

Abstract
Pathway inhibition of the RAS-driven MAPK pathway using small-molecule kinase inhibitors has been a key focus for treating cancers driven by oncogenic RAS, yet significant clinical responses are lacking. Feedback reactivation of ERK driven by drug-induced RAF activity has been suggested as one of the major drug resistance mechanisms, especially in the context of oncogenic RAS. To determine if additional adaptive resistance mechanisms may co-exist, we characterized global phosphoproteomic changes after MEK inhibitor selumetinib (AZD6244) treatment in KRAS-mutant A427 and A549 lung adenocarcinoma cell lines employing mass spectrometry-based phosphoproteomics. We identified 9,075 quantifiable unique phosphosites (corresponding to 3,346 unique phosphoproteins), of which 567 phosphosites were more abundant and 512 phosphosites were less abundant after MEK inhibition. Selumetinib increased phosphorylation of KSR-1, a scaffolding protein required for assembly of MAPK signaling complex, as well as altered phosphorylation of GEF-H1, a novel regulator of KSR-1 and implicated in RAS-driven MAPK activation. Moreover, selumetinib reduced inhibitory serine phosphorylation of MET at Ser985 and potentiated HGF- and EGF-induced AKT phosphorylation. These results were recapitulated by pan-RAF (LY3009120), MEK (GDC0623), and ERK (SCH772984) inhibitors, which are currently under early-phase clinical development against RAS-mutant cancers. Our results highlight the unique adaptive changes in MAPK scaffolding proteins (KSR-1, GEF-H1) and in RTK signaling, leading to enhanced PI3K-AKT signaling when the MAPK pathway is inhibited.
Introduction
Oncogenic KRAS mutations are major drivers of lung cancer growth and survival, occurring in nearly 25% of patients with lung adenocarcinoma (1). Oncogenic KRAS-driven lung cancers are often associated with poor prognosis and are notoriously refractory to conventional cytotoxic chemotherapies (2). Cancer cells are often addicted to aberrant activation of specific oncoproteins for their growth and survival, and targeting driver oncoproteins leads to better efficacy compared with conventional chemotherapies for some cases (3). For example, lung cancer cells exhibiting dysregulated epidermal growth factor receptor (EGFR) activity caused by somatic mutations are specifically sensitive to EGFR tyrosine kinase inhibitors (4–6). However, despite two decades of effort for target-based approaches to cancers derived from oncogenic KRAS activation, outcomes have not been satisfactory. One major reason is the inherent difficulty in blocking KRAS activity with small molecule inhibitors. As alternative strategies, targeting KRAS effectors such as RAF-MEK or phosphoinositide 3-kinase (PI3K) has been suggested. However, preclinical studies have shown MEK or PI3K inhibition in lung cancer leads to variable responses, and a subset of KRAS-mutant cancer cells are refractory to MEK or PI3K inhibitors (7–9).
Although many reasons have been suggested, one central feature may be related to a cancer cell’s ability to rapidly adapt to targeted agents, leading to adaptive or acquired drug resistance. Appreciation is growing for the role of adaptive resistance to targeted agents mediated by changes in feedback programs, leading to secondary activation of survival kinases (10, 11). Especially in the context of RAS-driven cancers, MAPK pathway reactivation after pharmacological MEK inhibition has been suggested as an important drug resistance mechanism. This feedback activation is regulated by drug-induced formation of new protein complexes, such as RAF homo/heterodimers or the RAF-MEK complex. One interesting feature of these drug-induced protein complexes is their relation to the molecular mechanism of action of the specific inhibitors used. A recent study reported that a subset of MEK inhibitors that are inactive in RAS-mutant cancers (AZD6244, GDC-0973) promotes BRAF-CRAF heterodimer formation allowing feedback activation of MEK and ERK, whereas RAS active MEK inhibitors (GDC-0623, G-573) stabilize a nonproductive RAF-MEK complex preventing MEK feedback activation (12). In contrast, another RAS active MEK inhibitor, trametinib, prevents MEK feedback reactivation through inhibition of MEK-RAF complex formation (13). However, the current understanding on this process has relied on focused and hypothesis-driven approaches, which could provide limited information depending on proteins or post-translational modifications examined and the availability of antibody reagents. Given the diversity of cancer signaling functioning in interconnected networks, a system-level understanding of this therapeutic escape process could unveil additional adaptive resistance mechanisms.
In this study, we used a mass spectrometry-based phosphoproteomics approach to delineate mechanisms of adaptive resistance in response to MEK inhibitor selumetinib (AZD6244) in KRAS-mutant lung cancer. Our mass spectrometry data combined with statistical and bioinformatic analyses offered a landscape of phosphoproteome response to pharmacological MEK inhibition, which serves as a valuable resource for understanding systems-level perturbations of MEK inhibition. Notably, our data showed selumetinib reduces inhibitory MET phosphorylation (Ser985). Follow-up studies revealed that pharmacological inhibition of MEK, as well as RAF and ERK, promotes EGFR- and MET-induced AKT phosphorylation. It has been reported that tumor microenvironment-driven receptor tyrosine kinase (RTK) signaling is involved in drug resistance (14, 15). A recent study indicated reduced proteolytic shedding of receptor tyrosine kinases by MEK inhibition is a new mechanism promoting RTK-driven drug resistance (16). Our results revealed another new mechanism by which MAPK inhibition leads to enhanced RTK signaling, which could promote microenvironment-driven RTK signaling and drug resistance.
Materials and Methods
Cell lines
Cells were maintained in RPMI 1640 medium supplemented with 10% FBS. Cells were confirmed to be free of mycoplasma using PlasmoTest (Invivogen).
Drugs
AZD6244 and MEK162 were purchased from ChemiTek. Drugs were reconstituted with DMSO at 50 mM, and aliquots were stored at −80°C.
Cell viability assay
Cell viability was analyzed by CellTiter-Glo (Promega) according to the manufacturer’s recommendations. Cells, plated at 1×103 per well in black-wall 384-well plates, were exposed to drugs for 72 hours before analysis.
Western blotting
Cells were washed with ice-cold PBS, and whole cell extracts were prepared using lysis buffer (0.5% NP-40, 50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitor (Roche) and phosphatase inhibitor cocktail (Sigma-Aldrich). Whole cell extracts were resolved on SDS-PAGE and transferred to nitrocellulose membrane. The membrane was blocked in 5% skim milk/PBST and then incubated in primary antibody at 4ºC overnight. Bound antibodies were visualized by horseradish peroxidase-conjugated secondary antibodies and SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Primary antibodies used for our study were purchased from Cell Signaling Technology (except for β-actin, which was from Sigma-Aldrich).
SILAC labeling
A427 and A549 cells were grown in media containing “heavy” [13C6]-L-Lys and [13C6,15N4]-L-Arg and “light” [12C6]-L-Lys and [12C6,14N4]-L-Arg for 7 days. At the end of 7 days, in biological replicate 1, light cells were treated with vehicle control (DMSO), and heavy cells were treated with selumetinib. In a parallel biological replicate 2, heavy cells were used as control and light cells were treated with drug. Twenty-four hours after treatment, cell pellets were collected in ice-cold PBS, washed twice, and then suspended in 300 μL of Lysis buffer (20 mM HEPES, pH 8.0, 9 M urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate). Using a microtip (Branson, Cell Discruptor 200), each suspension was sonicated at 15-W output with 3 bursts of 30 seconds each, cooling on ice for 10 seconds between each burst. Lysates were cleared by 20-minute centrifugation at 16,000 × g. Protein concentrations were determined with Bradford assay.
Trypsin digestion and basic pH reversed-phase liquid chromatography fractionation
In both biological replicates, equal amounts of protein from heavy and light extracts were mixed. The mixture (2 mg for A427, 1.5 mg for A549) was reduced with 4.5 mM DTT for 1 hour at 60ºC, cooled to room temperature, and then alkylated with 10 mM iodoacetamide at room temperature in the dark for 30 minutes. Reduced and alkylated proteins were diluted 4-fold with 20 mM HEPES buffer, pH 8.0, to a final concentration of 2 M urea and digested overnight at 37ºC with 1:40 enzyme-to-protein ratio of trypsin (Worthington). The resulting peptide solution was de-salted using C18 reversed-phase cartridges (Sep-Pak, Waters) and lyophilized. Dried peptides were dissolved in 20 mM aqueous ammonium formate, pH 10, for fractionation with basic pH reversed-phase liquid chromatography (17). The basic pH reversed-phase liquid chromatography separation was performed on a 4.6 mm × 100 mm column packed with 3-μm particle size, 300-Å pore size C18 resin (Xbridge, Waters). The gradient was as follows: 100% of solvent A (2% acetonitrile, 5 mM ammonium formate, pH 10) for 9 minutes, with concentration of solvent B (90% acetonitrile, 5 mM ammonium formate, pH 10) increased from 6% in 4 minutes, to 28.5% in 50 minutes, to 34% in 5.5 minutes, to 60% in 13 minutes, and then kept constant at 60% for 8.5 minutes. The flow rate was 0.6 mL/min, and 12 concatenated fractions were collected. Each fraction was lyophilized and re-dissolved in IMAC binding buffer (1% aqueous acetic acid and 30% acetonitrile) for phosphopeptide enrichment.
Phosphopeptide enrichment
Phosphopeptides in each fraction were enriched using IMAC resin (Sigma; PHOS-Select™ Iron Affinity Gel). Briefly, the tryptic peptides from each fraction were redissolved in IMAC binding buffer. IMAC resin (20 μL slurry/mg of digested protein) was washed twice with binding buffer. Phosphopeptides were incubated with the IMAC resin for 30 minutes at room temperature, with gentle agitation every 5 minutes. After incubation, the IMAC resin was washed twice with buffer 1 (0.1% acetic acid, 100 mM NaCl, pH 3.0), followed by 2 washes with buffer 2 (30% acetonitrile, 0.1% acetic acid, pH 3.0), and 1 wash with H2O. Phosphopeptides were eluted with 20% aqueous acetonitrile containing 200 mM ammonium hydroxide and concentrated by vacuum centrifugation. Concentrated peptides were diluted with 15 μL of 2% aqueous acetonitrile with 0.1% formic acid. LC-MS/MS and statistical analyses for MS data are described in Supplementary Materials and Methods.
Results
Characterization of phenotypic effect of MEK inhibitors in KRAS-mutant lung cancer cell lines
To determine how KRAS-mutant lung cancer cells respond to MEK inhibition, we assessed the efficacy of two clinical MEK inhibitors, selumetinib (AZD6244) and binimetinib (MEK162), in 11 lung cell lines harboring oncogenic KRAS mutations. We observed that maximum inhibition of cell viability was ~50%–60% at clinically achievable drug concentrations (1–2 μM) (18) (Figure 1A and Supplementary Figure S1). Next, four KRAS-mutant lung cell lines (A427, A549, H23, and H460) were treated with MEK inhibitors for 48 hours, and biochemical evidence of apoptosis and cell cycle arrest was examined. We observed no pronounced evidence of increased apoptosis (PARP cleavage) in all tested cell lines, and p27 induction, an indicator of cell cycle arrest, was observed in only H460 cells (Figure 1B). Collectively, these results recapitulated other preclinical studies and an early-phase clinical study that showed modest effects of MEK inhibition in KRAS-mutant lung cancer (19–22).
Figure 1. Phenotypic effect of MEK inhibitors on KRAS-mutant NSCLC cell lines.

A, Relative cell viability after MEK inhibitor AZD6244 and MEK162 treatment at 1 μM. Cells were incubated with MEK inhibitors for 72 hours, followed by cell viability assay (Promega). Representative triplicates ± SD are presented, which showed similar results at least 2 times. Dose-dependent effects are shown in Supplementary Figure 1. B, p27 expression, pERK, and PARP cleavage after MEK inhibitor treatment (1 μM, 48 hours).
Phosphoproteomic response to selumetinib in KRAS-mutant lung cancer cells
Although the modest response of MEK inhibitors in KRAS-mutant cancer could be attributed to feedback reactivation of ERK by drug-driven RAF activation, we hypothesized that additional adaptive resistance mechanisms could exist to support drug-treated cancer cell viability. To explore this possibility, we characterized altered global phosphorylation after exposing KRAS-mutant lung cancer cells with selumetinib to gain further insight into its drug action. Based on a previous pharmacokinetic study that identified a Cmax of selumetinib ranging from 528 to 952 ng/mL (1.15–2.08 μM) (18), 1 μM was chosen for phosphoproteomics experiments. Two KRAS-mutant lung cancer cell lines, A427 and A549, were employed and each cell line was analyzed in two biological replicates including label swap experiments. SILAC-labeled cells were exposed to selumetinib (1 μM, 24 hours) or vehicle control (DMSO), and then phosphorylated peptides (pS/pT/pY) were enriched, followed by liquid chromatography and tandem mass spectrometry (LC-MS/MS) analyses as performed in our previous study (23). A 24-hour time point was chosen based on a previous study on kinome-level response to a MEK inhibitor (24). Figure 2 shows the workflow of our phosphoproteomic strategy. Using this approach, we identified a total of 9,075 unique phosphosites; 6,094 and 7,281 unique phosphosites from A427 and A549, respectively (Supplementary Figure S3, A–C, and Supplementary Table S1). Histograms of the log2 drug-to-DMSO SILAC ratios are roughly normally distributed and centered at zero (Supplementary Figure S3, E–F), which is consistent with previous studies showing similar number of phosphopeptides could be both increased or decreased following kinase inhibitor treatment (25–27). We next focused on phosphosites whose abundance was affected by MEK inhibition. Log2 ratios for treatment vs. control calculated from four experiments (two experiments from A427 and A549) should meet the following criteria: i) at least two log2 ratios must be present, ii) the signs of all log2 ratios agree, iii) at least one log2 ratio changes by at least ±0.585 (1.5-fold), and iv) at least two log2 ratios change by at least ±0.1375 (1.1-fold). From this analysis, we defined 1079 selumetinib-regulated phosphosites, in which 567 phosphosites (435 phosphoproteins) were increased and 512 phosphosites (411 phosphoproteins) were decreased by the drug treatment (Supplementary Table S2). We named these phosphosites as “selumetinib-regulated phosphosites” and the corresponding phosphoproteins as “selumetinib-regulated phosphoproteins.”
Figure 2. Work flow of SILAC-based phosphoproteomics approach.

The SILAC-labeled KRAS-mutant NSCLC cell lines (A427 and A549) were treated with selumetinib (1 μM, 24 hours) or DMSO vehicle control. Target inhibition was confirmed by Western blotting for pERK (Supplementary Figure S2). Phosphopeptides were enriched from fractionated tryptic peptides, followed by LC-MS/MS analysis. Detailed methods for mass spectrometry and statistical analyses are described in Supplementary Materials and Methods.
Analysis of selumetinib-regulated phosphosites
We aimed to identify phospho-motifs specifically enriched in altered phosphosites, which could further predict kinases potentially regulated by selumetinib treatment from selumetinib-regulated phosphosites. Motif discovery analysis revealed that proline-directed motifs (pS/T-P) were most enriched in both increased and decreased phosphosites (Figure 3, A and B). Enriched proline-directed motifs in decreased phosphosites group are consistent with reduction of mitogen-activated kinase activity by selumetinib treatment. This analysis also suggested that activities of other proline-directed kinases such as cyclin-dependent kinase could be modulated by selumetinib treatment. In addition, phosphopeptides containing a basophilic motif with an arginine residue at −3 position (R-x-x-S) were found to be most enriched in the increased phosphosites group, suggesting selumetinib could activate basophilic kinases. KinomeXplorer analysis, which predicts kinase-substrate relationships (28), revealed CDK1 is the most enriched kinase by both increased and decreased phosphopeptides and also showed pronounced enrichment of PKCβ, a basophilic kinase (29), in the increased phosphosite group. These results are consistent with the motif analysis indicating enrichment of proline-directed motifs in both increased and decreased phosphosites and more enrichment of basophilic motif in increased phosphosites group, respectively (Figure 3, C and D).
Figure 3. Motif analysis and kinase prediction for selumetinib-regulated phosphosites.

A and B, Representative phospho-motifs enriched in up- (A) and down- (B) regulated phosphopeptides after selumetinib treatment using Motif-x. Motifs with significance of P<10−6 are shown. C and D, Pie chart for enriched kinase groups to match up- (C) and down- (D) regulated phosphopeptides after selumetinib treatment using NetworKIN.
Analysis of selumetinib-regulated phosphoproteome
Next, we set out to examine how selumetinib remodels the global phosphoproteome. First, we identified biological pathways significantly enriched by proteins whose phosphorylations were differentially regulated by MEK inhibition. GeneGO (Metacore, Thomson Reuters) pathway enrichment analysis of selumetinib-regulated phosphoproteins showed that the drug treatment led to alterations in pathways involved in control of cell growth/proliferation, translation, cytoskeleton remodeling (Supplementary Table S3). To gain a more comprehensive view of altered signaling networks by selumetinib treatment, all differentially expressed phosphoproteins (selumetinib-regulated phosphoproteins) were analyzed with GeneGo Metacore to identify all direct and nearby connecting phosphoproteome and transcriptional interactions (shown as thick and thin arrows, respectively) based on the literature. These interactions were combined with consensus direction of drug-induced change (decreased: green, increased: red) and then reduced to a set of self-consistent interactions that agree with the observed direction of change (such as decreased inhibitor levels leading to increased expression of the target gene). Altered phosphorylation of kinases and transcription factors was especially shown in hexagons and parallelograms, respectively, as the activity of these proteins is mostly regulated by phosphorylation. Figure 4 shows the resulting network view, and the selected kinases in which phosphorylation was altered by selumetinib are shown in Table 1. The network showed previously validated MEK inhibitor-induced alterations as connected nodes such as decreased phospho-ERK1 (green MAPK3 node) with decreased phospho-p90RSK1 (green RPS6KA3 node) and increased phospho-MEK1 and MEK2 (red MAP2K1 and MAP2K2 nodes, respectively) with increased phospho-BRAF and RAF1 as a result of feedback activation of RAF (12, 13). It also showed that selumetinib increased phosphorylation of a panel of kinases, including PAK1, MAP2K4 (MKK4), BRSK2, and PRKDC (DNAPK), which are involved in the cell cycle regulation, stress responses, and DNA damage responses. Whether selumetinib activates or inhibits these kinase activities remains to be elucidated since functions for these altered phosphosites have not been annotated. The network showed selumetinib increased β-catenin phosphorylation (red CTNNB1 node). The increased phosphosite, Ser675, has been known to be phosphorylated by cyclic AMP-dependent protein kinase (PKA), leading to its stabilization and transcriptional activation (30, 31). This suggests MEK inhibition could promote transcriptional activation of β-catenin via PKA. Increased histone methyltransferase EZH2 phosphorylation was also shown as a node with high connectivity like β-catenin. The increased phosphosite, Thr487, is regulated by CDK1, promoting its ubiquitin-mediated degradation by the proteasome (32, 33), and is consistent with our motif and kinase prediction analysis suggesting CDK1 activity being modulated by selumetinib treatment (Figure 3). This regulatory mechanism is important for cancer cell proliferation, warranting further investigation to study how MAPK pathway could be linked to CDK1-EZH2-mediated cell cycle regulation.
Figure 4. Self-consistent network composed of selumetinib-regulated phosphoproteins.

Red nodes indicate increased phosphoproteins, green nodes indicate decreased phosphoproteins, and dim yellow nodes indicate genes/complexes added by GeneGo Metacore to connect significantly changed nodes but were either not observed to be significantly changed or are complexes for which no data were available. Orange arrows indicate activation/phosphorylation, and blue lines indicate inhibition/dephosphorylation. Gray dashed lines indicate membership within a protein complex. Phosphorylation is indicated by thick lines, and transcriptional regulation is indicated by narrow lines. Node size corresponds to number of edges connecting to the node. Transcription factors are shown in parallelograms, kinases are shown in hexagons, and everything else is shown in ellipses.
Table 1.
Selected selumetinib-regulated phosphopeptides
| A427 | Phosphosite | Sequence | Log2FC |
|---|---|---|---|
| MEK-ERK pathway | |||
| ERK1 | pT202/pY204 | IADPEHDHTGFLT(ph)EY(ph)VATR | −1.61 |
| MEK1 | pS222 | LCDFGVSGQLIDSMANS(ph)FVGTR | 4.46 |
| BRAF | pS605 | SRWS(ph)GSHQFEQLSGSILWMAPEVIR | 1.76 |
| CRAF | pS296/pS301 | SHSESASPSALSSS(ph)PNNLS(ph)PTGWSQPK | 2.83 |
| RSK2 | pT365 | T(ph)PKDSPGIPPSANAHQLFR | −2.20 |
| RSK1 | pS221 | KAYS(ph)FCGTVEYMAPEVVNR | 2.72 |
| RTK | |||
| EPHA2 | pY772 | VLEDDPEATY(ph)TTSGGKIPIR | −0.42 |
| EPHA7 | pY791 | VIEDDPEAVY(ph)TTTGGKIPVR | 0.92 |
| Signaling kinase | |||
| AMPKA1 | pS486 | SIDDEITEAKS(ph)GTATPQR | 1.44 |
| PAK2 | pT169 | GTEAPAVVT(ph)EEEDDDEETAPPVIAPRPDHTK | 3.04 |
| MKK4 | pS90 | LKIS(ph)PEQHWDFTAEDLKDLGEIGR | 1.57 |
| A549 | Phosphosite | Sequence | Log2FC |
|---|---|---|---|
| MEK-ERK pathway | |||
| ERK1 | pT202/pY204 | IADPEHDHTGFLT(ph)EY(ph)VATR | −3.09 |
| MEK1 | pS222 | LCDFGVSGQLIDSMANS(ph)FVGTR | 2.38 |
| BRAF | pS605 | SRWS(ph)GSHQFEQLSGSILWMAPEVIR | 0.53 |
| CRAF | pS296/pS301 | SHSESASPSALSSS(ph)PNNLS(ph)PTGWSQPK | 0.34 |
| RSK2 | pT365 | T(ph)PKDSPGIPPSANAHQLFR | −2.40 |
| RSK1 | pS221 | KAYS(ph)FCGTVEYMAPEVVNR | 1.22 |
| RTK | |||
| MET | pT977/pS985 | VHT(ph)PHLDRLVS(ph)AR | −1.20 |
| EGFR | pS991/pS995 | MHLPS(ph)PTDS(ph)NFYR | 0.68 |
| EPHA2 | pY772 | VLEDDPEATY(ph)TTSGGKIPIR | −1.62 |
| Signaling kinase | |||
| AMPKA1 | pS486 | SIDDEITEAKS(ph)GTATPQR | 1.34 |
| PAK2 | pT169 | GTEAPAVVT(ph)EEEDDDEETAPPVIAPRPDHTK | 1.04 |
| MKK3 | pT218 | MCDFGISGYLVDS(ph)VAKTMDAGCKPYMAPER | −1.50 |
| MKK4 | pS90 | LKIS(ph)PEQHWDFTAEDLKDLGEIGR | 0.54 |
We next focused our attention on altered phosphorylations in MAPK signaling proteins and illustrated them in Figure 5, which shows decreased ERK phosphorylation (Thr202/Tyr204 in ERK1) and a feedback increase of phosphorylation in MEK (Ser222) and RAF isoforms (BRAF Ser605 and CRAF Ser296/Ser301). Furthermore, we found that selumetinib increased phosphorylation of KSR-1 (Thr268), a scaffolding protein required for assembly of MAPK signaling complex (34) and altered phosphorylations on GEF-H1 (Ser122/Ser151). GEF-H1 is been known as a guanine nucleotide exchange factor (GEF) for the Rho family small GTPase proteins (35); however, recently GEF-H1 was shown to promote RAS-driven MAPK via modulating KSR-1 phosphorylation (36). KSR-1 and BRAF or CRAF interaction is enhanced by pharmacological RAF inhibition, regulating drug-induced RAF dimer formation (37–39). Given these studies, selumetinib-regulated KSR-1 and GEF-H1 phosphorylations could modulate their scaffolding function for MAPK pathway activation. We also observed that selumetinib differentially regulated phosphorylation sites in p90RSK, a downstream kinase of ERK. The phosphorylation of Thr365, an ERK-dependent site, was decreased by MEK inhibitor treatment; however, we found that phosphorylation of another key phosphosite for kinase activation in N-terminal kinase domain Ser221, a PDK1 target site (40), was significantly increased, suggesting compensatory phosphorylation occurs in response to loss of ERK activity. Another notable aspect revealed from this network-based approach is that selumetinib altered serine phosphorylation of RTKs; i) selumetinib increased EGFR phosphorylation at Ser991/Ser995, which is involved in receptor trafficking (41), raising the possibility that MEK inhibitors could be involved in EGFR trafficking via modulating these phosphosites. ii) MET phosphorylation at Ser985 was decreased by selumetinib. The reduced expression of this phospho-serine residue is associated with increased MET kinase activity (42, 43), suggesting that MEK inhibition could lead to MET activation by relieving inhibitory serine phosphorylation.
Figure 5. Altered phosphorylations in MAPK signaling cascade by selumetinib.
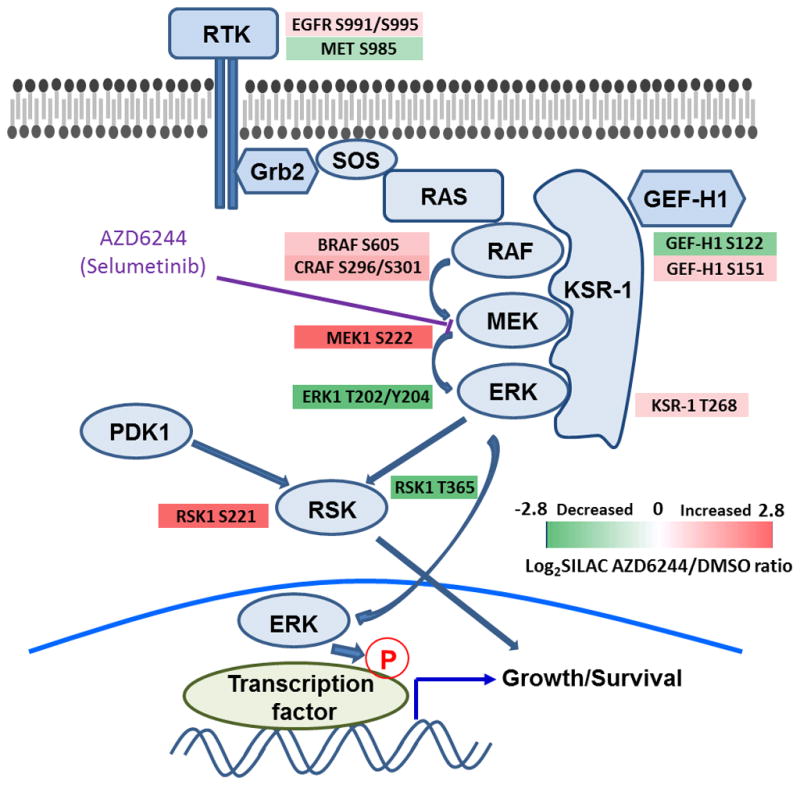
The log2-transformed fold-change of phosphopeptide abundance is shown in color (green: decreased, red: increased).
Selumetinib promotes MET- and EGFR-induced AKT phosphorylation
The decreased MET Ser985 phosphorylation was confirmed by visual inspection of extracted ion chromatogram, MS1 and MS2 spectra (Supplementary Figure S4). Next, we tested whether MEK inhibition could promote MET signaling. We also tested whether EGFR signaling activity could be enhanced by MEK inhibition given its role in providing bypass survival signaling in drug-treated cells (15). A549 cells were pretreated with selumetinib (1 μM, 24 hours) and exposed to MET ligand HGF or EGFR ligand EGF, then tyrosine phosphorylation of MET or EGFR and the activities of their downstream signals, ERK and AKT, were examined. MET Ser985 phosphorylation is negatively associated with its activating tyrosine phosphoryation (42, 43); however we observed that MEK inhibitor pretreatment did not increase MET tyrosine phosphorylation (Tyr1234/1235) in the A549 cell line. Both basal and ligand-induced ERK phosphorylation were suppressed by drug treatment. Interestingly, selumetinib promoted HGF- and EGF-induced AKT phosphorylation specifically at Thr308, a PDK1 target site, whereas mTOR-dependent Ser473 phosphorylation was marginally affected by selumetinib (Figure 6, A and B).
Figure 6. Effect of MAPK inhibition on MET and EGFR signaling.

A and B, A549 cells were incubated with selumetinib (1 μM, 24 hours) and then treated with HGF (A) or EGF (B) at 25 ng/mL for indicated hours. C and D, A549 cells were incubated with MAPK inhibitors (1 μM, 24 hours) and then treated with HGF (C) or EGF (D) at 25 ng/mL for 30 minutes. DM, DMSO; LY, LY3009120 (pan-RAFi); AZD, AZD6244 (MEKi); GDC, GDC0623 (MEKi); SCH, SCH772984 (ERKi). E and F, H23 cells were treated as described above. G and H, A549 cells were incubated with selumetinib (1 μM, 24 hours), and then endogenous MET (G) or integrin β4 (H) was immunoprecipitated. Co-immunoprecipitated integrin β4 (G) or MET (H) was detected by Western blotting.
Recently, several new-generation small-molecule kinase inhibitors targeting MAPK signaling cascade have shown promising pre-clinical efficacy and are in early-phase clinical trials. A new pan-RAF inhibitor, LY3009120, can block signaling of both RAF homo- and heterodimers, thus minimizing paradoxical reactivation of the ERK pathway, which is associated with drug resistance of selective BRAF inhibitors such as vemurafenib (44). This compound has activity in some KRAS-mutant models. A selective novel ERK inhibitor, SCH772984, was shown to overcome BRAF and MEK inhibitor resistance in KRAS-mutant cells in a preclinical study (45). We next asked whether these new MAPK inhibitors could promote RTK-driven AKT phosphorylation. A549 cells were pretreated with BRAF inhibitor vemurafenib, pan-RAF inhibitor LY3007120, RAS-inactive MEK inhibitor AZD6244, RAS-active MEK inhibitor GDC0623 (due to differential ability to prevent feedback MEK activation upon treatment) (12, 13), and ERK inhibitor SCH772984. Similar to selumetinib, we observed that the HGF- or EGF-induced AKT Thr308 phosphorylation was enhanced in cells pretreated with these MAPK inhibitors (except for vemurafenib), indicating that pathway inhibition of MAPK, not only MEK inhibition, can lead to enhanced ligand-induced MET and EGFR signaling (Figure 6, C and D). We next examined whether these observations could be recapitulated in another KRAS-mutant lung cancer cell line, H23. Similarly, we observed ligand-induced AKT phosphorylation was enhanced by MAPK inhibitors, except for GDC0623. Ligand-induced AKT Ser473 phosphorylation was also enhanced in this cell line. In contrast to A549, we observed that RAF, MEK, and ERK inhibitors promoted basal MET tyrosine phosphorylation in H23 cells (Figure 6, E and F).
We observed that these MAPK inhibitors failed to significantly increase MET autophosphorylation (Y1234/1235) in A549, in which we observed reduction of inhibitory serine phosphorylation, whereas MET tyrosine phosphorylation was enhanced by MAPK inhibition in H23 cells (Figure 6E). We hypothesized that perhaps these MAPK inhibitors could remodel RTK protein complexes instead of directly affecting MET kinase activity. It has been reported that MET forms a dimer complex with integrin β4. Tyrosine-phosphorylated integrin β4 potentiates HGF-induced MAPK and PI3K-AKT signaling to support anchorage-independent growth and invasion (46, 47); however, physiological conditions that induce this active MET complex have not been identified. We reasoned that the MEK inhibitor could promote MET-integrin β4 interaction, enhancing downstream signaling. To test this hypothesis, MET-bound integrin β4 was examined between control and selumetinib-treated cells, and we observed that selumetinib promoted MET-integrin β4 interaction in A549 cells (Figure 6, G and H) and lung cancer cell lines harboring MET amplification, EBC1 and H1648 (Supplementary Figure S5, A and B). Collectively, these results suggest that MEK inhibition could enhance MET activity or remodel the MET interactome.
Discussion
Here, we describe a large-scale mass spectrometry-based proteomics approach to delineate phosphoproteome responses to the MEK inhibitor selumetinib in the context of KRAS-mutant lung cancer. Previous studies showed global phosphoproteome responses to MEK and RAF inhibition in the context of BRAF-mutant melanoma (48, 49); to our knowledge, this study is the first global phosphoproteomics approach that addresses how KRAS-mutant lung cancer cells respond to pharmacological MEK inhibition. Importantly, we demonstrate widespread increases in protein phosphorylation following treatment with a MEK inhibitor, which at first glance seems counterintuitive. However, this result is consistent with other observations that kinase inhibitors can lead to increase phosphorylation of some substrate proteins. Previously, we revealed TBK1 knockdown leads to increased phosphorylation of EGFR, MET, and their downstream ERK→Jun, Myc in KRAS-mutant lung cancer cells (23). The tyrosine kinase inhibitor dasatinib induces compensatory activation of a panel of RTKs, including MET and IGF-1R in DDR2-mutant lung squamous cancer cells (25); likewise, EGFR-mutant lung cancer cells demonstrate RTK activation after EGFR TKI treatment, including MET, IGF1R, and AXL (27). We observed a similar number of proteins whose phosphorylation is elevated following selumetinib treatment, suggesting that these system-level changes could modulate the effects of kinase inhibitors by activating cell intrinsic compensatory mechanisms sustaining cell survival. Finally, our global phosphoproteomics database will be a useful resource for generating further hypotheses on both adaptive resistance mechansims and pathways affected by MEK inhibitors.
The most notable finding from our study is that the MEK inhibitor reduced inhibitory phosphorylation of MET (Ser985). The follow-up experiments showed enhanced AKT phosphorylation after HGF stimulation when MAPK pathway is inhibited. Although we did not see significant changes in inhibitory EGFR phosphorylation in our dataset such as p-Thr669 which promotes EGFR internalization (50), we observed MAPK inhibitors also could promote EGFR-induced AKT activation. Compensatory activation of AKT upon RAF or MEK inhibition has been observed in multiple tumors (51–54), which involves coexpressed RTKs including EGFR (52, 53, 55), IGF1R (51), and EGFR/HER3 (54). A recent combinatorial drug screening study showed targeting compensatory RTK or downstream PI3K-AKT pathway is synergistic with BRAF inhibition in the context of BRAF mutant melanoma (56); however, we could not observe pronounced synergy from targeted inhibtion of MET or EGFR with MEK in lung cancer cells from in vitro 72-hour cell viability asssay (Supplementary Figure S6). One possibility is that the phenotype of this enhanced RTK ligand stimulation following MAPK pathway inhibition could be better seen in other settings closer to in vivo such as 3D spheroid culture. The feedback RTK activation in the aformentioned studies are mediated by drug-induced autotyrosine phosphorylation; however, our study highlights a novel feedback activation mechanism of RTKs in response to MAPK inhibition, which involves altered Ser/Thr phosphorylation leading to hypersensitivity to ligand stimulation. We observed that the MEK inhibitor enhanced tyrosine phosphorylation of MET in H23 but not in A549 cells. Interestingly, the MEK inhibitor promoted formation of new MET-integrin β4 complex in A549. Besides its known role in cell-extracellular matrix communication, integrin β4 is a signaling adaptor of MET. Tyrosine phosphorylated integrin β4 by MET recruits Shc and PI3K for MAPK and AKT pathway activation, respectively (47). It is also known that integrin β4 activates HGF-induced SHP2-SRC pathway activation for oncogenic growth (46). Given these previous observations, our results raise the possibility that the MEK inhibitor could prime MET activation via relieving inhibitory phosphorylation in cancer cells and/or allow formation of a drug-induced MET-integrin β4 complex for full downstream activation in the presence of minute amounts of growth factor ligands.
The importance of tumor-stromal interaction in modulating drug sensitivity is increasingly being recognized (57). Stromal secretion of HGF reactivates MAPK and PI3K-AKT pathways and confers BRAF inhibitor resistance to BRAF-mutant melanoma (14), and increased plasma HGF was associated with poor prognosis in patients with BRAF-mutant melanoma who were treated with a BRAF inhibitor and in patients with KRAS wild-type colorectal cancer treated with an anti-EGFR antibody (15, 58). Based on our results, it is possible that cancer cells treated with MAPK pathway inhibitors are hypersensitive to stromal HGF, promoting stroma-induced drug resistance. Further investigation for phenotypic consequences of enhanced MET signaling in cancer cells treated with MAPK inhibitors will be an important next step.
The underlying mechanisms whereby MEK inhibition leads to decreased Ser985 of MET remains to be elucidated. The Ser985 site is located within the juxtamembrane region of MET, regulated by crosstalk between PKC-δ/ε and Ser/Thr protein phosphatase 2A (PP2A) (42). Although we did not observe altered phosphorylations in PKC-δ/ε or PP2A, it is still possible that MEK inhibition could lead to either increased PP2A or decreased PKC-δ/ε activity. Our results raise the possibility that modulating activities of these enzymes could be an alternative way to target stroma-induced MET activation in cancer cells treated with MEK inhibitors.
Collectively, our phosphoproteomics study provides a more comprehensive view of phosphoproteome responses to selumetinib in the context of KRAS-mutant lung cancer. Our study indicated that MEK inhibition could enhance MET and EGFR-induced AKT activation, presumably via modulation of inhibitory phosphorylation and/or forming new RTK protein complexes (e.g. MET-integrin β4). Newer RAS active MAPK inhibitors (e.g., the pan-RAF inhibitor LY3009120, the MEK inhibitor GDC0623) are being tested for RAS-mutant cancers in the clinic, and we show that these drugs also enhanced MET and EGFR-induced AKT activation. Given the importance of stromal contribution of drug resistance, our finding has high clinical significance in predicting potential drug resistance mechanisms and furthermore provides a rationale for co-targeting of MAPK and EGFR/MET signaling in KRAS-mutant lung cancer treatment. Our study warrents further study to investigate whether these MAPK inhibitors could modulate phosphorylation or protein-protein interaction of other RTKs (e.g., IGF-1R, ERBB2), and their subsequent role in kinase inhibitor resistance.
Supplementary Material
Implications.
This study highlights the unique adaptive changes in MAPK scaffolding proteins (KSR-1, GEF-H1) and in RTK signaling, leading to enhanced PI3K/AKT signaling when the MAPK pathway is inhibited.
Acknowledgments
We thank Rasa Hamilton (Moffitt Cancer Center) for editorial assistance, Dr. Yian Ann Chen (Moffitt Bioinformatics & Biostatistics Department) for her input on statistical analysis, and Dr. Eunjung Kim (Moffitt Integrated Mathematical Oncology Group) for her assistance with generating histograms in Supplementary Figure S3. Our study also received valuable assistance from the Proteomics and Cancer Informatics Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI-designated Comprehensive Cancer Center, supported in part under NIH grant P30-CA76292 and Moffitt Foundation.
Grant Support
This work was supported, in whole or in part, by National Institutes of Health SPORE Grant P50 CA119997, R21 CA181848-01A1 and by the Moffitt Lung Cancer Center of Excellence.
Footnotes
Conflicts of interest: No potential conflicts of interest were disclosed by the authors.
References
- 1.Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter? J Clin Oncol. 2013;31:1112–21. doi: 10.1200/JCO.2012.43.0454. [DOI] [PubMed] [Google Scholar]
- 2.Singh A, Settleman J. Oncogenic K-ras “addiction” and synthetic lethality. Cell Cycle. 2009;8:2676–7. doi: 10.4161/cc.8.17.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinstein IB. Cancer Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 4.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 5.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genom. 2010;3:26. doi: 10.1186/1755-8794-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matallanas D, Crespo P. New druggable targets in the Ras pathway? Curr Opini Mol Therap. 2010;12:674–83. [PubMed] [Google Scholar]
- 10.Chandarlapaty S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012;2:311–9. doi: 10.1158/2159-8290.CD-12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klinger B, Bluthgen N. Consequences of feedback in signal transduction for targeted therapies. Biochem Soc Trans. 2014;42:770–5. doi: 10.1042/BST20140130. [DOI] [PubMed] [Google Scholar]
- 12.Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–6. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 13.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller MA, Oudin MJ, Sullivan RJ, Wang SJ, Meyer AS, Im H, et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016;6:382–99. doi: 10.1158/2159-8290.CD-15-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Meth. 2013;10:634–7. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 20.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Therap. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 21.Lamba S, Russo M, Sun C, Lazzari L, Cancelliere C, Grernrum W, et al. RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep. 2014;8:1475–83. doi: 10.1016/j.celrep.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 22.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc Natl Acad Sci U S A. 2013;110:12414–9. doi: 10.1073/pnas.1220674110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai Y, Kim JY, Watters JM, Fang B, Kinose F, Song L, et al. Adaptive responses to dasatinib-treated lung squamous cell cancer cells harboring DDR2 mutations. Cancer Res. 2014;74:7217–28. doi: 10.1158/0008-5472.CAN-14-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, et al. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal. 2010;3:rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, Li J, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin Cancer Res. 2014;20:4059–74. doi: 10.1158/1078-0432.CCR-13-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, et al. KinomeXplorer: an integrated platform for kinome biology studies. Nat Meth. 2014;11:603–4. doi: 10.1038/nmeth.2968. [DOI] [PubMed] [Google Scholar]
- 29.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–60. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 30.Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol Cell Biol. 2005;25:9063–72. doi: 10.1128/MCB.25.20.9063-9072.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–6. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 32.Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–20. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511–9. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witzel F, Maddison L, Bluthgen N. How scaffolds shape MAPK signaling: what we know and opportunities for systems approaches. Front Physiol. 2012;3:475. doi: 10.3389/fphys.2012.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren Y, Li R, Zheng Y, Busch H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem. 1998;273:34954–60. doi: 10.1074/jbc.273.52.34954. [DOI] [PubMed] [Google Scholar]
- 36.Cullis J, Meiri D, Sandi MJ, Radulovich N, Kent OA, Medrano M, et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell. 2014;25:181–95. doi: 10.1016/j.ccr.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 37.Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, et al. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc Natl Acad Sci U S A. 2011;108:6067–72. doi: 10.1073/pnas.1102554108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKay MM, Freeman AK, Morrison DK. Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases. 2011;2:276–81. doi: 10.4161/sgtp.2.5.17740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKay MM, Ritt DA, Morrison DK. RAF inhibitor-induced KSR1/B-RAF binding and its effects on ERK cascade signaling. Curr Biol. 2011;21:563–8. doi: 10.1016/j.cub.2011.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–58. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 41.Tong J, Taylor P, Moran MF. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol Cell Proteom. 2014;13:1644–58. doi: 10.1074/mcp.M114.038596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hashigasako A, Machide M, Nakamura T, Matsumoto K, Nakamura T. Bi-directional regulation of Ser-985 phosphorylation of c-met via protein kinase C and protein phosphatase 2A involves c-Met activation and cellular responsiveness to hepatocyte growth factor. J Biol Chem. 2004;279:26445–52. doi: 10.1074/jbc.M314254200. [DOI] [PubMed] [Google Scholar]
- 43.Gandino L, Longati P, Medico E, Prat M, Comoglio PM. Phosphorylation of serine 985 negatively regulates the hepatocyte growth factor receptor kinase. J Biol Chem. 1994;269:1815–20. [PubMed] [Google Scholar]
- 44.Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell. 2015;28:384–98. doi: 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 45.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–50. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 46.Bertotti A, Comoglio PM, Trusolino L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J Cell Biol. 2006;175:993–1003. doi: 10.1083/jcb.200605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trusolino L, Bertotti A, Comoglio PM. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. 2001;107:643–54. doi: 10.1016/s0092-8674(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 48.Parker R, Clifton-Bligh R, Molloy MP. Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol Cancer Therap. 2014;13:1894–906. doi: 10.1158/1535-7163.MCT-13-0938. [DOI] [PubMed] [Google Scholar]
- 49.Stuart SA, Houel S, Lee T, Wang N, Old WM, Ahn NG. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol Cell Proteom. 2015;14:1599–615. doi: 10.1074/mcp.M114.047233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winograd-Katz SE, Levitzki A. Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene. 2006;25:7381–90. doi: 10.1038/sj.onc.1209737. [DOI] [PubMed] [Google Scholar]
- 51.Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–47. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565–72. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 54.Yoon YK, Kim HP, Han SW, Hur HS, Oh do Y, Im SA, et al. Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Therap. 2009;8:2526–36. doi: 10.1158/1535-7163.MCT-09-0300. [DOI] [PubMed] [Google Scholar]
- 55.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roller DG, Capaldo B, Bekiranov S, Mackey AJ, Conaway MR, Petricoin EF, et al. Combinatorial drug screening and molecular profiling reveal diverse mechanisms of intrinsic and adaptive resistance to BRAF inhibition in V600E BRAF mutant melanomas. Oncotarget. 2016;7:2734–53. doi: 10.18632/oncotarget.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12:217–28. doi: 10.1038/nrd3870. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi N, Yamada Y, Furuta K, Honma Y, Iwasa S, Takashima A, et al. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2014;110:2716–27. doi: 10.1038/bjc.2014.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.