

Abstract
The mosquito-borne dengue virus serotypes 1-4 (DENV1-4) and West Nile virus (WNV) cause serious illnesses worldwide associated with considerable morbidity and mortality. According to the World Health Organization (WHO) estimates, there are about 390 million infections every year leading to ~500,000 dengue haemorrhagic fever (DHF) cases and ~25,000 deaths, mostly among children. Antiviral therapies could reduce the morbidity and mortality associated with flaviviral infections, but currently there are no drugs available for treatment. In this study, a high-throughput screening assay for the Dengue protease was employed to screen ~120,000 small molecule compounds for identification of inhibitors. Eight of these inhibitors have been extensively analyzed for inhibition of the viral protease in vitro and cell-based viral replication using Renilla luciferase reporter replicon, infectivity (plaque) and cytotoxicity assays. Three of these compounds were identified as potent inhibitors of DENV and WNV proteases, and viral replication of DENV2 replicon and infectious RNA. Fluorescence quenching, kinetic analysis and molecular modeling of these inhibitors into the structure of NS2B-NS3 protease suggest a mode of inhibition for three compounds that they bind to the substrate binding pocket.
Keywords: Protease inhibitors, High-throughput screening, Renilla luciferase reporter replicon, virus infectivity assays, therapeutic index, fluorescence quenching
Introduction
Dengue virus serotypes 1-4 (DENV1-4) cause the most frequent mosquito-borne infections to humans. According to a recent estimate by the World Health Organization, DENVs cause 390 million infections annually (Mitka, 2013), and are primarily transmitted by the Aedes aegypti and A. albopictus mosquitos. Dengue virus (DENV) belongs to the flavivirus genus of the Flaviviridae family and is endemic throughout tropical and sub-tropical countries in the world (for reviews, see (Beasley, 2005; Gould and Solomon, 2008; Guzman et al., 2010; Lindenbach and Rice, 2003) causing frequent epidemics. Infection with any of the DENV serotypes may be asymptomatic in the majority of cases or may result in a mild flu-like syndrome (known as dengue fever (DF). However, secondary infections by a different DENV serotype can cause symptoms collectively known as “severe dengue”, characterized by coagulopathy, increased vascular fragility and permeability, thought to result from antibody-dependent enhancement (Halstead et al., 2005; Sierra et al., 2010). Currently, there is no antiviral drug available for human use.
The DENV genome consists of a ~11 kb, plus strand, RNA molecule, that upon entry into a host cell, is translated into a single polyprotein in the endoplasmic reticulum (ER) membrane. This polyprotein undergoes co- and post-translational cleavages by the host signal peptidase and furin, as well as the viral serine protease NS3 to form mature structural and non-structural proteins. The three structural proteins, capsid (C), precursor membrane (prM), and envelope (E), are generated from the N-terminal region of the polyprotein. The seven non-structural (NS) proteins are generated in the order of NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5, from the C-terminal region of the polyprotein (Henchal and Putnak, 1990; Kautner et al., 1997).
The NS2B-NS3 protease plays a crucial role in viral replication as it is essential for the processing of the polyprotein followed by the assembly of the viral replication complex (Padmanabhan and Strongin, 2010; Sampath and Padmanabhan, 2009). The requirement for the viral protease in the DENV life cycle makes it an excellent target for development of antiviral therapeutics (Lim et al., 2013; Noble et al., 2010). The NS2B-NS3 protease cleaves the polyprotein in cis at NS2B-NS3 site and trans at the C-terminal regions of C anchored to the ER membrane to form mature C as well as NS2A-2B, NS3-4A, NS4A-2K, and NS4B-5 sites to form mature NS2A, NS2B, NS3, NS4A, NS4A lacking 2K peptide (Miller et al., 2007), NS4B and NS5 proteins (reviewed in (Padmanabhan and Strongin, 2010)). The interaction of the NS3pro domain (1–185 aa of NS3 protein) with the conserved hydrophilic domain of ~48 aa residues of NS2B are sufficient for the in vitro protease activity (Chambers et al., 1993; Clum et al., 1997; Falgout et al., 1993). However, for replication of the virus in mammalian cells, full-length NS2B and NS3 proteins associated with ER membranes are required (Chambers et al., 1993; Chambers et al., 1995; Matusan et al., 2001). The NS2B-NS3 protease cleavage sites are comprised of two basic amino acids (RR, RK, KR, or occasionally QR) followed by an amino acid with a short side chain such as Gly, Ala, or Ser (for reviews, (Lindenbach et al., 2007; Padmanabhan and Strongin, 2010)). To assess the protease activity in vitro, the tetrapeptide substrate with two Arg residues at P1 and P2 positions linked to the fluorogenic group, 7-amino- 4-methyl coumarin moiety (AMC), benzoyl (Bz)-Nleu-Lys-Arg-Arg-AMC, was found to be an optimal substrate (Li et al., 2005). Incubation with the recombinant protease results in the release of AMC fluorescence which follows Michaelis-Menten kinetics allowing for the determination of IC50 and Ki values (Li et al., 2005; Mueller et al., 2008).
We have previously established a High-Throughput Screening (HTS) assay for the identification of small molecule inhibitors of the WNV protease in a 384-well format using a library of ~32,000 compounds. We identified compounds containing the 8-hydroxyquinoline (8- OHQ) scaffold as potent inhibitors of both DENV2 and WNV proteases (Ezgimen et al., 2012; Lai et al., 2013a; Mueller et al., 2008). Here, we report a HTS assay of ~120,000 commercially available compounds to identify additional scaffolds of DENV protease inhibitors. Using a slightly modified assay, only ~0.1% (<100) of the total compounds after cherry-picking, showed good inhibition of DENV2 protease in vitro. From this list, a select number of compounds showed potent Ki values in the range of 0.22 ± 0.03 μM to 6.9 ± 0.80 μM in the in vitro assays, and EC50 values for inhibition of viral replication in the range of 0.08 ± 0.01 μM to ~ 9.0 μM determined by plaque assays.
2. Materials and Methods
2.1. High-throughput screening (HTS)
HTS was performed at the The Institute of Chemistry and Cell Biology Screening Facility at Harvard University (ICCB-Longwood). The compound libraries used for screening in the study include: ActiMolTimTec 1, Asinex 1, BIOMOL ICCB Known Bioactives 3, Biomol 4 FDA Approved Drug Library, Chemical Diversity 2, Enamine 1, 2, 2a, ChemBridge 3 and 4, ChemDiv 3, 5, ICBG Fungal Extracts 11, Life Chemicals 1, Maybridge 4 and 5, MicroSource Discovery 1, MicroSource US Drug Collection, Mixed Commercial Plate 5, NIH Clinical Collection 1 and 2, NINDS Custom Collection 2, Prestwick Collection 1 and 2, Sigma LOPAC containing a total of 119,997 compounds. Briefly, into each well of 384-well plates (Corning 3575), 20 μL of DENV2 protease (30 nM) in protease assay buffer was dispensed using a Wellmate liquid dispenser (Matrix, Hudson, NH). Test compounds [5 mg/ml] in dimethyl sulfoxide [DMSO], 100 nL were pin-transferred into duplicate plates using a compound transfer robot (Epson, Carson, CA). The final inhibitor concentrations in the assays varied due to different molecular weights of the compounds screened in the primary HTS. Bovine pancreatic trypsin inhibitor, also known as aprotinin (BPTI; 7.5 μM) was used as a positive control for one column in each plate. Another column did not receive any compounds (negative control). The compound-enzyme solutions were incubated for 15 minutes at room temperature before 10 μL of the 7.5 μM substrate Bz-Nle-Lys-Arg-Arg-AMC (Bachem, Torrence, CA) was added. Plates were transferred to a 37 °C incubator for 15 minutes, and fluorescence intensity was measured using 380 nm and 460 nm excitation and emission wavelengths with EnVision plate reader (Perkin-Elmer, Waltham, MA). To avoid aggregation-based non-specific inhibition by the compounds (Ezgimen et al., 2009; Feng et al., 2005; Leung et al., 2001), 0.1% CHAPS was added in the protease assay buffer. Compounds which inhibited the cleavage of the substrate by at least 50% were considered as positive hits and selected for further analysis. Secondary analysis of compounds were performed at two concentrations (7 and 17 μg/mL) in 100 μL reactions using the cherry-picked aliquots received at concentrations (5mg/mL).
2.2. Protein expression, purification and active site titration
The DENV2 and WNV proteases were expressed from 2L cultures of TOP10F′ cells (Invitrogen) transformed with the expression plasmids pQE30-NS2BH(QR)-NS3pro (Yon et al., 2005) and WNV clone 107 (Mueller et al., 2007), respectively. The expression plasmids for the DENV1 and DENV4 NS2BH-NS3 proteases each containing the respective authentic C-terminal amino acids of NS2B followed by the NS3pro domain were constructed by PCR amplification of coding sequences from the full-length cDNA clones of DENV1 and DENV4 (Alcaraz-Estrada et al., 2010; Puig-Basagoiti et al., 2006). To construct the DENV3 NS2BH-NS3pro plasmid, a clone of DENV3 cDNA in pRS424 vector (YU4N) with a deletion of E and part of NS1 genes that could be stably propagated in E. coli (Gifted by Dr. Barry Falgout, FDA) was used for overlap extension PCR following standard molecular biology techniques. The plasmid clones for DENV1, DENV3 and DENV4 NS2BH-NS3pro domains contained the four amino acids from the C-terminus of NS2B linked to NS3pro that include the NS2B-3 cleavage site in pET-32a between NcoI and BamHI restriction sites (unpublished data). The DENV2 protease expression plasmid codes for the NS2B hydrophilic domain and the NS3pro domain flanking the two amino acid residues, QR, at the NS2B-3 site (Yon et al., 2005) (see above).
E. coli cells transformed with DENV1-4 and WNV NS2B-NS3pro expression plasmids were grown in LB medium supplemented with ampicillin (100 μg/mL) and 0.1% glucose at 37°C until the OD600 reached 0.6–0.7. The medium was then replaced with fresh LB-ampicillin with 1 mM IPTG. Expression of the recombinant proteins was induced for 4 h at 37°C. Cells were then pelleted and either frozen or used immediately for purification using a Talon metal affinity resin (Clontech) as reported previously (Mueller et al., 2008). Since E. coli-expressed proteins might contain a certain amount of misfolded molecules, we sought to determine the fraction of the proteases that are catalytically active. The fractions of active proteases in our enzyme preparations were thus determined by assessing Bz-Nle-Arg-Arg-AMC cleavage efficiency using 25 nM of protease and increasing amounts of BPTI. The Ki of BPTI against WNVpro is 24 nM (Aleshin et al., 2007) and against DENV2pro 26 nM (Mueller et al., 2007). Using a linear regression model of the percentage protease activity against BPTI concentration, we can extrapolate the concentration of active protease present in the sample as the x-intercept (Figure 1). The active site concentrations of all proteases were close to their respective protein concentration indicating that the enzyme preparations contained 98–99% active proteases (data not shown).
Figure 1.
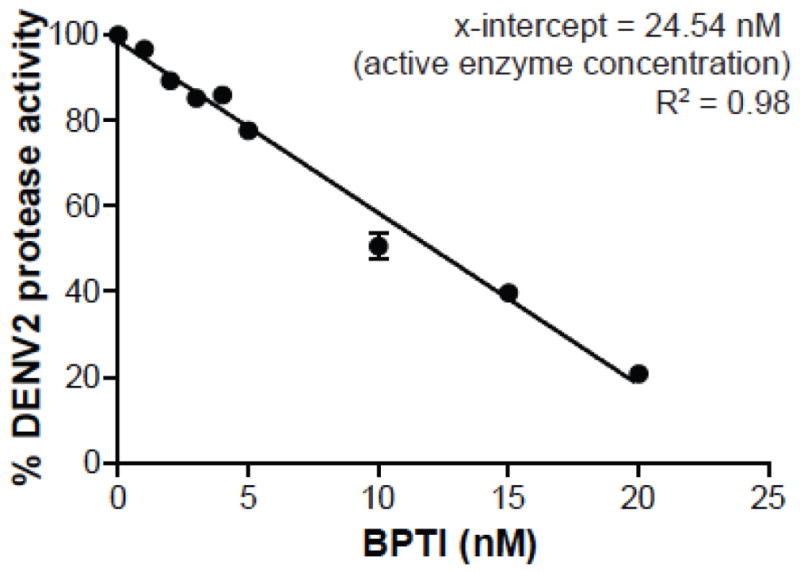
Active-Site titration of DENV2 protease with BPTI. X-intercept indicates the active concentration of DENV2 protease (25 nM protein was used based on protein concentration).
2.3. Further in vitro characterization of hits
From the list of positive compounds, the highest scoring hits (0.3% of the total number of compounds screened) were cherry-picked for validation and further analysis. Compounds which were previously classified as “promiscuous hits”, as they often occurred in multiple screens against diverse targets in the ICCB-Longwood database, were excluded for further analysis. Aliquots (1.2 μL) of the cherry-picked compounds in the same concentration as used in the primary HTS were provided by ICCB-Longwood for secondary validation assays. The activities of these compounds were tested against the DENV2 protease at two concentrations, 7 and 17 μg/mL, in 100 μL standard protease assay mixtures to examine dose-dependent inhibition. From this list of cherry-picked compounds, 29 commercially available compounds exhibiting good potency were purchased and screened in secondary assays (all commercially available compounds were assured to be minimum 95% pure by NMR and LC-MS). Only 22 were soluble in DMSO at a concentration of 5 mM. The activities of the 22 compounds were tested against DENV2 and WNV proteases at 10 and 25 μM (Table 1). Eight of those compounds which showed at least 50% inhibition at 10 μM were selected for determination of their IC50 values.
Table 1.
Percent Inhibition of DENV2 and WNV proteases by compounds (at 10 and 25 μM) selected from HTS and validation assays.
| Compound | Inhibition of DENV2pro (%) | Inhibition of WNVpro (%) | Further analyzed | ||
|---|---|---|---|---|---|
| At 10 μM | At 25 μM | At 10 μM | At 25 μM | ||
| 1477N17 (MW 344) | 51 | 89 | 54 | 95 | A |
| 1480C06 (MW 354) | 19 | 41 | 5 | 50 | |
| 1481E05 (MW 362) | 57 | 92 | 45 | 92 | B |
| 1508P19 (MW 381) | 30 | 63 | 1 | 59 | |
| 1578O21 (MW 246) | i.s. | i.s. | i.s. | i.s. | |
| 1579D07 (MW 264) | i.s. | i.s. | i.s. | i.s. | |
| 1580G21 (MW 262) | 7 | −2 | −9 | 1 | |
| 1585F07 (MW 298) | −5 | −6 | −14 | −13 | |
| 1585J20 (MW 409) | 2 | 5 | −15 | −8 | |
| 1586C04 (MW 390) | 96 | 94 | 98 | 98 | C |
| 1587K07 (MW 338) | 63 | 82 | 71 | 95 | D |
| 1588C13 (MW 298) | 17 | 42 | 16 | 82 | |
| 1589E03 (MW 396) | i.s. | i.s. | i.s. | i.s. | |
| 1593H11 (MW 406) | 75 | 86 | 75 | 95 | E |
| 1595F21 (MW 304) | 12 | 21 | −6 | 8 | |
| 1596L13 (MW 282) | 19 | 39 | −6 | 14 | |
| 1602A03 (MW 310) | 8 | −4 | −22 | −18 | |
| 1607H05 (MW 237) | 15 | 32 | −18 | 2 | |
| 1610E01 (MW 249) | 15 | 29 | −11 | 21 | |
| 1628D17 (MW 412) | i.s. | i.s. | i.s. | i.s. | |
| 1628E04 (MW 353) | 25 | 40 | 40 | 79 | |
| 1720I04 (MW 382) | i.s. | i.s. | i.s. | i.s. | |
| 1725P16 (MW 372) | 18 | 52 | 26 | 75 | |
| 1989F14 (MW 220) | 24 | 56 | 31 | 82 | |
| 1989F18 (MW 294) | i.s. | i.s. | i.s. | i.s. | |
| Ellagic acid (MW 302) | i.s. | i.s. | i.s. | i.s. | |
| Tolcapone (MW 273) | 90 | 92 | 99 | 95 | F |
| Tannic acid (MW1701) | 99 | 101 | 98 | 96 | G |
| Suramin (MW 1291) | 72 | 85 | 58 | 73 | H |
i.s.: insoluble even at 5 mM in DMSO. Compounds A-H were chosen for additional characterization of efficacy in vitro protease assays and in cell-based assays culture.
2.3.1. In Vitro Protease Assay
This assay measures the proteolytic activity of the viral NS3 protease domain complexed with the required NS2B hydrophilic domain by using the fluorogenic peptide substrate, Bz-Nle-Lys-Arg-Arg-AMC. The standard assay mixture (100 μL) contained 200 mM Tris-HCl, pH 9.5, 6 mM NaCl, 30% glycerol, 20 nM enzyme, 25 μM substrate, CHAPS 0.1% and in the presence or absence of protease inhibitor at 37°C in a 96-well blackwall microtiter plates. The reaction was monitored within a monochrometer-based spectroflurometer (Spectromax Gemini EM from Molecular Devices, Inc.) running on Softmax Pro software. Upon cleavage, the AMC fluorophore is released, thereby increasing fluorescence. This increase is dependent on the incubation time, and concentrations of enzyme, substrate, and inhibitor in a manner that obeys Michaelis-Menten kinetics. The fluorescence of AMC was measured using a fluorometer at excitation and emission wavelengths of λex=380nm and λem=460nm, respectively (for a detailed protocol, see (Lai et al., 2013b)). BPTI was used as a positive control and 1% DMSO (no inhibitor; 100% activity) served as a negative control. For determination of IC50 values, standard protease assays were performed in the presence of different inhibitor concentrations (0.05, 0.1, 0.5, 1, 2, 5, 10 and 25 μM) for 30 min. IC50 values were calculated using the GraphPad Prism v5.04 using the four-parameter nonlinear regression analysis (Hill slope method).
2.3.2. Determination of Ki of compounds
Four different concentrations (0–3 μM) of compounds and eight different concentrations of substrate (3.90, 7.81, 15.62, 31.25, 62.5, 125, 250, 500 μM) for each inhibitor concentration were tested in the in vitro DENV2 protease assay using 20 nM protein. Fluorescence was measured in duplicate wells at an interval of 90 sec. The fluorescence values (RFUs) were converted to molar amounts using a standard curve determined from known amounts of AMC and their corresponding RFU values. The velocity values (mole substrate cleaved/minute) were then calculated for each substrate:inhibitor pair. Ki values were calculated with GraphPad Prism software v5.04 with non-linear regression at competitive inhibition mode of enzyme-kinetics.
2.3.3. Renilla luciferase reporter replicon assay
BHK-21 cells (ATCC), stably expressing DENV2 replicon RNA (Alcaraz-Estrada et al., 2013; Boonyasuppayakorn et al., 2014; Manzano et al., 2011) encoding Renilla luciferase reporter (1×104 cells/well of a 96-well plate), were treated with varying concentrations (0, 0.5, 1, 2, 5, 10, and 25 μM) of different inhibitors. Following ~24 h of incubation, cells were lysed with 50μL 1X lysis buffer, followed by shaking for ~5min. The luciferase activity was measured using a luminometer (Berthold Tech., LB-960) as described previously (Boonyasuppayakorn et al., 2014).
2.3.4. Cytotoxicity of compounds to BHK-21 cells
BHK-21 (1×104 cells/well of a 96-well plate) were treated with 0.39, 0.78, 1.56, 3.12, 6.25, 12.50, 25, 50, 100 μM of each compound and incubated for ~24 h. The cytotoxicity was measured using the Cell Counting Kit-8 (Dojindo, Rockville, MD) following the manufacturer’s instructions. Briefly, 10 μL of a 1X CCK-8 solution/well was added to all the wells. The plates were placed back in an incubator at 37°C. After 2 h, the absorbance at 450 nm was measured. The concentration of each compound at which 50% loss of viability (CC50) occurred was determined.
2.3.5. Efficacy of compounds on infectivity and viral RNA replication by plaque assays and qRT PCR
BHK-21 cells (1×105) in MEM-alpha medium + 10% FBS were plated onto each well of 12-well plates. After 18 h, cells were infected with DENV2 (MOI 1) in MEM-alpha medium + 2% FBS for 90 min. Virus inocula were replaced with 2 mL OptiMEM (Gibco, Life Sciences) with increasing concentrations of drugs (0.03–10μM). After incubation for 12 h at 37°C, FBS was added to each well at a final concentration of 5%. After an additional 12 h, the supernatant was collected and the viral titers were quantified by plaque assays as previously described (Boonyasuppayakorn et al., 2014). Briefly, LLC-MK2 cells were plated at 1 × 105 cells per well in 24-well plates containing 1 ml DMEM + 10% FBS and incubated overnight. Serially diluted supernatants from infected BHK-21 cells treated with compounds described above were incubated with monolayers of LLC-MK2 cells in paired wells of 24-well plates for 2 h. Cells were overlaid with DMEM containing 1.0% carboxymethyl cellulose and were incubated for 7 days at 37°C. The plaques formed on monolayers were visualized by fixing the cells with 3.4% paraformaldehyde and staining with crystal violet solution (0.05%) as described (Boonyasuppayakorn et al., 2014).
To determine the viral RNA copy number by qRT-PCR, total intracellular RNAs were extracted from the infected cells (control and treated) by treatment with TRIzol reagent (Invitrogen, Life, Grand Island, NY) according to the manufacturer’s protocol. RNAs were quantified using BioSpec-nano (Shimadzu, Japan) and used as templates for cDNA synthesis by reverse transcriptase in 20 μL containing 1 μg RNA. Each qRT-PCR contained 12.5 μL of the iQ SYBR Green Supermix reagent (Bio-Rad, Hercules, CA), 0.5 μL of primers (20 nM) specifically designed to anneal to the DENV-2 NS5 gene (Manzano et al., 2011), 9.5 μL of DEPC water, and 2 μL of RNA to a final volume of 25 μL. The amplification protocol consisted of the following steps: 95°C for 2 min, followed by 50 cycles at 95°C for 10 seconds, 50°C for 30 seconds and 72°C for 30 seconds and melt curve analysis was carried out. The glyceraldehyde-3-phosphate gene (GAPDH) was used as a reference control.
2.3.7. Fluorescence quenching assay
A fluorescence quenching assay has been used to discriminate between specific and non-specific inhibition of DENV2 protease (Bodenreider et al., 2009). This particular assay is based on the intrinsic fluorescence of Trp50, which is located near the active site center of -DENV and WNV proteases. The fluorescence of this tryptophan is quenched by inhibitors that bind to the active site of protease and have absorbance near 330 nm. The absorbance of compounds C, F (100 μM), G and H (10μM) were determined and all four compounds absorb near 300–330 nm indicating they are suitable candidates to be tested using this assay.
Briefly, a competition assay was performed with 10 different concentrations of inhibitors (0.09, 0.18, 0.36, 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50 μM) and DENV2 protease (2μM). The autofluorescence of the samples was monitored within a monochrometer-based spectroflurometer (Spectromax Gemini EM from Molecular Devices, Inc.) running on Softmax Pro software with excitation wavelength of 280 nm and an emission wavelength of 340 nm. All determinations were performed in triplicates. Titration data were normalized by the formula (F0−F)/F0, where F is the measured fluorescence and F0 is the measured fluorescence in the absence of inhibitors.
2.3.7. Molecular modeling Studies
The crystal structure of DENV3 NS2B-NS3pro (3U1I) in the catalytically active closed conformation was used as a template for modeling studies. A homology model of DENV2 protease was built using SWISS- MODEL (Biasini et al., 2014). PatchDock server (Schneidman-Duhovny et al., 2005) was used to dock the molecules C, F and G into the active site of DENV2 protease. PatchDock is a geometric algorithm and it finds docking transformations that yields optimal molecular shape complementarity.
3. Results
3.1. Identification of DENV2 protease inhibitors using HTS
The flavivirus protease is essential for the processing of the polyprotein which generates the viral proteins required for viral replication and maturation of infectious virions. Therefore, the protease is an ideal target for the discovery of antivirals against Dengue. A previously established HTS for small molecule inhibitors of the WNV protease in a 384-well format (Mueller et al., 2008) identified several compounds from which we launched a detailed structure-activity relationship analysis using the 8-hydroxyquinoline scaffold (Ezgimen et al., 2012; Lai et al., 2013a). In this study, we performed HTS of ~120,000 compounds to expand our HTS campaign for discovery of more broad chemical scaffolds from which we could initiate a lead optimization process using slightly modified assay conditions. First, we used an optimal fluorogenic tetrapeptide substrate Bz-Nle-Lys-Arg-Arg-AMC (Li et al., 2005). We used a purified preparation of DENV2 NS2B-QR-NS3pro which contains the hydrophilic domain of NS2B ending with the last two amino acids (QR) and the N-terminal 185 amino acids of protease domain (Yon et al., 2005). The initial phase of HTS of ~25K compounds revealed a higher hit rate (1.75%) and false positives, and hence we included 0.1% CHAPS detergent in our assay buffers to avoid nonspecific aggregation-based inhibitors (Ezgimen et al., 2009; Feng and Shoichet, 2006; Leung et al., 2001).
Our primary screen resulted in 507 compounds (0.42%) that exhibited at least 50% inhibition of protease activity in the 384-well HTS format (Figure 2). We cherry picked 242 of the primary hits (Figure 2) to further validate hits based on their relative strengths of inhibition in 96-well format. Compounds that frequently appear in other unrelated HTS assays done in ICCB-Longwood were flagged as potentially highly reactive or “promiscuous” molecules and were excluded. Only 88 of the 242 compounds validated in our secondary assay, which were retested in 100 μL reaction at 7 and 17 μg/mL concentrations in 96-well format provided by ICCB-Longwood facility (Fig. 2, 36% validation rate). Of these, 29 compounds were purchased from commercially available sources based on their relative strengths of inhibition when reanalyzed in molar concentrations.
Figure 2.

Pie-chart of primary and secondary assays for selection of inhibitors.
3.2. Inhibition of DENV2 and WNV proteases by selected compounds in vitro
Twenty-nine validated compounds were purchased from commercial sources for detailed analysis and 10 mM stock solutions were prepared in DMSO. Seven of these did not completely dissolve even at 5 mM and were not further investigated (Table 1). The remaining hits were assayed at fixed molar concentrations of 10 and 25 μM to determine their relative potencies. Only 8 compounds (A to H) were shown to have =50% inhibition (Figure 3; Table 1). IC50 values of all the eight compounds were determined against DENV1-4 and WNV proteases. Compound G (tannic acid) was found to be the most potent inhibitor against all five flavivirus proteases tested (0.11–0.82 μM) followed by Compound F (tolcapone) (0.61–1.25 μM). Compounds H (suramin) and C had IC50s in the range of 2.5–4.1 μM which was followed by compounds E, A and B (Table 2). The Ki of these 8 compounds were determined against DENV2 protease (Table 3).
Figure 3.

Structures of 8 selected compounds identified by HTS
Table 2.
IC50 values of selected compounds against DENV1-4 proteases and West Nile virus protease.
| Compounds | A | B | C | D | E | F | G | H | |
|---|---|---|---|---|---|---|---|---|---|
| DENV 1 Pro | IC50 | 9.65 ±0.32 | 11.29±0.33 | 4.06±0.21 | 10.83±0.37 | 6.20±1.20 | 1.15±0.1 | 0.77±0.05 | 2.76±0.25 |
| Hill-slope | 2.01±0.16 | 1.55±0.10 | 1.51±0.18 | 3.92±0.27 | 0.71±0.13 | 1.26±0.10 | 1.79±0.09 | 0.92±0.05 | |
| DENV 2 Pro | IC50 | 8.22±0.24 | 10.01±0.28 | 4.05±0.18 | 10.45±0.40 | 4.84 ± 0.60 | 0.98±0.06 | 0.56±0.04 | 2.66±0.19 |
| Hill-Slope | 1.79±0.11 | 2.21±0.18 | 1.61±0.18 | 2.75±0.16 | 1.11±0.16 | 1.51±0.07 | 1.60±0.07 | 0.97±0.04 | |
| DENV 3 Pro | IC50 | 9.95±0.45 | 9.66±0.63 | 2.94±0.18 | 11.14±0.38 | 5.0±1.40 | 0.91±0.06 | 0.40±0.03 | 3.25±0.18 |
| Hill-slope | 1.62±0.15 | 1.33±0.17 | 0.95±0.09 | 2.70±0.13 | 1.03±0.16 | 1.60±0.08 | 1.53±0.06 | 1.05±0.04 | |
| DENV 4 Pro | IC50 | 8.95±0.34 | 12.20±0.47 | 3.40±0.11 | 11.04±0.37 | 5.32±0.60 | 0.64±0.03 | 0.13±0.01 | 3.73±0.14 |
| Hill-slope | 2.22±0.21 | 1.95±0.19 | 1.53±0.12 | 3.20±0.16 | 1.22±0.13 | 1.45±0.05 | 0.79±0.03 | 1.25±0.12 | |
| WNV Pro | IC50 | 6.43±0.38 | 10.06±0.33 | 2.53±0.10 | 6.56±0.24 | 4.31 ± 0.17 | 0.70±0.04 | 0.12±0.01 | 3.24±0.11 |
| Hill-slope | 1.63±0.18 | 1.92±0.18 | 1.38±0.22 | 2.64±0.13 | 1.12±0.11 | 1.58±0.05 | 0.88±0.05 | 1.29±0.03 | |
Values presented here are averages of duplicate experiments and reported with the standard errors of the mean.
Table 3.
Ki, EC50, CC50 and SI index of selected compounds.
| Compound ID | Kinetics of Inhibition | EC50 (μM) | CC50 (μM) | Therapeutic index (TI) | |||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Ki (μM) | R2 | DENV2 Plaque | DENV2 Replicon | TI-1 | TI-2 | ||
|
| |||||||
| A | 6.9±0.80 | 0.99 | >20 | >20 | >100 | * | * |
| B | 2.7±0.33 | 0.98 | >20 | >20 | 62.44 | * | * |
| C | 1.0±0.15 | 0.97 | 8.97 ± 0.05 | 6.9 ± 0.6 | 76.11 | 8.48 | 11.02 |
| D | 3.6±0.39 | 0.98 | 14.23 ± 0.06 | >20 | >100 | >7.02 | * |
| E | 1.6±0.16 | 0.99 | >20 | 10.4 ± 4.2 | 80.01 | * | 7.69 |
| F | 0.22±0.03 | 0.98 | 2.03 ± 0.1 | 2.29 ± 0.3 | 29.16 | 14.36 | 12.73 |
| G | 0.34±0.05 | 0.98 | 0.084 ± 0.010 | (N.D) | 68.08 | 810.47 | * |
| H | 0.87±0.14 | 0.99 | 8.62 ± 1.32 | (N.D) | >100 | >11.60 | * |
Indicates that the TI could not be determined due to lack of inhibitory effect in the tested concentration range (0–20 μM). N.D (not determined)
3.3. Determination of the efficacy of compounds in inhibition of DENV2 replication using the stable BHK-21/DENV2 Replicon cells
Initially, the eight compounds were screened for inhibition of DENV2 replicon replication at 3 different concentrations (5, 10 and 25 μM) using the stable BHK-21/DENV2 replicon cell line expressing Renilla luciferase (Boonyasuppayakorn et al., 2014). Compounds C, E and F were found to inhibit DENV2 replication as compared to the 1% DMSO-treated control cells. These compounds were further screened at six different concentrations as described under Materials and Methods to determine the EC50 values. The EC50 of compound F was 2.29 ± 0.3 μM, followed by compound C (6.9 ± 0.6 μM) and compound E (10.4 ± 4.2 μM) (Table 3). Compounds A, B, D, G and H did not inhibit the DENV2 replicon replication (Table 3).
3.4. Cytotoxicity assay
The cytotoxicity of the eight compounds was estimated by using the colorimetric CCK-8 cell viability kit on parental BHK-21 cells. Of the eight compounds assayed, compounds B, C, E, and G (CC50 60–80 μM), and F (CC50 ~30 μM), and H had cytotoxic effects after 24 h exposure. Compounds A, D and H did not exhibit any cytotoxicity at the highest concentration tested (~ 100 μM) (Table 3).
3.5. Plaque assay and qRT PCR
The inhibitory effects of these compounds on viral replication and infectivity were also examined by two independent methods. In the first method, the titers of virions produced at various concentrations of the compounds were measured by a plaque assay. This method probes whether any of the compounds has any effect on production of infectious virions. In the second method, the copy number of the viral RNA in the virus particles released from infected BHK-21 cells was determined by qRT-PCR after treatment with each compound. The copy number of RNA includes RNA from both infectious and non-infectious virus particles.
The results of plaque assays of eight compounds indicated that only compounds C, D, F, G and H were shown to be inhibitory with EC50s of 8.97 ± 0.05 μM, 14.23 ± 0.06 μM, 2.03 ± 0.1, 0.084 ± 0.010 and 8.62 ± 1.32 μM respectively (Figure 4, Table 3). The qRT-PCR was performed for cells treated with 2 μM (for compounds F and G) or 10 μM (for compounds C, D and H) of the drugs. Our results indicate that the compounds C, D, F, G and H showed good inhibition as compared to DMSO treated control (Figure 5). The therapeutic indices (TI) were calculated as the ratios of CC50/EC50. Only four of the 8 compounds (C, F, G and H) exhibited < 10 μM inhibition in plaque assays, indicating that these candidates have the potential to be developed into broad-spectrum anti-flaviviral drugs.
Figure 4.

EC50 values for inhibition of DENV2-infected BHK-21 cells determined by plaque assays. Compound C (A), Compound D (B), Compound F (C), Compound G (D), and Compound H (E).
Figure 5.
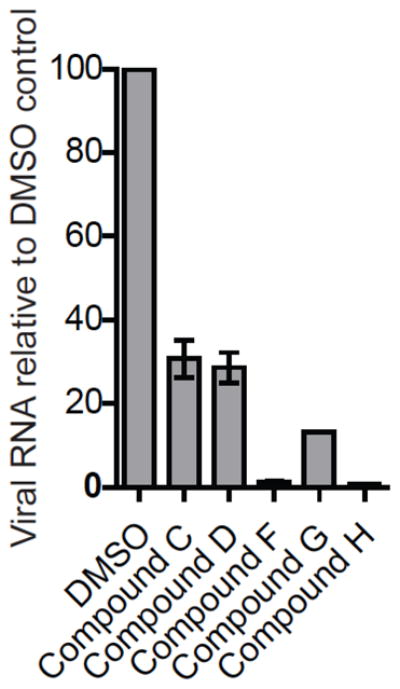
Inhibition of DENV2 replication analyzed by qPCR. BHK-21 cells were infected with DENV2 (MOI 1) and the viral RNA were isolated after 24h and the RNA copy number was determined as described under Materials and Methods. DMSO treated control is taken as 100% and percent reduction of viral RNA by treatment of DENV-2 infected BHK-21 cells with the compounds at the indicated concentrations is plotted. Compounds C, D and H at 10 μM; Compounds F and G at 2 μM concentrations.
3.6. Fluorescence quenching assay
The absorption spectra of four compounds (C, F, G and H) absorb near 300–330 nm indicating they are suitable candidates to be tested using this assay (Figure 6A). Compounds C, F and G quenched tryptophan fluorescence in a dose dependent manner (Figure 6B). Compound H failed to quench the tryptophan fluorescence indicating that it is a non-specific inhibitor. Addition of 10 μM BPTI to the reaction could partially restore the fluorescence of compounds C and G indicating their competitive mode of binding (Figure 6C). Compound F did not compete with BPTI (Figure 6C) but a caused significant decrease of the fluorescence with increasing inhibitor concentration, which confirms the binding of inhibitor within 10–15 Å (Förster distance for FRET) from Trp50 (Figure 6B).
Figure 6.

(A) Absorbance spectra of compounds C, F, G and H. (B) Fluorescence spectra of DENV2 protease (2μM) with various concentrations of compounds, C, F, G and H. (C) Competition of BPTI on the fluorescence quenching in the presence of 50 μM compound and 10 μM BPTI. (D) Kinetic analysis of compounds C, F and G. Lineweaver-Burk (LB) plots, of compounds, C, F, and G.
3.7. Kinetic Analysis of Inhibition by compounds C, F and H
To determine the biochemical basis of inhibition of compounds C, F, and G, we analyzed the kinetics of inhibition of DENV2 protease activity by deriving their Lineweaver-Burk plots (Figure 6D). In all three cases, the best fit lines for each inhibitor concentration converge at the origin indicating competitive inhibition (Whiteley, 2000).
3.8. Molecular modeling
Thus far, we have shown that compounds C, F, and G are competitive inhibitors of DENV2 protease. We thus sought to further examine the potential interactions between these compounds and the DENV protease active site by docking the structures of these inhibitors onto the homology model of DENV2 protease derived from DENV3 protease (3U1I). Molecular modeling suggests that all the three compounds were found to bind to the active site of protease (Figure 7). The binding efficiency, as indicated by PatchDock score, was the highest for compound G (6740) followed by compound C (3784) and compound F (3340). The atomic contact energy (ACE) was least for compound G (−467.16) followed by compound C (−236.11) and compound F (−181.95). High PatchDock scores and low ACE values are considered as a good indicator of successful docked conformation.
Figure 7.

Molecular modeling using the DENV3 NS2B-NS3 protease structure (3U1I) (Noble et al., 2012). (A): Compound C (magenta); (B): Compound F (cyan); (C): Compound G (blue). Active site triad (H51, D75 and S135) is highlighted in yellow.
4. Discussion
In this study, we performed in vitro HTS for small molecule inhibitors of the DENV2 protease. We initially performed our screen with 24,846 compounds without 0.1% CHAPS detergent in the reaction buffer. From this first round of screening, we obtained 436 (1.75%) hits which were mostly false positives (89%). We then modified our two succeeding rounds of HTS by adding 0.1% CHAPS in the reaction buffer. This yielded lower hit rates of 0.1% and 0.04%. Lower hit rates are likely due to several possible reasons. First, the presence of CHAPS reduces aggregation of compounds in solution, which decreases false positives (Ezgimen et al., 2009; Feng and Shoichet, 2006). Second, the in vitro protease assay requires a high pH (9.5) for optimal activity (Leung et al., 2001; Nall et al., 2004), which leads to protonation of some compounds resulting in false positives/negatives. Third, the assay buffers contain 20–30% glycerol which might have resulted in lower hit rates leading to pipetting errors in the automated liquid handling facilities (Noble et al., 2010). Fourth, the crystal structures of NS2B hydrophilic peptide-NS3 protease domain shows the active site to be relatively flat and negatively charged thus potentially contributing to lower hit rates observed (Erbel et al., 2006).
Crystal structures of WNV NS2B-NS3pro in the presence and absence of BPTI (Aleshin et al., 2007; Erbel et al., 2006), DENV2 (Erbel et al., 2006) and DENV1 (Chandramouli et al., 2010) without an inhibitor as well as that of DENV3 protease in complex with a peptide substrate-based inhibitor (Bz-Nleu-Lys-Arg-Arg-CHO) or BPTI (Noble et al., 2012) have been solved. These structures were solved using the cofactor NS2B hydrophilic peptide fused to the NS3 protease domain flanking the linker of 9 amino acids, GGGGSGGGG. In these structural models, the NS2B cofactor was found to adopt two distinct conformations. Without an inhibitor, the C-terminal region of NS2B is found dissociated from the active site of the NS3pro domain resulting in an “open” (inactive) conformation. In contrast, in the presence of a substrate-based inhibitor, NS2B wraps around the NS3pro domain interacting with residues in the active site in NS3 resulting in “closed” (active) conformation (Erbel et al., 2006). However, NMR structures of non-covalently associated NS2B and NS3pro domains of the “unlinked” form revealed that the protease assumed a “closed” conformation even in the absence of a substrate-based inhibitor in solution (Kim et al., 2013). This study suggests that NS2B linked to NS3pro via the GGGGSGGGG artificial linker could be the cause for conformational changes. In our case, the NS2B-NS3 precursor polypeptide consists of the hydrophilic NS2B cofactor linked to the NS3 protease domain via the two (QR in the case of DENV2) or four C-terminal amino acids of NS2B (in DENV1, 3 and 4) which undergoes cis cleavage resulting in the formation of a non-covalently associated NS2B and NS3 complex. This complex presumably exists in the active “closed” conformation.
Many flaviviral proteins have been targeted for drug discovery (Noble et al., 2010). Viral protease inhibitors have been developed into highly successful drugs (Patick and Potts, 1998). Protease inhibitors are used in treatment of non-infectious diseases like hypertension, myocardial infarction, peridontitis, thrombosis, pancreatitis and respiratory diseases. For example, angiotensin-converting enzyme (ACE) inhibitors are targeted to treat cardiovascular ailments (Smith and Vane, 2003). In addition, inhibitors of viral proteases, such as ritonavir, atazananvir tipranavir, Telaprevir and boceprevir (Chang et al., 2010; Flexner et al., 2005) have been used extensively in HIV and hepatitis C virus treatments (Pearlman, 2012).
The viral protease plays an important role in the life cycle of the DENV. In this study, using a purified preparation of DENV2pro as a target in a HTS campaign, we identified a class of compounds with catechol and polyphenolic scaffolds as potent inhibitors of DENV and WNV proteases. Eight compounds were selected for detailed analysis to determine the IC50 and Ki values using the in vitro protease assays. Six of these compounds are catechols (A–F), a polyphenolic compound (G) and a polyanionic compound (H). Only three compounds (C, F, tolcapone, and G, tannic acid) were found to be potent inhibitors of NS2B/NS3 proteases of four serotypes of DENV as well as WNV. Two of these compounds have been used to treat both viral and non-viral diseases. Yang et al. reported that 17 catechol derivatives are currently prescribe, FDA-approved drugs (Yang et al., 2007).
Tolcapone, a nitro-catechol derivative, was approved by FDA for the treatment of Parkinson’s disease and is a potent and selective inhibitor of catechol-O-methyl transferase (Antonini et al., 2008; Kaakkola, 2000). Tolcapone was introduced in Europe in 1997 and in the United States in 1998. However, the drug was withdrawn from the market (December 1998 to August 2004) due to hepatotoxicity in the patients who were taking the drug that resulted in three deaths (Borges, 2005; Colosimo, 1999). The hepatotoxicity was attributed due to elevated transaminases, and further studies showed that hepatoxicity was found only in people who had pre-existing liver conditions. Hence, Tolcapone treatment for Parkinson’s disease was re-approved under strict monitoring of serum enzymes (Borges, 2005; Colosimo, 1999).
Compound G, tannic acid, is a commercial form of Tannin and a polymer of gallic acid molecules and glucose. It is a phytochemical and found in tea, oak wood, berries, and Chinese galls. Tannic acid has been used for centuries as a natural home remedy for treatment of cold sores, fever blisters, diaper rash, sore throat, inflamed tonsils, spongy gums and acute dermatitis. It is also used orally for bleeding, chronic diarrhea, dysentery, bloody urine, painful joints and persistent coughs (Chung et al., 1998). Antiviral activities have been demonstrated for at least twelve different viruses: influenza virus H3N2, H5N3, herpes simplex virus-1, vesicular stomatitis virus, Sendai virus, Newcastle disease virus, poliovirus, coxsackievirus, adenovirus, rotavirus, feline calicivirus, human papillomavirus, and mouse norovirus (Buzzini et al., 2008; Theisen et al., 2014; Ueda et al., 2013). More importantly, previous studies have shown that hydrolysable tannins, chebulagic acid and punicalagin protect against dengue virus infections (Lin et al., 2013). Compound C is a novel scaffold which has not been reported earlier to have anti-DENV activity.
In summary, our studies suggest that compound C, tolcapone, tannic acid and are highly potent inhibitors of flaviviral proteases, DENV2 replication and infectivity. Their efficacy in combination with their high therapeutic indices make them potential lead compounds that could form the basis for development of more potent derivatives by structure-activity relationship studies in future.
Highlights.
A high-throughput screening against the dengue virus protease target was performed.
Of the eight selected compounds, three exhibited good therapeutic indices.
Fluorescence quenching, kinetic and modeling supported competitive mode of inhibition.
This study may lead to more potent inhibitors for treatment of viral infections.
Acknowledgments
This work was supported by This work was supported in whole or in part by National Institutes of Health grants, AI070791-03S1, U01-AI082068, R21AI109185, (to R.P.), and the NIH grant awarded to the New England Regional Center of Excellence (NSRB) (U54 AI057159), and a bridge funding from Biomedical Graduate Research Organization of Georgetown University, a Cosmos Club Foundation Young Scholars Award to M.M., and U54 AI057159 to ICCB-Longwood Screening Facility. We thank Dr. Priya Srinivasan at GUMC, and the faculty and staff of ICCB-Longwood for their assistance in the HTS done in their facility. We thank Sweta Batni, David Vogel and Jessica Connor for their assistance in construction of the Dengue virus protease expression plasmids.
Abbreviations
- DENV
dengue virus
- HTS
High-Throughput Screening
- NS
nonstructural protein
- WNV
West Nile virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alcaraz-Estrada SL, Manzano MI, Del Angel RM, Levis R, Padmanabhan R. Construction of a dengue virus type 4 reporter replicon and analysis of temperature-sensitive mutations in non-structural proteins 3 and 5. J Gen Virol. 2010;91:2713–2718. doi: 10.1099/vir.0.024083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz-Estrada SL, Reichert ED, Padmanabhan R. Construction of self-replicating subgenomic West Nile virus replicons for screening antiviral compounds. In: Vasudevan RPaS., editor. Methods Mol Biol. Humana Press, Springer Science +Business Media; 2013. pp. 283–299. 2013/07/04 ed. [DOI] [PubMed] [Google Scholar]
- Aleshin AE, Shiryaev SA, Strongin AY, Liddington RC. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007;16:795–806. doi: 10.1110/ps.072753207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini A, Abbruzzese G, Barone P, Bonuccelli U, Lopiano L, Onofrj M, Zappia M, Quattrone A. COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson’s disease (PD): relevance for motor and non-motor features. Neuropsychiatr Dis Treat. 2008;4:1–9. doi: 10.2147/ndt.s2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley DW. Recent advances in the molecular biology of west nile virus. Curr Mol Med. 2005;5:835–850. doi: 10.2174/156652405774962272. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenreider C, Beer D, Keller TH, Sonntag S, Wen D, Yap L, Yau YH, Shochat SG, Huang D, Zhou T, Caflisch A, Su XC, Ozawa K, Otting G, Vasudevan SG, Lescar J, Lim SP. A fluorescence quenching assay to discriminate between specific and nonspecific inhibitors of dengue virus protease. Anal Biochem. 2009;395:195–204. doi: 10.1016/j.ab.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Boonyasuppayakorn S, Reichert ED, Manzano M, Nagarajan K, Padmanabhan R. Amodiaquine, an antimalarial drug, inhibits dengue virus type 2 replication and infectivity. Antiviral Res. 2014;106:125–134. doi: 10.1016/j.antiviral.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges N. Tolcapone in Parkinson’s disease: liver toxicity and clinical efficacy. Expert Opin Drug Saf. 2005;4:69–73. doi: 10.1517/14740338.4.1.69. [DOI] [PubMed] [Google Scholar]
- Buzzini P, Arapitsas P, Goretti M, Branda E, Turchetti B, Pinelli P, Ieri F, Romani A. Antimicrobial and antiviral activity of hydrolysable tannins. Mini reviews in medicinal chemistry. 2008;8:1179–1187. doi: 10.2174/138955708786140990. [DOI] [PubMed] [Google Scholar]
- Chambers TJ, Nestorowicz A, Amberg SM, Rice CM. Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. J Virol. 1993;67:6797–6807. doi: 10.1128/jvi.67.11.6797-6807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers TJ, Nestorowicz A, Rice CM. Mutagenesis of the yellow fever virus NS2B/3 cleavage site: determinants of cleavage site specificity and effects on polyprotein processing and viral replication. J Virol. 1995;69:1600–1605. doi: 10.1128/jvi.69.3.1600-1605.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouli S, Joseph JS, Daudenarde S, Gatchalian J, Cornillez-Ty C, Kuhn P. Serotype-specific structural differences in the protease-cofactor complexes of the dengue virus family. J Virol. 2010;84:3059–3067. doi: 10.1128/JVI.02044-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MW, Giffin MJ, Muller R, Savage J, Lin YC, Hong S, Jin W, Whitby LR, Elder JH, Boger DL, Torbett BE. Identification of broad-based HIV-1 protease inhibitors from combinatorial libraries. Biochem J. 2010;429:527–532. doi: 10.1042/BJ20091645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KT, Lu Z, Chou MW. Mechanism of inhibition of tannic acid and related compounds on the growth of intestinal bacteria. Food Chem Toxicol. 1998;36:1053–1060. doi: 10.1016/s0278-6915(98)00086-6. [DOI] [PubMed] [Google Scholar]
- Clum S, Ebner KE, Padmanabhan R. Cotranslational membrane insertion of the serine proteinase precursor NS2B-NS3(Pro) of dengue virus type 2 is required for efficient in vitro processing and is mediated through the hydrophobic regions of NS2B. J Biol Chem. 1997;272:30715–30723. doi: 10.1074/jbc.272.49.30715. [DOI] [PubMed] [Google Scholar]
- Colosimo C. The rise and fall of tolcapone. J Neurol. 1999;246:880–882. doi: 10.1007/s004150050477. [DOI] [PubMed] [Google Scholar]
- Erbel P, Schiering N, D’Arcy A, Renatus M, Kroemer M, Lim SP, Yin Z, Keller TH, Vasudevan SG, Hommel U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol. 2006;13:372–373. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- Ezgimen M, Lai H, Mueller NH, Lee K, Cuny G, Ostrov DA, Padmanabhan R. Characterization of the 8-hydroxyquinoline scaffold for inhibitors of West Nile virus serine protease. Antiviral Res. 2012;94:18–24. doi: 10.1016/j.antiviral.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezgimen MD, Mueller NH, Teramoto T, Padmanabhan R. Effects of detergents on the West Nile virus protease activity. Bioorg Med Chem. 2009;17:3278–3282. doi: 10.1016/j.bmc.2009.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgout B, Miller RH, Lai CJ. Deletion analysis of dengue virus type 4 nonstructural protein NS2B: Identification of a domain required for NS2B-NS3 protease activity. Journal of Virology. 1993;67:2034–2042. doi: 10.1128/jvi.67.4.2034-2042.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner C, Bate G, Kirkpatrick P. Tipranavir. Nat Rev Drug Discov. 2005;4:955–956. doi: 10.1038/nrd1907. [DOI] [PubMed] [Google Scholar]
- Gould EA, Solomon T. Pathogenic flaviviruses. Lancet. 2008;371:500–509. doi: 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martinez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. Dengue: a continuing global threat. Nat Rev Microbiol. 2010;8:S7–16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead SB, Heinz FX, Barrett AD, Roehrig JT. Dengue virus: molecular basis of cell entry and pathogenesis, 25–27 June 2003, Vienna, Austria. Vaccine. 2005;23:849–856. doi: 10.1016/j.vaccine.2004.03.069. [DOI] [PubMed] [Google Scholar]
- Henchal EA, Putnak JR. The dengue viruses. Clin Microbiol Rev. 1990;3:376–396. doi: 10.1128/cmr.3.4.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakkola S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs. 2000;59:1233–1250. doi: 10.2165/00003495-200059060-00004. [DOI] [PubMed] [Google Scholar]
- Kautner I, Robinson MJ, Kuhnle U. Dengue virus infection: epidemiology, pathogenesis, clinical presentation, diagnosis, and prevention. J Pediatr. 1997;131:516–524. doi: 10.1016/s0022-3476(97)70054-4. [DOI] [PubMed] [Google Scholar]
- Kim YM, Gayen S, Kang C, Joy J, Huang Q, Chen AS, Wee JL, Ang MJ, Lim HA, Hung AW, Li R, Noble CG, Lee le T, Yip A, Wang QY, Chia CS, Hill J, Shi PY, Keller TH. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. The Journal of biological chemistry. 2013;288:12891–12900. doi: 10.1074/jbc.M112.442723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai H, Sridhar Prasad G, Padmanabhan R. Characterization of 8-hydroxyquinoline derivatives containing aminobenzothiazole as inhibitors of dengue virus type 2 protease in vitro. Antiviral Res. 2013a;97:74–80. doi: 10.1016/j.antiviral.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai H, Teramoto T, Padmanabhan R. Construction of dengue virus protease expression plasmid and in vitro protease assay for screening antiviral inhibitors. Methods in Molecular Biology. 2013b doi: 10.1007/978-1-4939-0348-1_21. in press. [DOI] [PubMed] [Google Scholar]
- Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- Li J, Lim SP, Beer D, Patel V, Wen D, Tumanut C, Tully DC, Williams JA, Jiricek J, Priestle JP, Harris JL, Vasudevan SG. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J Biol Chem. 2005;280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, Yokokawa F, Nilar S, Smith P, Beer D, Lescar J, Shi PY. Ten years of dengue drug discovery: progress and prospects. Antiviral Res. 2013;100:500–519. doi: 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Lin LT, Chen TY, Lin SC, Chung CY, Lin TC, Wang GH, Anderson R, Lin CC, Richardson CD. Broad-spectrum antiviral activity of chebulagic acid and punicalagin against viruses that use glycosaminoglycans for entry. BMC Microbiol. 2013;13:187. doi: 10.1186/1471-2180-13-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Adv Virus Res. 2003;59:23–61. doi: 10.1016/s0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- Lindenbach D, Thiel HJ, Rice C. Flaviviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Field’s Virology. 5. Lippincott-Raven Publishers; Philadelphia: 2007. pp. 1101–1152. [Google Scholar]
- Manzano M, Reichert ED, Polo S, Falgout B, Kasprzak W, Shapiro BA, Padmanabhan R. Identification of cis-acting elements in the 3′-untranslated region of the dengue virus type 2 RNA that modulate translation and replication. J Biol Chem. 2011;286:22521–22534. doi: 10.1074/jbc.M111.234302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusan AE, Kelley PG, Pryor MJ, Whisstock JC, Davidson AD, Wright PJ. Mutagenesis of the dengue virus type 2 NS3 proteinase and the production of growth-restricted virus. J Gen Virol. 2001;82:1647–1656. doi: 10.1099/0022-1317-82-7-1647. [DOI] [PubMed] [Google Scholar]
- Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem. 2007;282:8873–8882. doi: 10.1074/jbc.M609919200. [DOI] [PubMed] [Google Scholar]
- Mitka M. Dengue more prevalent than previously thought. JAMA. 2013;309:1882. doi: 10.1001/jama.2013.4903. [DOI] [PubMed] [Google Scholar]
- Mueller NH, Pattabiraman N, Ansarah-Sobrinho C, Viswanathan P, Pierson TC, Padmanabhan R. Identification and biochemical characterization of small-molecule inhibitors of west nile virus serine protease by a high-throughput screen. Antimicrob Agents Chemother. 2008;52:3385–3393. doi: 10.1128/AAC.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller NH, Yon C, Ganesh VK, Padmanabhan R. Characterization of the West Nile virus protease substrate specificity and inhibitors. Int J Biochem Cell Biol. 2007;39:606–614. doi: 10.1016/j.biocel.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Nall TA, Chappell KJ, Stoermer MJ, Fang NX, Tyndall JD, Young PR, Fairlie DP. Enzymatic characterization and homology model of a catalytically active recombinant West Nile virus NS3 protease. J Biol Chem. 2004;279:48535–48542. doi: 10.1074/jbc.M406810200. [DOI] [PubMed] [Google Scholar]
- Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. Strategies for development of Dengue virus inhibitors. Antiviral Res. 2010;85:450–462. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Noble CG, Seh CC, Chao AT, Shi PY. Ligand-bound structures of the dengue virus protease reveal the active conformation. J Virol. 2012;86:438–446. doi: 10.1128/JVI.06225-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan R, Strongin AY. Translation and processing of the dengue virus polyprotein. In: Hanley KA, Weaver SC, editors. Frontiers in Dengue Virus Research. Caister Academic Press; Norfolk, U.K: 2010. pp. 14–33. [Google Scholar]
- Patick AK, Potts KE. Protease inhibitors as antiviral agents. Clin Microbiol Rev. 1998;11:614–627. doi: 10.1128/cmr.11.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman BL. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect Dis. 2012;12:717–728. doi: 10.1016/S1473-3099(12)70060-9. [DOI] [PubMed] [Google Scholar]
- Puig-Basagoiti F, Tilgner M, Forshey BM, Philpott SM, Espina NG, Wentworth DE, Goebel SJ, Masters PS, Falgout B, Ren P, Ferguson DM, Shi PY. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob Agents Chemother. 2006;50:1320–1329. doi: 10.1128/AAC.50.4.1320-1329.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath A, Padmanabhan R. Molecular targets for flavivirus drug discovery. Antiviral Res. 2009;81:6–15. doi: 10.1016/j.antiviral.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra B, Perez AB, Vogt K, Garcia G, Schmolke K, Aguirre E, Alvarez M, Kern F, Kouri G, Volk HD, Guzman MG. Secondary heterologous dengue infection risk: Disequilibrium between immune regulation and inflammation? Cell Immunol. 2010;262:134–140. doi: 10.1016/j.cellimm.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Smith CG, Vane JR. The discovery of captopril. FASEB J. 2003;17:788–789. doi: 10.1096/fj.03-0093life. [DOI] [PubMed] [Google Scholar]
- Theisen LL, Erdelmeier CA, Spoden GA, Boukhallouk F, Sausy A, Florin L, Muller CP. Tannins from Hamamelis virginiana bark extract: characterization and improvement of the antiviral efficacy against influenza A virus and human papillomavirus. PloS one. 2014;9:e88062. doi: 10.1371/journal.pone.0088062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Kawabata R, Irie T, Nakai Y, Tohya Y, Sakaguchi T. Inactivation of pathogenic viruses by plant-derived tannins: strong effects of extracts from persimmon (Diospyros kaki) on a broad range of viruses. PloS one. 2013;8:e55343. doi: 10.1371/journal.pone.0055343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley CG. Enzyme kinetics: partial and complete uncompetitive inhibition. Biochem Educ. 2000;28:144–147. doi: 10.1016/s0307-4412(00)00029-7. [DOI] [PubMed] [Google Scholar]
- Yang DP, Ji HF, Tang GY, Ren W, Zhang HY. How many drugs are catecholics. Molecules. 2007;12:878–884. doi: 10.3390/12040878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yon C, Teramoto T, Mueller N, Phelan J, Ganesh VK, Murthy KH, Padmanabhan R. Modulation of the nucleoside triphosphatase/RNA helicase and 5′-RNA triphosphatase activities of dengue virus type 2 nonstructural protein 3 (NS3) by interaction with NS5, the RNA-dependent RNA polymerase. J Biol Chem. 2005;280:27412–27419. doi: 10.1074/jbc.M501393200. [DOI] [PubMed] [Google Scholar]