups to maleimide. The Cy3B-labeled P-mal conjugate was then conjugated to MORF2-SH in the presence of 10 mM TCEP in PBS, pH 7.2, for 24 h. The MORF2 content was determined using UV absorbance at 268 nm.
2.4. Synthesis of Fab′-MORF2-AF647 and Fab′-CCK-Cy5
The 1F5 monoclonal antibody was produced using a hybridoma cell line cultured in a Fibercell systems bioreactor, and purified on a protein G column [42]. The purified antibody was then digested to produce F(ab′)2. The F(ab′)2 was then labeled with Cy5-NHS by reaction with side-chain lysines. The degree of labeling was determined using UV– vis spectroscopy. The F(ab′)2-Cy5 was reduced using the reducing agent TCEP, and then conjugated to a maleimide-functionalized CCE to produce the Fab′-CCE-Cy5 conjugate (Fig. 1). Maleimide-functionalized MORF1 was synthesized by reacting SMCC with the amino group on the 3′ end of MORF1.
2.5. Synthesis and characterization of backbone degradable HPMA copolymer 2P-Cy3–Cy5
The synthesis of the diblock copolymer 2P-Cy3–Cy5 is described previously [33]. The 2P in the name of the conjugate represents that the conjugate is composed of two polymer blocks linked by the enzyme-degradable peptide sequence, GFLG. The monomers HPMA, MA-GG-TT, and MA-GFLG-NH-Boc were copolymerized in methanol at 40 °C using peptide2CTA as a RAFT agent (Fig. 2). The active CTA ends of the polymer were removed using V-65 (Figs. 2-1b) in excess as the CTA may be toxic to cells. The polymer was then dissolved in DMSO and Cy5-NH2 was attached to the polymer backbone by aminolysis of the thiazolidine-2-thione (TT) groups. Unreacted dye was removed using a PD10 column. The Boc protecting group was then removed and Cy3-NHS ester was attached to the enzyme degradable linker. The Cy3 and Cy5 content was determined using UV–vis spectroscopy.
2.6. Cell culture and treatment conditions
Human Burkitt’s B cell non-Hodgkin’s lymphoma Raji cells (European Collection of Cell Cultures, UK and ATCC) and A2780 human ovarian cancer cells (ATCC) were cultured in RPMI 1640 with 10% FBS at 37 °C in a humidified atmosphere with 5% CO2 (v/v). Media was supplemented with a mixture of antibiotics (100 units/mL penicillin, 0.1 mg/mL streptomycin). Cells with a passage number between 5 and 12 were used for experiments.
2.7. 3D dSTORM imaging and image analysis
For each test, 2.5 × 105 Raji cells were treated with 1 μM Fab′-MORF1-AF647 in 0.5 mL of culture media. The conjugate was incubated for 1 h, and then the cells were washed to remove unbound Fab′-MORF1-AF647. The cells were re-suspended in media, and the P-MORF2-Cy3B conjugate was added and incubated for either 1 h or 5 h (concentration = 2 μM). After incubation the cells were washed and fixed using 4 wt.% paraformaldehyde (PFA) in PBS pH 7.0.
The A2780 cells were seeded in a 6-well plate at a concentration of 104 cells/mL (3 mL/well) for 24 h prior to adding the 2P-Cy3–Cy5. The conjugate 2P-Cy3–Cy5 was dissolved in PBS and added to the cells at a concentration of 25 μg/mL. The cells were incubated with the conjugate for 4 h or 24 h after which the cells were washed and fixed using 4% PFA in PBS.
The cells were imaged immediately after fixing. Imaging buffer was prepared as previously described [27]. Briefly, the buffer was prepared using a 1 M Tris–HCL solution (pH 8), and had to following contents: 10 mM NaCl, 10 w/v.% glucose, 50 mM 2-mercaptoethylamine (MEA), 169 AU glucose oxidase and 1404 AU catalase. Imaging was done using a Vutara 200 microscope equipped with a Photometrics EMCCD camera (512 × 512 pixel, 16 μm pixel size for super-resolution imaging), a CCD camera (1392 × 1040 for widefield imaging) and a 60× water objective with numerical aperture 1.2. Two color channels (z-stacks) were detected sequentially at 50 frames/s. At each z step, 100 frames were collected. Laser powers were adjusted to provide sufficient fluorophore switching for localization and to minimize photobleaching. Data analysis was performed using the Vutara SRX software (ver. 5.21). Single molecules were identified by their brightness frame by frame after removing the background. Identified particles were then localized in three dimensions by fitting the raw data in a customizable region of interest (typically 16 × 16 pixels) centered on each particle in each plane with a 3D model function that was obtained from recorded bead data sets. Fit results were stored as data lists for further analysis. The image resolution capable of experimentally being achieved is 20 nm laterally (x and y) and 50 nm axially (in z).
2.8. Confocal microscopy
Cells were suspended in wells at a concentration of 1.5 × 106 cells/mL. A total of 290 μL of the cell suspension was placed in wells of a 48-well plate. Fab′-CCK-Cy5 was added to a final concentration of 1 μM, and P-(CCE)7-FITC was added to a final peptide concentration of 20 μM. An excess of 20× CCE was added based on preliminary work in the lab showing this ratio yielded maximum apoptosis induction [28]. After treatment, cells were washed with PBS to remove unbound conjugate and re-suspended in PBS for imaging. Confocal images were acquired using a Leica TCS-SP2-AOBS, equipped with 9 excitation lasers from 405 nm to 633 nm. The 488 and 633 lasers were used to detect FITC and Cy5 respectively. Cells were placed on a glass microscopy slide and covered with a glass coverslip.
3. Results and discussion
3.1. Characterization of HPMA copolymer conjugates and Fab′ conjugates
3.1.1. Drug-free macromolecular therapeutics
We synthesized HPMA copolymers by RAFT polymerization with low polydispersity (Fig. 3). The pendant amino groups in the backbone were converted to maleimide groups for conjugation to thiol-functional MORF2 or CCE. Previously, MORF2 was conjugated to HPMA copolymer by reaction of a primary amino group on the 3′ end of MORF2 with a TT moiety on the polymer backbone. However, the TT group is easily hydrolyzed and not easily accessible on the polymer, which then required TT groups in the backbone (~20) to graft three MORF2s to the polymer. Additionally, RAFT polymerization of the MA-GG-TT monomer required acidic conditions to prevent hydrolysis and the PDI of the polymer was 1.16. Instead of using aminolysis as a bioconjugation strategy we used a thiol–ene bioconjugation, which resulted in polymers with more narrow polydispersity and higher bioconjugation efficiency. The RAFT polymerization was conducted in water/methanol (80/20) with HPMA and APMA as comonomers and yielded polymers with PDI = 1.04. Thiol-functional MORF2 was conjugated to produce a P-MORF2 conjugate with a valence of three per polymer backbone. Additionally this synthetic group accommodates facile attachment of NHS ester-functional synthetic dyes to amino side groups. Hybridization of MORF1 and MORF2 morpholinos was demonstrated by UV–vis and CD measurements [6].
Fig. 3.
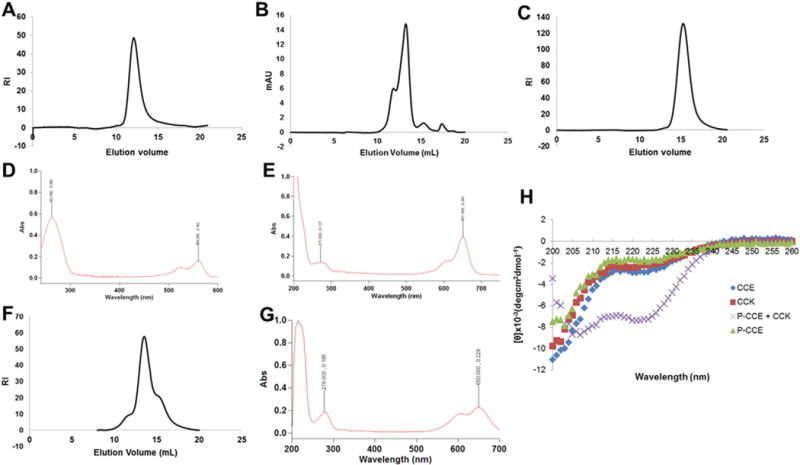
Characterization of polymer and protein conjugates. A) SEC chromatograph of P-NH2 used to construct P-MORF2-Cy3B; B) SEC chromatograph of Fab′-MORF1-AF647; C) SEC chromatograph of FITC-P-NH2 used to construct P-(CCE)7-FITC; D) UV spectra for pure P-(MORF2)3-Cy3B; E) UV spectra for pure Fab′-MORF1-AF647; F) SEC chromatograph of 2P-NH2; G) UV spectra of F(ab′)2-Cy5; H) circular dichroism measurements confirming coiled-coil formation.
3.1.2. Backbone degradable HPMA copolymer conjugates
We also synthesized a 2nd generation backbone-degradable polymer conjugate bearing a model drug, Cy3, tethered via an enzyme degradable linker. The backbone of the polymer was synthesized using Peptide2CTA—a di-functional chain transfer agent (Fig. 2) [35]. Peptide2CTA includes the peptide sequence GFLG flanked by two phenylcarbonothioylthio groups for RAFT polymerization. During polymerization two copolymer blocks grow to equal molecular weights joined by the lysosomally degradable peptide sequence GFLG. This design renders HPMA copolymers with Mw between 50 and 100 kDa biocompatible, as the backbone can be cleaved and the polymer blocks excreted by the kidneys. Degradation of the 2P-Cy3–Cy5 conjugate by papain was reported previously [33].
The polymer conjugate P-CCE and CCK formed a coiled-coil as evidenced by the characteristic minima at 222 nm and 208 nm of the ellipticity signal (Fig. 3). Peptide, oligonucleotide and fluorescent label content in each synthesized conjugate are listed in Table 1.
Table 1.
HPMA polymer conjugate and protein conjugate characteristics.
| Conjugate | Mw | PDI (Mw/Mn) | Dye content | MORF or peptide content |
|---|---|---|---|---|
| Fab′-MORF1-AF647 | ~60 kDa | N/A | ~3/Fab′-MORF1 | 1/Fab′ |
| P-(MORF2)3-Cy3B | 180 kDa | 1.04 | 23 nmol/mg | 3/macromolecule |
| P-(CCE)7-FITC | 71 kDa | 1.04 | ~2/polymer chain | 7/macromolecule |
| Fab′-CCK-Cy5 | ~55 kDa | N/A | 0.35/Fab′ | 1/Fab′ |
| 2P-Cy3–Cy5 | 86 kDa | 1.27 | Cy3: 112 nmol/mg Cy5: 143 nmol/mg |
N/A |
3.2. dSTORM imaging and analysis of CD20 organization after treatment with Fab′-MORF1 and P-(MORF2)3
We showed how dSTORM imaging could be used to evaluate release of a drug from a carrier, but dSTORM can also be used to investigate changes in membrane organization after exposure to a therapeutic. The Fab′-MORF1-AF647 binds to CD20 on the surface of B cells. The CD20 membrane protein is considered non-internalizing [43], however some internalization occurs in the Raji cell line. If P-(MORF2)3-Cy3B is added the morpholinos oligomerize and crosslink CD20 in the membrane.
Previously we showed that lipid raft distribution correlated with apoptosis induction, and that raft sizes greater than 100 nm resulted in effective apoptosis induction [27]. Here we investigated how the whole cell distribution of CD20 changed with time by imaging the cells after 2 h incubation (Fig. 4A and Movie 1) and 6 h incubation (Fig. 4B and Movie 2) with the conjugates. Little to no internalization of Fab′-MORF1-AF647 was observed at 2 h or 6 h, however more internalization may be seen at longer incubation times. It is known that receptor crosslinking can lead to internalization and localization to lysosomes in some cases such as when the anti-Her2 antibody trastuzumab is hyper-crosslinked [44]. Iron oxide nanoparticles targeted to CD20 have shown internalization in undifferentiated lymphoma B lymphocytes (MC116 cells) [45]. The pair-correlation function at 6 h (Fig. 4C), the histogram of localizations per cluster (Fig. 4E), and the cluster area (Fig. 4G) showed changes in protein distribution. It is possible that the difference would disappear if the whole population of cells were imaged and quantified. Previously, we showed that CD20 cluster size did not correlate strongly with apoptosis at 6 h [27].
Fig. 4.
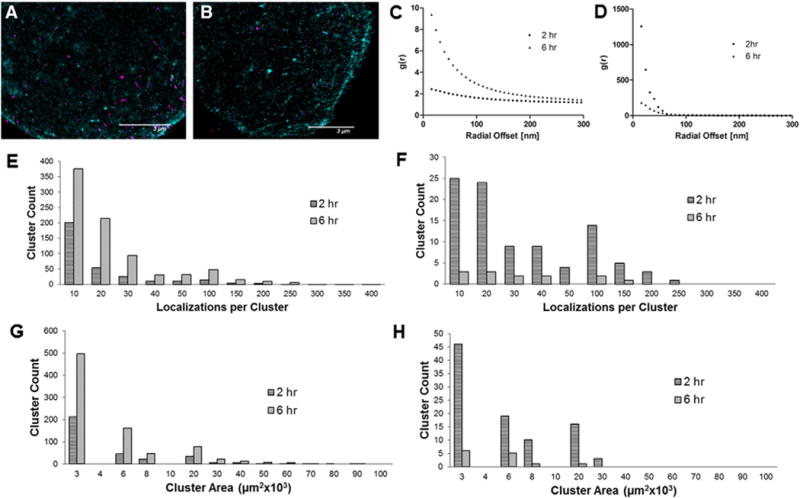
dSTORM images and quantitative analysis of Raji cells treated with Fab′-MORF1-AF647 and P-(MORF2)3-Cy3B. A) and B) dSTORM image of single Raji cell after 2 h and 6 h, respectively (magenta = P-(MORF2)3-Cy3B; teal/green = Fab′-MORF1-AF647); C) pair-correlation function (2 h); D) pair-correlation function (6 h); E) histogram of the number of localizations per cluster of Fab′-MORF1-AF647; F) histogram of the number of localizations per cluster of P-(MORF2)3-Cy3B; G) histogram of Fab′-MORF1-AF647 cluster areas; H) histogram of P-MORF2-Cy3B cluster areas. In graphs E and G there are several clusters above 400 localizations/cluster and several above 100 μm3 × 103 that are not shown in the graph.
Interestingly the total number and size of P-(MORF2)3-Cy3B clusters decreased at 6 h compared to the 2 h time point. For example, the number of clusters with ~100 localizations was 15 at 2 h but only 2 at 6 h. An explanation could be that not all MORF2 on the P-(MORF2)3-Cy3B conjugate bind MORF1 on the surface of the cell, which results in some conjugates being tightly bound (3 crosslinks) and some being more weekly bound (a single crosslink). Overtime the weekly bound conjugates more easily dissociate, which would account for the decrease in number and area of the clusters. Alternatively the decrease could be attributed to cell to cell variation.
There are two main advantages of dSTORM over diffraction limited optical imaging techniques: 1) resolution is increased by an order of magnitude [46]; and 2) single molecule coordinate information makes it possible to use biophysical mathematical tools to determine valuable information [25]. The enhanced image resolution obtained using dSTORM as compared to TIRFM is shown in Fig. S1. dSTORM images revealed nanoscale organization on the cell membrane that was previously obscured in diffraction limited optical imaging techniques. Furthermore, dSTORM provides information on single molecules that were activated and localized during imaging, which is not possible in traditional optical imaging techniques (Table S1).
3.3. Confocal and dSTORM imaging of cell crosslinking
An advantage of using the Vutara 3D microscope is that it can produce high resolution images of interactions between cells. Research groups are using super-resolution microscopy techniques to study T cell receptor (TCR)–peptide–MHC interactions at the immunological synapse [47]. An exciting application of nanomedicine is in immunoengineering where drugs, peptides, or proteins are delivered to immune cells to activate a particular kind of immune response [48]. In immunoengineering, drug delivery systems not only act on individual cells but also rely on other cells to deliver cargo or activate a particular kind of immune response. A better understanding gained through super-resolution imaging of the localization of nanomedicines in immune cells would aid in improving nanocarrier designs.
The original design of drug-free macromolecular therapeutics included the pair of oppositely charged peptides CCE/CCK that form an antiparallel coiled-coil heterodimer [28]. Recent studies have shown that the pair of oligonucleotides MORF1 and MORF2 self-assemble more quickly in vitro, have higher binding affinity, and induce apoptosis to higher levels in CD20+ B cells [6]. Early confocal microscopy studies in the lab hinted at cell–cell crosslinking so we further studied that possibility using CCE/CCK and MORF1/MORF2 using confocal imaging and dSTORM. Confocal imaging of P-(CCE)7-FITC and Fab′-CCK-Cy5 co-localization revealed large clusters at the interface between two cells (Fig. 5A–C). The Fab′-CCK-Cy5 binds to the surface of CD20+ cells, and then the P-(CCE)7-FITC conjugate with multiple copies of CCE form coiled-coils with surface CCK. It is supposed that when the density of CCE is higher than the polymers ability to accommodate more Fab′-CCK conjugates, CCE can then form coiled-coils with CCK bound to other cells. We also showed that the interaction between CCK and CCE was specific coiled-coil formation rather than peptide aggregation at the cell surface (Fig. S2).
Fig. 5.
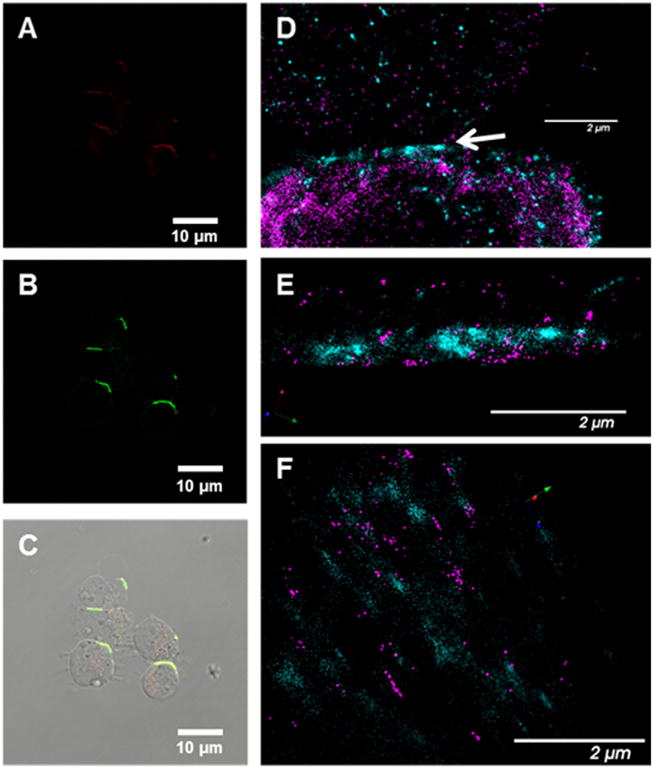
Confocal microscopy (A–C) (red = Fab′-CCK-Cy5; Green = P-(CCE)7-FITC) and dSTORM images (D–E) of crosslinked cells (magenta = P-(MORF2)3-Cy3B; Teal/green = Fab′-MORF1-AF647). Panels A, B, and C contain confocal images of Raji cells treated with Fab′-CCK-Cy5 and P-(CCE)7-FITC for 6 h (A = Cy5 channel; B = FITC channel; C = Overlay); D) dSTORM image of interface between cells where the arrow indicates point of contact between cells; E) cropped dSTORM image from D of only the interface; F) alternate view of image E.
We used dSTORM to investigate the interface between cells cross-linked with the self-assembling nanoconjugates Fab′-MORF1-AF647 and P-(MORF2)3-Cy3B (Fig. 5D–F). The contact area between the cells was approximately 200 nm where high levels of Fab′-MORF1-AF647 and P-(MORF2)3-Cy3B were detected.
3.4. dSTORM imaging and analysis of A2780 cells treated with 2P-Cy3–Cy5
High resolution images were acquired of A2780 ovarian cancer cells treated with 2P-Cy3–Cy5 for 4 h (Fig. 6A and Movie 3) and 24 h (Fig. 6B and Movie 4). Significant colocalization of Cy3 and Cy5 is observed at the 4 h time point giving a Pearson correlation coefficient of 0.58 and Manders’ overlap coefficient (MOC) of 0.59. At 24 h, the Pearson correlation coefficient and MOC decreased to 0.005 and 0.02, respectively. The pair-correlation functions for Cy3 and Cy5 show similar shape and characteristic decay lengths, which indicated that cluster sizes are similar (Fig. 6C). A histogram of cluster volumes for Cy3 (model drug) and Cy5 (polymer label) at 4 h had similar cluster distribution (Fig. 6G). At 24 h, the pair-correlation function for Cy3 approached a random distribution of molecules in the image whereas the Cy5 pair-correlation function showed the presence of clusters. Fig. 6E and F show estimates of the number of molecules per cluster at 4 h and 24 h respectively. At 24 h there was only a single cluster of Cy3 molecules with greater than 70 localizations per cluster, but at 4 h there were nearly 20 clusters of Cy3 molecules with greater than 70 localizations. The relative cluster volumes between Cy3 and Cy5 changed after 24 h incubation. As expected, the size and number of Cy3 clusters decreased relative to Cy5 clusters. In the lysosomes the enzyme cathepsin B cleaves the GFLG sequence linking Cy3 to the polymer allowing the model drug to be released from the polymer and to freely diffuse through the lysosomal membrane and into the cytoplasm. The Cy5 appeared to remain localized in clusters inside the cell as expected since the polymer cannot escape from the lysosomal compartment.
Fig. 6.
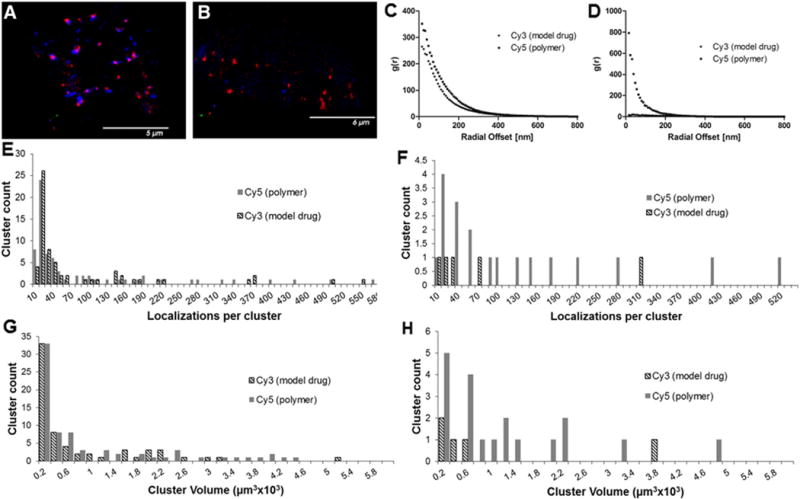
dSTORM images and quantitative analysis of A2780 cells treated with 2P-Cy3–Cy5. A) and B) dSTORM images of single A2780 cells after 4 h and 24 h, respectively (blue = Cy3 (model drug), red = Cy5 (polymer)); C) pair-correlation function (4 h); D) pair-correlation function (24 h); E) histogram of the number of localizations per cluster (4 h); F) histogram of the number of localizations per cluster (24 h); G) histogram of cluster volumes (4 h); H) histogram of cluster volumes (24 h).
In addition to determining the sizes of clusters and the spatial relationship between two different fluorescent probes, localization microscopy provides estimates of molecule density. At 4 h, the concentration of Cy3 inside the cell estimated from density calculations was approximately 13 molecules/μm3. The intracellular concentration at 24 h was 2.77 molecules/μm3 for Cy3 and 2.9 molecules/μm3 for Cy5. Since these measurements were performed for a single cell it is possible that the cells measured took up different amounts of polymer; however we should be able to compare the ratio of Cy5 and Cy3 in each cell to understand how the cell processes the individual components. The ratio of Cy5/Cy3 in the conjugate was 1.3. At 4 h, the ratio of Cy5/Cy3 was 1.5 indicating little release of Cy3. At 24 h, the ratio of Cy5/Cy3 was 1.04, which suggests that the model drug Cy3 remained preferentially inside the cell while the polymer bound to Cy5 was trafficked out of the cell.
4. Conclusions
HPMA is a versatile monomer that can be polymerized into biocompatible polymers for crosslinking proteins on the cell surface or for conjugation of cytotoxic drugs to decrease systemic adverse effects and trigger release in a specific organelle. The P-MORF2 conjugate was prepared effectively by grafting the oligonucleotides to the polymer via thiol–ene coupling rather than aminolysis of TT groups. The thiol–ene coupling strategy allowed for mild polymerization conditions, produced polymers with lower polydispersity, and achieved better bioconjugation efficiency.
Super resolution localization microscopy is a powerful new tool to quantitatively evaluate the localization and mechanism of nano drug delivery systems. dSTORM provided images revealing nanoscale organization of proteins and therapeutics bound to the surface of B cells. The increase in nanocluster size of Fab′-MORF1-AF647 and P-MORF2-Cy3B bound to the surface of the cell supports the designed functional intent of drug-free macromolecular therapeutics to crosslink cells in the membrane. However, in previous studies we have shown that apoptosis induction relied predominantly on lipid raft cluster formation rather than on nanoclusters of Fab′-MORF1-AF647 bound to CD20 [27]. dSTORM imaging and analysis of drug-free macromolecular therapeutics showed that protein distribution in the membrane changed over the course of incubation, and the distribution of the therapeutic components changed with time as well. The quantitative information inherent in dSTORM images provided useful information for understanding the mechanism of therapeutics, which could potentially aid in the design of improved nano drug delivery systems.
Evidence of cell crosslinking in confocal images of cells treated with Fab′-CCK/P-CCE and in dSTORM images of cells treated with Fab′-MORF1-AF647/P-MORF2-Cy3B indicated that cell–cell adhesion mediated through biorecognition may be a general phenomenon of drug-free macromolecular therapeutics. dSTORM provided further insight into the nanoscale organization of the therapeutics at the cell–cell contact point.
The spatial randomization after 24 h incubation with ovarian cancer cells indicated that Cy3 was released from the 2nd generation HPMA diblock copolymer, 2P-Cy3–Cy5. The Cy5 conjugated to the polymer via a stable non-degradable linker was spatially distributed as clusters inside the cell, presumably localized in lysosomes. However, at 24 h the Cy5/Cy3 ratio inside the cell decreased, which could be explained by some Cy5 polymer being trafficked outside the cell.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2016.02.005.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number F31CA186237 (to J.M.H.) and from the National Institute of General Medical Sciences grant R01GM95606 (to J.K.). Confocal microscopy work was supported by a Whitaker Foundation Summer Grant at the Centro de Investigación Príncipe Felipe (CIPF) (to J.M.H). We would like to thank Ana Armiñán (CIPF) who provided guidance in cell culture. Alberto Hernández (CIPF) helped acquire all the confocal images and provided advice on optimizing treatment conditions. María Jesus Vicent D’Ocon (CIPF) graciously lent lab space and cell culture materials for the confocal microscopy experiments. We would like to thank John Schreiner and Steve Callahan for access to the statistical analysis software. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and National Science Foundation.
Footnotes
Competing interests
J.K. and J.Y. are inventors on a pending US patent application (PCT/US2014/023784; assigned to the University of Utah) related to this work. J.K. is the Chief Scientific Advisor and J.Y. is the Scientific Advisor for Bastion Biologics. Otherwise, the authors declare no competing financial interests.
References
- 1.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 2.Kopeček J, Kopečková P. HPMA copolymers: origins, early developments, present, and future. Adv Drug Deliv Rev. 2010;62:122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 4.Rösler A, Vandermeulen GWM, Klok H-A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv Drug Deliv Rev. 2012;64:270–279. doi: 10.1016/s0169-409x(01)00222-8. (Supplement) [DOI] [PubMed] [Google Scholar]
- 5.Jain K, Kesharwani P, Gupta U, Jain NK. A review of glycosylated carriers for drug delivery. Biomaterials. 2012;33:4166–4186. doi: 10.1016/j.biomaterials.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 6.Chu T-W, Yang J, Zhang R, Sima M, Kopeček J. Cell surface self-assembly of hybrid nanoconjugates via oligonucleotide hybridization induces apoptosis. ACS Nano. 2013;8:719–730. doi: 10.1021/nn4053827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 9.Kiick KL. Polymer therapeutics. Science. 2007;317:1182–1183. doi: 10.1126/science.1145951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 11.Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 12.Linde S, Kasper R, Heilemann M, Sauer M. Photoswitching microscopy with standard fluorophores. Appl Phys B Lasers Opt. 2008;93:725–731. [Google Scholar]
- 13.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burnette DT, Sengupta P, Dai Y, Lippincott-Schwartz J, Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci. 2011;108:21081–21086. doi: 10.1073/pnas.1117430109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- 17.Simonson PD, Rothenberg E, Selvin PR. Single-molecule-based super-resolution images in the presence of multiple fluorophores. Nano Lett. 2011;11:5090–5096. doi: 10.1021/nl203560r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dempsey GT, Bates M, Kowtoniuk WE, Liu DR, Tsien RY, Zhuang X. Photoswitching mechanism of cyanine dyes. J Am Chem Soc. 2009;131:18192–18193. doi: 10.1021/ja904588g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenfield D, McEvoy AL, Shroff H, Crooks GE, Wingreen NS, Betzig E, Liphardt J. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 2009;7:e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biteen JS, Thompson MA, Tselentis NK, Bowman GR, Shapiro L, Moerner WE. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nat Methods. 2008;5:947–949. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linde Svd, Heilemann M, Sauer M. Live-cell super-resolution imaging with synthetic fluorophores. Annu Rev Phys Chem. 2012;63:519–540. doi: 10.1146/annurev-physchem-032811-112012. [DOI] [PubMed] [Google Scholar]
- 23.Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, Baird BA. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One. 2012;7:e31457. doi: 10.1371/journal.pone.0031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sengupta P, Jovanovic-Talisman T, Lippincott-Schwartz J. Quantifying spatial organization in point-localization superresolution images using pair correlation analysis. Nat Protoc. 2013;8:345–354. doi: 10.1038/nprot.2013.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honerkamp-Smith AR, Veatch SL, Keller SL. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. BBA-Biomembr. 2009;1788:53–63. doi: 10.1016/j.bbamem.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartley JM, Chu T-W, Peterson EM, Zhang R, Yang J, Harris J, Kopeček J. Super-resolution imaging and quantitative analysis of membrane protein/lipid raft clustering mediated by cell-surface self-assembly of hybrid nanoconjugates. Chembiochem. 2015;16:1725–1729. doi: 10.1002/cbic.201500278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu K, Liu JH, Johnson RN, Yang JY, Kopeček J. Drug-free macromolecular therapeutics: induction of apoptosis by coiled-coil-mediated cross-linking of antigens on the cell surface. Angew Chem Int Ed. 2010;49:1451–1455. doi: 10.1002/anie.200906232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu K, Yang J, Liu J, Kopeček J. Coiled-coil based drug-free macromolecular therapeutics: in vivo efficacy. J Control Release. 2012;157:126–131. doi: 10.1016/j.jconrel.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu T-W, Zhang R, Yang J, Chao MP, Shami PJ, Kopeček J. A two-step pretargeted nanotherapy for CD20 crosslinking may achieve superior anti-lymphoma efficacy to rituximab. Theranostics. 2015;5:834–846. doi: 10.7150/thno.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu T-W, Kosak K, Shami P, Kopeček J. Drug-free macromolecular therapeutics induce apoptosis of patient chronic lymphocytic leukemia cells. Drug Deliv Transl Res. 2014;4:389–394. doi: 10.1007/s13346-014-0209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu TW, Kopecek J. Drug-free macromolecular therapeutics — a new paradigm in polymeric nanomedicines. Biomater Sci. 2015;3:908–922. doi: 10.1039/C4BM00442F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Zhang R, Radford DC, Kopeček J. FRET-trackable biodegradable HPMA co-polymer–epirubicin conjugates for ovarian carcinoma therapy. J Control Release. 2015;218:36–44. doi: 10.1016/j.jconrel.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan H, Sima M, Yang J, Kopeček J. Synthesis of long-circulating, backbone degradable HPMA copolymer–doxorubicin conjugates and evaluation of molecular-weight-dependent antitumor efficacy. Macromol Biosci. 2013;13:155–160. doi: 10.1002/mabi.201200353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan HZ, Yang JY, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition– fragmentation chain transfer polymerization and thiol–ene coupling reaction. Biomacromolecules. 2011;12:247–252. doi: 10.1021/bm101254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopeček J, Bažilová H. Poly[N-(2-hydroxypropyl)methacrylamide]—I. Radical polymerization and copolymerization. Eur Polym J. 1973;9:7–14. [Google Scholar]
- 37.Šubr V, Ulbrich K. Synthesis and properties of new N-(2-hydroxypropyl) methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React Funct Polym. 2006;66:1525–1538. [Google Scholar]
- 38.Zhang R, Yang J, Sima M, Zhou Y, Kopeček J. Sequential combination therapy of ovarian cancer with degradable N-(2-hydroxypropyl)methacrylamide copolymer paclitaxel and gemcitabine conjugates. Proc Natl Acad Sci. 2014;111:12181–12186. doi: 10.1073/pnas.1406233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules. 2001;34:2248–2256. [Google Scholar]
- 40.Yang JY, Xu CY, Wang C, Kopeček J. Refolding hydrogels self-assembled from N-(2-hydroxypropyl)methacrylamide graft copolymers by antiparallel coiled-coil formation. Biomacromolecules. 2006;7:1187–1195. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scales CW, Vasilieva YA, Convertine AJ, Lowe AB, McCormick CL. Direct, controlled synthesis of the nonimmunogenic, hydrophilic polymer, poly(N-(2-hydroxypropyl)methacrylamide) via RAFT in aqueous media. Biomacromolecules. 2005;6:1846–1850. doi: 10.1021/bm0503017. [DOI] [PubMed] [Google Scholar]
- 42.Chu T-W, Yang J, Kopeček J. Anti-CD20 multivalent HPMA copolymer-Fab′ conjugates for the direct induction of apoptosis. Biomaterials. 2012;33:7174–7181. doi: 10.1016/j.biomaterials.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P, Thomas ED. Monoclonal antibody-1F5 (anti-CD20) serotherapy of human B-cell lymphomas. Blood. 1987;69:584–591. [PubMed] [Google Scholar]
- 44.Moody PR, Sayers EJ, Magnusson JP, Alexander C, Borri P, Watson P, Jones AT. Receptor crosslinking: a general method to trigger internalization and lysosomal targeting of therapeutic receptor:ligand complexes. Mol Ther. 2015;23:1888–1898. doi: 10.1038/mt.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang T, Kievit FM, Veiseh O, Arami H, Stephen ZR, Fang C, Liu Y, Ellenbogen RG, Zhang M. Targeted cell uptake of a noninternalizing antibody through conjugation to iron oxide nanoparticles in primary central nervous system lymphoma. World Neurosurg. 2013;80:134–141. doi: 10.1016/j.wneu.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 46.Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dustin ML, Depoil D. New insights into the T cell synapse from single molecule techniques. Nat Rev Immunol. 2011;11:672–684. doi: 10.1038/nri3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldberg Michael S. Immunoengineering: how nanotechnology can enhance cancer immunotherapy. Cell. 2015;161:201–204. doi: 10.1016/j.cell.2015.03.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.