

Abstract
The stress-induced chaperone protein Hsp70 enables the initiation and progression of many cancers, making it an appealing therapeutic target for development. Here we show that cancer cells resistant to Hsp70 inhibitors in vitro remain sensitive to them in vivo, revealing the pathogenic significance of Hsp70 in tumor stromal cells rather than tumor cells as widely presumed. Using transgenic mouse models of cancer, we found that expression of Hsp70 in host stromal cells was essential to support tumor growth. Furthermore, genetic ablation or pharmacological inhibition of Hsp70 suppressed tumor infiltration by macrophages needed to enable tumor growth. Overall, our results illustrate how Hsp70 inhibitors mediate the anti-cancer effects by targeting both tumor cells and tumor stromal cells, with implications for the broad use of these inhibitors as tools to ablate tumor-associated macrophages that enable malignant progression.
Introduction
Levels of the heat shock protein Hsp70 (HspA1A) have been implicated in cancer (1–5). Genetic ablation of Hsp70 facilitated oncogene-induced senescence in Her2-positive breast cancer (6), (7), (3), which defines effects of Hsp70 on cancer initiation. Using PyMT model of breast cancer it was demonstrated that Hsp70 also has profound effects on metastasis (8). The requirements for Hsp70 for cancer, prompted development of this protein as a drug target, and a number of Hsp70 inhibitors have been developed (see ref (2) for review).
Since micro-environment is emerging as a critical factor in tumor initiation and progression (9), (10), it is possible that Hsp70 expression in stroma may also be important for cancer development. For example, cancer-associated fibroblasts (CAF) can facilitate invasion and metastasis (9), and the importance of CAF for tumor development was linked to heat shock transcription factor Hsf1 (11). Tumor Associated Macrophages (TAM) can also supply cancer with EGF and angiogenic factors (12),(13), thus promoting invasion and metastasis (14), (12,15).
Given a critical role of stroma in cancer, several drugs have been developed which target tumor microenvironment (10,16), but our options to target CAF or TAM are still limited. Here we demonstrate that stromal Hsp70 is critical for tumor development and that stromal macrophages can be effectively targeted by our Hsp70 inhibitor series.
Materials and methods
Cell cultures
B16F10, MCF-7, HeLa, E0771 were from ATCC. Cells were obtained between 2003 and 2010. Cell authentication by ATCC is done by STR profiling. All cells were cultivated in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS at 37 C and 5% CO2. Cell survival was determined by CellTiter 96 Aqueous One Solution Assay (Promega) according to manufacture instructions. Cells were seeded in 96-well plates, incubated with different concentration of JG98 for 24 hr.
Macrophage isolation and cultivation
Bone marrow macrophages were isolated from wild type (C57BL6, Jackson lab), or Hsp70 knockout mice. Macrophages were grown in DMEM/F12 containing 10% FBS and 20% L-929 conditioned medium to form a monolayer of macrophages for 5 d.
Macrophages migration assays
A. “Wound healing” assay
Macrophages were seeded on a 6-well plate (0.25×106 cells/well), and 5 d later cell monolayer was scratched using p200 tip. Cells were treated with JG-98 or left untreated, and “wound healing” was recorded 24 h later. For quantification, pictures were taken of three random fields along the scratch, and identical rectangles with width corresponding to the width of original scratch were drawn in these fields. Cells migrated into the areas of these rectangles were counted, and data were normalized to the number of cells migrated in control scratch without JG-98 treatment.
B. Transwell assay
Macrophages were plated (2×104 cells/well) on transwell insert (8μM pore size) of 24 well-plate in 200 μL macrophage media supplemented with 1%FBS with or without JG-98. Bottom chamber was filled with the same media supplemented with 10%FBS. Migrated cells were counted under microscope, and data were normalized to the number of cells migrated in control transwell without JG-98 treatment.
Mice and tumors
For allograft tumors, B16F10 melanoma cells or E0071 carcinoma were injected s.c. into mouse right flanks either in PBS (0.5×106 or 1×106 cells, B16F10) or Matrigel at 1:1 ratio (1×106 cells, E0071). For tumor xenografts, MCF7 and HeLa cells were mixed at a 1:1 ratio with Matrigel, and 1×106 cells were injected s.c. into both left and right flanks female NCR nude mice (Taconic). JG-98 was injected i.p. at dose of 7 mg/kg every other day. Tumor growth was monitored using caliper and calculated according to the formula L × W2 × π/6, where L is length and W is width.
Histochemistry
Excised tumors from mice were fixed with 4% formaldehyde; immunostaining and quantification was performed by Premier Lab.
Results
Stromal cells play an important role in tumor sensitivity to the Hsp70 inhibitor JG-98
In vitro toxicity experiments showed that MCF7 cells were significantly more resistant to JG-98 than HeLa (Fig. 1A). On the other hand, when sensitivity to JG-98 was measured in vivo in xenograft models, tumors derived from HeLa cells were more resistant and tumors derived from MCF7 cells (Fig. 1B,C). These sensitivity differences in vitro and in vivo suggested that tumor stroma may significantly contribute to the anti-tumor effect of JG-98.
Fig. 1. Tumor stroma contributes to the anti-cancer effect of JG-98.

A. Sensitivity of cancer cell lines to JG-98 cell in culture. MCF7 and HeLa cells were treated with indicated concentrations of JG-98, and their viability was measured. Data shown are means+/− SEM of triplicates, here and below *p<0.05, **p<0.01, ***p<0.001 by Student t-test. B. C. Sensitivity of MCF7 (B) and HeLa cells (C) to JG-98 in vivo. Xenograft tumors were established in nude mice. When tumors reached 100mm3 JG-98 was administered. Data shown are means +/− SEM for 10 tumors per group (B) or 6 tumors per group (C). D. Expression of MDR1 leads to resistance of cells to JG-98. Upper panel: Intracellular fluorescence of JG-98 in HeLa cells and HeLa/MDR1 cells. Lower panel: Sensitivity to JG-98 of HeLa and HeLa/MDR1. Cells were treated with indicated concentrations of JG-98, and cell viability was measured. Data shown are means+/− SEM of triplicates. E. Sensitivity of HeLa and HeLa/MDR1 cells to JG-98 in vivo. When xenograft tumors reached 100mm3, JG-98 was administered. Data shown are means +/− SEM for 5 tumors per group.
To dissect the role of stroma in tumor sensitivity to JG-98, we sought to establish cancer cells resistant to JG-98 in vitro, and test whether these cells remain sensitive to JG-98 in vivo due to the stromal effects. Our recent screen for genes that modulate the response of cells to JG-98 provided the clue towards this goal, since knockdowns of ABCB1 (MDR1), ABCC1 and ABCC2 significantly sensitized cells to the compound (data not shown), suggesting that MDR pumps provide the resistance. Therefore, we compared responses to JG-98 of parental HeLa cells and HeLa overexpressing MDR1. Since JG-98 is fluorescent, its accumulation in cells was monitored microscopically after treatment with 0.3μM JG-98, and washing out the excess of the compound. Lower levels of JG-98 were accumulated in HeLa/MDR1 compared to parental HeLa cells (Fig. 1D). In line with this observation, HeLa/MDR1 cells were significantly more resistant to JG-98 compared to the parental line (Fig. 1D).
To investigate effects of JG-98 on tumor growth in vivo, xenograft tumors were established from these cell lines. Surprisingly, JG-98 had profound growth inhibitory effects on both control HeLa-derived xenografts (Fig. 1C) and HeLa/MDR1 xenografts (Fig. 1E). Effects on HeLa/MDR1 tumors were apparently stronger than effects on parental HeLa tumors, probably because of the overall slower tumor growth (Fig. 1E). Therefore, JG-98 can inhibit growth of tumors derived even from JG-98 resistant cells, indicating that its effects on host cells significantly contribute to anti-cancer activities of this class of Hsp70 inhibitors.
JG-98 inhibits infiltration of macrophages into tumors
To address what types of stromal cells are affected by JG-98, tumors from control and drug-treated mice were immunostained for markers of macrophages (F4/80 antibody), fibroblasts (SMA-1 antibody) and endothelial cells (CD-31 antibody). JG-98 treatment significantly reduced the number of macrophages in tumors (Fig. 2A,B), while fibroblasts and endothelial cells were not affected (Figs. 2B and S1).
Fig. 2. JG-98 treatment reduces infiltration of macrophages into tumors.
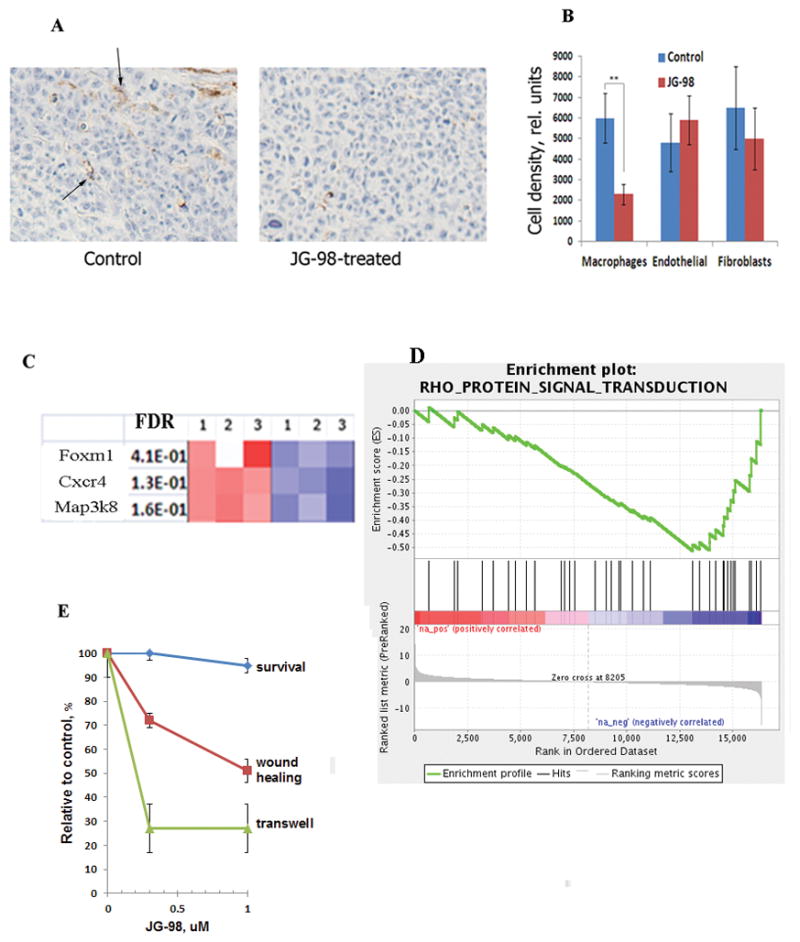
A. JG-98 suppresses macrophage infiltration. Tumors established with HeLa/MDR1 cells were collected from control and JG-98-treated animals on day 6 after the start of drug administration, sectioned and stained with F4/80 antibodies. Brown color (arrows) indicates macrophages. B. Quantification of infiltration of macrophages, endothelial cells and fibroblasts in tumors. The sections were stained with F4/80, anti-CD31 (endothelium) and SMA (fibroblasts) antibody. Quantification was done in 4 fields from 3 tumors each. Data shown are mean of pixels per field +/−SD. C. Treatment of macrophages with JG-98 affects genes that regulate migration. RNA was isolated from macrophages that were treated with 1μM JG-98 or left untreated, and gene expression was assessed by microarrays. Genes related to motility/migration among 100 top hits are shown. The figure presents data from three independent experiments. Color coding show False Discovery Rate (FDR), which is the estimated probability that a gene with a given normalized enrichment score represents a false positive finding. In other words, blue colors reflect statistically significant downregulation of each gene compared to red colors. D. Enrichment profile of the RHO signaling pathway according to Gene Set Enrichment Analysis. Enrichment plot reflects FDR in a gene set. For more detailed explanation see http://software.broadinstitute.org/gsea/doc/GSEAUserGuideTEXT.htm E. Effects of JG-98 on macrophage migration. Macropahges were treated with indicated concentrations of JG-98, and migration was measured by the “wound healing” and transwell assays as described in Materials and Methods. Data shown are means +/− SEM of triplicates.
Therefore inhibition of Hsp70 by JG-98 may affect macrophage migration, resulting in reduced infiltration into tumors. To address this possibility, mouse bone marrow macrophages were treated with JG-98 for 16 h, mRNA isolated from control and treated cells, and global gene expression analysis was assessed by microarray (GEO Series GSE82311). Among downregulated genes, we identified genes specifically implicated in the macrophage migration, including MAP3K8 (17), CXCR4 (18), and FOXM1 (19) (Fig. 2C and S2). In addition, gene set enrichment analysis has identified that the RHO signaling pathway was also suppressed (Fig. 2D). Among 36 components of the pathway, 15 were significantly downregulated (Table S1).Therefore, inhibition of Hsp70 affects multiple pathways implicated in migration of macrophages, and ability of the latter to infiltrate tumors might be defined by cumulative defects in these pathways.
To evaluate effects of Hsp70 inhibition, macrophages were treated with JG-98, and their migration was assessed by “would healing” and transwell assays. In both assays JG-98 significantly suppressed macrophage migration (Figs. 2E). Importantly, effects on migration were seen even at 300 nM concentration while toxicity was observed only at concentrations 3 μM (Fig. 2E). Therefore, inhibition of Hsp70 has a profound effect on migration of macrophages and their infiltration into tumors.
Knockout of Hsp70 in host suppresses infiltration of macrophages and tumor growth
Results described above indicate that chemical inhibition of Hsp70 in tumor stroma cells significantly contributes to the tumor control. Because of possible off-target effects of the pharmacological agent, we sought to test the role of stromal Hsp70 in tumor support in genetic experiments. We used WT and Hsp70 knockout (KO) mice as recipients of allogenic tumor cells. Since these mice are not immune compromised, we had to utilize mouse cancer cells compatible with this strain, e.g. B16 melanoma cells. Cells, developed visible tumors in almost 100% of WT animals within 6–8 days (Table 1). However, when injected in Hsp70KO mice, these same cells did not produce tumors within at least 2 months (the period of observation) (Table 1). Therefore, host cells must carry active Hsp70 gene to allow development of allograft tumors with B16 melanoma. Similarly, Hsp70KO block tumor development with another syngeneic E0771 breast carcinoma line (Table 1). Therefore the ability to form allograft tumors at least with these cancer lines depended on the presence of Hsp70 in host cells, which was consistent with the data obtained with JG-98 and HeLa xenograts.
Table 1.
Emergence of allogenic tumors in Hsp70 knockout mice
| WT | K/O | Chi- squared test | |
|---|---|---|---|
| Exp 1: B16 melanoma (0.5×106cells in PBS) | 6/6 | 0/6 | P<0.001 |
| Exp 2: B16 melanoma (0.5×106 in PBS) | 7/8 | 0/8 | P<0.001 |
| Exp 3 E0771 breast carcinoma (0.5×106 in matrigel) | 4/6 | 0/6 | P<0.02 |
We further tested if the absence of Hsp70 in host cells suppresses macrophage infiltration in forming tumors at the injection sites. B16 melanoma cells (10 million in matrigel) were injected into WT and Hsp70KO animals. Mice were sacrificed every other day after injection and tissue in the injection site was isolated, fixed and sectioned. As expected, in the WT animals the cells gave rise to tumors, while in the Hsp70 knockout animals cancer cells steadily disappeared from the injection site, and by day 9 became practically undetectable (not shown). Infiltration of macrophages in the mass of injected cells was assessed by immunostaining with F4/80 antibodies. While massive infiltration was seen in the WT animals by day 5 following injection, much lower number of macrophages migrated into the cancer cell mass in the Hsp70KO (Fig. 3A). Therefore, Hsp70KO impairs macrophage infiltration, similar to JG-98 treatment. Notably, unlike in WT animals, there was a rim of macrophages surrounding tumors in Hsp70KO (Fig. S3) indicating that Hsp70KO affects macrophage infiltration into the tumor rather than macrophage number.
Fig. 3. Hsp70 knockout in host cells suppresses infiltration of macrophages in tumors.
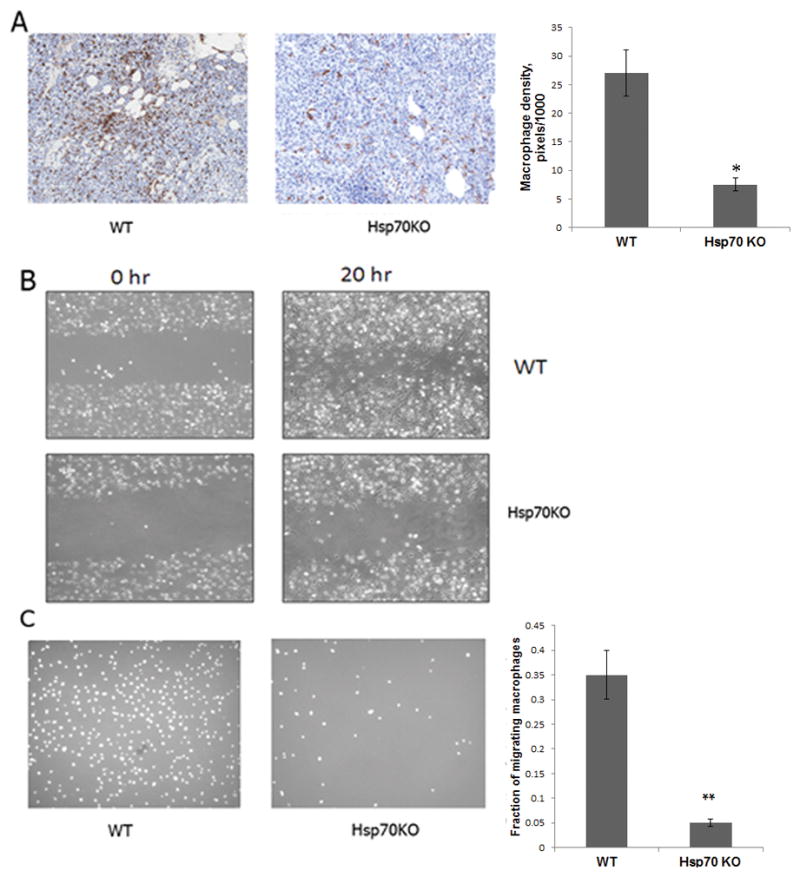
A. Hsp70 knockout in host suppresses macrophage infiltration into the tumor cell injection sites. WT and Hsp70KO mice were injected s.c. with 106 B16 melanoma cells in matrigel. On day 5 post-injection, the material at the injection site was collected, sliced and stained for the presence of macrophages with F4/80 antibodies Macrophages are in brown color and their number was quantified as in Fig. 2A. B and C. Hsp70KO suppress migration of macrophages. Bone marrow macrophages were isolated from WT and Hsp70KO mice and their migration was monitored by the “wound healing” (B) and transwell (C, left panel) assay as described in Fig. 2E. C. Left panel – picture of cells migrated in transwell assay, right panel – quantification of the transwell migration. Data shown are means+/SEM of triplicates.
Following experiments with JG-98, we evaluated effects of the genetic ablation of Hsp70 on in vitro migration of macrophages isolated from WT and Hsp70KO mice. The knockout led to a significant suppression of macrophage migration in in vitro wound healing and transwell assays (Figs. 3B and 3C). Therefore, genetic ablation of Hsp70 has a profound effect on suppression of migration of macrophages and their infiltration into tumors, which corresponds to the effect of Hsp70 inhibitor JG-98.
Discussion
There have been multiple attempts to target stroma for cancer treatment, since targeting stroma provide benefits due to different sensitivity of stromal cells to drugs compared to cancer cells, and since targeting stroma reduces chances to develop drug resistance due to selection of resistant clones (20). Recently drug targeting TAM and CAF have been developed (21, 14,16), but our arsenal of treatments is still very limited.
Here we provide evidence that targeting Hsp70 in stroma can suppress tumor growth. This effect was associated with inhibition of infiltration of macrophages into the tumor site. Of note, effects of Hsp70 knockout are limited to certain cancer models, e.g. allografts of B16 melanoma and E0771 breast tumor. On the other hand, in the PyMT model breast tumors emerged in Hsp70KO (though grew slower than in WT), and similarly in the model of Ras-transformed fibroblasts tumors emerged and grew (22), suggesting that in these models the requirement for TAM is less strict.
Previously we and others demonstrated inhibition of migration of tumor cells and metastasis in Hsp70KO (8). Here, we found that similarly Hsp70 is critical for migration of macrophages. These effects were associated with inhibition of important pathways, e.g. p38, FoxM1, Rho and probably other pathways. Regulation of these pathways by Hsp70 may also involve a co-chaperone Bag3 (4). Interestingly, JG-98 blocks Hsp70-Bag3 interaction (23), suggesting, that Bag3 may also be involved in suppression of macrophage motility. This possibility is consistent with previous finding that Bag3 depletion suppresses cells’ migration (24). By inhibiting macrophage motility, JG-98 series of compounds may affect immune functions in the organism, which requires focused investigation.
A dramatic evidence for the importance of Hsp70 in stroma for tumor development came from the experiment with HeLa cells clone resistant to the Hsp70 inhibitor JG-98. Despite the resistance in vitro, when xenograft tumors established with these cells demonstrated high sensitivity to JG-98. Therefore effects on stroma significantly contribute to the anti-tumor effects of this Hsp70 inhibitor. This finding is highly important because it demonstrates that intrinsic cancer cells’ resistance to this compound cannot protect tumors, and therefore development of resistance due to the clonal selection is very unlikely. Accordingly, this class of compounds could be effective against cancers with high probability of selection of resistant clones due to amplification of MDR1, like lung cancer. This novel strategy of targeting macrophages could have a wide range of applications. For example, it could target macrophages to suppress the restoration of vasculature following radiation therapy (25), and thus could be effectively combined with radiation or other chemotherapies. Overall, this work describes requirements of Hsp70 in stromal macrophages for tumor development, and suggests a series of chemical compounds that target this system.
Supplementary Material
Acknowledgments
Grant Support: MYS - R01 CA176326, JG - R01 NS059690
Footnotes
Authors do not have conflict of interest.
References
- 1.Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31(3):164–72. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Sherman MY, Gabai VL. Hsp70 in cancer: back to the future. Oncogene. 2015;34(32):4153–61. doi: 10.1038/onc.2014.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng L, Hunt C, Yaglom JA, Gabai VL, Sherman MY. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene. 2011;30(25):2836–45. doi: 10.1038/onc.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colvin TA, Gabai VL, Gong J, Calderwood SK, Li H, Gummuluru S, et al. Hsp70-Bag3 module regulates cancer-related signaling networks. Cancer Research. 2014;74(17):4731–40. doi: 10.1158/0008-5472.CAN-14-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gabai VL, Sherman MY, Yaglom JA. HSP72 depletion suppresses gamma H2AX activation by genotoxic stresses via p53/p21 signaling. Oncogene. 2010;29(13):1952–62. doi: 10.1038/onc.2009.480. [DOI] [PubMed] [Google Scholar]
- 6.Yaglom JA, Gabai VL, Sherman MY. High Levels of Heat Shock Protein Hsp72 in Cancer Cells Suppress Default Senescence Pathways. Cancer Res. 2007;67(5):2373–81. doi: 10.1158/0008-5472.CAN-06-3796. [DOI] [PubMed] [Google Scholar]
- 7.Gabai VL, Yaglom JA, Waldman T, Sherman MY. Heat Shock Protein Hsp72 Controls Oncogene-Induced Senescence Pathways in Cancer Cells. Mol Cell Biol. 2009;29(2):559–69. doi: 10.1128/MCB.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong J, Weng D, Eguchi T, Murshid A, Sherman MY, Song B, et al. Targeting the hsp70 gene delays mammary tumor initiation and inhibits tumor cell metastasis. Oncogene. 2015;34(43):5460–71. doi: 10.1038/onc.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pietras K, Östman A. Hallmarks of cancer: Interactions with the tumor stroma. Experimental Cell Research. 2010;316(8):1324–31. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 10.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–9. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scherz-Shouval R, Santagata S, Mendillo Marc L, Sholl Lynette M, Ben-Aharon I, Beck Andrew H, et al. The Reprogramming of Tumor Stroma by HSF1 Is a Potent Enabler of Malignancy. Cell. 2014;158(3):564–78. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen P, Huang Y, Bong R, Ding Y, Song N, Wang X, et al. Tumor-Associated Macrophages Promote Angiogenesis and Melanoma Growth via Adrenomedullin in a Paracrine and Autocrine Manner. Clinical Cancer Research. 2011;17(23):7230–39. doi: 10.1158/1078-0432.CCR-11-1354. [DOI] [PubMed] [Google Scholar]
- 14.Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends in Immunology. 2015;36(4):229–39. doi: 10.1016/j.it.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Nowicki A, Szenajch J, Ostrowska G, Wojtowicz A, Wojtowicz K, Kruszewski AA, et al. Impaired tumor growth in colony-stimulating factor 1 (CSF-1)-deficient, macrophage-deficient op/op mouse: evidence for a role of CSF-1-dependent macrophages in formation of tumor stroma. Int J Cancer. 1996;65(1):112–9. doi: 10.1002/(SICI)1097-0215(19960103)65:1<112::AID-IJC19>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 16.Ries Carola H, Cannarile Michael A, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell. 2014;25(6):846–59. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 17.Rowley SM, Kuriakose T, Dockery LM, Tran-Ngyuen T, Gingerich AD, Wei L, et al. Tumor Progression Locus 2 (Tpl2) Kinase Promotes Chemokine Receptor Expression and Macrophage Migration during Acute Inflammation. Journal of Biological Chemistry. 2014;289(22):15788–97. doi: 10.1074/jbc.M114.559344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawig L, Klasen C, Weber C, Bernhagen J, Noels H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Frontiers in Immunology. 2015;6 doi: 10.3389/fimmu.2015.00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balli D, Ren X, Chou FS, Cross E, Zhang Y, Kalinichenko VV, et al. Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene. 2012;31(34):3875–88. doi: 10.1038/onc.2011.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klement GL. Eco-evolution of cancer resistance. Science Translational Medicine. 2016;8(327):327fs5–27fs5. doi: 10.1126/scitranslmed.aaf3802. [DOI] [PubMed] [Google Scholar]
- 21.Goruppi S, Dotto GP. Mesenchymal stroma: primary determinant and therapeutic target for epithelial cancer. Trends in Cell Biology. 2013;23(12):593–602. doi: 10.1016/j.tcb.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dodd K, Nance S, Quezada M, Janke L, Morrison JB, Williams RT, et al. Tumor-derived inducible heat-shock protein 70 (HSP70) is an essential component of anti-tumor immunity. Oncogene. 2014 doi: 10.1038/onc.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Colvin T, Rauch JN, Acosta-Alvear D, Kampmann M, Dunyak B, et al. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol Cancer Ther. 2015;14(3):642–8. doi: 10.1158/1535-7163.MCT-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwasaki M, Homma S, Hishiya A, Dolezal SJ, Reed JC, Takayama S. BAG3 Regulates Motility and Adhesion of Epithelial Cancer Cells. Cancer Research. 2007;67(21):10252–59. doi: 10.1158/0008-5472.CAN-07-0618. [DOI] [PubMed] [Google Scholar]
- 25.Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, et al. Blockade of Tumor Necrosis Factor α Signaling in Tumor-Associated Macrophages as a Radiosensitizing Strategy. Cancer Research. 2010;70(4):1534–43. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.