

Abstract
Biological N2 fixation to NH3 may proceed at one or more Fe sites in the active-site cofactors of nitrogenases. Modeling individual e−/H+ transfer steps of iron-ligated N2 in well-defined synthetic systems is hence of much interest but remains a significant challenge. While molecular Fe species have been recently demonstrated to catalyze the formation of NH3 from N2, mechanistic details of these processes remain elusive. Herein, we report the synthesis and isolation of a diamagnetic, 5-coordinate formally iron(IV) Fe═NNH2+ species supported by a tris(phosphino)silyl ligand via the direct protonation of a terminally bound Fe-N2− complex. The Fe═NNH2+ complex is redox-active, and low-temperature spectroscopic data and DFT calculations evidence an accumulation of significant radical character on the hydrazido ligand upon one-electron reduction to S = 1/2 Fe═ NNH2. At warmer temperatures, Fe═NNH2 rapidly converts to an iron hydrazine complex, Fe-NH2NH2+, via the additional transfer of proton and electron equivalents in solution. Fe-NH2NH2+ can liberate ammonia, and the sequence of reactions described here demonstrates that an iron site can shuttle from a distal intermediate (Fe═NNH2+) to an alternating intermediate (Fe-NH2NH2+) en route to NH3 liberation from N2. It is interesting to consider the possibility that similar “hybrid” mechanisms for N2 reduction may be operative in biological N2 fixation.
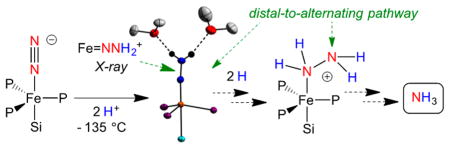
Graphical Abstract
INTRODUCTION
The proton-coupled reduction of dinitrogen (N2) to ammonia (NH3) by nitrogenase enzymes sustains life and has been under study for decades. Known nitrogenases employ a cofactor comprised of seven Fe-atoms and one additional metal site (Mo, V, or Fe).1 Despite a suite of crystallographic, theoretical, and spectroscopic studies,2 the mechanistic details of N2 reduction and the metallic site(s) of N2 coordination are uncertain.
The feasibility of N2 reduction at an Fe or Mo site has been tested with synthetic model complexes.3 Well-defined Mo systems have been reported to catalyze the direct reduction of N2 to NH3 in the presence of proton and electron equivalents,4 and our laboratory has recently disclosed molecular Fe complexes that furnish catalytic yields of NH3 from N2.5 While synthetic studies of the Mo systems have revealed a number of isolable Mo(NxHy) species that inform likely mechanistic scenarios of N2 activation and overall reduction,6,4c similar studies on the Fe catalyzed systems are challenged by the high reactivity of the putative Fe(NxHy) intermediates and their varied spin states.
An iron hydrazido(2−) complex, Fe═NNH2+, has been invoked as a likely intermediate in Fe-catalyzed reaction mixtures with the tris(phosphine)borane (TPB) system (TPB = tris(2-(diisopropylphosphino)phenyl)borane)).5,7 Its detection in operando when both strong acid and reductant are present is not feasible; the species is far too reactive under such conditions. We therefore generated {[TPB]Fe═NNH2}+ at low temperature by double protonation of {[TPB]Fe(N2)}− in the absence of exogenous reductant and characterized this species with a suite of spectroscopic techniques including EPR/ ENDOR, XAS, and Mössbauer spectroscopies.7 {[TPB]Fe═ NNH2}+ decays rapidly at temperatures above −78 °C, frustrating our attempts to purify and study it by X-ray crystallography and to map its further reactivity patterns.
The complex {[SiPiPr3]Fe(N2)}− ([SiPiPr3] = tris(2-(diisopropylphosphino)phenyl)silyl(−)) is isostructural to the {[TPB]Fe(N2)}− catalyst. While catalytic amounts of NH3 (7.0(1) equiv of NH3 per Fe) are generated when {[TPB]Fe-(N2)}− is exposed to the originally reported catalytic conditions (−78 °C in Et2O, 1 atm N2, 50 equiv of {H(OEt2)2}{BArF24}, 60 equiv of KC8), {[SiPiPr3]Fe(N2)}− liberates substoichiometric amounts of NH3 (0.8(1) equiv of NH3 per Fe) under the same conditions.5 We surmised that the doubly protonated form of this species, {[SiPiPr3]Fe═NNH2}+, might be more readily characterized than {[TPB]Fe═NNH2}+ owing to its predicted diamagnetism (18-electron species). Herein, we report its synthesis and high-resolution crystal structure. This isolable Fe═NNH2+ species is derived from protonation of its Fe(N2)− congener. We additionally explore the redox pairs Fe═NNH2+/Fe═NNH2 and Fe═NNMe2+/Fe═NNMe2 and demonstrate the overall conversion of Fe NNH2+ to NH3 via an Fe-NH2NH2+ intermediate. These observations in sum establish that a molecular iron system can traverse both distal (Fe═NNH2) and alternating (Fe-NH2NH2) intermediates en route to NH3 formation from N2, providing synthetic precedent for a new “hybrid” pathway for Fe-mediated N2 reduction (Scheme 1).7,8
Scheme 1.

Distal and Alternating Pathways for N2 Reduction, and the Hybrid N2 Reduction Pathway Emphasized Herein
RESULTS AND DISCUSSION
As for {[TPB]Fe(N2)}−,7 the successful protonation of Fe(N2) complexes supported by the [SiPiPr3] ligand required very low temperatures.9 For example, the addition of 1 or 2 equiv of the acid {H(OEt2)2}{BArF24} to {K(Et2O)}{[SiPiPr3]Fe(N2)} (1) at −78 °C resulted in the immediate formation of mixtures containing both one-electron oxidized [SiPiPr3]Fe(N2) (2) and two-electron oxidized {[SiPiPr3]Fe(N2)}{BArF24} (3). These proton-induced oxidation reactions likely proceed via an unstable and as yet unobserved iron diazenido species, [SiPiPr3]Fe(NNH) (4), structurally and electronically related to the previously reported and stable silyldiazenido complex, [SiPiPr3]Fe(NNSiMe3).9 An alternative hydride product, [SiPiPr3]Fe(N2)(H), that would derive from protonation at iron instead of N2 is not observed; [SiPiPr3]Fe(N2)(H) is a very stable complex that has been characterized,10 and were it produced as the kinetic product of protonation, we would anticipate observing it, as it should also be the thermodynamically preferred isomer.
Combination of 1 with 5 equiv of {H(OEt2)2}{BArF24} in thawing 2-MeTHF at −135 °C instead produced a pale lavender solution (Figure 1A) with UV–visible features that are distinct from the oxidation products Fe-N2 2 and Fe-N2+ 3. The in situ 57Fe Mössbauer spectrum (Figure 1B) collected on similarly prepared solutions derived from 57Fe-enriched 1 evidences a new integer-spin Fe complex (δ = 0.126 mm/s and ΔEQ = 1.484 mm/s) assigned as Fe═NNH2+ 5 (vide infra), that constitutes ~90% of the Fe in solution; Fe-N2+ 3 is present as a minor (~10%) component. Compound 5 is persistent for hours in solution at temperatures of −78 °C once prepared in this manner but is increasingly unstable as the solution is warmed further.
Figure 1.

Spectroscopic data collected in situ on compound 5. (A) UV–visible absorbance spectra of 3, 2, and 5. Spectra were collected in 2-MeTHF at −80 °C. (B) Zero-field 57Fe Mössbauer spectra of 57Fe-enriched 5 as a 3 mM solution in 2-MeTHF prepared from 1 and collected at 80 K. The minor component (10%) was identified as complex 3 derived from competitive oxidation.
The isolation of 5 as a crystalline solid free of Fe-containing impurities was facilitated by the substitution of the BArF24 counteranion with a less-soluble analogue. The reaction of 1 with 3 equiv of trifluoromethanesulfonic acid (HOTf) proceeded similarly at −135 °C, but this compound (5′) could be effectively precipitated out of solution in 49% yield by the addition of pentane at temperatures of −78 °C or lower (Scheme 2). The 57Fe Mössbauer spectrum of solid 5′ (Supporting Information) reveals a single Fe-containing species with parameters that are similar to those of 5 in frozen solution (Figure 1B). Solid 5′ displays intense vibrational features at 3207 and 3039 cm−1 that shift to 2380 and 2241 cm−1 in 5′-d2 (prepared from the reaction of 1 with DOTf) assigned to N–H stretching frequencies with strong hydrogen bonding interactions.11,12 These vibrational features persist in solid samples of 5′ that have been stored for days at −30 °C in the absence of air and moisture.
Scheme 2.

Functionalization of [SiPiPr3]Fe(N2) Complexes
Characterization of {[SiPiPr3]Fe═NNH2}+
The stability of 5 and 5′ in solution at −78 °C permitted growth of single crystals suitable for X-ray diffraction, and their respective structures are depicted in Figure 2. The structures differ in that 5 features an independent Et2O molecule hydrogen bonded to each of the protons of the NNH2 ligand, and in 5′ the NNH2 protons feature tight hydrogen bonding interactions with the triflate anion, and these interactions form the basis of dimeric (5′)2 units in the crystal lattice (Supporting Information). The structures are nonetheless highly similar with respect to the Fe═NNH2+ subunit; short Fe–N distances (~1.67 Å) are found that reflect substantial Fe–N multiple bond character (Table 1), a characteristic feature of most terminal metal hydrazido(2−) complexes.6,11 The αN-atoms display linear geometries, and the location of the nitrogen-bound protons in the Fourier difference map divulges trigonal-planar βN-atoms. The N–N distances (~1.27 Å) are markedly increased from that displayed by {Na(12-crown-4)2}{[SiPiPr3]Fe(N2)} (1.132(4) Å) and 1 (1.146(4) Å) (Supporting Information).9a The N–N distance in 5′ correlates with a broad feature centered at 1443 cm−1 in the IR spectrum that shifts to 1401 cm−1 in 15N-5′ and is assigned to the ν(NN) stretching frequency. IR features of similar energy have been observed in Mo- and W═NNH2 complexes.11 While a number of X-ray diffraction studies on mononuclear13 and dinuclear14 ═ Fe complexes that support the isomeric diazene ligand (HN NH) have been disclosed, 5 and 5′ are the first crystallographically characterized complexes that contain a terminal Fe═NNH2 unit. The structural parameters they reveal are consistent with those recently deduced from XAS and ENDOR spectroscopies for the catalytically relevant species, [TPB]Fe═NNH2+.7
Figure 2.

X-ray diffraction crystal structure of 5 and core-atom structures of 5′, 6, and 8 with thermal ellipsoids drawn at 50% probability. Hydrogen atoms (excepting the N–H′s), the BArF24 counteranion of 5, the triflate counteranion of 6, and cocrystallized solvent molecules have been removed for clarity. Refer to the Supporting Information for complete crystallographic details.
Table 1.
Crystallographic Bond Metrics
| 5 | 5′ | 6 | 8 | |
|---|---|---|---|---|
| Fe–Na | 1.672(2) | 1.668(2) | 1.691(2) | 1.773(1) |
| N–Na | 1.272(3) | 1.273(3) | 1.270(3) | 1.276(2) |
| Fe–N–Nb | 175.3(2) | 175.0(2) | 174.7(2) | 158.63(9) |
| Si–Fe–Nb | 173.99(6) | 170.82(7) | 164.64(9) | 174.19(4) |
Bond distances in Å.
Bond angle in degrees.
Although Fe═NNH2+ 5′ is a stable solid, solutions of 5′ decompose at temperatures of 0 °C and higher to an intractable mixture of Fe-containing species. Seeking to prepare a more stable analogue of 5′, we reacted 1 with excess MeOTf at −78 °C which, upon warming the reaction mixture to room temperature, precipitated {[SiPiPr3]Fe(NNMe2)}{OTf} (6) as a purple solid. Unlike the isoelectronic Fe═NNH2+ species, Fe═NNMe2+ 6 is quite stable both in the solid state and in solution. The relevant metrical data derived from the solid-state crystal structure of 6 (Figure 2) are similar to those of 5′ (Table 1).
Compounds 5′ and 6 exhibit diamagnetic ground states, permitting their further characterization by multinuclear NMR spectroscopies (Figure 3).15 A single broad resonance is found in the 31P{1H} NMR spectrum of 5′, consistent with averaged 3-fold symmetry in solution. Compound 15N-5′ exhibits two resonances in the 15N NMR spectrum at δ = 518 and 198 ppm, corresponding to the αN- and βN-atoms, respectively (Figure 3A).6b,16 The resonance at δ = 198 ppm appears as a triplet of doublets (1JNH = 96 Hz, 1JNN = 11 Hz) whereas the feature at 518 ppm is broadened due to unresolved coupling to the phosphine ligands. In the 1H NMR spectrum, 15N-5′ displays a broad doublet (1JNH = 97 Hz) at δ = 9.5 ppm assigned to the NNH2 protons. The magnitude of the 1JNH coupling constant in 5′ is consistent with sp2 hybridization at the βN-atom17 and similar to that found in other terminal metal-hydrazido(2−) complexes.6b,16 These data confirm that the structure of 5′ found in the solid state is maintained in solution. Related NMR data for 6 reproduces the salient features exhibited by 5′ (Supporting Information).
Figure 3.

NMR spectra of 5′ recorded at −60 °C in 9:1 THF-d8:CD3CN. (A) 15N NMR spectrum of 15N-5′. (B) 31P{1H} NMR spectrum of 5′. (C) Overlaid 1H and 1H{15N} spectra of 15N-5′. The central feature in the 1H spectrum results from contamination of 15N-5′ with the natural abundance 5′.
Redox Chemistry of [SiPiPr3]Fe═NNR2+
The intermediacy of Fe═NNH2+ 5′ in the formation of NH3 requires additional proton or electron equivalents. Both 5 and 5′ were found to be stable at −78 °C to the presence of additional proton equivalents; we therefore explored the one-electron reduction chemistry of 5′ and Fe═NNMe2+ 6 to generate neutral [SiPiPr3]Fe═NNH2 (7) and its methylated derivative [SiPiPr3]Fe═NNMe2 (8), respectively. Cyclic voltammetry measurements on THF electrolytes of Fe═NNMe2 6 reveal a reversible reduction event at −1.73 V (Supporting Information). The chemical reduction of 6 with 1 equiv of Na(Hg) (Scheme 2) and subsequent workup furnished paramagnetic 8, whose crystal structure (Figure 3) reveals a lengthened Fe–N distance (from 1.69 to1.77 Å) concomitant with substantial bending at the αN-atom from 175° to 159° (Table 1). The βN-atom retains sp2 hybridization, and the N–N bond length is essentially unchanged. The X-band EPR spectrum of 8 indicates an S = 1/2 ground state (Figure 4A, gavg = 2.04), consistent with its room temperature magnetic moment in C6D6 (μeff = 1.7 μB). Magnetically perturbed 57Fe Mössbauer studies of 8 (Figure 4B) demonstrate strong 57Fe hyperfine coupling and much slower relaxation properties compared to Fe(N2) 2 (Supporting Information); distinctive features that span a range of 5 mm/s at temperatures of 80 K and lower are observed.
Figure 4.

(A) X-band EPR spectra of Fe═NNH2 7 and 7-d2, derived from the in situ reduction of 5′ or 5′-d2, respectively, with Cp*2Co; Fe═NNMe2 8 and 15N-8 collected at 77 K in 2-MeTHF glasses. Signals derived from S = 1/2 Fe-N2 2 have been subtracted from the displayed spectra of 7 and 7-d2 for clarity. (Inset) Prominent features of 8 that differ in 15N-8. These features arise from hyperfine coupling to single 31P and single 14/15N nuclei of comparable magnitude. (B) 57Fe Mössbauer spectra of in situ-prepared 7 and 8 obtained by subtracting out quadrupole doublet impurities from the raw data. A 50 mT magnetic field was applied (left) perpendicular and (right) parallel to the propagation of γ-beam. The solid lines are theoretical fits to an S = 1/2 spin Hamiltonian operating in the slow relaxation regime. Refer to the Supporting Information for a detailed discussion and the derived spin Hamiltonian parameters.
Fe═NNH2 7 is far less stable than 8 and required characterization at cryogenic temperatures. Compound 5′ reacted with Cp*2Co in 2-MeTHF at −135 °C to produce dark brown solutions that rapidly bleached when warmed to −78 °C or higher temperatures (vide infra). EPR (Figure 4A) and 57Fe Mössbauer spectra (Figure 4B) collected on similarly prepared frozen reaction mixtures derived from 5′ confirmed the generation of a new S = 1/2 species (gavg = 2.04) as the major Fe-containing component. Notably, the 57Fe Mössbauer spectrum of this complex is nearly identical to that displayed by Fe═NNMe2 8, allowing us to assign it as the isoelectronic species 7. Accordingly, the theoretically predicted gas-phase optimized geometry and electronic structure of 7 are very similar to those of 8 (Supporting Information). Compounds 7 and 8 are predicted to have substantial radical character on the NNR2 and phosphine ligands, as also evident from their respective EPR data. Differences between the X-band EPR spectra of 15N-8 and 8 establish strong hyperfine coupling (ca. 30 MHz) to a single N-atom (Inset of Figure 4A). In addition, marked differences in the EPR spectra of Fe═NNH2 7 and Fe═NND2 7-d2 demonstrate significant hyperfine coupling to one or both nitrogen-bound H-atom(s); this value is estimated to be as high as 25 MHz through analysis of the second derivative EPR spectrum at g2 and g3 (Supporting Information. The related S = 1/2 {[TPB]Fe(NNH2)}+ exhibited 1H hyperfine coupling as large as 18 MHz between g2 and g3.7
Conversion of [SiPiPr3]Fe═NNH2+ to [SiPiPr3]Fe-NH2NH2+
Upon warming to temperatures of −78 °C and higher, solutions that contain Fe═ NNH2 7 and Fe═NNH2+ 5′ undergo a spontaneous disproportionation to a mixture of Fe species that include the previously reported iron hydrazine complex, {[SiPiPr3]Fe(NH2NH2)}{OTf} (9),9a as a major component. Thawing THF solutions of 5′ were combined with stoichiometric Cp*2Co and allowed to warm slowly to room temperature over 10 min. After minimal workup, NMR analyses of the resulting mixtures (Supporting Information) revealed the formation of roughly equal amounts of Fe-NH2NH2+ 9 and Fe-N2 2 as major products, alongside small amounts of [SiPiPr3]Fe(OTf) (10) and {[SiPiPr3]Fe(NH3)}− {OTf} (11).9 9 was also detected in a one-pot reaction from Fe-N2− 1 via the sequential addition of 2 equiv of HOTf and 0.5 equiv of Cp*2Co to 1 in 2-MeTHF at −135 °C. Significant quantities of free N2H4 (0.53(6) equiv per Fe) and NH3 (0.16(2) equiv) were detected when these reaction mixtures were quenched with HCl 10 min after warming. This product distribution was found to be time dependent: reaction mixtures quenched after standing at room temperature for 24 h revealed the presence of 0.27(6) equiv of N2H4 and 0.39(5) equiv of NH3, establishing further conversion of N2H4 to NH3 in these mixtures.
The overall formation of both N2H4 and NH3 from an Fe═NNH2+/Fe═NNH2 redox pair is interesting, given that the complex [SiPPh3]Fe(N2) was observed to liberate significant amounts of N2H4 upon exposure to HBF4(Et2O) and CrCl2 some years ago,18 whereas the anion {[SiPiPr3]Fe(N2)}− was instead observed to liberate NH3 in the presence of KC8 and {H(OEt2)2}{BArF24};5 N2H4 is anticipated to gradually degrade to NH3 under the latter conditions,5 and hence, even if formed as an intermediate product, its concentration may not build up.
The formation of the Fe-containing products Fe-NH2NH2+ 9 and Fe-N2 2 can most simply be rationalized by the reaction sequence shown in Scheme 3. As discussed above, 57Fe Mössbauer and EPR studies indicate that 5′ is reduced by Cp*2Co to generate neutral Fe═NNH2 7 at temperatures as low as −135 °C. At higher temperatures, we speculate that in situ-generated 7 reacts bimolecularly with remaining 5′ in solution. Exchange of H+ and e− equivalents between these two compounds results in the formation of neutral Fe-N2 2 and cationic Fe-NH2NH2+ 9 as the overall reaction products. Whereas 2 is stable to the presence of Cp*2Co, 9 is slowly reduced by Cp*2Co to afford detectable quantities of NH3 and thereby a mixture of 2 and Fe-OTf 10. DFT studies predict that the conversion of 5′ + 7 → 2 + 9 is highly exergonic (−45 kcal/ mol) (Supporting Information).
Scheme 3.

Comparison of the Reaction Products Observed in the Reduction of (A) Fe═NNH2+ 7 and (B) Mo═NNH2 Supported by the Tri(amido)amine [HIPTN3N]3− Ligand Framework19
CONCLUSIONS
The present study has described the thorough characterization, including the first crystallographic evidence, of a terminally bonded Fe═NNH2 species; this formally iron(IV) complex is derived from the activation and protonation of N2 coordinated to iron. Numerous examples of iron oxos, nitrides, and imides featuring strong, covalent iron-to-ligand multiples bonds have been characterized in the past 15 years.20 The possibility to use such covalency as a strategy for N2 reduction to NH3 is a plausible one, and the stoichiometric chemistry established with the present tris(phosphino)silyl iron system underscores this point. Our ability to isolate diamagnetic [SiPiPr3]Fe═NNH2+ and its more stable methylated analogue, [SiPiPr3]Fe═ NNMe2+, enables their thorough characterization and also a study of their one-electron reduction chemistry. The stable S = 1/2 complex [SiPiPr3]Fe═NNMe2 has been structurally characterized, and its other spectroscopic parameters are very similar to those of the far less stable S = 1/2 species [SiPiPr3]Fe═NNH2, which must instead be characterized at very low temperature. Each S = 1/2 species evidences significant spin-leakage onto the hydrazido ligand.
A fascinating transformation occurs as solutions containing in situ-generated [SiPiPr3]Fe═NNH2 and [SiPiPr3]Fe═NNH2+ are allowed to warm, disproportionating to [SiPiPr3]Fe-NH2NH2+ and [SiPiPr3]Fe-N2. Some [SiPiPr3]Fe-NH3+ is also produced in this process; we had previously shown that [SiPiPr3]Fe-NH2NH2+ can liberate [SiPiPr3]Fe-NH3+ and free NH3 in solution. Hence, these collective observations show that iron-bound N2 can be protonated to generate a distal8 intermediate, [SiPiPr3]Fe═NNH2+, and further reduced/ disproportionated to an alternating intermediate,8 [SiPiPr3]Fe-NH2NH2+, that serves as a source of NH3 via late-stage N–N cleavage. The conversion of N2 to NH3 via an N2H4 intermediate therefore does not require an alternating pathway; it can instead be initiated along a distal pathway. Such a scenario is distinct from the early stage N–N cleavage pathway to generate terminal nitrides that is thought to occur in the molybdenum N2 reduction catalysts of Schrock and Nishibayashi, respectively.4
The catalytically relevant [TPB]Fe-N2− system is thought to proceed via a distal S = 1/2 [TPB]Fe═NNH2+ intermediate.7 This species cannot be isolated and independently studied owing to its greater instability and the presence of additional iron components, and it remains unclear whether NH3 production in this case derives from similar late-stage cleavage to first produce N2H4, akin to [SiPiPr3]Fe═NNH2, or if an early-stage cleavage pathway instead generates a terminal iron-bound nitride, such as [TPB]Fe═N or (TPB)Fe═N+. The greater flexibility of the Fe–B bond relative to the Fe–Si bond may afford access to different intermediates. However, that [SiPiPr3]Fe-N2− generates appreciable amounts of NH3 under the same conditions as [TPB]Fe-N2−, and that its isostructural carbon analogue [CPiPr3]Fe-N2− is a catalyst for N2-to-NH3 conversion but is not nearly as flexible as the [TPB]Fe system,5b suggest the possibility and perhaps even likelihood of a unifying distal-to-alternating mechanistic sequence en route to NH3 for these three iron systems.
While we have here demonstrated the viability of a hybrid distal-to-alternating reaction pathway for NH3 generation via N2H4, we still caution that different Fe-mediated N2 reduction systems, with variable reaction conditions, may sample alternative pathways.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM 070757) and the Gordon and Betty Moore Foundation. J.R. was additionally supported by a fellowship from the Caltech Center for Environmental Microbial Interactions.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b01230.
Detailed experimental, spectroscopic, and theoretical data (PDF)
X-ray crystallographic data for 1, 5, 5′, 6, and 8 (CIF)
References
- 1.Howard JB, Rees DC. Chem Rev. 1996;96:2965–2982. doi: 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seefeldt LC, Hoffman BM, Dean DR. Annu Rev Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC. Science. 2014;345:1620–1623. doi: 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hinnemann B, Norskov JK. Top Catal. 2006;37:55–70. [Google Scholar]
- 3.(a) Schrock RR. Acc Chem Res. 2005;38:955–962. doi: 10.1021/ar0501121. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nishibayashi Y. Inorg Chem. 2015;54:9234–9247. doi: 10.1021/acs.inorgchem.5b00881. [DOI] [PubMed] [Google Scholar]; (c) McWilliams SF, Holland PL. Acc Chem Res. 2015;48:2059–2065. doi: 10.1021/acs.accounts.5b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Yandulov DV, Schrock RR. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]; (b) Arashiba K, Kinoshita E, Kuriyama S, Eizawa A, Nakajima K, Tanaka H, Yoshizawa K, Nishibayashi Y. J Am Chem Soc. 2015;137:5666–5669. doi: 10.1021/jacs.5b02579. [DOI] [PubMed] [Google Scholar]; (c) Arashiba K, Miyake Y, Nishibayashi Y. Nat Chem. 2011;3:120–125. doi: 10.1038/nchem.906. [DOI] [PubMed] [Google Scholar]
- 5.(a) Anderson JS, Rittle J, Peters JC. Nature. 2013;501:84–87. doi: 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Creutz SE, Peters JC. J Am Chem Soc. 2014;136:1105–1115. doi: 10.1021/ja4114962. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ung G, Peters JC. Angew Chem, Int Ed. 2015;54:532–535. doi: 10.1002/anie.201409454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Chatt J, Dilworth JR, Richards RL. Chem Rev. 1978;78:589–625. [Google Scholar]; (b) Yandulov DV, Schrock RR. J Am Chem Soc. 2002;124:6252–6253. doi: 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]; (c) Hidai M, Mizobe Y. Chem Rev. 1995;95:1115–1133. [Google Scholar]
- 7.Anderson JS, Cutsail GE, Rittle J, Connor BA, Gunderson WA, Zhang L, Hoffman BM, Peters JC. J Am Chem Soc. 2015;137:7803–7809. doi: 10.1021/jacs.5b03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Acc Chem Res. 2013;46:587–595. doi: 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mankad NP, Muller P, Peters JC. J Am Chem Soc. 2010;132:4083–4085. doi: 10.1021/ja910224c. [DOI] [PubMed] [Google Scholar]; (c) Lee Y, Peters JC. J Am Chem Soc. 2011;133:4438–4446. doi: 10.1021/ja109678y. [DOI] [PubMed] [Google Scholar]
- 10.Fong H, Moret ME, Lee Y, Peters JC. Organometallics. 2013;32:3053–3062. doi: 10.1021/om400281v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehnert N, Tuczek F. Inorg Chem. 1999;38:1659–1670. doi: 10.1021/ic980939+. [DOI] [PubMed] [Google Scholar]
- 12.Steiner T. Angew Chem, Int Ed. 2002;41:48–76. [Google Scholar]
- 13.Field LD, Li HL, Dalgarno SJ, Turner P. Chem Commun. 2008:1680–1682. doi: 10.1039/b802039f. [DOI] [PubMed] [Google Scholar]
- 14.(a) Sellmann D, Soglowek W, Knoch F, Moll M. Angew Chem, Int Ed Engl. 1989;28:1271–1272. [Google Scholar]; (b) Saouma CT, Muller P, Peters JC. J Am Chem Soc. 2009;131:10358–10359. doi: 10.1021/ja903967z. [DOI] [PubMed] [Google Scholar]; (c) Li Y, Li Y, Wang B, Luo Y, Yang D, Tong P, Zhao J, Luo L, Zhou Y, Chen S, Cheng F, Qu J. Nat Chem. 2013;5:320–326. doi: 10.1038/nchem.1594. [DOI] [PubMed] [Google Scholar]
- 15.Solutions of 6 display temperature-dependent paramagnetism consistent with a low-lying triplet state (see Supporting Information for additional details).
- 16.Donovan-Mtunzi S, Richards RL, Mason J. J Chem Soc, Dalton Trans. 1984:1329–1332. [Google Scholar]
- 17.Binsch G, Lambert JB, Roberts BW, Roberts JD. J Am Chem Soc. 1964;86:5564–5570. [Google Scholar]
- 18.Mankad NP, Whited MT, Peters JC. Angew Chem, Int Ed. 2007;46:5768–5771. doi: 10.1002/anie.200701188. [DOI] [PubMed] [Google Scholar]
- 19.(a) Yandulov DV, Schrock RR. Inorg Chem. 2005;44:1103–1117. doi: 10.1021/ic040095w. [DOI] [PubMed] [Google Scholar]; (b) Schrock RR. Angew Chem, Int Ed. 2008;47:5512–5522. doi: 10.1002/anie.200705246. [DOI] [PubMed] [Google Scholar]; (c) Schenk S, Le Guennic B, Kirchner B, Reiher M. Inorg Chem. 2008;47:3634–3650. doi: 10.1021/ic702083p. [DOI] [PubMed] [Google Scholar]
- 20.(a) Saouma CT, Peters JC. Coord Chem Rev. 2011;255:920–937. doi: 10.1016/j.ccr.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hohenberger J, Ray K, Meyer K. Nat Commun. 2012;3:720. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.