

In addition to catalyzing N2-to-NH3 conversion under ambient conditions,[1] nitrogenase (N2-ase) enzymes facilitate the multi-electron reduction of a wide range of other substrates, such as azide, acetylene, carbon dioxide, and nitrous oxide.[2] Cyanide (CN−) is perhaps most noteworthy in this context as it is isoelectronic with N2 and can be reduced in two-, four-, or six-electron processes to furnish H2C≡NH, H3C-NH2, or CH4 and NH3, respectively.[3] To date there is scant synthetic precedent for the overall reduction of CN− to CH4 and NH3, either stoichiometrically or catalytically, by well-defined coordination complexes; beyond N2-ases, CN− conversion to CH4 and NH3 is thus far limited to extracted nitrogenase cofactors, or via reductive electrolysis by Ni electrodes.[4, 5]
We have had an ongoing interest in the study of single-site iron complexes that mediate N2-to-NH3 conversion as functional inorganic models of biological nitrogen fixation at the iron rich cofactors of N2-ases.[6] We naturally became interested in whether such single-site precursors might functionally model the enzymatic reduction of CN− to liberate CH4 and NH3. Such a process is likely to proceed through classic organometallic intermediates[7] with appreciable Fe-to-C covalency, and this possibility, in addition to the recent discovery that N2-ases can also mediate Fischer–Tropsch type CO reduction,[8] motivates functional organometallic model studies.
Herein we report an Fe(CN) complex supported by a tris(phosphine)silyl ligand ([SiPiPr3]Fe(CN), 1, Scheme 1) that releases significant amounts of NH3 and CH4 on exposure to excess acid and reductant. We also report the isolation and structural characterization of terminal Fe(CNH) and Fe-(CNH2) complexes that are plausible intermediates of the overall reduction process.
Scheme 1.
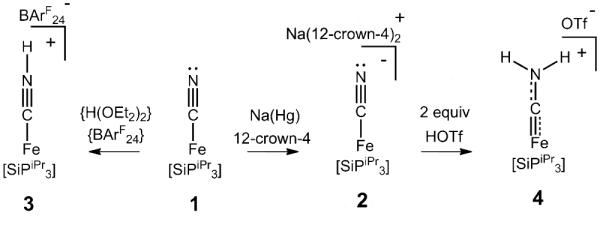
Synthesis of Fe(CNHx) complexes.
Entry into this CN− reduction system was made via the reaction of [SiPiPr3]Fe(Cl)[9a] ([SiPiPr3]=[(2-iPr2PC6H4)3Si]−) with a slight excess of either NaCN, K13CN, or KC15N to furnish the three isotopomers of [SiPiPr3]Fe(CN) (1) as pink solids. Compound 1 was found to display rich electrochemical behavior (see the Supporting Information) and reduction with Na(Hg) in the presence of 12-crown-4 cleanly afforded the one-electron reduced salt {Na(12-crown-4)2}{[SiPiPr3]Fe-(CN)} (2) as a dark brown solid. Complexes 1 and 2 are paramagnetic with solution spin states of S=1 (μeff=2.6 μB) and S=1/2 (μeff=2.0 μB), respectively, observed at room temperature. Anionic 2 additionally displays a quasi-axial EPR spectrum (g∥=2.23, g⊥≈2.05) in a 2-MeTHF glass at 20 K. The solid-state structures[10] reveal trigonal-bipyramidal Fe geometries (τ5=1.02 and 0.85 for 1 and 2, respectively; see Figure 1 and the Supporting Information) with the CN ligand positioned trans to the apical Si atom. These Fe–CN complexes are nearly isostructural to the similarly-charged Fe–N2 and Fe–CO complexes supported by the [SiPiPr3] platform.[9]
Figure 1.

X-ray diffraction crystal structures of 1, 3, 4, and 5 with thermal ellipsoids drawn at 50 % probability. Hydrogen atoms (excepting the N-H’s), the BArF24 counteranion of 3 and co-crystallized solvent molecules have been removed for clarity. Two independent molecules of 4 are found in the unit cell and only one is shown.
Fe(CN) 1 is readily protonated by the acid {H(OEt2)2}-{BArF24} (BArF24=(3,5-(CF3)2C6H3)4B−) to afford the cationic hydrogen-isocyanide complex, {[SiPiPr3]Fe(CNH)}{BArF24} (3) (Scheme 1). Compound 3 adopts an S=1 spin state in solution (μeff=2.6 μB), complicating the assignment of the acid-derived proton by NMR techniques. Fortunately, this proton is unambiguously located in the Fourier difference map of the solid-state structure of 3 (Figure 1) as a component of a C≡N-H ligand. A hydrogen bond is observed between the isocyanide proton and a co-crystallized molecule of diethyl ether (d(N…O): 2.639(3) Å, ∡](O…H-N): 173(4)°). The KBr IR spectrum of polycrystalline 3 displays very broad absorbances that span the range of 3100 to 1900 cm−1 (Supporting Information), a feature that may arise from the coupling of N–H and C≡N vibrational modes and a continuum of hydrogen bonding interactions in the polycrystalline state.[11] Overall, the bond metrics of the {Fe(CNH)} unit compare well to those of the few previously characterized examples.[7a, 12] By comparison, we have to date been unable to directly characterize related {Fe(NNH)} species supported by either tris(phosphino)silyl or tris(phosphino)borane ligands despite their presumed intermediacy in N2-to-NH3 conversion reactions.[6]
Further activation of the CN ligand was achieved via double protonation of the anionic Fe(CN) 2. Combining solutions of 2 with 2.5 equivalents of trifluoromethanesulfonic acid (HOTf) in thawing 2-MeTHF affords the cationic iron aminocarbyne complex, {[SiPiPr3]Fe(CNH2)}{OTf} (4) (Scheme 1). Aminocarbyne 4 displays averaged 3-fold symmetry in THF solution, as judged by its 1H NMR spectrum at 193 K; its EPR spectrum is quasi-axial (g∥=2.51, g⊥≈1.98, Figure 2B) with a larger observed g-anisotropy than that of Fe(CN) 2. The solid-state structure of 4 reveals a shortened Fe−C bond (1.800(4) Å) relative to those found in compounds 1–3 (1.97–1.91 Å) and is indicative of significant Fe-to-C multiple bond character. While the CNH2 hydrogen atoms were not directly located in the Fourier difference map, their presence is indicated by close contacts between the CN-derived N atom, the triflate counteranion and a co-crystallized molecule of THF (d(N…O) range from 2.68 Å–3.20 Å). Solid IR spectra of 4 (Figure 2A) also reveal strong absorptions near 3100 cm−1 that shift to 2300 cm−1 in 4-d2 (prepared from 2 and DOTf) which are assigned to N-H(D) stretching frequencies engaged in strong hydrogen bonding interactions.[11] Similar features are noted in the recently-described terminal Fe=NNH2 complex, {[SiPiPr3]Fe≡NNH2}{OTf},[6c] consistent with their similar structures. While synthetic Fe complexes have previously been shown to support terminal CNH ligands or bridging CNH2 functionalities,[13] compound 4 is the first example of a terminal Fe(CNH2) complex.[14]
Figure 2.

Pertinent spectroscopic data for Fe(CNH2) 4 and related complexes. A) Solid IR spectra of 4 (black) and 4-d2 (gray). B) X-band EPR spectra of frozen 2-MeTHF solutions of 6 (top), 4 (middle) and 2 (bottom) collected at 20 K. C) Zero field, 80 K 57Fe Mössbauer spectra of solid 3 (top), 57Fe-enriched 4 contaminated with 24 % 3' collected as a frozen 2-MeTHF solution (middle) and 57Fe-enriched 6 (bottom) collected as a frozen 2-MeTHF solution.
We next examined reductive cleavage of the coordinated cyanide unit from Fe(CN) 1 (Scheme 2). After canvassing several conditions we found that stirred Et2O solutions containing 1, 24 equiv of Cp*2Co and 24 equiv [2,5-Cl2-PhNH3][OTf] maintained at −78°C furnished an average of 0.33(4) equiv/Fe of NH3 upon warming to room temperature overnight. Isotopically-enriched [15NH4][Cl] is the sole nitrogen-containing product detected by 1H and 15n NMR spectroscopy when 15N-1 is subjected to these conditions, hence establishing that the NH3 is derived from the cyanide N-atom. The use of 50 instead of 24 equivalents of both Cp*2Co and [2,5-Cl2-PhNH3][OTf] does not increase the yield of NH3 (0.31(2) equiv/Fe). Control reactions that replace 1 with [TBA][CN] as a soluble source of CN(−) do not generate detectable quantities of NH3, implicating a likely role of the [SiPiPr3]Fe platform in the activation of cyanide towards cleavage.
Scheme 2.

Reductive protonolysis of Fe(CN) 1. Ar=2,5-Cl2-C6H3. Refer to the Supporting Information for experimental details.
To ascertain the C-containing product(s) of these reactions, the headspaces were analyzed by gas chromatography (Figure 3) and found to contain 0.41(8) equiv/Fe of CH4. Exposure of 13C-1 to these reaction conditions furnishes 13CH4 as the dominant isotopomer detected by GC-MS (Figure 3C). Finally, replacing [2,5-Cl2-PhNH3][OTf] with partially-enriched DOTf as the proton source furnishes a mixture of deuterated methane products with masses up to and including 20 (Figure 2D) as is expected for CD4. Very little CH4 is detected (0.007 equiv) when [TBA][CN] is used in place of 1 in these reactions (Figure 2A). Taken together, these analyses indicate that the [SiP3iPr]Fe fragment facilitates the proton-coupled six-electron reduction of coordinated cyanide to CH4 and NH3 in moderate overall yields.
Figure 3.

A) Representative GC-FID chromatograms of sampled headspaces in the reaction of (dashed gray) 1 or (dashed black) [TBA][CN] with 24 equiv Cp*2Co and 24 equiv [2,5-Cl2-PhNH3][OTf ] in Et O, and (black) 5 with 10 equiv KC8 and 10 equiv HOTf. B) Mass spectrum of CH4 produced from 1. C) Mass spectrum of CH4 produced from compound 13C-1. D) Mass spectrum of CHxD4−x produced from 1, 24 equiv Cp*2Co and 24 equiv partially-enriched DOTf.
We also studied the stoichiometric reactivity of the Fe(CNH) and Fe(CNH2) complexes to assess the intermediacy of these species in a cyanide cleavage process. In the absence of additional proton or electron equivalents, Fe-(CNH2) 4 is unstable in solution (τ1/2 (293 K)=24 min), decaying to a mixture of Fe-containing products that include {[SiPiPr3]Fe(CNH)}{OTf} (3') and [SiPiPr3]Fe(OTf)[6c] as readily-identified species. Independent reactions reveal that both NH3 (0.09(2) equiv/Fe) and H2 (0.24 equiv/Fe) are released on leaving solutions of 4 to stand overnight. Furthermore, Fe(CNH) 3' slowly converts to [SiPiPr3]Fe(OTf) in THF solutions (τ1/2 (298 K)=4 h), presumably with the loss of the CNH ligand. A similar ligand displacement reaction was observed for the hydrazine adduct, {[SiPiPr3]Fe(N2H3}{OTf}.[9a] This ligand exchange is likely irreversible under the relevant reaction conditions as free hydrogen isocyanide readily converts to hydrogen cyanide.[7a] Non-productive decomposition of an Fe(CNHx) complex may therefore partially account for the moderate yields (< 41 %) of CH4 and NH3 formed upon exposure of 1 to excess proton and electron equivalents.
Given the challenges arising from the thermal instability of 4 and 3 in solution, we pursued more robust Fe(CNR2) species to model the intermediacy of a terminal Fe carbyne complex in the reductive C–N bond cleavage described above. The neutral and diamagnetic dialkylaminocarbyne complex [SiPiPr3]Fe(CNMe2) (5), can be prepared in a one pot reaction via the sequential addition of Na(Hg) and MeOTf to solutions of 1 (82 % yield) (Figure 1). Fe(CNMe2) 5 is isoelectronic to the previously reported iron siloxycarbyne complex[9b] and accordingly displays a short Fe-C distance (1.710(1) Å), a long C-N distance (1.328(1) Å), and a down-field resonance (δ=279.6 ppm) in the 13C{1H} NMR spectrum assigned to the carbyne carbon. Complex 5 is readily oxidized by {Cp2Fe}{BArF24} to afford {[SiPiPr3]Fe(CNMe2)}{BArF24} (6). The salient spectroscopic features of Fe(CNMe2)+ 6 closely match those of the isoelectronic Fe(CNH2)+ 4 (Figure 2). Compounds 5 and 6 are very stable in solution and in the solid state when stored at room temperature under an inert atmosphere.
Fe(CNMe2) 5 was found to react with proton and electron equivalents to afford a mixture of CH4, Me2NH and small amounts of Me3N. Whereas Fe(CN) 1 is reduced to CH4 and NH3 with Cp*2Co and [2,5-Cl2-PhNH3][OTf] (vide supra), exposure of Et2O solutions of 5 to these reagents only furnishes small quantities of CH4 (ca. 0.01 equiv/Fe). Apparently, more reactive proton and electron sources are required for C–N bond scission in the case of 5, and 0.47 equiv of CH4 are detected when 5 is exposed to 10 equiv of KC8 and HOTf (Scheme 3). Reactions that employ 13C-5 generate 13CH4 as the predominant isotopomer, confirming the carbyne carbon as the source of this hydrocarbon. Analysis of the reaction volatiles obtained when 15N-5 is exposed to these conditions reveals two detectable 15n-containing products (see the Supporting Information). Resonances assigned to the N-H protons of [H15NMe3][Cl] and [H215NMe2][Cl] are present at δ=10.84 and 9.05 ppm, respectively, in the 1H NMR spectrum and display well-resolved coupling to adjacent 15n- and 1H nuclei. Comparison of the features present in the 1H, 13C, and 15n NMR spectra to authentic samples of these ammonium salts solidifies their assignments. [SiPiPr3]Fe(OTf) was identified as the major Fe-containing product of these reactions (see the Supporting Information).
Scheme 3.

Reductive protonolysis of Fe(CNMe2) 5. Refer to the Supporting Information for experimental details.
The organic products of these reactions lend some insight into possible pathways for Fe-mediated cyanide reduction. Upon exposure of Fe(CNMe2) 5 to proton and electron equivalents, dimethylamine is found to be the dominant (90 %) N-containing product. Its formation is consistent with a mechanism whereby an early cleavage of the cyanide-derived C≡N bond occurs and is followed by the formation of CH4. Scheme 4 outlines scenarios by which such a process might in principle proceed, invoking unusual terminal carbide intermediates [path (i)] or terminal methylidyne intermediates [path (ii)-to-(iii)]. Such pathways are conceptually related to the distal pathway of N2 reduction.[1b] While only observed as a minor product, the NMe3 product cannot be derived from a distal pathway, and may indicate a competing pathway wherein H-atom equivalents are delivered to the Fe-bound carbyne carbon of 5 without C–N bond rupture [Scheme 4, paths (iv) and (v)]. Conceptually related crossover distal-to-alternating mechanisms have been proposed for Fe-mediated N2 activation,[6c] and these scenarios will warrant further consideration in the present context of cyanide reduction. The fact that different reaction conditions/reagents are required for the activation of the cyanide-derived C–N bond in Fe(CNMe2) 5 and Fe(CN) 1 necessitates caution in mechanistically relating the reaction profile of 5 to generate CH4 and HNMe2 (or NMe3) with that of 1 to generate CH4 and NH3. Nonetheless, reactions pathways that bypass C–N bond cleavage, such as that represented by (ν) of Scheme 4, do not appear to be competent in the case of the proton-coupled reduction of 1 as MeNH2 is not observed.
Scheme 4.

Possible routes to the formation of CH4 and amine products.
In conclusion, we have disclosed the synthesis of a trigonal bipyramidal Fe(CN) species whose C≡N bond is cleaved upon exposure to proton and electron equivalents. An unusual Fe(CNH2) complex has been characterized and may serve as a plausible intermediate en route to the CH4 and NH3 products derived from this 6-electron/proton C≡N cleavage reaction. This work lends indirect support to the notion that one (or more) Fe centers within the FeMoco (or FeVco) can serve to activate myriad substrates, including N2, CO and cyanide. The catalytic reduction of cyanide by a synthetic molecular system remains a worthwhile challenge, and the complexes described here serve as promising starting points.
Supplementary Material
Acknowledgements
This work was supported by the NIH (grant number GM 070757) and the NSF (GRFP to J.R.). We thank Lawrence Henling and Michael Takase for assistance with XRD studies, and Kathryn Perez and Nathan Dalleska for assistance with GC experiments.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under http://dx.doi.org/10.1002/anie.201606366.
References
- [1].a) Howard JB, Rees DC. Chem. Rev. 1996;96:2965–2982. doi: 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]; b) Seefeldt LC, Hoffman BM, Dean DR. Annu. Rev. Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Seefeldt LC, Yang Z-Y, Duval S, Dean DR. Biochim. Biophys. Acta Bioenerg. 2013;1827:1102–1111. doi: 10.1016/j.bbabio.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bur gess BK, Lowe DJ. Chem. Rev. 1996;96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- [3].Li J, Burgess BK, Corbin JL. Biochemistry. 1982;21:4393–4402. doi: 10.1021/bi00261a031. [DOI] [PubMed] [Google Scholar]
- [4].a) Lee CC, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2015;54:1219–1222. doi: 10.1002/anie.201410412. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015;127:1235–1238. [Google Scholar]; b) Tanifuji K, Lee CC, Ohki Y, Tatsumi K, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2015;54:14022–14025. doi: 10.1002/anie.201507646. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015;127:14228–14231. [Google Scholar]
- [5].a) Fedurco M, Sartoretti CJ, Augustynski J. J. Am. Chem. Soc. 1999;121:888–889. [Google Scholar]; b) Fedurco M, Sartoretti CJ, Augustynski J. J. Electrochem. Soc. 2001;148:D19–D23. [Google Scholar]
- [6].a) Anderson JS, Rittle J, Peters JC. Nature. 2013;501:84–87. doi: 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anderson JS, Cutsail G, III, Rittle J, Connor B, Gunderson W, Zhang L, Hoffman B, Peters JC. J. Am. Chem. Soc. 2015;137:7803–7809. doi: 10.1021/jacs.5b03432. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rittle J, Peters JC. J. Am. Chem. Soc. 2016;138:4243–4248. doi: 10.1021/jacs.6b01230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Fehlhammer WP, Fritz M. Chem. Rev. 1993;93:1243–1280. [Google Scholar]; b) Pombeiro AJL, da Silva MFCG, Michelin RA. Coord. Chem. Rev. 2001;218:43–74. [Google Scholar]
- [8].Hu Y, Lee CC, Ribbe MW. Science. 2011;333:753–755. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Lee Y, Mankad NP, Peters JC. Nat. Chem. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee Y, Peters JC. J. Am. Chem. Soc. 2011;133:4438–4446. doi: 10.1021/ja109678y. [DOI] [PubMed] [Google Scholar]
- [10]. CCDC 1488907, 1488908, 1488909, 1488910, 1488911 and 1488912 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- [11].Steiner T. Angew. Chem. Int. Ed. 2002;41:48–76. [Google Scholar]; Angew. Chem. 2002;114:50–80. [Google Scholar]
- [12].a) Amrhein PI, Drouin SD, Forde CE, Lough AJ, Morris RH. J. Chem. Soc. Chem. Commun. 1996:1665–1666. [Google Scholar]; b) Rieger D, Hahn FE, Fehlhammer PW. J. Chem. Soc. Chem. Commun. 1990:285–286. [Google Scholar]
- [13].Fehlhammer PW, Schoder F, Beck G, Schrçlkamp S. Z. Anorg. Allg. Chem. 1993;619:1171–1176. [Google Scholar]
- [14].a) Pombeiro AJL, Hughes DL, Pickett CJ, Richards RL. J. Chem. Soc. Chem. Commun. 1986:246–247. Terminally-bound W(CNH2) and Re(CNH2) complexes have been previously described. [Google Scholar]; b) Hughes DL, Ibrahim SK, Ali HM, Pickett CJ. J. Chem. Soc. Chem. Commun. 1994:425–427. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.