

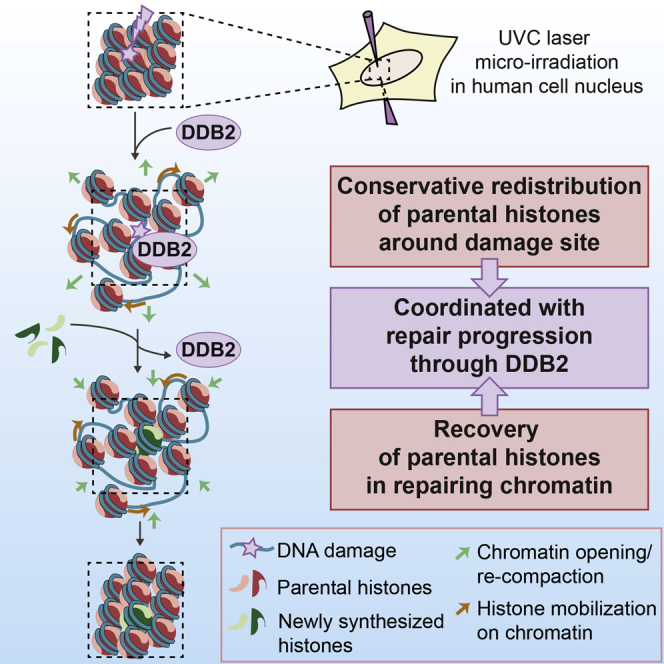
Summary
Chromatin integrity is critical for cell function and identity but is challenged by DNA damage. To understand how chromatin architecture and the information that it conveys are preserved or altered following genotoxic stress, we established a system for real-time tracking of parental histones, which characterize the pre-damage chromatin state. Focusing on histone H3 dynamics after local UVC irradiation in human cells, we demonstrate that parental histones rapidly redistribute around damaged regions by a dual mechanism combining chromatin opening and histone mobilization on chromatin. Importantly, parental histones almost entirely recover and mix with new histones in repairing chromatin. Our data further define a close coordination of parental histone dynamics with DNA repair progression through the damage sensor DDB2 (DNA damage-binding protein 2). We speculate that this mechanism may contribute to maintaining a memory of the original chromatin landscape and may help preserve epigenome stability in response to DNA damage.
Graphical Abstract
Highlights
-
•
Parental H3 histones redistribute to the periphery of UVC-damaged regions
-
•
The redistribution involves histone mobilization on chromatin and chromatin opening
-
•
Parental histones recover massively during repair progression
-
•
Parental histone dynamics may help coordinate DNA repair with epigenome integrity
Adam et al. identify a conservative process coordinated with UVC damage repair progression, whereby parental histones rapidly redistribute away from damaged chromatin regions and subsequently recover. Parental histone dynamics coupled to DNA repair may contribute to maintaining epigenome integrity upon DNA damage.
Introduction
Cellular genomes are constantly exposed to various sources of DNA damage (Ciccia and Elledge, 2010, Hanawalt, 2015, Hoeijmakers, 2009, Jackson and Bartek, 2009), threatening not only genome stability, but also the integrity of chromatin organization (Adam et al., 2015, Lukas et al., 2011, Smeenk and van Attikum, 2013). The basic unit of chromatin is the nucleosome core particle, in which DNA is wrapped around a histone protein octamer comprising an (H3-H4)2 tetramer flanked by two H2A-H2B dimers (Kornberg, 1974, Oudet et al., 1975, Luger et al., 1997). Variations at the level of this repetitive unit, through histone variants and post-translational modifications (Bannister and Kouzarides, 2011, Maze et al., 2014, Talbert and Henikoff, 2010), as well as further chromatin compaction, constitute a major source of information that regulates gene expression and cell identity (Filipescu et al., 2014, Probst et al., 2009). How chromatin is reorganized in response to DNA damage and to which extent the information that it carries can be preserved is thus of fundamental importance.
Our current view of chromatin dynamics following DNA damage in human cells is based on the Access-Repair-Restore (ARR) model (Polo and Almouzni, 2015, Smerdon, 1991). This model postulates that chromatin is first disorganized in response to DNA damage, which facilitates access to repair factors, followed by restoration of chromatin structure. Yet, it is still unclear whether, when, and by which mechanisms the pre-damage chromatin state is restored (Dabin et al., 2016). Notably, chromatin restoration after damage involves the deposition of newly synthesized histones (Adam et al., 2013, Dinant et al., 2013, Luijsterburg et al., 2016, Polo et al., 2006), which could potentially replace damaged histones and leave a mark of the damage experience. Thus, assuming that nucleosome density remains at a steady state, a subset of parental histones should be evicted from chromatin during the Access step, which would limit the capacity to recover the original epigenetic information at damaged sites (Figure 1A). Consistent with this idea, recent reports provide evidence for nucleosome destabilization and histone eviction during DNA double-strand break repair (Goldstein et al., 2013, Li and Tyler, 2016, Xu et al., 2010) and in response to UVC irradiation (Lan et al., 2012, Wang et al., 2006, Zavala et al., 2014). UVC damage sites also show reduced histone density, promoted by the UV damage sensor DDB2 (DNA damage-binding protein 2) (Luijsterburg et al., 2012). However, a massive loss of parental histones from damaged chromatin would certainly threaten epigenome maintenance.
Figure 1.

Rapid Decrease in Parental H3 Histone Density in UVC-Damaged Chromatin Regions
(A) Left: current model for histone dynamics in UVC-damaged chromatin (adapted from Adam et al., 2015). The incorporation of new histones (green) raises questions about the fate of parental histones (red). Right: experimental strategy for tracking parental histone dynamics at DNA damage sites.
(B) Distribution of parental histones H3.3 (red) at the indicated time points after UVC laser damage in U2OS cells stably expressing H3.3-SNAP and GFP-XPC.
(C) Dynamics of parental H3.3 (red) at early time points after local UVC damage in U2OS H3.3-SNAP cells. White arrowheads, illuminated areas. Scale bars, 10 μm. Red fluorescence measured in irradiated areas is normalized to before laser. Error bars, SD from n cells scored in two independent experiments. Labeling parental histones in green instead of red gave similar results (data not shown).
See also Figures S1 and S2 and Movie S1.
To fully understand how chromatin integrity is preserved or altered in response to genotoxic stress, it is critical (1) to examine the fate of parental histones present in chromatin before damage and which carry the original epigenetic information and (2) to track them long enough after damage to examine their contribution to repaired chromatin together with newly deposited histones. We developed two complementary approaches for specific tracking of parental histones following UVC damage in human cells. Challenging our current view of damaged chromatin rearrangements, we show that rather than being massively evicted and lost from damaged chromatin, parental histones redistribute in a conservative manner and subsequently recover. Our mechanistic studies demonstrate that both the redistribution and recovery of parental histones are tightly coordinated with repair progression through the UVC damage sensor DDB2. We thus propose a conservative model by which parental histone dynamics coupled to DNA damage repair contribute to the maintenance of epigenome integrity during the response to UVC damage.
Results
Rapid Reduction of Parental Histone Density in UVC-Damaged Chromatin Regions
We developed two complementary approaches combining UVC laser micro-irradiation with specific tracking of parental histones in living cells. We first took advantage of the SNAP-tag technology, which we exploited previously to follow new histone deposition at UVC damage sites (Adam et al., 2013), to fluorescently label old histones 48 hr prior to irradiation (Figure 1A). We used U2OS cells that stably express H3.3-SNAP histones (Dunleavy et al., 2011) and a GFP-tagged version of the repair factor XPC (xeroderma pigmentosum C) for visualizing damage sites in live cells (Figure S1A). Real-time imaging of parental H3.3 dynamics after local UVC irradiation revealed a rapid reduction of the red fluorescence associated with parental H3.3, which was restricted to the damaged chromatin area marked by GFP-XPC and detectable at least for 1 hr after irradiation (Figure 1B). We observed a similar decrease of parental H3.3 signal in cells that do not express GFP-XPC (Figure 1C; Movie S1), thus showing that exogenous expression of the repair factor XPC does not alter the histone response to UVC. UVC damage led to a progressive reduction of the red fluorescence associated with parental histones, with a maximum of 40% loss 10 min after DNA damage infliction (Figure 1C). We verified that the region of low histone density observed after damage did not correspond to a nucleolus (Figure S1B). We also ruled out the possibility that the decrease in parental H3.3 signal could result from photo-bleaching of the red fluorescence by the UVC laser, as irradiating paraformaldehyde-fixed cells with UVC did not reduce the red signal intensity (Figure 1C). Thus, the observed decrease in old H3.3 signal in UVC-irradiated chromatin regions actually reflects enhanced dynamics of parental H3.3 histones in response to genotoxic stress.
Considering that SNAP-tag-based labeling may interfere with parental H3 dynamics, we also developed a complementary method to track parental histones based on photo-activation of PA-GFP (photoactivatable GFP) in U2OS cells engineered to stably express H3.3-PA-GFP and RFP-XPC (Figures S1A and S1C). Consistent with our previous findings, we observed a marked reduction of parental H3.3 signal in UVC-damaged chromatin regions within minutes after laser micro-irradiation in live cells and not in paraformaldehyde-fixed cells (Figures S1D and S1E). Thus, using two distinct methods to monitor old histone dynamics, our data reveal a local loss of parental H3.3 histone signal in damaged chromatin regions. Furthermore, we observed an altered distribution of parental histones upon UVC damage in all irradiated cells throughout interphase (Figure S2A), and this was not restricted to the H3.3 variant since parental H3.1 signal was also reduced at UVC damage sites (Figures S2A and S2B). We recapitulated these results in MCF7 cancer cells and in BJ primary fibroblasts and observed similar dynamics for parental histone H4 (Figures S2C–S2F).
Collectively, these results reveal a rapid reduction of parental H3 and H4 histone density in UVC-damaged chromatin.
Conservative Redistribution of Parental Histones to the Periphery of UVC-Damaged Regions
We next sought to determine whether the local reduction in parental H3 staining upon UVC irradiation actually reflects parental histone loss from damaged chromatin. For this, we measured changes in old H3.3 fluorescence within the entire cell nucleus after local UVC damage. To maximize sensitivity in this assay, we reduced the size of the area of interest by photo-bleaching H3.3-SNAP-associated fluorescence in the whole nucleus apart from a small patch (Figure 2A). Next, photo-bleaching 40% of the fluorescence inside this patch led to a 20% fluorescence reduction in the entire nucleus (Figure 2B). In contrast, targeting UVC irradiation to the fluorescent patch, while leading to a comparable 40% loss of signal in the irradiated area, did not result in a detectable loss of fluorescence from the entire nucleus (Figure 2B). From these results, we conclude that parental H3 histones mobilized early after genotoxic stress remain in the damaged nucleus and do not undergo massive degradation, as also supported by biochemical analyses of parental H3.3 levels after UVC damage (Figure S2G).
Figure 2.

Conservative Redistribution of Parental Histones to the Periphery of UVC-Damaged Regions
(A) Experimental procedure for measuring parental histone loss and redistribution around the UVC-damaged zone. Microscopy images show fluorescent patches of parental H3.3 (red) before and 15 min after local UVC damage (top) or photo-bleaching (bottom) in U2OS cells stably expressing H3.3-SNAP and GFP-XPC. Scale bar, 5 μm.
(B) Parental H3.3 fluorescence measured in the entire nucleus and in the bleached (red) or damaged zone (purple) 15 min post-laser micro-irradiation is normalized to before laser (dotted line). n, number of cells.
(C) Parental H3.3 fluorescence measured in concentric regions around the UVC (top graph) or bleaching laser impact (bottom graph) at the indicated time points is normalized to the fluorescence in the patch before laser.
(D) Difference in red fluorescence distribution obtained by subtracting 0 min from 15 min values quantified in (C). The positive and negative areas under the UVC curve (purple) are equivalent. The position of the repair zone is based on GFP-XPC accumulation at 15 min. Error bars, SD from n cells scored in two independent experiments.
(E) Interpretation of the results shown in (D): redistribution of old H3.3 histones to the periphery of UVC-damaged regions. Areas were converted to distances.
See also Figures S2 and S3.
We then refined our analysis by measuring the spatial distribution of parental H3.3 histones around the damaged area (Figure 2C). Notably, we observed that the reduction of parental histone signal in the damaged region was counterbalanced by an increase of signal in the surrounding area (Figures 2D and 2E). Importantly, this conservative redistribution of parental histones did not occur upon local photo-bleaching of parental H3.3 fluorescence (Figures 2C and 2D), and we obtained similar results by photo-activation-based tracking of parental histones (Figures S3A–S3D). Furthermore, the area showing reduced histone density gradually increased over time post-irradiation (Figure 3A), which reveals that parental histone dynamics proceed radially outward. Such redistribution of parental histones argues against their eviction from damaged chromatin, because, if solubilized in the nucleoplasm, they would not be retained in a defined space in the periphery of the irradiated region. Supporting the fact that mobilized histones remain chromatin associated, detergent extraction of live cells after UVC irradiation did not alter the redistribution pattern of parental histones (Figure S3E). Altogether, these data demonstrate that parental H3 histones redistribute to the periphery of damaged chromatin. Such redistribution can involve parental nucleosomes sliding away from DNA lesions and/or an expansion of chromatin in the irradiated region, pushing away surrounding undamaged chromatin fibers.
Figure 3.

Chromatin Expansion and Histone Mobilization on Chromatin Contribute to Parental H3.3 Redistribution after UVC Damage
(A) Distribution of parental H3.3 (red) at the indicated time points after UVC laser damage in U2OS H3.3-SNAP cells. The area of parental histone loss is measured as a function of time post-UVC. Error bars, SEM from n cells scored in two independent experiments.
(B) Distribution of parental H3.3 (red) and DNA (blue, stained with Hoechst) before and 15 min after local UVC damage in U2OS H3.3-SNAP cells. The reduced Hoechst staining observed at damage sites is not due to photo-bleaching by the UVC laser (data not shown) or to DNA denaturation during UVC damage repair (Figure S3F). White arrowheads, irradiated areas. The boxplot shows quantifications of histone (red) and DNA (blue) fluorescence loss in irradiated areas (n cells scored in two independent experiments). Staining parental H3.3 in green and DNA in red with NUCLEAR-ID Red DNA stain gave similar results (data not shown).
(C) UVC-damaged chromatin areas visualized by staining for cyclobutane pyrimidine dimers (CPD, purple) after UVC laser damage in U2OS cells stably expressing H3.3-SNAP and GFP-XPC. The initial damaged chromatin area was measured in paraformaldehyde-fixed cells (no expansion, no GFP-XPC recruitment) and compared to 15 min after irradiation in live cells (n cells scored in two independent experiments). Insets: zoomed-in views (×3) of dashed line boxes. Scale bars, 10 μm.
(D) Quantification of parental H3.3 (red) and DNA (blue) fluorescence loss in irradiated areas as a function of UVC damage. The saturations of the two mechanisms contributing to parental histone loss (dotted lines) are obtained from the graphs in (F).
(E) Working model for parental H3.3 redistribution around UVC damage sites.
(F) Top: difference between parental H3.3 and DNA signal loss in irradiated areas as a function of UVC damage, which reflects histone mobilization on chromatin. Bottom: expansion of the damaged area marked by GFP-XPC as a function of UVC damage, indicative of chromatin opening. Dotted lines indicate the saturation of each mechanism. Error bars, SEM from at least 35 cells scored in three independent experiments.
See also Figures S3 and S5.
To test these hypotheses and gain further insights into the mechanisms underlying parental histone dynamics in UVC-damaged regions, we monitored, in parallel, histone and DNA densities. Thus, we found that parental H3.3 redistribution was accompanied by a decrease in DNA density within UVC-damaged regions (Figures 3B and S3F), indicative of chromatin opening. The area of UVC-damaged DNA increased by a factor of 1.26 within 15 min post-damage (Figure 3C), consistent with the 20% DNA density loss measured in the same experimental conditions (Figure 3B). This strongly supports the idea that the reduced DNA density in the damaged region results from chromatin opening. Interestingly, while histone and DNA signal loss both increased with the exposure time to UVC laser, histone signal loss exceeded DNA signal loss in all conditions examined (Figure 3D). This observation cannot be accounted for by chromatin opening only, which would lead to an equal reduction in histone and DNA densities in the damaged area. Since we already ruled out the possibility of histone dissociation from chromatin and extensive histone degradation (Figures 2 and S3), a possible explanation is that the extra loss in histone density reflects histone mobilization on chromatin away from damage sites (Figure 3E). The biphasic shape of the curves for histone and DNA signal loss as a function of UVC damage (Figure 3D) also points to a possible dual mechanism driving parental histone redistribution, each plateau corresponding to the saturation of a distinct process (Figure 3F). Our findings suggest that chromatin opening along with histone mobilization on damaged chromatin drive parental histone redistribution to the periphery of UVC-damaged regions.
The UVC Damage Sensor DDB2 Is Critical for Parental Histone Redistribution
To search for molecular determinants of parental H3 redistribution upon UVC damage, we examined the connection with UVC damage repair by knocking down factors involved at different steps in the NER (nucleotide excision repair) pathway (Figure 4A and S4A; reviewed in Alekseev and Coin, 2015, Marteijn et al., 2014). Decreasing the expression of the late repair factor XPG (xeroderma pigmentosum G), required for excision of the damaged oligonucleotide before repair synthesis, only moderately reduced parental H3.3 redistribution upon UVC irradiation (Figure S4B). Similarly, knocking down the early repair factor ERCC6 (excision repair cross-complementing 6) involved in damage recognition within transcribed genes did not markedly impair parental H3.3 dynamics (Figure S4B). Consistent with this, cells treated with a transcription inhibitor prior to UVC irradiation did not show major defects in old H3.3 redistribution (data not shown). These results indicate a relatively minor contribution of transcription-coupled repair and late repair steps to the reduced parental H3 histone density at damaged sites.
Figure 4.

Parental Histone Redistribution Is Controlled by the Repair Factor DDB2
(A) Scheme representing the main repair factors involved in UVC damage detection in the global genome NER pathway. Microscopy images show the distribution of parental H3.3 (red) 15 min after UVC laser damage in U2OS cells stably expressing H3.3-SNAP and GFP-XPC treated with the indicated siRNAs (siLUC: control). siRNA efficiencies were verified by western blot. The red fluorescence loss measured in damaged areas is normalized to before laser (n cells scored in two independent experiments).
(B) Distribution of parental H3.3 (red) 15 min after UVC laser damage in U2OS cells stably expressing H3.3-SNAP and GFP-XPC or GFP-DDB2. Expression levels of exogenous XPC and DDB2 relative to the endogenous proteins are shown on the western blot. Red fluorescence loss is measured in irradiated areas relative to before laser and the area of fluorescence loss is marked by GFP-tagged NER factors.
(C) Distribution of parental H3.3 (green) upon tethering of mCherry-LacR (LacR) or mCherry-LacR-DDB2 (LacR-DDB2) to the LacO array in U2OS LacO cells stably expressing H3.3-SNAP. The area of the LacO array and green fluorescence at the LacO array are displayed on the boxplots (n cells scored in two independent experiments). White arrowheads, irradiated areas or LacO array. Scale bars, 10 μm.
See also Figures S4 and S5.
In contrast, when knocking down the UVC damage sensor DDB2 involved in global genome repair, we observed a striking impairment in parental H3.3, H3.1, and H4 density loss at UVC damage sites (Figures 4A and S4C–S4E). We did not observe comparable defects in parental H3.3 dynamics upon downregulation of XPC, another early repair factor involved in global genome repair (Figure S3F), or upon knockdown of the DDB2 partners DDB1 and CUL4A (Cullin 4A) (Figure 4A), which are part of an E3-ubiquitin ligase complex that modifies various substrates at sites of UVC damage (Nouspikel, 2011). Consistent with a minor contribution of DDB1 and CUL4A to parental H3.3 density loss, we found that preventing de novo ubiquitylation reactions by treating cells with a proteasome inhibitor or with a neddylation inhibitor did not markedly alter the redistribution of old H3.3 in damaged chromatin (Figures S4F and S4G). These data indicate that the ubiquitylation activity of the DDB1-DDB2-CUL4A-containing complex does not play a major role in parental histone dynamics following UVC damage. Parental histone redistribution to the periphery of damaged chromatin regions is thus coupled to the earliest steps of global genome NER with a prominent role for DDB2.
To further characterize the contribution of DDB2 to parental histone redistribution upon UVC damage, we tested the effect of DDB2 overexpression. Expressing exogenous DDB2 significantly increased the area of parental histone signal loss after local UVC irradiation: this area was 50% larger in cells overexpressing DDB2 than in cells overexpressing another early NER factor, XPC (Figure 4B). These results thus indicate that DDB2 levels are limiting for parental histone redistribution in UVC-irradiated chromatin regions. Furthermore, artificial tethering of DDB2 to a LacO (lactose operator) array in the absence of DNA damage led to a marked reduction of parental H3.3 histone density at the LacO array (Figure 4C). DDB2 tethering also triggered an expansion of the LacO array, as previously reported (Luijsterburg et al., 2012). These results reveal that DDB2 binding to chromatin is sufficient for parental histone redistribution even in the absence of DNA damage.
To gain mechanistic insights into DDB2 effect on parental histone dynamics, we tested the potential contribution of chromatin remodelers with reported connections to the DDB2 complex, namely ALC1 (amplified in liver cancer 1) and INO80 (inositol-requiring 80) (Jiang et al., 2010, Pines et al., 2012). Knocking down these remodelers did not recapitulate the effect of DDB2 downregulation (Figures S5A and S5B). Similarly, interfering with poly(ADP-ribosyl)ation, which has been associated with DDB2 and UV-damaged chromatin decompaction (Luijsterburg et al., 2012, Pines et al., 2012), did not impact parental histone redistribution at UVC damage sites (Figures S5C and S5D). However, our analyses of DNA and histone signal loss at UVC sites upon DDB2 knockdown (Figure S5E) indicate a major contribution of DDB2 to chromatin opening only, consistent with previous observations (Luijsterburg et al., 2012), suggesting that additional factors come into play to promote histone mobilization on chromatin.
Collectively, our data put forward the early repair factor DDB2 as a master regulator of parental histone redistribution by chromatin opening at UVC sites.
Parental H3.3 Redistribution Is Independent of New H3.3 Deposition
As we previously demonstrated that DDB2 controls the recruitment of the histone chaperone HIRA (histone regulator A), which promotes the deposition of newly synthesized histones H3.3 at UVC damage sites (Adam et al., 2013), we investigated the potential coupling between parental and new H3.3 dynamics in response to UVC irradiation. For this, we labeled parental and new histones in different colors within the same sample and compared the kinetics of parental histone density loss and new histone deposition upon local UVC damage (Figure 5A; Movies S2 and S3). While parental H3.3 histones are redistributed within minutes after damage induction, new histone H3.3 accumulation at damage sites becomes detectable only 30 min after local UVC irradiation (Figure 5A). We obtained similar results when we swapped the SNAP reagents, labeling old H3.3 in green and new H3.3 in red (data not shown). Thus, our assay reveals that parental histone density loss precedes new histone deposition at UVC damage sites.
Figure 5.

Parental H3.3 Redistribution Is Independent of New H3.3 Deposition
(A) Dynamics of parental (red) and new H3.3 (green) at the indicated time points after UVC laser damage in U2OS H3.3-SNAP cells. Red and green signals measured in damaged areas are normalized to before laser. Error bars, SD from n cells scored in two independent experiments.
(B) Distribution of parental and new H3.3 as in (A) in cells treated with the indicated siRNAs (siLUC: control). HIRA knockdown is verified by western blot and by the inhibition of new H3.3 deposition at damage sites.
(C) Distribution of parental and new H3.3 as in (B). siRNA efficiency was verified by qRT-PCR. Error bars, SD from two independent experiments. Red fluorescence loss is measured in damaged areas at 15 min compared to before laser (n cells scored in two independent experiments). White arrowheads, irradiated areas. Scale bars, 10 μm.
Given that the histone chaperone HIRA promotes the deposition of newly synthesized H3.3 at UVC damage sites (Adam et al., 2013), we tested whether the same chaperone was responsible for parental H3.3 dynamics in UVC-damaged regions. Interestingly, HIRA downregulation did not impair old H3.3 signal loss at UVC sites (Figure 5B), showing that this histone chaperone does not participate in any significant manner in parental H3.3 dynamics after UVC damage. These findings indicate that parental H3.3 redistribution likely occurs independently of new H3.3 deposition. Consistent with this, preventing the synthesis of new H3.3 by siRNA (small interfering RNA) (Figure 5C, green) did not interfere with parental H3.3 displacement from UVC-damaged regions (Figure 5C, red; note that this treatment did not affect parental H3.3 levels).
Altogether, these data demonstrate that parental histone H3.3 redistribution in UVC-damaged regions is functionally independent of new H3.3 deposition.
Recovery of Parental Histones Coupled to Repair Progression through DDB2 Release
Since parental histones are still detected in the periphery of UVC-damaged regions early after DNA damage, we investigated whether and to which extent they contribute to chromatin restoration after damage. For this purpose, we examined both parental and new H3.3 dynamics in parallel with repair progression in U2OS cells stably expressing H3.3-SNAP and CFP-XPC (Figure S6A). This analysis revealed that parental histones recovered in chromatin undergoing UVC damage repair, reaching almost complete recovery (90% of the initial signal) 9 hr after UVC irradiation, which mirrors the slow kinetics of UVC damage repair (Figure 6A). A similar extent of parental histone recovery at UVC damage sites was measured in U2OS cells stably expressing H3.3-PA-GFP (Figure S6B) and in MFC7 cells transiently expressing H3.3-SNAP (Figure S6C). Noteworthy, parental histone recovery was delayed compared to new histone incorporation (Figure 6A), suggesting that they involve distinct mechanisms. To gain insight into how parental histones recover, we compared their dynamics to basal histone mobility assessed by FRAP (fluorescence recovery after photo-bleaching). While parental histone recovery in UVC-damaged regions reached 80% within 12 hr after irradiation, it did not exceed 20% in an undamaged chromatin region of the same nucleus within the same time frame (Figure S6D), consistent with a previous report (Kimura and Cook, 2001). This demonstrates that parental H3.3 recovery in chromatin undergoing repair does not result from basal histone turnover but actually reflects the relocation of parental histones that were mobilized away from UVC-damaged regions. Notably, when measuring the area of parental histone density loss over time after UVC damage, we observed that parental histone recovery proceeded radially inward (Figure 6B), suggesting that displaced histones were moving back by an opposing mechanism to their initial redistribution. The area of new histone accumulation by contrast did not shrink (Figure S6E), arguing that chromatin restoration does not proceed solely via re-compaction. As a result, recovered parental histones mix with newly synthesized histones in repairing chromatin (Figure S6F).
Figure 6.

Recovery of Parental Histones Coupled to Repair Progression
(A) Dynamics of parental (red) and new H3.3 (green) at the indicated time points after UVC laser damage in U2OS cells stably expressing H3.3-SNAP and CFP-XPC. Green and red signals are quantified in irradiated areas relative to before laser. Cells that did not repair efficiently (based on CFP-XPC retention) were excluded from the analysis. Error bars, SD from n cells scored in two independent experiments. Staining parental histones in green and new histones in red gave similar results (not shown).
(B) Area of parental H3.3 histone loss as a function of time post-UVC irradiation, reflecting opening and closure of the damaged zone. Error bars, SEM from n cells scored in two independent experiments.
(C) Dynamics of parental H3.3 (red) at the indicated time points after UVC laser damage in U2OS cells stably expressing H3.3-SNAP and GFP-DDB2 treated with the indicated siRNAs (siLUC: control). The efficiency of XPG depletion is indicated by sustained retention of GFP-DDB2 at UV sites. Red fluorescence measured in damaged areas is normalized to before laser. Error bars, SEM from n cells scored in two independent experiments.
(D) Distribution of parental H3.3 (green) upon IPTG-mediated release of mCherry-LacR-DDB2 from the LacO array in U2OS LacO cells stably expressing H3.3-SNAP. The green fluorescence at the LacO array and LacR-DDB2 area (red) measured after IPTG addition are normalized to before IPTG. Error bars, SD from n cells scored in two independent experiments. White arrowheads, irradiated areas or LacO array. Scale bars, 10 μm.
We next sought to determine whether parental H3.3 recovery in damaged chromatin was coupled to repair progression. For this, we depleted the late repair factor XPG, which interferes with repair progression with no major effect on the early redistribution of parental histones in damaged chromatin (Figure S3B). Parental H3.3 recovery, however, was markedly impaired in XPG-depleted cells (Figure 6C; Movies S4 and S5) down to a rate similar to basal histone turnover (Figures S6D and S6G). These results indicate that parental histone recovery in damaged chromatin is dependent on repair progression. Given that XPG depletion also significantly delayed DDB2 release from chromatin, we assessed more directly the role of DDB2 in parental histone recovery. For this, we triggered LacR-DDB2 release from the LacO array by IPTG addition (Figure 6D). This resulted in rapid recovery of old H3.3 at the LacO array, which highlights the key role of DDB2 in controlling parental histone dynamics in UVC-damaged chromatin.
Collectively, our results underline the major contribution of parental histones to chromatin restoration coupled to repair and establish that parental histone recovery is coordinated with repair progression through DDB2 release from damaged chromatin.
Discussion
By exploiting real-time tracking of parental H3 and H4 histones after local UVC damage in human cells, we provide novel insights into epigenome maintenance in response to DNA damage. Our study indeed identifies a conservative process, tightly coordinated with repair progression, whereby parental histones rapidly redistribute away from UVC-damaged chromatin regions and subsequently recover (Figure 7). We propose that parental histones are kept in the periphery of the damaged areas to help restore chromatin organization after DNA repair, which may contribute to preserving a memory of chromatin identity in response to DNA damage.
Figure 7.

Model for Parental Histone Dynamics Coupled to Repair in UVC-Damaged Chromatin
UVC damage leads to the redistribution of parental histones to the periphery of damaged chromatin regions. This occurs via a decompaction of damaged chromatin, which pushes away the surrounding undamaged chromatin fibers, along with a mobilization of parental histones on chromatin away from damage sites, making room for new histone incorporation. Restoration of the overall chromatin organization proceeds by chromatin re-compaction and sliding back the nucleosomes bearing parental histones. The whole process is tightly coordinated with DNA repair progression through binding and release of the damage sensor DDB2.
Parental Histone Redistribution Away from Damaged Chromatin Regions
Chromatin rearrangements coupled to the early stages of the DDR, although considered to be critical for efficient DNA repair, still remained poorly characterized. Here, we provide evidence for a redistribution of parental histones to the periphery of UVC-damaged chromatin regions, which prompt us to re-evaluate our views on the Access-Repair-Restore model. Even though histone solubilization has been reported in response to genotoxic stress (Goldstein et al., 2013, Wang et al., 2006, Xu et al., 2010), our data support a model where parental H3 histones do not massively dissociate from chromatin but are redistributed away from UVC-damaged sites, possibly via nucleosome sliding and chromatin opening. This is consistent with a recent study suggesting that H2B histones are not solubilized following UV irradiation in human cells (Morisaki and McNally, 2014).
The extent of chromatin rearrangements in response to local DNA damage is a matter of debate. While one study indicates that chromatin destabilization affects the whole nucleus upon local UVC irradiation (Rubbi and Milner, 2003), several lines of evidence rather support the idea that chromatin is locally disorganized upon genotoxic stress (Dinant et al., 2013, Goldstein et al., 2013, Hinde et al., 2014, Kruhlak et al., 2006, Luijsterburg et al., 2012, Smeenk et al., 2013). Here, we reconcile these data by demonstrating that a local loss of parental histone density in UVC-damaged areas has a long-range impact on chromatin, potentially affecting the whole nucleus, because parental histone redistribution actually spreads over long distances away from the damaged regions (Figure 2E).
Whether and how histone redistribution facilitates access to damaged DNA and repair progression are not entirely clear. The recruitment of the damage sensor DDB2 to UVC lesions precedes and orchestrates parental histone dynamics. Mobilizing parental histones may be necessary for recruiting downstream NER factors to damaged chromatin. It is tempting to speculate that this may also contribute to protect parental histones from modifications by histone-modifying enzymes recruited to regions of ongoing repair, thereby promoting the maintenance of the original epigenetic information.
Mechanistically, both histone mobilization on chromatin and chromatin opening potentially contribute to chromatin disorganization in response to UVC damage. Whether they use similar regulatory factors as those promoting chromatin mobility in response to DNA breaks (Dion and Gasser, 2013) is an attractive possibility. In terms of molecular players, we identify the damage sensor DDB2 as a master regulator of parental histone dynamics at sites of UVC lesions, mostly via its ability to promote chromatin opening. Interestingly, while the ubiquitylation activity of DDB2-containing complex is required for new histone deposition in UVC-damaged chromatin (Adam et al., 2013), it is largely dispensable for parental histone dynamics. Consistent with our findings, ubiquitylation-deficient mutants of DDB2 induce chromatin expansion like wild-type DDB2 when artificially tethered to chromatin (Luijsterburg et al., 2012). Thus, DDB2-mediated chromatin expansion is largely independent of the other members of the UVC damage recognition complex. Whether DDB2 acts alone or in association with other factors to fulfill this activity will be important to investigate in future studies. Remarkably, DDB2 has very strong affinity for UVC-damaged DNA (Kulaksiz et al., 2005, Wittschieben et al., 2005), and DDB2 binds damaged DNA on nucleosomes (Osakabe et al., 2015). We can speculate that DDB2 binding to damaged chromatin may on its own push away surrounding nucleofilaments leading to chromatin opening. Given that DDB2 does not display ATPase/helicase or histone-binding domains, we rather favor a model where it works in concert with other factors directly involved in chromatin dynamics—such as histone chaperones and/or chromatin remodeling complexes that remain to be identified—to promote chromatin disorganization in damaged regions.
Epigenome Maintenance after DNA Damage
Our study identifies a pathway that may contribute to preserving the integrity of chromatin architecture in response to DNA damage. We unraveled that damaged chromatin reorganization is a two-step process with new histone incorporation preceding parental histone recovery. The biphasic nature of chromatin restoration is consistent with an early model based on the accessibility to nucleases of chromatin undergoing NER (reviewed in Smerdon, 1991).
While we cannot formally exclude that a limited amount of parental histones are ultimately degraded after UVC damage as reported for hyperacetylated histones in response to ionizing radiation (Qian et al., 2013), the conservative nature of parental histone redistribution around UVC damage sites and the almost complete recovery of parental histones during repair progression argue against massive histone degradation. Furthermore, the retention of parental histones proximal to the site of damage offers a possible redirection to the original site to promote a conservative restoration of chromatin architecture. It will be important to develop higher-resolution approaches to determine whether parental histones retrieve their original positions on the DNA sequence upon recovery and whether the topological organization of parental chromatin is fully re-established.
The major contribution of parental histones to the composition of repaired chromatin is crucial to envision mechanisms for epigenome maintenance after DNA damage. Indeed, it opens up the possibility that parental marks can be preserved and transferred to the new histones, which initially carry their own set of post-translational modifications (PTMs) (Loyola et al., 2006). In this respect, a parallel can be drawn between the restoration of chromatin after DNA damage and DNA replication (Alabert and Groth, 2012, Groth et al., 2007, MacAlpine and Almouzni, 2013).
At the replication fork, the maintenance of chromatin identity is achieved by old histone recycling with their PTMs and by subsequent modifications of new histones to mirror the parental ones (Alabert et al., 2015). Whether similar mechanisms operate in damaged chromatin is an attractive possibility. Noteworthy, while cells have to cope with 50% histone renewal during replication, most parental histones recover in damaged chromatin regions, which could facilitate the re-establishment of the original chromatin landscape. It is tempting to speculate that the presence of parental and new histones in neighboring nucleosomes may allow old PTM transmission to newly deposited histones after repair.
Finally, our data open up new avenues for understanding the etiology of several human diseases, including H3 mutant-associated cancers (reviewed in Kallappagoudar et al., 2015, Yuen and Knoepfler, 2013) and NER disorders (reviewed in DiGiovanna and Kraemer, 2012, Marteijn et al., 2014). Considering that DDB2 dynamics strongly impact the fate of parental histones in response to UVC damage, the phenotype of XPE patients harboring DDB2 mutations that prevent its binding to damaged DNA may not only reflect a DNA repair defect but also altered chromatin plasticity in response to genotoxic stress. Similarly, H3 mutations could contribute to the development of human cancers by affecting the resetting of the epigenome following DNA damage.
In conclusion, our work sheds new light to our current view of DNA damage-induced chromatin rearrangements and suggests that parental histone dynamics are critical to the maintenance of epigenome integrity in response to genotoxic stress. Our findings also pave the way for the identification of new factors that contribute to restoring damaged chromatin identity and for understanding how this protects cells against pathological conditions.
Experimental Procedures
Cell Culture and Drug Treatments
All cell lines and drug treatments are described in the Supplemental Experimental Procedures.
SNAP Labeling and Photo-activation of Histones
SNAP labeling of histone proteins was done as described (Bodor et al., 2012). Photo-activation experiments were performed in U2OS cells stably expressing H3.3-PA-GFP on a Zeiss LSM710 confocal microscope using the 30 mW 405 nm laser focused through an LD LCI Plan-Apochromat 25×/0.8 oil objective. See Supplemental Experimental Procedures for details.
UVC Laser Micro-irradiation and FRAP
UVC laser micro-irradiation was done as described (Adam et al., 2013, Dinant et al., 2007). Details for the FRAP procedure and for image acquisition and analysis are in Supplemental Experimental Procedures.
siRNA and Plasmid Transfections
siRNAs (Supplemental Experimental Procedures) were transfected into cells using Lipofectamine RNAiMAX (Invitrogen). Cells were transiently transfected with plasmid DNA (1 μg/mL final, Supplemental Experimental Procedures) using Lipofectamine 2000 (Invitrogen) 48 hr before subsequent cell treatment.
Cell Extracts and Western Blot, Immunofluorescence, and qRT-PCR
These procedures, including lists of antibodies and primer pairs, are described in Supplemental Experimental Procedures.
Statistical Analysis
p values for mean comparisons between two groups were calculated with a Student’s t test, including Welch’s correction when necessary, using R software. Multiple comparisons were performed by one-way ANOVA with Bonferroni post-test using GraphPad Prism.
Author Contributions
S.A., J.D. (equal contribution), and S.E.P. designed and performed experiments, analyzed the data, and wrote the manuscript. O.C. provided technical assistance. C.B. designed the UVC laser microscope set-up for G.A.’s lab. O.L. and O.R. implemented the UVC laser technology and helped with image analyses. A.C. produced the BJ H3.1-SNAP cell line in P.L.’s lab. S.E.P. and G.A. co-directed S.A.’s work. G.A. provided inputs for experimental design using the UVC laser technology and SNAP tagging and for data analysis and writing. S.E.P. supervised the project.
Acknowledgments
We thank our colleagues for fruitful discussions and critical reading of the manuscript. N. Dantuma, E. Dunleavy, S. Jackson, L. Jansen, J. Lippincott-Schwartz, R. Nishi, and H. van Attikum shared reagents. The ImagoSeine core facility (Institut Jacques Monod, France BioImaging) assisted with confocal microscopy. This work was supported by the European Research Council (ERC-2013-StG-336427 “EpIn”), the French National Research Agency (ANR-12-JSV6-0002-01, “Who am I”? LabEx ANR-11-LABX-0071, ANR-11-IDEX-0005-01, France-BioImaging ANR-10-INSB-04), EDF Radiobiology program RB 2014-01, and the Fondation ARC. Research in G.A. group is supported by la Ligue Nationale contre le Cancer (Equipe labellisée), the European Commission Network of Excellence EpiGeneSys (HEALTH-F4-2010-257082), ERC-2009-AdG_20090506 “Eccentric,” ANR-11-LABX-0044_DEEP, and ANR-10-IDEX-0001-02 PSL∗. P.L.’s group is funded by VIRUCEPTION (ANR-13-BSV3-0001-01) and the LabEx DEVweCAN (ANR-10-LABX-61). S.A. received PhD fellowships from University Pierre et Marie Curie and La Ligue contre le Cancer. J.D. received a PhD fellowship from University Paris Diderot.
Published: September 15, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and five movies and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.08.019.
Supplemental Information
Dynamics of parental histones H3.3 (red) during the first 10 min after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP. 21 images were captured at 30 s intervals and are displayed at 2 frames/s. The resulting motion picture is shown with a superimposed white arrowhead pointing to the laser irradiation site.
{kind=link}
Dynamics of newly synthesized histones H3.3 (green) during the first 2 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP. 25 images were captured at 5 min intervals and are displayed at 2 frames/s. The resulting motion picture is shown with a superimposed white arrowhead pointing to the laser irradiation site.
{kind=link}
Dynamics of parental histones H3.3 (red) during the first 2 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP. 25 images were captured at 5 min intervals and are displayed at 2 frames/s. The resulting motion picture is shown with a superimposed white arrowhead pointing to the laser irradiation site.
{kind=link}
Dynamics of parental histones H3.3 (red) during the first 12 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP and GFP-DDB2 treated with siRNA control siLUC. 49 images were captured at 15 min intervals and are displayed at 4 frames/s. The resulting motion picture (red channel only) is shown with a superimposed white arrowhead pointing to the laser irradiation site.
{kind=link}
Dynamics of parental histones H3.3 (red) during the first 12 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP and GFP-DDB2 treated with siXPG. 49 images were captured at 15 min intervals and are displayed at 4 frames/s. The resulting motion picture (red channel only) is shown with a superimposed white arrowhead pointing to the laser irradiation site.
{kind=link}
References
- Adam S., Polo S.E., Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Adam S., Dabin J., Polo S.E. Chromatin plasticity in response to DNA damage: the shape of things to come. DNA Repair (Amst.) 2015;32:120–126. doi: 10.1016/j.dnarep.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C., Groth A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Alabert C., Barth T.K., Reverón-Gómez N., Sidoli S., Schmidt A., Jensen O.N., Imhof A., Groth A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–590. doi: 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseev S., Coin F. Orchestral maneuvers at the damaged sites in nucleotide excision repair. Cell. Mol. Life Sci. 2015;72:2177–2186. doi: 10.1007/s00018-015-1859-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A.J., Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor D.L., Rodríguez M.G., Moreno N., Jansen L.E.T. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr. Protoc. Cell Biol. 2012;Chapter 8:Unit8.8. doi: 10.1002/0471143030.cb0808s55. [DOI] [PubMed] [Google Scholar]
- Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabin J., Fortuny A., Polo S.E. Epigenome maintenance in response to DNA damage. Mol. Cell. 2016;62:712–727. doi: 10.1016/j.molcel.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna J.J., Kraemer K.H. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C., de Jager M., Essers J., van Cappellen W.A., Kanaar R., Houtsmuller A.B., Vermeulen W. Activation of multiple DNA repair pathways by sub-nuclear damage induction methods. J. Cell Sci. 2007;120:2731–2740. doi: 10.1242/jcs.004523. [DOI] [PubMed] [Google Scholar]
- Dinant C., Ampatziadis-Michailidis G., Lans H., Tresini M., Lagarou A., Grosbart M., Theil A.F., van Cappellen W.A., Kimura H., Bartek J. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell. 2013;51:469–479. doi: 10.1016/j.molcel.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Dion V., Gasser S.M. Chromatin movement in the maintenance of genome stability. Cell. 2013;152:1355–1364. doi: 10.1016/j.cell.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Dunleavy E.M., Almouzni G., Karpen G.H. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G1 phase. Nucleus. 2011;2:146–157. doi: 10.4161/nucl.2.2.15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipescu D., Müller S., Almouzni G. Histone H3 variants and their chaperones during development and disease: contributing to epigenetic control. Annu. Rev. Cell Dev. Biol. 2014;30:615–646. doi: 10.1146/annurev-cellbio-100913-013311. [DOI] [PubMed] [Google Scholar]
- Goldstein M., Derheimer F.A., Tait-Mulder J., Kastan M.B. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc. Natl. Acad. Sci. USA. 2013;110:16874–16879. doi: 10.1073/pnas.1306160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A., Rocha W., Verreault A., Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Hanawalt P.C. Historical perspective on the DNA damage response. DNA Repair (Amst.) 2015;36:2–7. doi: 10.1016/j.dnarep.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinde E., Kong X., Yokomori K., Gratton E. Chromatin dynamics during DNA repair revealed by pair correlation analysis of molecular flow in the nucleus. Biophys. J. 2014;107:55–65. doi: 10.1016/j.bpj.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Wang X., Bao S., Guo R., Johnson D.G., Shen X., Li L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc. Natl. Acad. Sci. USA. 2010;107:17274–17279. doi: 10.1073/pnas.1008388107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallappagoudar S., Yadav R.K., Lowe B.R., Partridge J.F. Histone H3 mutations—a special role for H3.3 in tumorigenesis? Chromosoma. 2015;124:177–189. doi: 10.1007/s00412-015-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H., Cook P.R. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg R.D. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- Kruhlak M.J., Celeste A., Dellaire G., Fernandez-Capetillo O., Müller W.G., McNally J.G., Bazett-Jones D.P., Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaksiz G., Reardon J.T., Sancar A. Xeroderma pigmentosum complementation group E protein (XPE/DDB2): purification of various complexes of XPE and analyses of their damaged DNA binding and putative DNA repair properties. Mol. Cell. Biol. 2005;25:9784–9792. doi: 10.1128/MCB.25.22.9784-9792.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L., Nakajima S., Kapetanaki M.G., Hsieh C.L., Fagerburg M., Thickman K., Rodriguez-Collazo P., Leuba S.H., Levine A.S., Rapić-Otrin V. Monoubiquitinated histone H2A destabilizes photolesion-containing nucleosomes with concomitant release of UV-damaged DNA-binding protein E3 ligase. J. Biol. Chem. 2012;287:12036–12049. doi: 10.1074/jbc.M111.307058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Tyler J.K. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. eLife. 2016;5:5. doi: 10.7554/eLife.15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola A., Bonaldi T., Roche D., Imhof A., Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell. 2006;24:309–316. doi: 10.1016/j.molcel.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Luijsterburg M.S., Lindh M., Acs K., Vrouwe M.G., Pines A., van Attikum H., Mullenders L.H., Dantuma N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 2012;197:267–281. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M.S., de Krijger I., Wiegant W.W., Shah R.G., Smeenk G., de Groot A.J.L., Pines A., Vertegaal A.C.O., Jacobs J.J.L., Shah G.M., van Attikum H. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end joining. Mol. Cell. 2016;61:547–562. doi: 10.1016/j.molcel.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J., Lukas C., Bartek J. More than just a focus: rhe chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- MacAlpine D.M., Almouzni G. Chromatin and DNA replication. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a010207. a010207–a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn J.A., Lans H., Vermeulen W., Hoeijmakers J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014;15:465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- Maze I., Noh K.-M., Soshnev A.A., Allis C.D. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014;15:259–271. doi: 10.1038/nrg3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T., McNally J.G. Photoswitching-free FRAP analysis with a genetically encoded fluorescent tag. PLoS ONE. 2014;9:e107730. doi: 10.1371/journal.pone.0107730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T. Multiple roles of ubiquitination in the control of nucleotide excision repair. Mech. Ageing Dev. 2011;132:355–365. doi: 10.1016/j.mad.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Osakabe A., Tachiwana H., Kagawa W., Horikoshi N., Matsumoto S., Hasegawa M., Matsumoto N., Toga T., Yamamoto J., Hanaoka F. Structural basis of pyrimidine-pyrimidone (6-4) photoproduct recognition by UV-DDB in the nucleosome. Sci. Rep. 2015;5:16330. doi: 10.1038/srep16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudet P., Gross-Bellard M., Chambon P. Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell. 1975;4:281–300. doi: 10.1016/0092-8674(75)90149-x. [DOI] [PubMed] [Google Scholar]
- Pines A., Vrouwe M.G., Marteijn J.A., Typas D., Luijsterburg M.S., Cansoy M., Hensbergen P., Deelder A., de Groot A., Matsumoto S. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012;199:235–249. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S.E., Almouzni G. Chromatin dynamics after DNA damage: the legacy of the access-repair-restore model. DNA Repair (Amst.) 2015;36:114–121. doi: 10.1016/j.dnarep.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S.E., Roche D., Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127:481–493. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Probst A.V., Dunleavy E., Almouzni G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- Qian M.-X., Pang Y., Liu C.H., Haratake K., Du B.-Y., Ji D.-Y., Wang G.-F., Zhu Q.-Q., Song W., Yu Y. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell. 2013;153:1012–1024. doi: 10.1016/j.cell.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi C.P., Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003;22:975–986. doi: 10.1093/emboj/cdg082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk G., van Attikum H. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu. Rev. Biochem. 2013;82:55–80. doi: 10.1146/annurev-biochem-061809-174504. [DOI] [PubMed] [Google Scholar]
- Smeenk G., Wiegant W.W., Marteijn J.A., Luijsterburg M.S., Sroczynski N., Costelloe T., Romeijn R.J., Pastink A., Mailand N., Vermeulen W., van Attikum H. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell Sci. 2013;126:889–903. doi: 10.1242/jcs.109413. [DOI] [PubMed] [Google Scholar]
- Smerdon M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol. 1991;3:422–428. doi: 10.1016/0955-0674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- Talbert P.B., Henikoff S. Histone variants—ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- Wang H., Zhai L., Xu J., Joo H.-Y., Jackson S., Erdjument-Bromage H., Tempst P., Xiong Y., Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell. 2006;22:383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- Wittschieben B.Ø., Iwai S., Wood R.D. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J. Biol. Chem. 2005;280:39982–39989. doi: 10.1074/jbc.M507854200. [DOI] [PubMed] [Google Scholar]
- Xu Y., Sun Y., Jiang X., Ayrapetov M.K., Moskwa P., Yang S., Weinstock D.M., Price B.D. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol. 2010;191:31–43. doi: 10.1083/jcb.201001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen B.T.K., Knoepfler P.S. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavala A.G., Morris R.T., Wyrick J.J., Smerdon M.J. High-resolution characterization of CPD hotspot formation in human fibroblasts. Nucleic Acids Res. 2014;42:893–905. doi: 10.1093/nar/gkt912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dynamics of parental histones H3.3 (red) during the first 10 min after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP. 21 images were captured at 30 s intervals and are displayed at 2 frames/s. The resulting motion picture is shown with a superimposed white arrowhead pointing to the laser irradiation site.
Dynamics of newly synthesized histones H3.3 (green) during the first 2 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP. 25 images were captured at 5 min intervals and are displayed at 2 frames/s. The resulting motion picture is shown with a superimposed white arrowhead pointing to the laser irradiation site.
Dynamics of parental histones H3.3 (red) during the first 2 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP. 25 images were captured at 5 min intervals and are displayed at 2 frames/s. The resulting motion picture is shown with a superimposed white arrowhead pointing to the laser irradiation site.
Dynamics of parental histones H3.3 (red) during the first 12 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP and GFP-DDB2 treated with siRNA control siLUC. 49 images were captured at 15 min intervals and are displayed at 4 frames/s. The resulting motion picture (red channel only) is shown with a superimposed white arrowhead pointing to the laser irradiation site.
Dynamics of parental histones H3.3 (red) during the first 12 hr after local damage with the UVC laser in U2OS cells stably expressing H3.3-SNAP and GFP-DDB2 treated with siXPG. 49 images were captured at 15 min intervals and are displayed at 4 frames/s. The resulting motion picture (red channel only) is shown with a superimposed white arrowhead pointing to the laser irradiation site.