

Abstract
In this study, genome-wide expression analyses were used to study the response of Saccharomyces cerevisiae to stress throughout a 15-day wine fermentation. Forty per cent of the yeast genome significantly changed expression levels to mediate long-term adaptation to fermenting grape must. Among the genes that changed expression levels, a group of 223 genes was identified, which was designated as fermentation stress response (FSR) genes that were dramatically induced at various points during fermentation. FSR genes sustain high levels of induction up to the final time point and exhibited changes in expression levels ranging from four- to 80-fold. The FSR is novel; 62% of the genes involved have not been implicated in global stress responses and 28% of the FSR genes have no functional annotation. Genes involved in respiratory metabolism and gluconeogenesis were expressed during fermentation despite the presence of high concentrations of glucose. Ethanol, rather than nutrient depletion, seems to be responsible for entry of yeast cells into the stationary phase.
Keywords: gene expression, fermentation, stress response, wine, HXK2
Introduction
Strains of Saccharomyces cerevisiae are industrially important due to extensive use in baking, brewing, wine-making, and in the production of fuel ethanol. Saccharomyces cerevisiae has complex regulatory networks to sense, respond, and adapt to changing environments, but the regulation of metabolic pathways and mechanisms for adapting to the extreme conditions in industrial processes, such as fermentation, have not been well studied. In the natural environment of grape must fermentation, S. cerevisiae is subject to high osmotic pressure, hypoxia, high concentrations of sugar, and low nitrogen levels. In addition, as the fermentation progresses, ethanol levels can reach 16% (v/v); in standard laboratory media the ethanol concentration never exceeds 1% (v/v). During fermentation, stationary-phase growth is reached within c. 48h, but the yeast actively ferments and then survives for months in wine. Many of the 1253 genes that have not been characterized (Pena-Castillo & Hughes, 2007) may well play an important role during the later adaptive stages of fermentation.
Perturbation of environmental conditions has led to the identification of a multitude of stress responses (see Hohmann & Mager, 2003 for a review). While numerous genomics studies have addressed specific responses, a few studies have examined the common components of generalized environmental stress. Large-scale transcriptome analyses of short-term responses have identified the ~868 gene ‘environmental stress response’ (ESR) (Gasch et al., 2000) and the ~499 gene ‘common environmental response’ (CER) (Causton et al., 2001), which overlap by ~337 genes. Functions of genes induced by stresses in both studies include carbohydrate metabolism, protein degradation, response to reactive oxygen species, protein folding, and genes with stress response elements (STREs) in their promoters. Repressed genes include those involved in translation and protein synthesis, cytoplasmic ribosomal proteins, tRNA synthesis, and translation. Both the ESR and CER studies identified many genes with unknown functions that respond to stress.
In contrast to the transient stress responses, S. cerevisiae is capable of making adaptive changes to grow both aerobically or anaerobically depending on the environmental conditions. In the presence of glucose at concentrations that exceed 0.5% (w/v), the yeast will ferment regardless of the presence of oxygen. Genes involved in respiratory metabolism and gluconeogenesis can also be regulated by a carbon source (Schuller, 2003). When glucose is depleted, the cells undergo a diauxic shift and respire ethanol produced during fermentation (DeRisi et al., 1997). During the transition, the transcriptional repressor Mig1p is exported from the nucleus to the cytoplasm. Mig1p is a key regulator of carbon catabolite repression; the nuclear export of Mig1p results in derepression of genes required for the utilization of alternative carbon sources (Nehlin & Ronne, 1990; De Vit et al., 1997). The exact mechanism of signal transduction in response to glucose is not entirely understood. Furthermore, little is known about how yeast metabolizes carbon when both glucose and ethanol are in excess.
The incorporation of transcript profiling technologies into enology research has led to a series of observations about gene expression during the early stages of fermentation. Several publications have demonstrated a dramatic change in gene expression patterns during the transition to stationary growth (Puig & Perez-Ortin, 2000; Devantier et al., 2005; Varela et al., 2005). Differences in gene expression patterns between commercial strains of wine yeast revealed a common pattern of stress response between wine strains at the beginning of vinification (Zuzuarregui & del Olmo, 2004; Zuzuarregui et al., 2006). Rossignol et al. (2003) explored the genomic expression patterns of S. cerevisiae during a 3-day fermentation in a synthetic medium. In this latter study, stationary-phase changes were noted, along with the observation of a plentitude of stress response processes being active. To identify how S. cerevisiae responds adaptively to the long-term environmental stresses present during grape must fermentation, the yeast transcriptome was profiled over a period of 15 days.
Materials and methods
Strains, media, and growth conditions
An industrial wine strain of S. cerevisiae, Vin13 (Anchor Yeast, South Africa), was used. The yeast was inoculated into 1 L filter-sterilized Riesling grape juice (Marks et al., 2003) with a final cell count of 4×106mL−1. The grape juice contained 214 g L−1 sugar (equimolar amounts of glucose and fructose), 70 mg L−1 ammonia, and 140 mg L−1 free α-amino nitrogen (Marks et al., 2003). Incubation was stationary at 20 °C without the addition of oxygen. Fermentations were conducted in triplicate, and cells were harvested from fermentations after 0.5%, 2%, 3.5%, 7%, and 10% (v/v) ethanol was produced.
Analysis of fermenting wine
Enzymatic kits were used to determine glucose and fructose concentrations according to the manufacturer’s instructions (Roche Molecular Biochemicals, Laval, QC). Ethanol was measured by HPLC as described previously (Erasmus et al., 2003). Water activity was measured using an Aqualab Series 3 water activity meter (Decagon Devices, Pullmann, WA). All measurements were performed in triplicate.
Microarray analysis
Total RNA was extracted, and isolation of mRNA and cDNA synthesis was performed according to Causton et al. (2001). Double-stranded cDNA purification, cRNA synthesis, and fragmentation were performed according to Marks et al. (2003). Affymetrix Yeast Genome S98 Chips were used (Affymetrix, Santa Clara, CA). Preparation of hybridization solution, hybridization, and washing, staining, and scanning of yeast arrays were performed as described by the manufacturer (Eukaryotic Arrays GeneChip Expression Analysis and Technical Manual, Affymetrix, Santa Clara, CA). Washing and staining were performed as described previously (Marks et al., 2003).
Analyses of gene expression data
Feature intensities from Affymetrix S98 chips were preprocessed using the robust multi-array average procedure (Irizarry et al., 2003) to obtain probe-set-summarized, normalized gene expression data. Of the 9335 probesets, 6299 probesets were retained that map to verified, uncharacterized, or dubious ORFs from the Saccharomyces Genome Database (SGD) (Cherry et al., 1998). Differences in gene expression patterns between commercial wine yeast strains at the beginning of vinification are well characterized (Zuzuarregui & del Olmo, 2004; Zuzuarregui et al., 2006) and the first two time points were not included in the analysis. However, data obtained for these two time points were also deposited in ArrayExpress (GSE8536). Therefore, for each of the 6299 probesets, the primary data consist of three independent biological replicates at each of five time points (the ethanol concentrations of 0.5%, 2%, 3.5%, 7%, and 10% (v/v) were nominally coded as 24, 48, 60, 120, and 340 h, respectively). For the purposes of temporal modeling, the predictor t was defined as the logarithm of the sampling time (in hours), centered around the associated log-time midpoint. The 15 expression measurements for any probeset g were summarized with the following quadratic polynomial:
The intercept β0g captures the overall expression level, whereas the more interesting pair of temporal parameters (β1g, β2g) provides a probeset-specific summary of the expression change observed during fermentation; the linear term β1g reflects a general trend (e.g. up vs. down) and the quadratic term reflects overall shape (e.g. concave vs. convex).
A statistical test for differential expression was conducted with the null hypothesis of (β1g, β2g) = (0,0). The P-values from this F-test were converted to q-values (Storey & Tibshirani, 2003). A probeset was classified and the estimated proportion of null genes was ~10%. A probeset was classified as differentially expressed if it had a q-value of 0.001 or less and a predicted fold-change of 2 or more at at least one fermentation time point, based on the model.
The temporal parameters (β1g, β2g) were used as the input features for supervised clustering (Bryan, 2004), in order to identify groups of genes with similar temporal trends. The clusters were anchored by 20 genes, selected to span the observed (β1g, β2g) values. A probeset was assigned to cluster k if the squared Euclidean distance to cluster anchor k was smaller than that to any other anchor. All analyses were conducted within the R statistical software environment (R Development Core Team, 2006) and relied on the libraries affy (Gautier et al., 2004; Gentleman et al., 2004) and q-value (Dabney & Storey, 2006).
Functional enrichment of genes in clusters
The frequency of individual Gene Ontology (GO) annotation terms was used as a basis to assess biological significance of the gene expression data (Ashburner et al., 2000). Owing to the fact that the vast majority of GO terms are associated with very few genes (i.e. most terms are highly specific), a truncated version of the GO database was used. The truncated GO database consisted of 475 terms, selected for being associated with more than 10 genes. The level, or number of parents in the branching GO hierarchy, was also used as a tool in the analysis. The association between the clusters and GO terms was studied using contingency tables. The χ2 statistic (and its conventional P-value) was used to summarize evidence for enrichment or depletion of genes having a functional annotation within gene clusters.
Identification of regulatory elements overrepresented in gene clusters
Promoter sequences corresponding to the 5′ untranslated region 500 base pairs upstream of the initial ATG for each ORF were downloaded from SGD. A collection of yeast-specific transcription factor-binding site (TFBS) motifs was compiled from the yeast regulatory sequence analysis (YRSA) system (Sandelin et al., 2003) and from the literature (supplementary Table S1). Using the TFBS suite of Perl regulatory analysis modules (Lenhard & Wasserman, 2002), patterns matching the set of 44 TFBS weight matrix profiles in the collection, were searched for and the locations and scores for hits with matrix match scores exceeding 80% of the normalized score range were noted. For each of the clusters, and two ‘combined clusters’ comprising clusters 1–6 and clusters 18–20, two statistical measures were calculated: a Z-score and Fisher’s exact probability to determine which, if any, of the TFBSs were over-represented in the gene clusters relative to a background set comprised of all yeast promoter sequences (Ho Sui et al., 2005).
The Z-score measures how frequently a particular TFBS occurs in the promoters of coexpressed genes in a cluster and compares it with the frequency of occurrence in the background set, determining overrepresentation of the motif at the nucleotide level. Briefly, , where x is the observed number of binding site nucleotides for a given TFBS in the coexpressed set, μ is the expected number of binding site nucleotides based on the background sequences, and σ is the SD. The score is based on the normal approximation to the binomial distribution.
In contrast, the Fisher exact probability compares the proportion of genes in a cluster that contain a particular TFBS to the proportion of genes in the background that contain the TFBS, determining overrepresentation at the gene level. The Fisher’s exact test computes the probability, given the observed marginal frequencies, of obtaining exactly the frequencies observed and any configuration more extreme (Fleiss, 1981).
Results and discussion
To investigate how yeast adapts to the harsh conditions during the fermentation of grape must, genome-wide transcription was assayed at five time points during alcoholic fermentation. Correlation coefficients for the biological replicates at individual time points ranged from 0.9575 to 0.9950. The full data set is available in supplementary Table S2.
Clustering of genes based on temporal expression profiles
For each gene, the temporal trend throughout the fermentation process was summarized with two parameter values that describe the overall change and shape of the expression profile. A seeded clustering algorithm was applied in order to group the genes around the expression patterns exhibited by 20 genes whose temporal trends span the range of patterns observed across the entire transcriptome (Fig. 1). For each gene, a test was conducted for temporal trend, based on the null hypothesis that the true expression pattern is a flat line (i.e., no change at any time), and these gene-specific P-values were converted to q-values to control the false discovery rate (Benjamini & Hochberg, 1995; Storey & Tibshirani, 2003). Differentially expressed genes were identified as genes having a q-value <0.001 and exhibiting a minimum of twofold change in expression at one or more time points during fermentation. Therefore, the definition of differential expression requires a statistically significant temporal trend as well as an expression change that is large in absolute magnitude; 2550 genes were identified that met these criteria, which correspond to 42% of the yeast genome (Table 1). Induction of 1123 genes was sustained until the end of fermentation (clusters 1–10), while 1279 genes were repressed (clusters 14–20). The expression of 148 genes was transiently induced and returned to steady-state levels by the final time point (clusters 11 and 12) (Fig. 1). The expression of 1876 genes was unaffected (cluster 13, Fig. 1).
Fig. 1.

Clustering of genes based on temporal expression profiles. The center graph shows the entire transcriptome plotted with the coefficients calculated from the expression model. The clusters are anchored by the genes numbered in black. The temporal expression of these genes is displayed in the 20 line graphs on the periphery. The dotted lines indicate twofold change in the expression levels. The number of genes in each cluster is circled in the cluster.
Table 1.
Summary of gene counts
| Cluster | Genes | Statistically significant temporal trend (q-value ≤ 0.001) | Minimum of twofold change between any two time points (FC≥2) | Differentially expressed (q-value ≤0.001 and FC≥2) | Sustained response (minimum of twofold change at final time point) | Strength of long-term response (smallest final expression change for the cluster) | Description of the response | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Count | Proportion | Count | Proportion | Count | Proportion | |||||
| 1 | 5 | 5 | 1.00 | 5 | 1.00 | 5 | 1.00 | 5 | 22.73 | Sustained induction |
| 2 | 8 | 8 | 1.00 | 8 | 1.00 | 8 | 1.00 | 8 | 13.62 | Sustained induction |
| 3 | 11 | 11 | 1.00 | 11 | 1.00 | 11 | 1.00 | 11 | 20.03 | Sustained induction |
| 4 | 63 | 63 | 1.00 | 63 | 1.00 | 63 | 1.00 | 63 | 4.83 | Sustained induction |
| 5 | 37 | 37 | 1.00 | 37 | 1.00 | 37 | 1.00 | 37 | 4.03 | Sustained induction |
| 6 | 100 | 100 | 1.00 | 100 | 1.00 | 100 | 1.00 | 100 | 4.38 | Sustained induction |
| 7 | 356 | 337 | 0.95 | 350 | 0.98 | 331 | 0.93 | 350 | 1.87 | Sustained induction |
| 8 | 345 | 307 | 0.89 | 250 | 0.72 | 239 | 0.69 | 191 | 1.11 | Sustained induction |
| 9 | 336 | 239 | 0.71 | 190 | 0.57 | 162 | 0.48 | 114 | 1.37 | Sustained induction |
| 10 | 182 | 178 | 0.98 | 182 | 1.00 | 178 | 0.98 | 132 | 1.43 | Sustained induction |
| 11 | 50 | 48 | 0.96 | 50 | 1.00 | 48 | 0.96 | 3 | 1.01 | Transient induction |
| 12 | 538 | 285 | 0.53 | 121 | 0.22 | 100 | 0.19 | 0 | 1.00 | Transient induction |
| 13 | 1876 | 161 | 0.09 | 1 | 0.00 | 0 | 0.00 | 0 | 1.00 | No change |
| 14 | 311 | 222 | 0.71 | 177 | 0.57 | 157 | 0.50 | 131 | 1.01 | Sustained repression |
| 15 | 372 | 311 | 0.84 | 241 | 0.65 | 226 | 0.61 | 137 | 1.36 | Sustained repression |
| 16 | 1123 | 760 | 0.68 | 473 | 0.42 | 450 | 0.40 | 473 | 1.33 | Sustained repression |
| 17 | 62 | 61 | 0.98 | 62 | 1.00 | 61 | 0.98 | 60 | 1.97 | Sustained repression |
| 18 | 368 | 366 | 0.99 | 368 | 1.00 | 366 | 0.99 | 368 | 3.45 | Sustained repression |
| 19 | 7 | 7 | 1.00 | 7 | 1.00 | 7 | 1.00 | 7 | 4.61 | Sustained repression |
| 20 | 41 | 41 | 1.00 | 41 | 1.00 | 41 | 1.00 | 41 | 8.66 | Sustained repression |
| Simple total | 6191 | 3547 | 0.57 | 2737 | 0.44 | 2590 | 0.42 | 2231 | ||
| Unique total | 6088 | 3493 | 0.57 | 2688 | 0.44 | 2549 | 0.42 | 2186 | ||
Genes in the FSR are shaded. As some genes occur in multiple clusters, the total number of unique genes in the data set in addition to a simple total of the counts in each cluster is provided.
Fermentation of grape must induces a novel and adaptive response
Under enological conditions, the yeast cell is exposed to changing nutrient concentrations and diverse forms of stress. Saccharomyces cerevisiae responds transcriptionally to stress by general and/or stimuli-specific response mechanisms. In contrast to the transient responses observed in laboratory studies (Gasch et al., 2000; Causton et al., 2001), sustained global changes were observed in transcript abundance for the duration of the fermentation process (15 days), indicating an adaptive response to fermentation stress (Fig. 1). The differentially expressed genes in clusters 1–6, i.e. genes exhibiting sustained and dramatic induction that persists until the final time point (Table 1), are defined as fermentation stress response (FSR) genes. Genes that are down-regulated from the FSR are excluded, as they are primarily involved in protein biosynthesis and ribosomal processing, functions known to be repressed under stress and in the stationary growth phase. A list of the 223 FSR genes is shown in Table 2. These genes show a fourfold or more change in expression at some point during fermentation.
Table 2.
FSR genes, GO process annotations, and the overlap with the ESR, the CER, salt/sorbital-induced osmotic stress (OSM1), sugar-induced osmotic stress (OSM2), short-term ethanol stress (E), genes involved in nitrogen limitation (N), and genes involved in oxidative stress (O)
| Cluster | Gene | Maximum fold change | GO biological process | ORF status | Other response
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ESR | CER | OSM1 | OSM2 | E | N | O | |||||
| 1 | HSP30 | 80.11 | Response to stress | V | x | x | |||||
| 1 | GAC1 | 44.21 | Meiosis* | V | |||||||
| 1 | GAT1 | 29.10 | Transcription initiation from RNA polymerase II promoter* | V | x | x | |||||
| 1 | CSR2 | 24.68 | Cell wall organization and biogenesis* | V | x | ||||||
| 1 | MCH5 | 22.73 | Riboflavin transport | V | |||||||
| 2 | CYB2 | 68.30 | Electron transport | V | x | x | |||||
| 2 | INO1 | 39.14 | Inositol metabolic process | V | |||||||
| 2 | PHM8 | 28.99 | Biological process unknown | V | x | x | x | x | |||
| 2 | PUT4 | 27.37 | Proline catabolic process* | V | x | x | |||||
| 2 | HSP26 | 24.45 | Response to stress* | V | x | x | x | x | |||
| 2 | MEP2 | 24.36 | Pseudohyphal growth* | V | |||||||
| 2 | SPI1 | 23.51 | Biological process unknown | V | x | x | x | x | |||
| 2 | SHC1 | 20.00 | Sporulation (sensu Fungi)* | V | x | ||||||
| 3 | YMR244W | 32.27 | biological process unknown | U | |||||||
| 3 | IRC8 | 32.13 | Biological process unknown | U | |||||||
| 3 | YJL150W | 31.22 | Biological process unknown | D | |||||||
| 3 | YOL047C | 29.44 | Biological process unknown | U | |||||||
| 3 | MBR1 | 27.90 | Aerobic respiration | V | x | ||||||
| 3 | YDL068W | 26.56 | Biological process unknown | D | |||||||
| 3 | YOR318C | 25.51 | Biological process unknown | D | |||||||
| 3 | SPS1 | 24.93 | Protein amino acid phosphorylation* | V | |||||||
| 3 | RDH54 | 23.27 | Meiotic recombination* | V | |||||||
| 3 | PES4 | 21.96 | Biological process unknown | V | |||||||
| 3 | APJ1 | 20.03 | Biological process unknown | V | x | x | |||||
| 4 | RPI1 | 19.20 | Thiamin biosynthetic process* | V | |||||||
| 4 | PUT1 | 19.06 | Glutamate biosynthetic process* | V | x | ||||||
| 4 | RSB1 | 18.74 | Response to toxin* | V | x | ||||||
| 4 | HPA2 | 18.35 | Histone acetylation | V | x | ||||||
| 4 | MFA1 | 17.69 | Pheromone-dependent signal transduction during conjugation with cellular fusion | V | |||||||
| 4 | SUE1 | 17.68 | Protein catabolic process | V | x | ||||||
| 4 | ATG1 | 17.61 | Autophagy* | V | x | x | |||||
| 4 | YFR017C | 17.51 | Biological process unknown | U | x | x | x | ||||
| 4 | VHS1 | 16.06 | Protein amino acid phosphorylation* | V | x | ||||||
| 4 | GAP1 | 15.99 | Amino acid transport* | V | |||||||
| 4 | PDR15 | 15.85 | Transport | V | x | ||||||
| 4 | YLR168C | 15.83 | Biological process unknown | U | |||||||
| 4 | YBR284W | 15.70 | Telomere maintenance | U | |||||||
| 4 | VHR1 | 14.77 | Regulation of transcription from RNA polymerase II promoter* | U | |||||||
| 4 | AGX1 | 14.62 | Glycine biosynthetic process, by transamination of glyoxylate | V | x | x | |||||
| 4 | GSC2 | 13.38 | Cell wall organization and biogenesis* | V | x | ||||||
| 4 | STR3 | 12.50 | Methionine biosynthetic process | V | |||||||
| 4 | RIM8 | 11.21 | Meiosis* | V | x | ||||||
| 4 | YBL065W | 11.15 | Biological process unknown | D | x | ||||||
| 4 | ACS1 | 10.79 | Histone acetylation* | V | |||||||
| 4 | JID1 | 10.41 | Biological process unknown | V | |||||||
| 4 | YLL020C | 10.18 | Biological process unknown | D | x | ||||||
| 4 | HXT6 | 9.76 | Hexose transport | V | |||||||
| 4 | YKL071W | 9.71 | Biological process unknown | U | x | ||||||
| 4 | DAL80 | 9.43 | Transcription* | V | x | ||||||
| 4 | PTR2 | 8.62 | Peptide transport* | V | |||||||
| 4 | TSL1 | 8.11 | Response to stress* | V | x | x | x | x | x | ||
| 4 | PIN3 | 7.88 | Actin cytoskeleton organization and biogenesis | V | x | ||||||
| 4 | DOT6 | 7.81 | Regulation of transcription from RNA polymerase II promoter* | V | |||||||
| 4 | QNQ1 | 7.69 | Biological process unknown | U | x | ||||||
| 4 | PDE1 | 7.65 | cAMP-mediated signaling | V | x | x | x | ||||
| 4 | GSY2 | 7.58 | Glycogen biosynthetic process | V | x | x | x | ||||
| 4 | UPS1 | 7.57 | Mitochondrial protein processing | V | |||||||
| 4 | YBR085C-A | 7.52 | Biological process unknown | U | |||||||
| 4 | YCR061W | 7.50 | Regulation of cell size | U | x | x | x | ||||
| 4 | YER158C | 7.43 | Biological process unknown | U | x | ||||||
| 4 | RGT1 | 7.26 | Glucose metabolic process* | V | |||||||
| 4 | CRC1 | 7.16 | Fatty acid metabolic process | V | |||||||
| 4 | IRC9 | 7.11 | Biological process unknown | D | x | ||||||
| 4 | DIA3 | 7.07 | Pseudohyphal growth* | V | |||||||
| 4 | GPX1 | 6.71 | Response to oxidative stress | V | x | x | |||||
| 4 | YNL194C | 6.63 | Sporulation (sensu Fungi) | V | x | x | x | ||||
| 4 | CTA1 | 6.48 | OX and reactive OX species metabolic process* | V | x | x | |||||
| 4 | MOD5 | 6.27 | tRNA modification | V | |||||||
| 4 | ATG8 | 6.25 | Protein targeting to vacuole* | V | x | x | x | x | |||
| 4 | HSP78 | 6.24 | Response to stress* | V | x | x | x | x | |||
| 4 | VID24 | 6.22 | Vesicle-mediated transport* | V | |||||||
| 4 | TPO4 | 6.11 | Polyamine transport | V | x | ||||||
| 4 | YLL056C | 5.97 | Response to toxin | U | |||||||
| 4 | SSD1 | 5.87 | Response to drug* | V | |||||||
| 4 | YIR016W | 5.85 | Biological process unknown | U | x | ||||||
| 4 | AVT4 | 5.80 | Amino acid export from vacuole | V | |||||||
| 4 | MGA1 | 5.78 | Filamentous growth | V | x | ||||||
| 4 | SSE2 | 5.66 | Protein folding* | V | x | x | x | x | |||
| 4 | HXT7 | 5.55 | Hexose transport | V | |||||||
| 4 | YMR244C-A | 5.54 | Biological process unknown | U | |||||||
| 4 | KRE1 | 5.51 | Cell wall organization and biogenesis | V | |||||||
| 4 | IRC15 | 5.42 | Biological process unknown | U | |||||||
| 4 | YOR1 | 5.16 | Telomere maintenance* | V | |||||||
| 4 | OPY2 | 5.11 | Cell cycle arrest in response to pheromone* | V | |||||||
| 4 | YMR253C | 5.10 | Biological process unknown | U | x | ||||||
| 4 | MRP8 | 4.88 | Translation | V | x | x | x | ||||
| 4 | VBA2 | 4.83 | Basic amino acid transport | V | |||||||
| 5 | MTR4 | 21.43 | Ribosome biogenesis and assembly* | V | |||||||
| 5 | DIP5 | 19.61 | Amino acid transport | V | |||||||
| 5 | PLB3 | 14.78 | Phosphatidylserine catabolic process* | V | |||||||
| 5 | YJL149W | 14.43 | Biological process unknown | U | x | ||||||
| 5 | BTN2 | 12.82 | Retrograde transport, endosome to Golgi* | V | x | x | |||||
| 5 | MUD1 | 12.72 | Nuclear mRNA splicing, via spliceosome | V | |||||||
| 5 | ADR1 | 12.61 | Transcription* | V | |||||||
| 5 | YDR042C | 12.49 | Biological process unknown | U | |||||||
| 5 | ULP2 | 11.21 | Mitotic spindle checkpoint* | V | |||||||
| 5 | YBR099C | 10.95 | Biological process unknown | D | |||||||
| 5 | YDL012C | 10.54 | Biological process unknown | U | |||||||
| 5 | URN1 | 10.40 | Biological process unknown | U | |||||||
| 5 | PHO80 | 9.88 | Telomere maintenance* | V | |||||||
| 5 | HAP1 | 9.63 | Positive regulation of transcription from RNA polymerase II promoter* | V | |||||||
| 5 | SNG1 | 9.58 | Response to drug | V | |||||||
| 5 | SRT1 | 9.43 | Protein amino acid glycosylation | V | |||||||
| 5 | MKS1 | 9.30 | Regulation of nitrogen utilization* | V | |||||||
| 5 | SLD2 | 8.98 | DNA strand elongation during DNA replication | V | |||||||
| 5 | HRP1 | 8.45 | mRNA cleavage* | V | |||||||
| 5 | NRD1 | 8.36 | Transcription termination from Pol II promoter, RNA polymerase(A)-independent | V | |||||||
| 5 | CHS1 | 8.35 | Cytokinesis, completion of separation* | V | x | ||||||
| 5 | SIS1 | 7.59 | Protein folding* | V | |||||||
| 5 | ISA1 | 7.58 | Telomere maintenance* | V | |||||||
| 5 | AQR1 | 7.14 | Drug transport* | V | |||||||
| 5 | VID27 | 7.09 | Biological process unknown | V | |||||||
| 5 | RCL1 | 6.63 | Ribosome biogenesis and assembly* | V | |||||||
| 5 | ECM3 | 6.32 | Cell wall organization and biogenesis | V | |||||||
| 5 | YML089C | 6.29 | Biological process unknown | D | |||||||
| 5 | MPC54 | 6.28 | Spore wall assembly (sensu Fungi) | V | |||||||
| 5 | YOR378W | 5.79 | Biological process unknown | U | |||||||
| 5 | TOR2 | 5.78 | Ribosome biogenesis and assembly* | V | |||||||
| 5 | DST1 | 5.66 | RNA elongation from RNA polymerase II promoter* | V | |||||||
| 5 | STE5 | 5.60 | Invasive growth (sensu Saccharomyces)* | V | |||||||
| 5 | YGL041C | 5.25 | Biological process unknown | D | |||||||
| 5 | SRC1 | 5.08 | Mitotic sister chromatid segregation | V | |||||||
| 5 | SSY5 | 5.02 | Protein processing* | V | |||||||
| 5 | STU1 | 4.93 | Microtubule nucleation | V | |||||||
| 6 | TPO2 | 17.87 | Polyamine transport | V | |||||||
| 6 | MUP3 | 17.09 | Amino acid transport | V | x | ||||||
| 6 | NRG1 | 16.02 | Regulation of transcription from RNA polymerase II promoter* | V | |||||||
| 6 | ARG82 | 14.69 | Response to drug* | V | |||||||
| 6 | RTS3 | 14.44 | Protein amino acid dephosphorylation | U | x | ||||||
| 6 | HRK1 | 14.38 | Cell ion homeostasis | V | |||||||
| 6 | IRC20 | 12.40 | Biological process unknown | U | x | ||||||
| 6 | GIP2 | 11.83 | Protein amino acid dephosphorylation* | V | x | x | |||||
| 6 | YKL070W | 11.82 | Response to toxin | U | |||||||
| 6 | GAT2 | 11.58 | Transcription | V | x | ||||||
| 6 | SAP155 | 11.45 | G1/S transition of mitotic cell cycle | V | |||||||
| 6 | PMC1 | 11.16 | Calcium ion homeostasis* | V | x | x | |||||
| 6 | XBP1 | 10.83 | Response to stress | V | x | x | x | ||||
| 6 | MEP1 | 10.78 | Nitrogen utilization* | V | |||||||
| 6 | YIL066W-A | 10.30 | Biological process unknown | D | |||||||
| 6 | PKH2 | 10.15 | Protein amino acid phosphorylation* | V | |||||||
| 6 | YMR102C | 9.70 | Biological process unknown | U | |||||||
| 6 | YDL010W | 9.37 | Biological process unknown | U | |||||||
| 6 | SWI1 | 9.31 | Regulation of transcription from RNA polymerase II promoter* | V | |||||||
| 6 | GIS1 | 9.12 | Spore wall assembly (sensu Fungi)* | V | x | ||||||
| 6 | YGR146C | 9.09 | Biological process unknown | U | x | x | |||||
| 6 | YJL144W | 8.99 | Response to desiccation | U | x | x | |||||
| 6 | RPN4 | 8.77 | Telomere maintenance* | V | x | x | |||||
| 6 | YIL152W | 8.51 | Biological process unknown | U | |||||||
| 6 | TPO1 | 8.35 | Polyamine transport | V | |||||||
| 6 | YLR194C | 8.28 | Chitin- and β-glucan-containing cell wall organization and biogenesis | U | x | ||||||
| 6 | FRT1 | 8.25 | Response to stress | U | |||||||
| 6 | KNS1 | 8.25 | Protein amino acid phosphorylation | V | x | x | |||||
| 6 | YDR186C | 7.77 | Biological process unknown | U | |||||||
| 6 | AHC1 | 7.72 | Histone acetylation | V | x | ||||||
| 6 | SKN7 | 7.62 | Response to oxidative stress* | V | x | ||||||
| 6 | MET4 | 7.62 | Positive regulation of transcription from RNA polymerase II promoter* | V | |||||||
| 6 | PDR5 | 7.58 | Response to drug* | V | |||||||
| 6 | YPL230W | 7.51 | Biological process unknown | U | x | ||||||
| 6 | YLL020C | 7.41 | Biological process unknown | D | x | ||||||
| 6 | TOS3 | 7.39 | Protein amino acid phosphorylation* | V | x | ||||||
| 6 | ZEO1 | 7.32 | Telomere maintenance* | V | |||||||
| 6 | MUB1 | 7.23 | Regulation of cell budding | V | |||||||
| 6 | RIM15 | 6.99 | Protein amino acid phosphorylation* | V | x | x | |||||
| 6 | ROG3 | 6.91 | Biological process unknown | U | |||||||
| 6 | HAC1 | 6.88 | Regulation of transcription from RNA polymerase II promoter* | V | |||||||
| 6 | KTR2 | 6.87 | Protein amino acid N-linked glycosylation* | V | x | ||||||
| 6 | FLO10 | 6.82 | Flocculation via cell wall protein-carbohydrate interaction | V | |||||||
| 6 | OPI9 | 6.67 | Biological process unknown | D | |||||||
| 6 | SKS1 | 6.58 | Protein amino acid phosphorylation* | V | |||||||
| 6 | YPL136W | 6.53 | Biological process unknown | D | |||||||
| 6 | TPO3 | 6.52 | Polyamine transport | V | |||||||
| 6 | SUR1 | 6.39 | Sphingolipid biosynthetic process* | V | |||||||
| 6 | ISF1 | 6.36 | Aerobic respiration | V | x | x | |||||
| 6 | MPH1 | 6.19 | DNA repair | V | |||||||
| 6 | YOL036W | 6.10 | Biological process unknown | U | |||||||
| 6 | YNL144C | 6.07 | Biological process unknown | U | |||||||
| 6 | NAB6 | 6.04 | RNA metabolic process | U | |||||||
| 6 | AFG3 | 6.01 | Translation* | V | |||||||
| 6 | PGA1 | 6.00 | Secretory pathway | U | |||||||
| 6 | YOR390W | 5.99 | Biological process unknown | U | |||||||
| 6 | SPG1 | 5.97 | Biological process unknown | U | x | ||||||
| 6 | MCH1 | 5.91 | Transport | V | x | ||||||
| 6 | YML116W-A | 5.91 | Biological process unknown | D | |||||||
| 6 | SSL2 | 5.86 | Transcription from RNA polymerase II promoter* | V | |||||||
| 6 | FSP2 | 5.85 | Biological process unknown | V | |||||||
| 6 | EDC2 | 5.83 | Deadenylation-dependent decapping | V | x | x | x | ||||
| 6 | OSW2 | 5.75 | Spore wall assembly (sensu Fungi) | U | x | ||||||
| 6 | PSR2 | 5.74 | Response to stress | V | |||||||
| 6 | PSK1 | 5.66 | Protein amino acid phosphorylation* | V | |||||||
| 6 | YAK1 | 5.51 | Protein amino acid phosphorylation | V | x | x | x | x | |||
| 6 | OAF1 | 5.49 | Peroxisome organization and biogenesis* | V | |||||||
| 6 | PDR1 | 5.47 | Response to drug* | V | x | ||||||
| 6 | GPI18 | 5.38 | GPI anchor biosynthetic process | U | |||||||
| 6 | SNQ2 | 5.37 | Response to drug* | V | x | ||||||
| 6 | IXR1 | 5.34 | DNA repair | V | |||||||
| 6 | MDN1 | 5.27 | rRNA processing* | V | |||||||
| 6 | YLR297W | 5.26 | Biological process unknown | U | |||||||
| 6 | PAU21 | 5.17 | Biological process unknown | U | |||||||
| 6 | YER093C-A | 5.17 | Biological process unknown | U | |||||||
| 6 | RAD54 | 5.17 | Chromatin remodeling* | V | |||||||
| 6 | RAD7 | 5.16 | Nucleotide–excision repair, DNA damage recognition | U | |||||||
| 6 | UBC8 | 5.01 | Protein monoubiquitination* | V | x | x | x | x | |||
| 6 | VPS72 | 5.01 | Protein targeting to vacuole* | V | |||||||
| 6 | MGR1 | 5.00 | Mitochondrial genome maintenance | U | x | ||||||
| 6 | YOR152C | 5.00 | Biological process unknown | U | x | ||||||
| 6 | TSC11 | 4.99 | Cell wall organization and biogenesis* | V | |||||||
| 6 | ISU1 | 4.97 | Iron ion homeostasis* | V | |||||||
| 6 | YMR252C | 4.96 | Biological process unknown | U | |||||||
| 6 | BUL1 | 4.96 | Mitochondrion inheritance* | V | x | ||||||
| 6 | RPO21 | 4.81 | Transcription from RNA polymerase II promoter | V | |||||||
| 6 | SPG5 | 4.80 | Biological process unknown | U | x | x | |||||
| 6 | KSP1 | 4.78 | Protein amino acid phosphorylation | V | |||||||
| 6 | INP1 | 4.77 | Peroxisome inheritance | U | |||||||
| 6 | LEE1 | 4.75 | Biological process unknown | V | x | ||||||
| 6 | PEP12 | 4.75 | Golgi to vacuole transport | V | x | x | |||||
| 6 | YPS3 | 4.75 | Chitin- and β-glucan-containing cell wall organization and biogenesis* | V | x | ||||||
| 6 | YMR085W | 4.62 | Biological process unknown | U | |||||||
| 6 | YML081W | 4.59 | Biological process unknown | U | |||||||
| 6 | PTK1 | 4.56 | Polyamine transport | V | |||||||
| 6 | YIL067C | 4.55 | Biological process unknown | U | |||||||
| 6 | YMR291W | 4.53 | Biological process unknown | U | x | x | x | ||||
| 6 | ASG1 | 4.43 | Biological process unknown | U | |||||||
| 6 | ATO3 | 4.41 | Nitrogen utilization* | V | |||||||
| 6 | SRX1 | 4.41 | Response to oxidative stress | V | x | x | |||||
Boldface indicates genes that have unknown biological process. An asterisk (*) adjacent to the GO term indicates that the gene product has more than one associated GO term, and only the term most commonly used for annotation is shown.
Within the 223 FSR genes, 20% overlap with the transient global stress responses characterized in the ESR or CER (Fig. 2, Table 2). A further 18% overlap with genes that respond to stress related to osmotic pressure, nitrogen depletion, ethanol increase, and oxidative stress (Table 2). By comparison, more than half (55%) of the repressed genes in clusters 18–20 overlap with the ESR or CER (supplementary Fig. S1).
Fig. 2.

FSR. Venn diagram showing the number of genes associated with two well-known transcriptionally characterized stress responses. ESR – genes induced in the environmental stress response (Gasch et al., 2000), CER – genes induced in the common environmental response (Causton et al., 2001), FSR – fermentation stress response genes. Refer also to supplementary Fig. S1.
Of the FSR genes, 28% lack a GO biological process annotation – they are uncharacterized. Of those that are characterized, more than half are associated with cellular and metabolic processes that facilitate the adaptation of the yeast to the continuously changing nutrient environment and the consequences thereof. Specifically, examination of the GO biological processes for these genes reveals that prevalent functional associations include transport (18%), organelle organization and biogenesis (15%), protein modification processes (13%), RNA metabolic processes (12%), response to stress (11%), and transcription (11%) (Fig. 3). The large set of transport-related proteins are primarily involved in glucose uptake, nitrogen regulation, vacuolar function [important for growth under ethanol stress (van Voorst et al., 2006)] and detoxification, reflecting the integrated response to nutrients in the environment, multiple stresses, and toxins produced during vinification. These results show that many of the FSR genes have not been associated with stress previously, and that a sizeable proportion of these genes required for long-term adaptation to fermentation conditions have yet to be characterized.
Fig. 3.

Functional annotations of genes in the FSR using biological process GO Slim Terms from SGD. The distribution of terms for the rest of the yeast genome is shown for comparison.
The stress response of a single industrial wine yeast strain, Vin13, in fermenting Riesling grape must has been examined; 40% of the yeast genome significantly changed expression levels to mediate long-term adaptation to fermenting grape must. Among the genes that changed expression levels, 223 FSR genes were identified that are permanently induced at various points during fermentation. However, it must be noted that the composition of grape musts can vary significantly with respect to the sugar concentration, and lipid, nitrogen, and vitamin compositions and concentrations. The transcriptional response of other industrial wine yeast strains in different grape musts should therefore be investigated to determine whether any additional FSR genes exist.
Attenuated glucose repression
Saccharomyces cerevisiae can grow oxidatively on many nonfermentative carbon sources such as pyruvate, lactate, acetate, and ethanol. These compounds are oxidized in the citric acid cycle and ATP is generated by reoxidation of reduced coenzymes in the mitochondrial electron transport system by oxidative phosphorylation. Several genes needed for oxidative energy metabolism, mitochondrial function, and the catabolism of nonfermentable carbon sources are repressed by high concentrations of glucose (Gancedo, 1998; Johnston, 1999).
At the onset of fermentation, the sugar concentration in the grape must was 21.4% (w/v) (equimolar amounts of glucose and fructose). These data show an increase in expression of multiple glucose-repressed genes, indicating a partial attenuation of classic glucose repression during fermentation. Young et al. (2003) identified 40 of the most highly glucose repressed genes based on expression ratios from derepressed vs. repressed growth conditions. Twenty-three of these genes are up-regulated during fermentation (supplementary Table S3A). Furthermore, genes involved in mitochondrial respiration/oxidative phosphorylation (supplementary Table S3B), hypoxic genes involved in heme, sterol and unsaturated fatty acid biosynthesis (supplementary Table S3C), and genes associated with oxidative stress (supplementary Table S3D) were all induced during fermentation while the glucose concentration in the media should still be repressive. Several genes annotated to ‘carbohydrate metabolism’ in clusters 9 and 10 (supplementary Table S3E) are reported to be glucose repressed (Dennis & McCammon, 1999; Lodi et al., 1999; Schuller, 2003); many of these genes are involved in glycolysis and gluconeogenesis. The induction of numerous glucose-repressed and oxygen-regulated genes indicates that cellular respiration may not be fully repressed during fermentation. Statistical analysis of the GO process terms shows an enrichment of the terms ‘electron transport’, ‘oxidative phosphorylation’, and ‘cellular respiration’ among the induced genes (Fig. 4).
Fig. 4.

Heat map of the association of GO terms to clusters. White represents depletion and enrichment increases with darker squares (based on the χ2 statistic).
The regulators Mig1p, Adr1p, and Cat8p play a pivotal role in the transcription of glucose-repressed genes. Mig1p binds to the promoter sequences and represses the expression of many genes in yeast cells growing in high concentrations of glucose. Conversely, Cat8p and Adr1p encode carbon source-responsive transcription factors shown to activate the expression of genes for metabolism of non-fermentative carbon sources when glucose is depleted (Schuller, 2003). Statistically enriched TFBSs in the promoters of FSR genes were ranked using Z-scores; the enrichment was then further evaluated with Fisher exact probabilities to determine enrichment of the sites at the gene level (Ho Sui et al., 2005). Binding sites for Mig1p and Adr1p, as well as the carbon source-responsive element (CSRE) bound by Cat8p ranked among the 10 most abundant TFBS motifs in the FSR genes (Table 3), lending further support to the hypothesis that glucose repression is not fully functional over the course of fermentation. A full listing of motif scores for each of the 20 clusters is available in supplementary Table S4.
Table 3.
Highest ranked transcription factor binding site motifs for FSR genes
| Rank | TFBS motif | Functional annotation | Z-score | Fisher score | Genes containing site | Logo |
|---|---|---|---|---|---|---|
| 1 | ADR1P | Carbohydrate metabolism, glucose repression | 19.33 | 8.21E-02 | 166 |
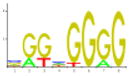
|
| 2 | PDR3* | Chemical agent resistance; detoxification | 18.35 | 7.26E-04 | 65 |

|
| 3 | STRE* | Stress response element | 17.82 | 7.57E-04 | 48 |

|
| 4 | CSRE* | Carbon source responsive element; gluconeogenesis | 14.81 | 1.40E-03 | 136 |
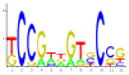
|
| 5 | UASPHR | DNA repair; DNA damage response | 13.78 | 4.60E-02 | 189 |

|
| 6 | LEU3* | Amino acid biosynthesis | 11.74 | 9.74E-04 | 47 |

|
| 7 | MIG1c* | Carbohydrate metabolism; glucose repression | 11.70 | 4.37E-03 | 132 |

|
| 8 | MIG1b | Carbohydrate metabolism; glucose repression | 10.53 | 6.29E-02 | 73 |
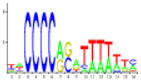
|
| 9 | UME6* | Amino acid metabolism; nitrogen metabolism, mitotic cycle and cell cycle control | 10.20 | 8.75E-03 | 114 |
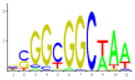
|
| 10 | CAR1_r | Nitrogen metabolism | 8.38 | 1.23E-01 | 114 |

|
TF motifs with Z-score>10 and Fisher scores <0.01. This combination of empirically-derived thresholds has been used to discriminate relevant binding sites in reference sets of genes while reducing the false positive rate to <10% in simulations using random promoter sequences (Ho Sui et al., 2005).
The HXK2 gene is highly expressed during growth on glucose (Moreno & Herrero, 2002). Hxk2p is involved in both hexose phosphorylation and the regulation of glucose repression (Entian et al., 1984; Rose et al., 1991). Hxk2 interacts with Mig1 to form a complex located in the nucleus that mediates glucose repression (Ahuatzi et al., 2007). Deletion of HXK2 leads to expression of a number of genes normally subject to carbon catabolite repression (Diderich et al., 2002). The data showed that HXK2 expression was down-regulated 18-fold during fermentation (supplementary Table S2; Cluster 20 in Fig. 1). The significant down-regulation of HXK2 expression during fermentation of grape must, may, therefore, contribute to the expression of genes that are normally repressed by glucose.
Proposed models for alleviation of glucose repression
A number of explanations are proposed for the observed attenuation of glucose repression. Firstly, there could be genetic differences between the industrial yeast strain used (Vin13) and the laboratory strains, resulting in an altered response to glucose. There is no evidence to suggest this is the case as Vin13 has been shown to respond comparably to laboratory strains to osmotic stress and nitrogen catabolite repression (Erasmus et al., 2003; Marks et al., 2003). Secondly, stress, particularly increasing ethanol concentration, could disrupt the structure of membranes affecting membrane-bound glucose sensors and/or could alter protein structure of other proteins involved in signaling. Thirdly, the increase in expression of glucose-repressed genes might be due to the activation of the retrograde response pathway. Because active dry yeast is prepared in a highly aerobic environment, the yeast cells contain large numbers of respiratory-efficient mitochondria at inoculation. Prolonged fermentative metabolism and aging of the cells would cause mitochondrial dysfunction, potentially inducing the retrograde response. This is unlikely, as the induction of no peroxisomal PEX genes was found. Fourthly, a yet unidentified ethanol-sensing mechanism that functions in the presence of excess glucose might exist. Finally, the significant down-regulation (18-fold) of HXK2 might contribute to relieve glucose repression during wine fermentations (Entian et al., 1984; Rose et al., 1991; Diderich et al., 2002; Ahuatzi et al., 2007). The down-regulation of HXK2 might also be partially responsible for stuck alcoholic wine fermentations.
The most noticeable change during wine fermentation is the decrease in fermentable sugars and the accompanied rapid increase in ethanol concentration. Ethanol’s dual role as a stressor to the cell and potential carbon source during respiration that may follow fermentation, along with our results, suggest that the yeast cell has adapted a unique mechanism to respond to the presence of ethanol independent of the glucose concentration. The response of yeast to ethanol is not detectable in classic laboratory studies because low concentrations of glucose (2% w/v) yield low concentrations of ethanol <1% (v/v). To further complicate matters, oxygen is limiting during grape must fermentations, thereby preventing the effective utilization of ethanol as a carbon source. Furthermore, down-regulation of HXK2 expression is not observed during short-term laboratory fermentations. It is likely that glucose repression is alleviated in response to both ethanol and oxygen in the environment and limited or no Hxk2p.
Ethanol as a regulator of fermentation
The main product of fermentation is ethanol, and wines contain as much as 16% (v/v) of this compound. Ethanol inhibits yeast growth and viability as it negatively affects membrane integrity as well as intracellular and membrane-related processes (Ingram & Buttke, 1984; Leao & Van Uden, 1984; Lloyd et al., 1993; Piper, 1995; Alexandre et al., 2001). Ethanol, at concentrations affecting growth and fermentation rates [3–10% (v/v)], causes potent activation of the plasma membrane H+-ATPase – a probable mechanism to regulate the yeast cell’s internal pH (Rosa & Sa-Correia, 1991). Although ethanol is the main stress factor during fermentation, relatively little is known of its effect on the transcriptome and proteome of yeast.
The short-term response of a laboratory strain of S. cerevisiae to a 7% (v/v) ethanol spike during the early exponential phase was investigated by global expression analysis (Alexandre et al., 2001). The yeast responded by increasing expression of genes involved in energy metabolism, stress response, protein trafficking, and ionic homeostasis. Comparison with the Alexandre data set reveals that 41 ethanol stress genes were induced during fermentation; included within the overlapping genes are the heat shock genes HSP104, HSP26, HSP30, HSP42, HSP78, SSA1, SSA4, and SSE1, and the ethanol stress gene GRE3. However, other ethanol stress genes were not activated in in this study, including HSP12, GPD1, ALD2, HSP82, HOR2, and DAK1. Ten short-term ethanol stress genes are present in the FSR, comprising 5% of the FSR (Table 2).
A genome-wide screen of the yeast deletion collection for ethanol-sensitive mutants on complex medium supplemented with 2% glucose and 6% ethanol identified 46 mutants with impaired growth (van Voorst et al., 2006). Genes that were required for growth included those involved in the general stress pathway, cell integrity pathway, vacuolar function, and mitochondrial function. Two of these (BEM2, SIT4) exhibited slow growth when further tested in fermentations supplemented with 20% glucose. None of the identified genes are part of the FSR; eight were induced in clusters 7–12 and 10 were repressed in clusters 15–20 (Fig. 1). However, other genes with similar functional profiles are present in the FSR, such as genes involved in vacuolar transport and mitochondrial function (Table 2), suggesting the activation of these pathways to regulate the response to ethanol.
Osmotic stress
The high sugar concentration (21.4% w/v) in the Riesling grape must is a source of osmotic stress to the yeast cell. This stress is partially relieved during fermentation due to the conversion of sugar to ethanol and CO2, as evidenced by an increase in water activity from an initial value of 0.965–0.987 at the last sampling point when ethanol is c. 10% (v/v). The osmo-regulatory response of S. cerevisiae results in the enhanced production and intracellular accumulation of glycerol as the main compatible solute to counter-balance osmotic pressure (Hohmann, 2002). This is mediated by the high osmolarity glycerol (HOG) pathway (Brewster et al., 1993). Genes induced by high salt or high sorbitol were compared with the genes in this data set; a total of 104 of 186 genes induced by high salt/sorbitol (Rep et al., 2000) were induced during the course of fermentation. 22 of these genes were present in the FSR (Table 2).
Analysis of gene expression patterns in a wine yeast strain subjected to 40% (w/v) sugar stress identified 589 genes with altered expression patterns (Erasmus et al., 2003). It was found that 232 of the sugar-induced stress genes are also induced during fermentation; 50 of these are in the FSR (Table 2). Osmotic stress-responding genes, based on the union of the Rep et al. (2000) and Erasmus et al. (2003) data sets, comprise 27% of the FSR, indicating that osmotic stress plays a role in the FSR.
Nature of the signal for entry into stationary growth
Entry into the stationary phase is common to adverse conditions and is reflected by the dramatic reduction of ribosome biogenesis (Warner, 1999; Nomura, 2001; Miyoshi et al., 2001, 2003; Jorgensen et al., 2004). Consistently, it is observed that the majority of repressed genes are preferentially annotated to protein metabolism, translation, and ribosome biogenesis and assembly (Fig. 4). Analysis of TFBS motifs in repressed genes shows enrichment of the rRNA processing element (RRPE), polymerase A and C (PAC) and repressor activator protein (RAP1) motifs (supplementary Table S4). These motifs are conserved within the promoters of many genes implicated in cell growth and ribosome synthesis (Moehle & Hinnebusch, 1991; Li et al., 1999; Hughes et al., 2000;Wade et al., 2001; Fingerman et al., 2003).
Under laboratory conditions (i.e. low concentrations of fermentable carbon sources such as glucose that is present at 2% in standard media), yeast cells enter diauxic growth when glucose is depleted, and subsequently enter into the stationary phase when ethanol is depleted (Johnston & Carlson, 1992; DeRisi et al., 1997). Carbon sources never become depleted during vinification, and carbon depletion, therefore, cannot be responsible for entry of yeast cells into the stationary phase during fermentation. Nitrogen sources, however, can become limiting during fermentation and is the main cause of problematic fermentations. These data show that yeast cells in fermenting grape musts enter the stationary phase as early as 32 h after inoculation when glucose is in excess and the ethanol concentration is ~2.0% v/v (Fig. 5). This is consistent with reports by Rossignol et al. (2003).
Fig. 5.

Growth of Vin13 in Riesling grape juice containing either 191, 300, 600, or 900mgL−1 N. The nitrogen content of Riesling grape juice containing 191mgL−1 N was adjusted to either 300, 600, or 900mgL−1 N using DAP. ADY of Vin13 was inoculated to 3×106 cells mL−1 in 250- mL Kimax bottles fitted with vapour locks containing 200 mL Riesling grape juice. Ethanol concentrations (% v/v) are indicated on the graph.
To test the hypothesis that nitrogen limitation is responsible for yeast cells entering into the stationary phase during fermentation, fermentations were conducted that contained increasing concentrations of di-ammonium phosphate (DAP); yeast growth, nitrogen utilization, and ethanol production were monitored during the course of the fermentation. Surprisingly, yeast cells entered into stationary phase at the same time point in all the fermentations, independent of nitrogen or carbon depletion (Fig. 5). However, the common factor in all instances was an ethanol concentration of c. 2.0% (v/v). No further growth occurred after 50 h when the ethanol concentration reached ~5.0% (v/v) despite the fact that nitrogen was in excess. After 32 h, the ammonium concentration in the control grape must, and must supplemented with 300, 600, or 900 mg L−1 DAP was 0, 63, 262, and 480 mg L−1, respectively. The free alpha amino nitrogen content was 13, 23, 39, and 59 mg L−1, respectively. This experiment was repeated in yeast nitrogen base (Difco) containing 21.4% sugars (equimolar amounts of glucose and fructose); nitrogen was adjusted with DAP to 191, 300, 600, or 900 mg L−1. Yeast cells entered the stationary phase after 48 h when the ethanol concentration reached c. 2% (v/v) (data not shown). Ethanol produced during fermentation thus seems to be the trigger for entry into the stationary phase.
Conclusions
Wine fermentations subject yeast to a barrage of stressors, including osmotic pressure, hypoxia, nitrogen depletion, and increasing ethanol concentrations. Global genomic expression patterns over the duration of fermentation revealed an integrated compendium of stress responses as well as a novel long-term adaptive response, which is referred to as the FSR. Approximately 28% of FSR genes have not yet been characterized. FSR genes exhibit sustained and dramatic induction under fermentation conditions and further studies will be required to elucidate their roles under wine-making conditions, which differ considerably from standard laboratory conditions. This study hypothesized that ethanol acts as a signal that activates a hitherto unidentified ethanol signal transduction pathway regulating genes in the FSR. The identification and characterization of regulatory circuits that govern the FSR will provide an insight into the remarkable ability of S. cerevisiae to flourish and ferment grape must and then survive the hostile environment of wine for months. A concerted study of yeast during wine fermentation will also lead to annotation of many of the orphan genes in the FSR. We cannot, however, exclude the possibility that fermentation of different grape musts that vary in composition might reveal more FSR genes. Finally, the results indicate that contrary to previous reports, growth arrest of yeast cells was not due to depletion of nitrogen in fermenting grape must; ethanol seems to be the trigger for entry into the stationary phase. These data suggest that studies restricted to standard laboratory conditions are inadequate to understand the regulation of yeast metabolism in industrial fermentations and the regulatory role of ethanol during wine fermentation should be explored.
Supplementary Material
Acknowledgments
H.J.J.v.V. thanks the National Sciences and Engineering Research Council of Canada (NSERC) (grant 217271-04) and the American Vineyard Foundation for financial support. J.B. is a Scholar of the Michael Smith Foundation for Health Research (MSFHR) and is also funded by NSERC. W.W.W. acknowledges support from the Canadian Institutes of Health Research (CIHR), the Canada Foundation for Innovation (CFI), and the MSFHR. S.H.S. is funded by NSERC and is MSFHR Senior Research Trainee. J.B. is supported by CIHR. G.K.v.d.M. acknowledges support from NSERC grant 262276-03.
Footnotes
Authors’ contribution
V.D.M. and S.J.H.S. contributed equally to this paper.
The following supplementary material is available for this article:
Table S1. Position weight matrix models for transcription factor binding sites used in the regulatory analysis.
Table S2. Probe-set-summarized, normalized expression measures and data analysis for RNA hybridized to Affymetrix S98 arrays. Cells were harvested during fermentations at 0.5%, 2%, 3.5%, 7% and 10% (v/v) ethanol, corresponding to approximately 24 h, 48 h, 60 h, 120 h, and 340 h time points, respectively.
Table S3. (A) Forty most highly glucose repressed genes (derived from Young et al., 2003). (B) Induced genes involved in mitochondrial respiration/oxidative phosphorylation. (C) Induced genes involved in sterol biosynthesis. (D) Induced genes involved in oxidative stress. (E) Induced genes annotated to carbohydrate metabolism.
Table S4. Identification of over-represented transcription factor binding site motifs in each gene cluster. Scores are also shown for genes in larger groupings of the clusters.
Fig. S1. Venn diagram showing the number of induced or repressed genes in fermentation that are associated with transcriptionally characterized stress responses. Environmental Stress Response (ESR), Common Environmental Response (CER), Fermentation Stress Response (FSR).
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1567-1364.2007.00338.x (This link will take you to the article abstract).
Please note: Blackwell Publishing is not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ahuatzi D, Riera A, Pelaez R, Herrero P, Moreno F. Hxk2 regulates the phosphorylation state of Mig1 and therefore its nucleocytoplasmic distribution. J Biol Chem. 2007;282:4485–4493. doi: 10.1074/jbc.M606854200. [DOI] [PubMed] [Google Scholar]
- Alexandre H, Ansanay-Galeote V, Dequin S, Blondin B. Global gene expression during short-term ethanol stress in Saccharomyces cerevisiae. FEBS Lett. 2001;498:98–103. doi: 10.1016/s0014-5793(01)02503-0. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Bryan J. Problems in gene clustering based on gene expression data. J Multivariate Anal. 2004;90:44–66. [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney A, Storey JD. R package version 1.1. 2006. q-value: Q-value estimation for false discovery rate control. [Google Scholar]
- Dennis RA, McCammon MT. Acn9 is a novel protein of gluconeogenesis that is located in the mitochondrial intermembrane space. Eur J Biochem. 1999;261:236–243. doi: 10.1046/j.1432-1327.1999.00267.x. [DOI] [PubMed] [Google Scholar]
- DeRisi JL, Vishwanath RI, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Devantier R, Scheithauer B, Villas-Boas SG, Pedersen S, Olsson L. Metabolite profiling for analysis of yeast stress response during very high gravity ethanol fermentations. Biotechnol Bioeng. 2005;90:703–714. doi: 10.1002/bit.20457. [DOI] [PubMed] [Google Scholar]
- De Vit MJ, Waddle JA, Johnston M. Regulated nuclear translocation of the Mig1 glucose repressor. Mol Biol Cell. 1997;8:1603–1618. doi: 10.1091/mbc.8.8.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diderich JA, Raamsdonk LM, Kuiper A, Kruckeberg AL, Berden JA, Teixeira de Mattos MJ, Van Dam K. Effects of a hexokinase II deletion on the dynamics of glycolysis in continuous cultures of Saccharomyces cerevisiae. FEMS Yeast Res. 2002;2:165–172. doi: 10.1111/j.1567-1364.2002.tb00081.x. [DOI] [PubMed] [Google Scholar]
- Entian KD, Kopetzki E, Frohlich KU, Mecke D. Cloning of hexokinase isoenzyme PI from Saccharomyces cerevisiae: pI transformants confirm the unique role of hexokinase isoenzyme PII for glucose repression in yeasts. Mol Gen Genet. 1984;198:50–54. doi: 10.1007/BF00328699. [DOI] [PubMed] [Google Scholar]
- Erasmus DJ, van der Merwe GK, van Vuuren HJJ. Genome-wide expression analyses: metabolic adaptation of Saccharomyces cerevisiae to high sugar stress. FEMS Yeast Res. 2003;3:375–399. doi: 10.1016/S1567-1356(02)00203-9. [DOI] [PubMed] [Google Scholar]
- Fingerman I, Nagaraj V, Norris D, Vershon AK. Sfp1 plays a key role in yeast ribosome biogenesis. Eukaryot Cell. 2003;2:1061–1068. doi: 10.1128/EC.2.5.1061-1068.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleiss JL. Statistical Methods for Rates and Proportions. John Wiley; New York: 1981. [Google Scholar]
- Gancedo JM. Yeast carbon catabolite repression. Microbiol Mol Biol Rev. 1998;62:334–361. doi: 10.1128/mmbr.62.2.334-361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy – analysis of Affymetrix genechip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev. 2002;66:300–372. doi: 10.1128/MMBR.66.2.300-372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S, Mager WH. Yeast Stress Responses. Springer-Verlag; Berlin: 2003. [Google Scholar]
- Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005;33:3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Ingram LO, Buttke TM. Effects of alcohols on micro-organisms. Adv Microb Physiol. 1984;25:253–300. doi: 10.1016/s0065-2911(08)60294-5. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Johnston M. Feasting, fasting and fermenting. Glucose sensing in yeast and other cells. Trends Genet. 1999;15:29–33. doi: 10.1016/s0168-9525(98)01637-0. [DOI] [PubMed] [Google Scholar]
- Johnston M, Carlson M. The Molecular Biology of the Yeast Saccharomyces: Gene Expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1992. [Google Scholar]
- Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004;18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao C, Van Uden N. Effects of ethanol and other alkanols on passive proton influx in the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 1984;774:43–48. doi: 10.1016/0005-2736(84)90272-4. [DOI] [PubMed] [Google Scholar]
- Lenhard B, Wasserman WW. TFBS: computational framework for transcription factor binding site analysis. Bioinformatics. 2002;18:1135–1136. doi: 10.1093/bioinformatics/18.8.1135. [DOI] [PubMed] [Google Scholar]
- Li B, Nierras CR, Warner JR. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol Cell Biol. 1999;19:5393–5404. doi: 10.1128/mcb.19.8.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd D, Morrell S, Carlsen HN, Degn H, James PE, Rowlands CC. Effects of growth with ethanol on fermentation and membrane fluidity of Saccharomyces cerevisiae. Yeast. 1993;9:825–833. doi: 10.1002/yea.320090803. [DOI] [PubMed] [Google Scholar]
- Lodi T, Alberti A, Guiard B, Ferrero I. Regulation of the Saccharomyces cerevisiae DLD1 gene encoding the mitochondrial protein D-lactate ferricytochrome c oxidoreductase by HAP1 and HAP2/3/4/5. Mol Gen Genet. 1999;262:623–632. doi: 10.1007/s004380051125. [DOI] [PubMed] [Google Scholar]
- Marks VD, van der Merwe GK, van Vuuren HJJ. Transcriptional profiling of wine yeast in fermenting grape juice: regulatory effect of diammonium phosphate. FEMS Yeast Res. 2003;3:269–287. doi: 10.1016/S1567-1356(02)00201-5. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Miyakawa T, Mizuta K. Repression of rRNA synthesis due to a secretory defect requires the C-terminal silencing domain of Rap1p in Saccharomyces cerevisiae. Nucleic Acids Res. 2001;29:3297–3303. doi: 10.1093/nar/29.16.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Shirai C, Mizuta K. Transcription of genes encoding trans-acting factors required for rRNA maturation/ribosomal subunit assembly is coordinately regulated with ribosomal protein genes and involves Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:1969–1973. doi: 10.1093/nar/gkg278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle CM, Hinnebusch AG. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:2723–2735. doi: 10.1128/mcb.11.5.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno F, Herrero P. The hexokinase 2-dependent glucose signal transduction pathway of Saccharomyces cerevisiae. FEMS Microbiol Rev. 2002;26:83–90. doi: 10.1111/j.1574-6976.2002.tb00600.x. [DOI] [PubMed] [Google Scholar]
- Nehlin JO, Ronne H. Yeast MIG1 repressor is related to the mammalian early growth response and Wilms’ tumour finger proteins. EMBO J. 1990;9:2891–2898. doi: 10.1002/j.1460-2075.1990.tb07479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M. Ribosomal RNA genes, RNA polymerases, nucleolar structures, and synthesis of rRNA in the yeast Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol. 2001;66:555–565. doi: 10.1101/sqb.2001.66.555. [DOI] [PubMed] [Google Scholar]
- Pena-Castillo L, Hughes TR. Why are there still over 1000 uncharacterized yeast genes? Genetics. 2007;176:7–14. doi: 10.1534/genetics.107.074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper PW. The heat shock and ethanol stress responses of yeast exhibit extensive similarity and functional overlap. FEMS Microbiol Lett. 1995;134:121–127. doi: 10.1111/j.1574-6968.1995.tb07925.x. [DOI] [PubMed] [Google Scholar]
- Puig S, Perez-Ortin JE. Stress response and expression patterns in wine fermentations of yeast genes induced at the diauxic shift. Yeast. 2000;16:139–148. doi: 10.1002/(SICI)1097-0061(20000130)16:2<139::AID-YEA512>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2006. URL http://www.R-project.org. [Google Scholar]
- Rep M, Krantz M, Thevelein JM, Hohmann S. The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J Biol Chem. 2000;275:8290–8300. doi: 10.1074/jbc.275.12.8290. [DOI] [PubMed] [Google Scholar]
- Rosa MF, Sa-Correia I. In vivo activation by ethanol of plasma membrane ATPase of Saccharomyces cerevisiae. Appl Environ Microbiol. 1991;57:830–835. doi: 10.1128/aem.57.3.830-835.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M, Albig W, Entian KD. Glucose repression in Saccharomyces cerevisiae is directly associated with hexose phosphorylation by hexokinases PI and PII. Eur J Biochem. 1991;199:511–518. doi: 10.1111/j.1432-1033.1991.tb16149.x. [DOI] [PubMed] [Google Scholar]
- Rossignol T, Dulau L, Julien A, Blondin B. Genome-wide monitoring of wine yeast gene expression during alcoholic fermentation. Yeast. 2003;20:1369–1385. doi: 10.1002/yea.1046. [DOI] [PubMed] [Google Scholar]
- Sandelin A, Hoglund A, Lenhard B, Wasserman WW. Integrated analysis of yeast regulatory sequences for biologically linked clusters of genes. Funct Integr Genomics. 2003;3:125–134. doi: 10.1007/s10142-003-0086-6. [DOI] [PubMed] [Google Scholar]
- Schuller HJ. Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Curr Genet. 2003;43:139–160. doi: 10.1007/s00294-003-0381-8. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Voorst F, Houghton-Larsen J, Jonson L, Kielland-Brandt MC, Brandt A. Genome-wide identification of genes required for growth of Saccharomyces cerevisiae under ethanol stress. Yeast. 2006;23:351–359. doi: 10.1002/yea.1359. [DOI] [PubMed] [Google Scholar]
- Varela C, Cardenas J, Melo F, Agosin E. Quantitative analysis of wine yeast gene expression profiles under winemaking conditions. Yeast. 2005;22:369–383. doi: 10.1002/yea.1217. [DOI] [PubMed] [Google Scholar]
- Wade C, Shea KA, Jensen RV, McAlear MA. EBP2 is a member of the yeast RRB regulon, a transcriptionally coregulated set of genes that are required for ribosome and rRNA biosynthesis. Mol Cell Biol. 2001;21:8638–8650. doi: 10.1128/MCB.21.24.8638-8650.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Young ET, Dombek KM, Tachibana C, Ideker T. Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J Biol Chem. 2003;278:26146–26158. doi: 10.1074/jbc.M301981200. [DOI] [PubMed] [Google Scholar]
- Zuzuarregui A, del Olmo ML. Expression of stress response genes in wine strains with different fermentative behavior. FEMS Yeast Res. 2004;4:699–710. doi: 10.1016/j.femsyr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Zuzuarregui A, Monteoliva L, Gil C, del Olmo M. Transcriptomic and proteomic approach for understanding the molecular basis of adaptation of Saccharomyces cerevisiae to wine fermentation. Appl Environ Microbiol. 2006;72:836–847. doi: 10.1128/AEM.72.1.836-847.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.