

Abstract
Santacruzamate A (SCA) is a natural product isolated from a Panamanian marine cyanobacterium, previously reported to have potent and selective histone deacetylase (HDAC) activity. To optimize the enzymatic and cellular activity, 40 SCA analogues were synthesized in a systematic exploration of the zinc-binding group (ZBG), cap terminus, and linker region. Two cap group analogues inhibited proliferation of MCF-7 breast cancer cells, with analogous increased degranulation of cytotoxic T cells (CTLs), while one cap group analogue reduced CTL degranulation, indicative of suppression of the immune response. Additional testing of these analogues resulted in reevaluation of the previously reported SCA mechanism of action. These analogues and the resulting structure-activity relationships will be of interest for future studies on cell proliferation and immune modulation.
Keywords: Santacruzamate A, Natural product analogues, Enzyme assays, Anti-proliferative activity, Immune modulation
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
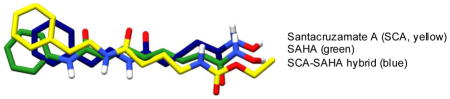
1. Introduction
Development of new cancer chemotherapeutic compounds is a fundamentally challenging endeavor requiring considerations at the in vitro, in vivo, preclinical, and clinical stages. Ideal compounds for drug discovery and development target cancerous cells while maintaining low toxicity to normal cells, with an ongoing shift away from discovery of new compounds with broad cytotoxicity to those with selective activity, including compounds that specifically regulate gene expression via epigenetic modulation, that target signaling pathways, or that enhance immune system function.1,2 Recently, epigenetic modulators such as inhibitors of DNA methyltransferase (DNMT) and histone deacetylase (HDAC) have received increased attention as anti-cancer agents, although their clinical utility has thus far been limited to leukemias and lymphomas.3,4 Likewise, there has been considerable attention towards the development of small molecule cancer therapeutics that affect immune system targets, although there are still very few currently available agents.5–8
HDACs and histone acetyltransferases (HATs) are key enzymes regulating the packaging of DNA around histones, having extensive impacts on gene transcription. HDAC enzymes remove acetyl groups from histone lysine residues, resulting in enhanced binding affinity with DNA and leading to repression of gene transcription. Regulation of acetylation and deacetylation patterns allows for acute control of differentiation, growth arrest, and apoptosis.9–11 Moreover, hyper-deacetylation can result in silencing of tumor suppression genes, loss of apoptosis, and initiation and propagation of tumorogenesis.12 Four HDAC inhibitors are currently approved by the United States Food and Drug Administration (US FDA) including suberoylanilide hydroxamic acid (SAHA, Vorinostat®, Fig. 1), romidepsin (Istodax®), panobinostat (Farydak®), and belinostat (Beleodaq®), all of which are approved for patients with refractory cancers (lymphoma or myeloma).13 HDACs exist in several classes and with many isozymes, with current research focused on the discovery and development of isozyme selective epigenetic modulators to avoid the side effects inherent in non-selective inhibitors.14–16
Figure 1.

Santacruzamate A (SCA, 1), SAHA, and the SCA-SAHA hybrid compound reported previously,18 and their overlaid 3D structures (SCA in yellow, SAHA in green, and SCA-SAHA hybrid in blue).
Targeted therapies that induce anti-cancer immunity have been the focus of intensive research efforts, with recent efforts to discover new small molecule cancer immunotherapeutics.8 Components of the adaptive and innate immune systems are capable of recognizing and killing cancer cells, initiated by tumor antigens that activate cytotoxic T lymphocytes (CTLs) through T cell receptors or by cell-surface molecules that activate natural killer cells.17 Although there are several clinically available monoclonal antibodies that modulate immune function, imiquimod (Aldara®) is one of the only small molecule immunomodulators approved for cancer patients in the US and is limited to cutaneous malignancies.7
We initially identified santacruzamate A (SCA, 1, Fig. 1) as a bioactive natural product isolated from the marine cyanobacterium, cf. Symploca sp.18 SCA consists of three structural moieties including an ethyl carbamate terminus, a modified γ-aminobutyric acid (GABA) linker, and a phenethylamine cap group and has several structural features in common with SAHA (Fig. 1), which led to an initial investigation of SCA for HDAC inhibition. SCA, SAHA, and most HDAC inhibitors contain three basic structural motifs including a zinc-binding group (ZBG), an aliphatic linker, and a surface recognition cap group (Fig. 1). In contrast to SCA, SAHA ends in a hydroxamate terminus and has a longer 6-carbon linker directly attached to the phenylamine cap. SAHA binds to HDAC with the phenyl cap group resting on top of the enzyme pocket, the aliphatic linker traversing through the small channel, and the hydroxamate binding to the zinc at the bottom.19 Because most hydroxamic acids suffer from pan-isozyme activity and several clinical liabilities (e.g., poor oral absorbance, rapid hydrolysis yielding poor pharmacokinetics, and strong non-specific affinity for metalloproteins),20 we were enthusiastic about SCA given the carbamate terminus. With its relatively small, linear modified peptide structure, SCA is easily amenable to synthetic modifications and advanced biological evaluation to fully explore structure-activity relationships. In an effort to optimize the enzyme activity and to enhance the cellular activity, we performed a systematic exploration of the three chemical regions of the SCA structure including modification of the zinc-binding group (ZBG), cap terminus, and linker region, resulting in 40 derivatives reported herein that were evaluated for anti-proliferative and immunomodulatory activity.
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of santacruzamate A zinc-binding group analogues
Given the known importance of ZBG binding for HDAC inhibition, the first structural modifications we made were to the carbamate terminus of SCA (Schemes 1–5). With SAHA, the hydroxamate allows for bi-dentate binding of zinc between the deprotonated terminal hydroxyl and the carbonyl oxygen with the catalytic pocket (see Fig. 3 and Molecular Modeling section for more details). This bi-dentate binding facilitates the strong metal binding characteristics of hydroxamate bearing compounds. Herein, SCA is hypothesized to exhibit mono-dentate coordination to the zinc and thus modification centered on adjusting the electronics around the carbonyl oxygen of the SCA carbamate. For this first phase we completed the synthesis of 20 compounds that differ in their ZBG terminus, leaving the three carbon aliphatic linker and phenethylamine cap group unaltered.
Scheme 1.

Synthesis of santacruzamate A N-carbamate analogues. Reagent and conditions: (i) ClCO2-R1, K2CO3 or NaOH, THF, 0 °C to rt, 70–90%; (ii) Boc2O, NaOH, THF, 97%; (iii) phenethylamine, EDC-HCl, TEA, cat. DMAP, CH2Cl2, 43–93%.
Scheme 5.

Synthesis of santacruzamate A inverted ethyl carbamate analogue. Reagents and Conditions: (i) TEA, MeOH, reflux, 92%; (ii) methyl-4-hydroxybutanoate, N,N′-disuccinimdyl carbonate, TEA 79%; (iii) NHS-methyl-4-hydroxybutanoate, 68% ethylamine, TEA, THF; (iv) phenethylamine, EDC-HCl, TEA, DMAP, CH2Cl2 56%.
Figure 3.

X-ray crystal structure of SAHA (green, left) bound to HDAC2 (gray, PDB: 4LXZ40), with santacruzamate A (1) (yellow, right) overlaid over the binding conformation of SAHA with HDAC2. The zinc ion (dark gray) within the HDAC2 active site is shown with an idealized octahedral metal geometry (idealized metal geometries of the zinc ion are shown as yellow arrows and geometries not filled by HDAC2 are labeled G′, G″, G″′ and proceed clockwise starting from left) using the Chimera program.45 SAHA binds to the zinc ion via chelation with G′ and G″, while santacruzamate A likely binds only to G′. Hydrogen bonds are depicted as orange lines. The surface of the HDAC2 active site, within 4.1Å of the ligands, was generated via Chimera and colored according to the corresponding heteroatom on enzyme surface.
N-Carbamate modifications (Scheme 1) included changing the ethyl carbamate to a methyl (5), a larger tert-butyl (8), elongating to a propyl (2) and butyl (3), elongating and adding unsaturation (6 and 9), an even larger phenyl (4), fluorination (7), and incorporating a thiocarbamate (10 and 11). Carbamate intermediates (Scheme 1, 2a–5a, 7a–11a) were all prepared via analogous synthetic methodology involving coupling of GABA with the respective chloroformate.21–23 Compound 8a was formed using standard di-tert-butyl dicarbonate (Boc2O) protection.24 To prepare compound 10a, prior to coupling with GABA, sodium ethoxide was added to thiophosgene and reacted to obtain the ethyl chlorothioformate 10a. All resulting carbamates were coupled to phenethylamine using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), trimethylamine (TEA), and catalytic 4-dimethylaminopyridine (cat. DMAP) yielding analogues 2–11.
Ten additional ZBG analogues were also prepared. To explore the possibility of incorporation of bi-dentate binding, compounds 13 and 14 were synthesized (Scheme 2). A simpler urea analogue was also prepared (12). In another series, the carbamate was exchanged for amide bonds with various termini (Scheme 3, 15–19). In addition, due to the potent zinc chelating affinity of the terminal thioester of largazole,25,26 we replaced the carbamate with an acetylated thiol (Scheme 4, 20). Lastly, in an effort to investigate the spatial orientation of the ethyl carbamate in the binding pocket an inverted ethyl carbamate, 21, was synthesized (Scheme 5).
Scheme 2.

Synthesis of santacruzamate A urea analogues. Reagents and conditions: (i) CS2, NaOH, H2O; (ii) 30% H2O2, KOH, H2O; (iii) KNO2, diluted H2SO4, 0 °C to rt; (iv) 68% ethylamine/H2O, 100 °C; (v) phenethylamine, EDC-HCl, TEA, cat. DMAP, CH2Cl2, 0 °C to rt; (vi) 12a or 12b, cat. p-TsOH, NEAT 180 °C, 30 torr; (vii) 13a or 13b, phenethylamine, EDC-HCl, TEA, cat. DMAP, CH2Cl2, 0 °C to rt.
Scheme 3.

Synthesis of santacruzamate A N-amide analogues. Reagents and conditions: (i) Ac2O, AcOH, Et2O; (ii) propionic anhydride or butyric anhydride, cat H2SO4, 100 °C; (iii) R2-COCl, NaOH, THF/H2O, 0 °C to rt; (iv) phenethylamine, EDC-HCl, TEA, cat. DMAP, CH2Cl2.
Scheme 4.

Synthesis of santacruzamate A thiol analogue. Reagents and conditions: (i) thioacetic acid, 0 °C to rt; (ii) thionyl chloride, DCM, refluxed for 2 h at 40 °C; (iii) 20a, H2O, Na2CO3, 0 °C to rt; (iv) phenethylamine, EDC-HCl, TEA, cat. DMAP, CH2Cl2.
The three urea analogues (Scheme 2, 12–14) were synthesized via 12a starting from GABA which was dimerized using CS2 to form a thiourea.27 This thiourea intermediate was either carried through directly or oxidized to the urea intermediate upon exposure to 30% H2O2 and KOH to form 12b.28 Intermediate 12b was then converted through to the ethyl urea following literature procedure,27 which was subsequently coupled to phenethylamine to afford 12. Compounds 13 and 14 were prepared by heating intermediates 12a or 12b with cat. p-TsOH at 30 mm Hg to form the cyclized pyrrolidone compounds.27
For synthesis of N-amide compounds 15a-19a, each new functionality was introduced as the acyl chloride via a modified Schotten–Baumann reaction (Scheme 3).29–31 Formation of the N-amide bond in compounds 16a and 17a were generated via reaction with propionic or butyric anhydride with cat. H2SO4, respectively. As with the carbamates, the phenethylamine moiety was incorporated via EDC coupling to yield compounds 15–19.
To form compound 20 (Scheme 4), S-2-(chlorocarbonyl)ethyl ethanethioate 20a was prepared by adding thioacetic acid to acrylic acid, followed by addition of thionyl chloride following literature procedure.32 Intermediate 20a was then reacted with GABA followed by subsequent coupling to phenethylamine as described above. The inverted carbamate 21 (Scheme 5) was made via opening γ-butyrolactone with methanol and TEA to form a terminal hydroxyl which was then converted to the active carbonate via reaction with N,N′-disuccinimidyl carbonate and subsequently reacted with aqueous ethylamine to form the inverted ZBG terminus. The final step involved routine peptide coupling with phenethylamine as described above.
2.1.2. Synthesis of santacruzamate A cap group analogues
The cap group has been theorized to be the initial sight of docking with the linker fitting into a hydrophobic groove that then orients the ZBG into its proper chelation position.4 On the cap end of SCA several modifications were prepared (Scheme 6), while keeping the GABA linker and ethyl carbamate of SCA unaltered. Two of these modifications removed the phenyl ring altogether (23 and 24). Three compounds with chlorination or fluorination at various positions were expected to provide electron withdrawing capacity (29, 30, 33). Additional modifications included replacement of the amide bond with an ester (31), replacement of the phenethyl with a thiazole ring (25), and replacement of the phenethylamine with a methylated phenylalanine (32). Compound 26 was prepared to provide a larger terminus that was previously shown to have antimalarial activity and HDAC inhibition.33 Compounds 27 and 28 provided information regarding the effects of an OTBS group either directly on the phenyl ring or one carbon away.
Scheme 6.

Synthesis of santacruzamate Acap group analogues. Reagent and conditions: (i) ethyl chloroformate, K2CO3, H2O, 0 °C to rt; (ii) cap-group R1, EDC-HCl, TEA, cat. DMAP, CH2Cl2; (iii) EDC-HCl, HOBt, TEA, phenethyl-OH, DCM, 0 °C to rt.
Cap derivatives were all synthesized starting from GABA, with conversion to the linked ethyl carbamate following standard acylation conditions via reaction with ethyl chloroformate using excess potassium carbonate in water to afford intermediate 22. All cap derivatives except 31 were coupled to their respective free acids via previously described EDC coupling procedures. Preparation of compound 31 utilized a PyBOP/HOBt mediated-coupling.34
2.1.3. Synthesis of santacruzamate A linker analogues
Structure-activity investigation of the linker region involved two specific types of modification (Scheme 7). The first of these modified the length of the linker from 1–5 carbons (34–37), while keeping the phenethylamine cap group and the ethyl carbamate linker unaltered. For the other type of linker modifications, the overall chain length of the molecule was kept constant while placement of the amide bond was varied from directly adjacent to the phenyl (39) to one carbon from the carbamate (41) as well as variations in between (38, 40). In addition, an analogue was prepared without the GABA linker, shortening the compound to the phenethylamine directly coupled to the ethyl carbamate (42). Synthesis of intermediates 34a–41a was accomplished using commercially available linker acids coupled with ethyl chloroformate following standard acylation conditions using excess potassium carbonate, followed by EDC coupling of the phenyl amines of various chain lengths to afford final compounds 34–41. For compound 42, the phenethylamine was coupled to ethyl chloroformate using the standard acylation conditions described above.
Scheme 7.

Synthesis of santacruzamate A linker analogues. Reagents and conditions: (i) ethyl chloroformate, K2CO3, H2O, 0 °C to rt; (ii) phenethylamine, aniline, benzenemethanamine, or benzenebutanamine, EDC-HCl, TEA, cat. DMAP, CH2Cl2, 0 °C to rt; (iii) ethylchloroformate, TEA, CH2Cl2.
2.2. Biological evaluation
All analogues were tested in our laboratory for activity against HDAC2 using newly purchased human recombinant enzyme (BPS Bioscience, Inc., San Diego, CA) with fluorogenic HDAC assay kits from Active Motif (Carlsbad, CA). Samples of SCA (1) natural product isolate and the synthetic compound from our previous report18 were analyzed for compound stability and used for biological testing. Due to limited sample availability, SCA-SAHA hybrid was re-synthesized to afford sufficient material for biological evaluation. During our HDAC2 testing of these new analogues we were unable to repeat the previous levels of activity (data not shown) due to high variability in the assay results, possibly due to variations in enzyme preparations.35 Therefore, we sent our compounds to Reaction Biology Corp. (Malvern, PA) for testing in their HDAC2 assay. Compounds were all screened in duplicate at 1 μM and found to be inactive (Table 1), in contrast to our original report of HDAC2 inhibition by SCA (1).18
Table 1.
Biological evaluation of santacruzamate A (1) analogues using enzymatic and cellular assays.
| Compd | Enyzmea | Solid Tumor Cellsb | Lymphoma Cellsb | PBMCb | CTLc % CD3 | |||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| HDAC2 | HCT116 | MCF-7 | MDA-MB-231 | HuT-78 | Molt-4 | |||
| 2 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 94 |
| 3 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 101 |
| 4 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 79 |
| 5 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 81 |
| 6 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 87 |
| 7 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 92 |
| 8 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 82 |
| 9 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 105 |
| 10 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 105 |
| 11 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 76 |
| 12 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 106 |
| 13 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 94 |
| 14 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 95 |
| 15 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 83 |
| 16 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 91 |
| 17 | >1 | >50 | >50 | ntd | >50 | >50 | >50 | 101 |
| 18 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 73 |
| 19 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 85 |
| 20 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 77 |
| 21 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 100 |
| 23 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 88 |
| 24 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 95 |
| 25 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 91 |
| 26 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 58 |
| 27 | >1 | >50 | 23.7 | >50 | >50 | >50 | >50 | 126 |
| 28 | >1 | >50 | 13.8 | >50 | >50 | >50 | >50 | 171 |
| 29 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 85 |
| 30 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 75 |
| 31 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 88 |
| 32 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 90 |
| 33 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 90 |
| 34 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 86 |
| 35 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 91 |
| 36 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 83 |
| 37 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 98 |
| 38 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 93 |
| 39 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | ntd |
| 40 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 83 |
| 41 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 81 |
| 42 | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 90 |
| SCA (1)e | >1 | 17.2 | ntd | ntd | 36.0 | 7.8 | ntd | ntd |
| SCA (1)f | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 77 |
| SCA-SAHAg | >1 | >50 | >50 | >50 | >50 | >50 | >50 | 113 |
| SAHAh | 29.1 | |||||||
| Doxorubicinh | 21.4 | 10.7 | ||||||
IC50 values [μM].
GI50 values [μM].
% CD3-stimulated anti-LAMP fluorescence in cytotoxic T lymphocytes (CTLs), screened at 100 μM.
nt = not tested.
Santacruzamate A (1), natural product. Previously reported values: HDAC2 IC50 0.119 nM; HCT116 GI50 29.4 μM; HuT-78 GI50 1.4 μM.18
Santacruzamate A (1), synthetic. Previously reported values: HDAC2 IC50 0.112 nM; HCT116 GI50 28.3 μM; HuT-78 GI50 1.3 μM.18
SCA-SAHA hybrid. Previously reported values: HDAC2 IC50 3.5 nM; HCT116 GI50 2.3 μM; HuT-78 GI50 0.7 μM.18
SAHA and doxyrubicin were utilized as positive controls for solid tumor cell assays.
These unexpected results led to further testing to attempt to understand the discrepancies, including testing both synthetic SCA (1) and the SCA-SAHA hybrid (Fig. 1) for activity on all of the HDAC isozymes (Table 2). Both compounds were tested by BPS Biosciences against HDACs 1–11 at two concentrations (10 μM and 5 μM), with either SAHA or TSA (trichostatin A, a known HDAC inhibitor) as positive controls. Santacruzamate A (1, synthetic) inhibited all 11 HDACs at 10 μM, with little to no inhibition at 5 μM, indicating IC50 values for these isozymes all lie in the 5–10 μM range. The SCA-SAHA hybrid was also found to inhibit most of HDAC isozymes at 10 μM, with the exception of HDAC5 and HDAC 7. At 5 μM, the hybrid compound still had an inhibitory effect on HDAC6 (59% inhibition), but was inactive against the other HDAC isozymes, indicative of IC50 values between 5–10 μM for HDACs 1–4 and 8–11, IC50 values >10 μM for HDACs 5 and 7, and an IC50 value < 5 μM for HDAC6. To explore other common epigenetic targets, SCA (1) was also tested for inhibition of sirtuins SIRT1–3 and SIRT5 (NAD+-dependent deacetylases) as well as inhibition of DNA-methyltransferases DNMT1, DNMT3A/3L, and DNMT3B/3L and found to be inactive at both 10 μM and 5 μM.
Table 2.
Inhibition of the Zn2+ dependent HDACs 1–11by santacruzamate A (1) and the SCA-SAHA hybrid.
| Isozyme | SCA | SCA-SAHA Hybrid | SAHAa | TSAa | ||
|---|---|---|---|---|---|---|
|
| ||||||
| 10 μM | 5 μM | 10 μM | 5 μM | 10 μM | 100 μM | |
| HDAC1 | 90 ± 0 | 7 ± 0.5 | 86 ± 0.5 | 31 ± 0.5 | 100 ± 0.3 | |
| HDAC2 | 80 ± 1.5 | 6 ± 0.5 | 76 ± 0.5 | 17 ± 1.0 | 99 ± 0.5 | |
| HDAC3 | 89 ± 1.0 | 10 ± 2.5 | 86 ± 0 | 36 ± 1.0 | 99 ± 0.7 | |
| HDAC4 | 86 ± 2.0 | 4 ± 2.5 | 80 ± 1.0 | 6 ± 0 | 94 ± 1.0 | |
| HDAC5 | 66 ± 0 | 0 ± 0 | 49 ± 3.0 | 0 ± 0.5 | 95 ± 1.5 | |
| HDAC6 | 83 ± 1.5 | 16 ± 2.5 | 85 ± 0 | 59 ± 1.0 | 100 ± 0 | |
| HDAC7 | 74 ± 1.0 | 0 ± 8.5 | 47 ± 0.5 | 29 ± 0 | 88 ± 0.9 | |
| HDAC8 | 73 ± 1.5 | 5 ± 8.0 | 70 ± 2.5 | 0 ± 3.0 | 90 ± 1.9 | |
| HDAC9 | 75 ± 0 | 10 ± 0 | 70 ± 0.5 | 14 ± 0.5 | 92 ± 1.8 | |
| HDAC10 | 93 ± 0 | 16 ± 0 | 82 ± 0.5 | 35 ± 0 | 100 ± 0 | |
| HDAC11 | 78 ± 0.5 | 0 ± 2.5 | 72 ± 0.5 | 24 ± 0.5 | 98 ± 0.5 | |
SAHA and TSA were utilized as positive controls. Values expressed as a % inhibition ± SEM.
Due to the differences in enzymatic activity from our initial reports to these current results, we elected to perform more biologically relevant, Western blot analyses to determine the ability of SCA (1) to cause changes in the histone acetylation status of H3K9, H3K23, and H4K12 (Fig. 2), chosen as a representative small panel of histone acetylation sites. Using the Molt-4 cell line, synthetic SCA was not found to have an effect on acetylation of these histones at 200 μM, as compared with SAHA, which induced significant histone acetylation at 2.5 μM. Thus, although there was enzymatic inhibition of HDACs by SCA at 10 μM, it appears that this does not translate to cellular inhibition of HDAC activity.
Figure 2.

Effect of santacruzamate A and SAHA on the levels of acetylated histones H3K9, H3K23, and H4K12 in Molt-4 cells.
As a complement to HDAC screening, all analogues were also screened for activity against several solid tumor and lymphoma cell lines (Table 1). Initial screening was performed at 50 μM with GI50 values obtained for active compounds. Compounds 27 and 28 were found to have activity against MCF-7 cells with GI50 values of 23.7 and 13.8 μM, respectively (Table 1). Both compounds are tert-butyldimethylsilyloxy (OTBS) cap group analogues with intact SCA ethyl carbamate and GABA-derived linker groups. No activity was found for synthetic compounds using HCT166 colon cancer cells or MDA-MB-231 triple-negative breast cancer cells. Synthetic analogues were also found to be inactive against HuT-78 cutaneous T lymphoma cells, Molt-4 acute lymphoblastic leukemia cells, and against non-cancerous peripheral blood mononuclear cells (PBMC). Interestingly, the SCA natural product isolate (1) was found to have activity against HCT116, HuT-78, and Molt-4 cells with GI50 values of 17.2, 36.0, and 7.8 μM, respectively, indicative of a possible contaminant in the natural product sample, since the synthetic SCA did not possess similar activity (this is in contrast to our previous report in which the synthetic and natural product SCA were equipotent, possibly due to experimental variations such as cell density and/or media type). This contaminant would likely possess potent anti-proliferative activity, given that the SCA isolate was found to be 98.5% pure, as measured via LC-MS (Figure S4A in Supplementary Material), and thus the contaminant would have to be present in very low quantities, but with sufficient potency to be able to cause biological activity. Given that the potent cytotoxic dolastatins have previously been reported from several Symploca species,36,37 we investigated the SCA natural product LC-MS and found the main impurity to have an [M + H]+ of 774.5 (Figure S4B in Supplementary Material), possibly consistent with dolastatin 17,38 although insufficient sample is available to confirm.
Based on the MCF-7 activity, we were interested in determining possible mechanisms for the anti-proliferative effects. Therefore, we tested all synthetic analogues in a phenotypic screen of cytotoxic T lymphocyte (CTL) lytic granule exocytosis that measures immunological activity of small molecules [insufficient material of the SCA natural product isolate (1) was available for this screen] (Table 1).39 Both anti-proliferative analogues, 27 and 28, were found to increase lytic granule exocytosis (26% and 71% augmentation of CD3-stimulated anti-LAMP fluorescence increases, respectively), corresponding to their levels of anti-proliferative activity and indicative of enhancement of the immune response. Compound 26, a cap group analogue with an extended terminus and an electron-withdrawing nitro group, was found to decrease granule exocytosis by 58%, indicative of suppression of the immune response.
2.3. Molecular modeling
Due to the structural similarity of SCA and SAHA but the divergence in enzymatic activity, we investigated the chemical differences in HDAC2 binding via molecular modeling. The primary structural difference between SAHA and SCA involves the incorporation of a hydroxamate ZBG terminus for SAHA, which includes carbonyl and hydroxyl oxygen moieties allowing for bidentate chelation of the zinc ion via an octahedral metal geometry. Our molecular model utilizes a previously published X-ray crystal structure of HDAC2 with SAHA (PDB: 4LXZ)40 and overlays SCA (1) within the existing binding pocket (Fig. 3). The model depicts the optimal octahedral binding metal geometries of the zinc ion in the HDAC2 binding pocket shown as yellow arrows (Fig. 3). The zinc ion is displayed as a 6-coordinated ion with three binding geometries filled by His183, Asp269 and Asp181, starting from left and proceeding counter clockwise. The remaining geometries are labeled as potential ligand binding geometries, denoted as G′, G″, and G′″, starting from left and proceeding clockwise.
In this model, SAHA is able to fill G′ and G″ but the zinc ion would require an addition electron density donor. The binding pocket is accessible to water molecules, which are believed to play a role during HDAC2 catalytic deacetylation,41 suggesting that a water molecule may occupy the remaining ligand binding location. The ability of SAHA to fill G′ and G″ is facilitated in part by the planar geometry of the hydroxamate moiety and the rotatable bond between C8 and C9. In addition to SAHA chelating the zinc ion, potential hydrogen bonding further stabilizes the interaction with HDAC2, including between Asp104 and the amide nitrogen nearest to the phenyl cap, between His146 and the terminal hydroxamate nitrogen, and between His145 and the terminal hydroxyl oxygen (Fig. 3). The two terminal hydrogen bonds may form hydrogen bonding and hydrogen shuttling networks by sharing or fully transferring the hydrogen species between themselves and Asp186 and Asp179 adjacent to His146 and His145 respectively (see Figure S2 in Supplementary Material).42 The delocalization of electron density via hydrogen bonding and hydrogen shuttling within this network may also further stabilize and enable the terminal oxygen species to donate electron density to the zinc ion.
Unlike SAHA, the terminal oxygen species of SCA (1) appears to only allow for monodentate chelation (Fig. 3). For the metal geometry to remain octahedral during SCA binding, an additional water molecule would be required. This additional water molecule may prevent SCA from obtaining a favorable binding orientation to the zinc ion, although it may also be possible that without a bidentate chelator, the zinc ion may exist in a 5-coordination state.42 The inability of SCA to form a favorable orientation to facilitate chelation may be further inhibited by the extended planar geometry of the terminal ethyl carbamate, potentially impeding the carbonyl oxygen from obtaining optimal orientation. Another issue with SCA binding involves the inability to stabilize the molecule in the binding pocket via hydrogen bonding with Asp104, His146, and His 145, since these residues are not situated properly when the carbamate terminus is coordinated with the zinc ion (Fig. 3). In addition, since the terminal carbamate moiety and linker of SCA resembles that of acetylated lysine, it may be possible that SCA acts as a substrate and undergoes catalysis,41 and thus does not bind as a traditional HDAC inhibitor.
3. Conclusion
To optimize the enzymatic and cellular activity of santacruzamate A (1), we systematically altered the scaffold including changing the zinc-binding group, cap terminus, and linker regions. In the process, we found enzymatic data that contrasted with our previously reported data, thus necessitating additional exploration of biological activity and resulting in new data on this scaffold. Previous studies have shown that enzyme assays can be complicated by numerous variables including source of enzyme, pH, temperature, buffers, solvents, substrates, preparation of enzyme, concentrations of assay components, time, among other variables.35 In addition, the need for fluorogenically labeled substrates can also affect results, potentially leading to false-positive and/or false-negative results.43 Also, HDAC assays rely on recombinant enzymes that are isolated from their partner proteins and thus unlikely to exist in their native conformations, resulting in an inability to translate the results from these enzymatic assays to physiologically relevant effects.44
We therefore followed up with substantial cellular testing in solid tumor and lymphoma cell lines as well as molecular biological tools to determine the effects of santacruzamate A and analogues on anti-proliferative and immunomodulatory activity. In these follow-up assays, several analogues were determined to be biologically active including two silated derivatives, compounds 27 and 28, which were found to exhibit both anti-proliferative and immune stimulation activity, as well as compound 26 which showed immune suppression activity. These compounds and the resulting structure-activity relationships are of interest for future studies and the molecular models that were reported herein will be useful in guiding development of additional analogues.
4. Experimental section
4.1. General
All reagents and chemicals purchased from Sigma-Aldrich or Acros and used directly. Hexane, tetrahydrofuran (THF), diethyl ether (Et2O), and dichloromethane (CH2Cl2) were used directly from a Baker cycle-tainer system. All glassware was flame-dried under vacuum, and all reactions performed under an argon atmosphere, unless otherwise noted. Flash chromatography performed using a 60 Å porosity, 32–63 μM silica gel. NMR spectra were obtained with a Bruker AVANCE 500 MHz spectrometer and analysis was performed on ACD/Spectrus processor 2012. HRMS performed at the University of Connecticut Mass Spectrometry Facility on an AccuTOF (JEOL) and using DART (IonSense) ionization source. Melting point were collected on a Mel-temp digital melting point apparatus. LC-MS data collected on an Agilent ESI single quadropole mass spectrometer coupled to an Agilent 1260 HPLC system with a G1311 quaternary pump, G1322 degasser, and a G1315 diode array detector using an Eclipse XDB-C18 (4.6 × 150 mm, 5 μm) column using a gradient of 40–100% acetonitrile with 0.1% formic acid over 30 min at a flow rate of 0.7 mL/min. Structural integrity of final target compounds were determined by 1H and 13C NMR, melting point, and HRMS. Purity of final target compounds were determined to be >95% by LC-MS.
4.2. Synthesis
4.2.1. General Procedure for the Preparation of Final Target Compounds
The intermediate free acid (1 equivalent) was dissolved in CH2Cl2 (7 mL) followed by the addition of triethylamine (2.2 equivalents). The reaction was cooled to 0 °C and stirred for 30 min. Then phenethylamine (1.1 equivalents) and 4-dimethylaminopyridine (DMAP) (cat., 0.1 equivalents) were added to the solution followed by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC-HCl) (1.1 equivalents) in five portions over 20 min. The solution stirred at 0 °C for 1 h then overnight at rt. The resulting solution was dissolved in an additional CH2Cl2 (25 mL), and washed with sat. NH4Cl (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL), organic layers combined, and sequentially washed with 20 mL of each of the following: sat. NaHCO3, H2O, brine. The organic layer was dried over Na2SO4 and concentrated to give a residue which was further purified with Celite (100% ethyl acetate). Then, the sample was dissolved in EtOAc, solution was heated, and cooled to 0 °C. The solution was filtered to yield desired product.
4.2.2. Zinc-binding group analogues
4.2.2.1. Ethyl 3-(phenethylcarbamoyl)propylcarbamate (1)
Compound 1 was synthesized following previously reported procedure.18
4.2.2.2. Propyl (4-oxo-4-(phenethylamino)butyl)carbamate (2)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 2a (0.33 g, 1.71 mmol), TEA (0.58 mL, 3.78 mmol), phenethylamine (0.24 mL, 1.89 mmol), DMAP (0.021 g, 0.17 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white solid (yield: 0.42 g, 84%). Mp 98–99 °C. 1H NMR (500 MHz, CDCl3) δ 7.12–7.25 (m, 5H), 6.00 (br s, 1H), 4.95 (br s, 1H), 3.92 (t, J=6.7 Hz, 2H), 3.44 (q, 2H), 3.09 (q, J=6.3 Hz, 2H), 2.75 (t, J=7.0 Hz, 2H), 2.19 (t, J=7.0 Hz, 2H), 1.72 (pentet, 2H), 1.55 (sextet, J=7.0 Hz, 2H), 0.85 (t, J=7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.5, 157.2, 138.8, 128.7, 128.5, 126.4, 66.4, 40.6, 40.1, 35.6, 33.6, 26.1, 22.3, 10.3. DART-HRMS (m/z) calculated for C16H25N2O3 [M+H]+ 293.1865, found 293.1861.
4.2.2.3. Butyl (4-oxo-4-(phenethylamino)butyl)carbamate (3)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 3a (0.35 g, 1.71 mmol), TEA (0.58 mL, 3.78 mmol), phenethylamine (0.24 mL, 1.89 mmol), DMAP (0.02 g, 0.17 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white solid (yield: 0.41 g, 78%). Mp 88–89 °C. 1H NMR (500 MHz, CDCl3) δ 7.12–7.26 (m, 5H), 5.96 (br s, 1H), 4.91 (br s, 1H), 3.96 (t, J=6.7 Hz, 2H), 3.44 (q, 2H), 3.10 (q, J=6.2 Hz, 2H), 2.75 (t, J=7.1 Hz, 2H), 2.10 (t, 2H), 1.72 (pentet, J=6.8 Hz, 2H), 1.51 (pentet, 2H), 1.30 (sextet, J=7.5 Hz, 2H), 0.85 (t, J=7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.7, 157.5, 139.1, 128.9, 128.8, 126.7, 64.9, 40.8, 40.3, 35.8, 33.9, 31.3, 26.4, 19.3, 13.9. DART-HRMS (m/z) calculated for C17H27N2O3 [M+H]+ 307.2022, found 307.2044.
4.2.2.4. Benzyl 3-(phenethylcarbamoyl)propylcarbamate (4)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 4a (0.1 g, 0.42 mmol) dissolved in CH2Cl2 (2 mL), TEA (0.12 mL, 0.83 mmol), phenethylamine (0.06 mL, 0.48 mmol), DMAP (0.005 g, 0.04 mmol), and EDC-HCl (0.09 g, 0.48 mmol). Product was obtained as a white solid (yield: 0.14 g, 95%). Mp 115–116 °C.1H NMR (500 MHz, CDCl3) δ 7.20–7.38 (m, 10H), 5.91 (br s, 1H), 5.10 (s, 2H), 5.09 (br s, 1H), 3.52 (q, J=6.7 Hz, 2H), 3.22 (q, J=6.3 Hz, 2H), 2.83 (t, J=7.1 Hz, 2H), 2.18 (m, 2H), 1.82 (pentet, J=6.8 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 172.6, 156.9, 138.9, 136.6, 128.8, 128.6, 128.5, 128.1, 128.1, 126. 5, 66.7, 40.7, 40.4, 35.7, 33.7, 26.1. DART-HRMS (m/z) calculated for C20H25N2O3 [M+H]+ 341.1865, found 341.1832.
4.2.2.5. Methyl 3-(phenethylcarbamoyl)propylcarbamate (5)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 5a (0.23 g, 1.43 mmol) dissolved in CH2Cl2 (10 mL), phenethylamine (0.2 mL, 1.64 mmol), TEA (0.4 mL, 2.12 mmol), DMAP (0.01 g, 0.08 mmol), EDC-HCl (0.31 g, 1.64 mmol). Product was obtained as a white solid (yield: 0.35 g, 94%). Mp 109–110 °C.1H NMR (500 MHz, CDCl3) δ 7.21–7.36 (m, 5H), 6.27 (br s, 1H), 5.34 (br s, 1H), 3.66 (s, 3H), 3.53 (q, 2H), 3.19 (q, J=5.9 Hz, 2H), 2.84 (t, J=7.2 Hz, 2H), 2.20 (t, J=7.1 Hz, 2H), 1.82 (pentet, J=6.8 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 172.7, 157.6, 138.9, 128.7, 128.6, 126.5, 52.0, 40.7, 40.3, 35.7, 33.7, 26.1. DART-HRMS (m/z) calculated for C14H21N2O3 [M+H]+ 265.1552, found 265.1543.
4.2.2.6. Allyl (4-oxo-4-(phenethylamino)butyl)carbamate (6)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 6a (0.37 g, 2.0 mmol) dissolved in CH2Cl2 (5 mL), TEA (0.56 mL, 4.0 mmol), phenethylamine (0.28 mL, 2.2 mmol), DMAP (0.02 g, 0.2 mmol), EDC-HCl (0.42g, 2.2 mmol), concentrated in vacuo. The sample was recrystallized using diethyl ether and hexane. Product was obtained as a white solid (yield: 0.47 g, 81%). Mp 90–91 °C. 1H NMR (500 MHz, CDCl3) δ 7.19–7.34 (m, 5H), 6.04 (br s, 1H), 5.88–5.97 (m, 1H), 5.21–5.34 (m, 2H), 5.17 (br s, 1H), 4.56 (d, J=5.4 Hz, 2H), 3.54 (q, 2H), 3.21 (q, J=6.5 Hz, 2H), 2.84 (t, J=7.1 Hz, 2H), 2.19 (t, J=6.9 Hz, 2H), 1.82 (pentet, J=6.8 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 172.5, 156.7, 138.9, 132.9, 128.7, 128.6, 126.5, 117.6, 65.5, 40.6, 40.3, 35.6, 33.7, 26.1. DART-HRMS (m/z) calculated for C16H23N2O3 [M+H]+ 291.1709, found 291.1717.
4.2.2.7. 2-Fluoroethyl (4-oxo-4-(phenethylamino)butyl)carbamate (7)
Converted to the N-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 7a (0.33 g, 1.71 mmol), TEA (0.58 mL, 3.78 mmol), phenethylamine (0.24 mL, 1.89 mmol), DMAP (0.02 g, 0.17 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white solid (yield: 0.4 g, 79%). Mp 95 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.35 (m, 5H), 5.86 (br s, 1H), 5.17 (br s, 1H), 4.54–4.65 (dt, 2H), 4.27–4.32 (dt, 2H), 3.52–3.58 (q, 2H), 3.22 (q, J=6.4 Hz, 2H), 2.85 (t, J=7.0 Hz, 2H), 2.20 (t, J=7.0 Hz, 2H), 1.84 (pentet, J=6.8 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 172.6, 156.6, 139.0, 128.9, 128.8, 126.7, 82.7, 64.1, 40.8, 40.5, 35.8, 33.8, 26.0. DART-HRMS (m/z) calculated for C15H22FN2O3 [M+H]+ 297.1614, found 297.1638.
4.2.2.8. tert-Butyl (4-oxo-4-(phenethylamino)butyl)carbamate (8)
Converted to the N-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 8a (0.25 g, 1.23 mmol) dissolved in CH2Cl2 (3 mL), TEA (0.34 mL, 2.46 mmol), phenethylamine (0.17 mL, 1.35 mmol), DMAP (0.01 g, 0.08 mmol), EDC-HCl (0.26 g, 1.35 mmol). Product was obtained as a white solid (yield: 0.31g, 75%). Mp 94–95 °C. 1H NMR (500 MHz, CDCl3) δ 7.13–7.25 (m, 5H), 5.97 (br s, 1H), 4.66 (br s, 1H), 3.45 (q, J=6.9 Hz, 2H), 3.04 (d, J=6.3 Hz, 2H), 2.76 (t, J=6.9 Hz, 2H), 2.09 (t, J=7.3 Hz, 2H), 1.70 (pentet, 2H), 1.36 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 172.5, 158.2, 138.9, 128.8, 128.6, 126.5, 79.3, 40.6, 39.7, 35.6, 33.7, 28.4, 26.4. DART-HRMS (m/z) calculated for C17H27N2O3 [M+H]+ 307.2022, found 307.2012. Data matched that reported previously.46
4.2.2.9. Prop-2-yn-1-yl (4-oxo-4-(phenethylamino)butyl)carbamate (9)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 9a (0.32 g, 1.71 mmol), TEA (0.58 mL, 3.78 mmol), phenethylamine (0.24 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white solid (yield: 0.32 g, 65%). Mp 86 °C. 1H NMR (500 MHz, CDCl3) δ 7.10–7.26 (m, 5H), 5.86 (br s, 1H), 5.21 (br s, 1H), 4.59 (d, 2H), 3.44 (q, 2H), 3.13 (q, 2H), 2.75 (t, J=7.0 Hz, 2H), 2.38 (t, 1H), 2.11 (t, 2H), 1.74 (pentet, 2H). 13C NMR (125 MHz, CDCl3) δ 172.7, 156.1, 139.0, 128.9, 128.8, 126.7, 78.5, 74.7, 52.6, 40.8, 40.6, 35.8, 33.8, 26.0. DART-HRMS (m/z) calculated for C16H21N2O3 [M+H]+ 289.1552, found 289.1578.
4.2.2.10. O-ethyl (4-oxo-4-(phenethylamino)butyl)carbamothioate (10)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 10a (0.5 g, 2.62 mmol) dissolved in CH2Cl2 (10 mL), TEA (0.73 mL, 5.23 mmol), phenethylamine (0.38 mL, 3.0 mmol), DMAP (0.03 g, 0.25 mmol), EDC-HCl (0.57 g, 3.0 mmol). Crude material was washed sequentially with 20 mL of the following solutions: 1M HCl, H2O, NaHCO3, and brine. Resulting material was purified using SiO2 plug and titrated overnight with hexanes. Product was obtained as a shimmering white solid (yield: 0.7 g, 91%). 1H NMR (500 MHz, CDCl3) δ 7.20–7.35 (m, 5H), 7.12 (br s, 1H), 5.79 (br s, 1H), 4.47 (q, 2H), 3.52–3.60 (m, 2H), 3.31 (q, J=6.5 Hz, 2H), 2.84 (t, J=7.0 Hz, 2H), 2.25 (t, 2H), 1.94 (pentet, 2H), 1.32 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 190.7, 172.8, 138.9, 128.9, 128.6, 126.8, 66.4, 45.11, 40.89, 35.77, 34.15, 24.2, 14.5. DART-HRMS (m/z) calculated for C15H23N2O2S [M+H]+ 295.1480, found 295.1506.
4.2.2.11. S-ethyl (4-oxo-4-(phenethylamino)butyl)carbamothioate (11)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 11a (0.4 g, 2.1 mmol) dissolved in CH2Cl2 (6 mL), TEA (0.58 mL, 4.2 mmol), phenethylamine (0.32 mL, 2.51 mmol), DMAP (0.026 g, 0.21 mmol), EDC-HCl (0.48 g, 2.51 mmol). Product was obtained as a white solid (yield: 0.5 g, 83%). Mp 118–119 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.35 (m, 5H), 5.99 (br s, 1H), 5.94 (br s, 1H), 3.55 (q, 2H), 3.31 (q, J=5.7 Hz, 2H), 2.92 (q, 2H), 2.85 (t, 2H), 2.20 (t, J=6.8 Hz, 2H), 1.84 (pentet, J=6.62 Hz, 2H), 1.30 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 191.0, 172.5, 138.8, 128.7, 128.6, 126.5, 44.6, 40.7, 35.6, 33.8, 25.5, 24.3, 15.7 DART-HRMS (m/z) calculated for C15H23N2O2S [M+H]+ 295.1480, found 295.1464.
4.2.2.12. 4-(3-Ethylureido)-N-phenethylbutanamide (12)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 12d (0.012g, 0.072 mmol) dissolved in CH2Cl2 (0.4 mL), TEA (0.022 mL, 0.16 mmol), phenethylamine (0.01 mL, 0.082 mmol), DMAP (0.8 mg, 0.007 mmol), EDC-HCl (0.016 g, 0.082 mmol). Product was obtained as a white solid (yield: 0.015 g, 76%). 1H NMR (500 MHz, CDCl3) δ 7.13–7.26 (m, 5H), 6.07 (br s, 2H), 4.78 (br s, 1H), 3.45 (q, 2H), 3.13 (m, 4H), 2.76 (t, 2H), 2.13 (t, 2H), 1.71 (pentet, 2H), 1.07 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 173.3, 158.9, 139.0, 128.9, 128.8, 126.7, 40.9, 39.9, 35.8, 35.7, 33.8, 26.4, 15.6. DART-HRMS (m/z) calculated for C15H24N3O2 [M+H]+ 278.1869, found 278.1896. Data matched that reported previously.46
4.2.2.13. N-phenethyl-4-(2-oxo-pyrrolidine-1-thiocarbonylamino)butanamide (13)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 13a (0.1 g, 0.43 mmol) dissolved in CH2Cl2 (3 mL), TEA (0.13 mL 1.0 mmol), phenethylamine (0.06 mL, 0.5 mmol), DMAP (0.005 g, 0.043 mmol), EDC-HCl (0.1 g, 0.5 mmol). The sample was dissolved in CH2Cl2, solution was heated, and product precipitated from solution with the addition of hexane. Solution cooled to −20 °C overnight and filtered. Product was obtained as a white solid (yield: 0.075 g, 53%). Mp 98–99 °C. 1H NMR (500 MHz, CDCl3) δ 10.79 (br s, 1H), 7.21–7.35 (m, 5H), 5.71 (br s, 1H), 4.23 (t, J=7.1 Hz, 2H), 3.69 (q, 2H), 3.55 (q, 2H), 2.85 (t, 2H), 2.72 (t, 2H), 2.23 (t, J=7.1 Hz, 2H), 1.98–2.09 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 180.8, 176.7, 171.8, 138.8, 128.8, 128.7, 126.5, 51.1, 44.6, 40.6, 35.7, 34.5, 33.7, 24.2, 16.9. DART-HRMS (m/z) calculated for C17H24N3O2S [M+H]+ 334.1589, found 334.1604.
4.2.2.14. N-phenethyl-4-(2-oxo-pyrrolidine-1-carbonylamino)butanamide (14)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 14a (0.085 g, 0.4 mmol) dissolved in CH2Cl2 (1 mL), TEA (0.11 mL, 0.8 mmol), phenethylamine (0.055 mL, 0.44 mmol), DMAP (0.005 g, 0.04 mmol), EDC-HCl (0.08 g, 0.44 mmol). The sample was dissolved in CH2Cl2, solution was heated, and product precipitated from solution with the addition of hexane. Solution cooled to −20 °C overnight and filtered. Product was obtained as a white solid (yield: 0.05 g, 40%). Mp 68–69 °C. 1H NMR (500 MHz, CDCl3) δ 8.48 (br s, 1H), 7.22–7.34 (m, 5H), 6.03 (br s, 1H), 3.86 (t, J=7.1 Hz, 2H), 3.55 (q, J=6.8 Hz, 2H), 3.32 (q, J=6.6 Hz, 2H), 2.85 (t, J=7.1 Hz, 2H), 2.62 (t, 2H), 2.19 (t, J=7.1 Hz, 2H), 2.05 (pentet, 2H), 1.87 (pentet, 2H). 13C NMR (125 MHz, CDCl3) δ 177.1, 172.2, 153.4, 139.0, 128.8, 128.6, 126.4, 45.7, 40.6, 38.8, 35.7, 33.7, 33.4, 26.2, 17.0. DART-HRMS (m/z) calculated for C17H24N3O3 [M+H]+ 318.1818, found 318.1823.
4.2.2.15. 4-Acetamido-N-phenethylbutanamide (15)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 15a (0.2 g, 1.38 mmol), TEA (0.38 mL, 2.74 mmol), phenethylamine (0.2 mL, 1.58 mmol), DMAP (0.01 g, 0.082 mmol), EDC-HCl (0.3 g, 1.58 mmol). Product was obtained as a white solid (yield: 0.25 g, 73%). Mp 112–113 °C. 1H NMR (500 MHz, CDCl3) δ 7.21–7.35 (m, 5H), 6.06 (br s, 2H), 3.56 (q, 2H), 3.28 (q, 2H), 2.86 (t, J=7.3 Hz, 2H), 2.21 (t, 2H), 1.82 (pentet, J=6.6 Hz, 2H), 1.65 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 172.9, 170.8, 138.9, 128.7, 128.6, 126.5, 40.8, 39.0, 35.7, 33.9, 25.5, 23.2. DART-HRMS (m/z) calculated for C14H21N2O2 [M+H]+ 249.1603, found 249.1581.
4.2.2.16. 4-Butyramido-N-phenethylbutanamide (16)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 16a (0.5 g, 2.89 mmol), TEA (0.81 mL, 5.78 mmol), phenethylamine (0.43 mL, 3.44 mmol), DMAP (0.035 g, 0.289 mmol), EDC-HCl (0.65 g, 3.44 mmol). Product was obtained as a white solid (yield: 0.66 g, 83%). Mp 119–120 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.34 (m, 5H), 6.35 (br s, 1H), 6.22 (br s, 1H), 3.54 (q, 2H), 3.27 (q, 2H), 2.85 (t, J=7.3 Hz, 2H), 2.15–2.21 (m, 4H), 1.81 (pentet, 2H), 1.67 (sextet, 2H), 0.96 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 173.7, 172.8, 151.7, 138.9, 128.7, 128.6, 126.5, 40.7, 38.9, 38.7, 35.7, 33.9, 25.6, 19.2, 13.8. DART-HRMS (m/z) calculated for C16H25N2O2 [M+H]+ 277.1916, found 277.1948.
4.2.2.17. N-phenethyl-4-propionamidobutanamide (17)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 17a (0.50 g, 2.89 mmol), TEA (0.81 mL, 5.78 mmol), phenethylamine (0.44 mL, 3.5 mmol), DMAP (0.03 g, 0.29 mmol), EDC-HCl (0.66 g, 3.46 mmol). Product was obtained as a white solid (yield: 0.52 g, 68%). Mp 117–118 °C. 1H NMR (500 MHz, CDCl3) δ 7.19–7.33 (m, 5H), 6.32 (br s, 1H), 6.23 (br s, 1H), 3.54 (q, 2H), 3.27 (q, J=6.0 Hz, 2H), 2.85 (t, J=7.1 Hz, 2H), 2.21 (m, 4H), 1.81 (pentet, J=6.6 Hz, 2H), 1.16 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 174.4, 172.8, 138.9, 128.7, 128.6, 126.5, 40.7, 38.9, 35.7, 33.9, 29.8, 25.5, 9.9. DART-HRMS (m/z) calculated for C15H23N2O2 [M+H]+ 263.1760, obs. 263.1784.
4.2.2.18. 3,3-dimethyl-N-(4-oxo-4-(phenethylamino)butyl)butanamide (18)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 18a (1.04 g, 4.85 mmol) dissolved in CH2Cl2 (30 mL), TEA (1.35 mL, 9.7 mmol), phenethylamine (0.7 mL, 5.6 mmol), DMAP (0.06 g, 0.49 mmol), EDC-HCl (1.06 g, 5.56 mmol). Product was obtained as an off white wet solid (yield: 1.32 g, 89%). Mp 87–88 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.32 (m, 5H), 6.44 (br s, 1H), 6.17 (br s, 1H), 3.53 (q, 2H), 3.24 (q, J=6.2 Hz, 2H), 2.85 (t, 2H), 2.20 (t, J=6.8 Hz, 2H), 1.80 (pentet, J=6.6 Hz, 2H), 1.04 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 172.8, 172.4, 138.9, 128.7, 128.6, 126.5, 50.6, 40.7, 38.8, 35.7, 34.0, 30.8, 29.8, 25.7. DART-HRMS (m/z) calculated for C18H29N2O2 [M+H]+ 305.2229, found 305.2214.
4.2.2.19. N-(4-oxo-4-(phenethylamino)butyl)benzamide (19)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 19a (0.21 g, 1.0 mmol), TEA (0.3 mL, 2.12 mmol), phenethylamine (0.14 mL, 1.14 mmol), DMAP (0.012 g, 0.1 mmol), EDC-HCl (0.22 g, 1.14 mmol). Product was obtained as a white solid (yield: 0.26 g, 83%). Mp 106–107 °C. 1H NMR (500 MHz, CDCl3) δ 7.85 (d, 2H), 7.16–7.50 (m, 8H), 6.43 (br s, 2H), 3.44–3.51 (m, 4H), 2.79 (t, J=7.3 Hz, 2H), 2.27 (t, 2H), 1.93 (pentet, 2H). 13C NMR (125 MHz, CDCl3) δ 173.2, 167.8, 138.8, 134.4, 131.4, 128.7, 128.6, 128.5, 127.0, 126.5, 40.75, 39.9, 35.6, 34.2, 25.0. DART-HRMS (m/z) calculated for C19H23N2O2 [M+H]+ 311.1760, found 311.1740.
4.2.2.20. S-(3-oxo-3-((4-oxo-4-(phenethylamino)butyl)amino)propyl)ethanethioate (20)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 20b (0.45 g, 1.93 mmol) dissolved in CH2Cl2 (5 mL), TEA (0.54 mL, 3.86 mmol), phenethylamine (0.29 mL, 2.3 mmol), DMAP (0.024 g, 0.19 mmol), EDC-HCl (0.44 g, 2.3 mmol). Product was obtained as a white solid (yield: 0.58g, 89%). Mp 100–101 °C. 1H NMR (500 MHz, CDCl3) δ 7.21–7.24 (m, 3H), 7.31–7.34 (m, 2H), 6.14 (br s, 1H), 5.95 (br s, 1H), 3.55 (m, 2H), 3.29 (m, 2H), 3.16 (t, 1H), 2.86 (t, 2H), 2.49 (t, 2H), 2.34 (s, 3H), 2.20 (m, 2H), 1.82 (pentet, 2H). 13C NMR (125 MHz, CDCl3) δ 195.7, 172.6, 170.9, 138.8, 128.7, 128.6, 126.5, 40.6, 39.1, 36.4, 35.6, 34.0, 30.5, 25.4, 25.0. DART-HRMS (m/z) calculated for C17H25N2O3S [M+H]+ 337.1586, found 337.1583.
4.2.2.21. 4-oxo-4-(phenethylamino)butyl ethylcarbamate (21)
21c (0.6 g, 3.15 mmol) was dissolved in THF (10 mL) and to this was added aqueous LiOH (0.75 g, 3.15 mmol, 1M) at rt for 90 min. The reaction mixture was neutralized with 6N HCl and concentrated. The aqueous layer was extracted with EtOAc (4 × 20 mL), washed with brine, dried over Na2SO4, and concentrated. Resulting solid was dissolved in EtOAc, solution was heated, and cooled to 0 °C. The solution was filtered to yield desired product and used in the next reaction without further purification. Material was converted to the N-phenylacetamide derivative using the general procedure described and was obtained as a white solid (yield: 0.21 g, 56%). Mp 110–111 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.35 (m, 5H), 6.07 (br s, 1H), 5.04 (br s, 1H), 4.11 (q, J=7.0 Hz, 2H), 3.54 (q, 2H), 3.20 (q, J=6.3 Hz, 2H), 2.84 (t, J=7.1 Hz, 2H), 2.19 (t, J=7.0 Hz, 2H), 1.81 (pentet, J=6.8 Hz, 2H), 1.25 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 172.7, 157.3, 139.1, 128.9, 128.8, 126.7, 61.0, 40.8, 40.3, 35.8, 33.9, 26.3, 14.8. DART-HRMS (m/z) calculated for C15H23N2O3 [M+H]+ 279.1709, found 279.1736.
4.2.3. Cap group analogues
4.2.3.1. Ethyl (4-(isopentylamino)-4-oxobutyl)carbamate (23)
Converted to the N-isopentylacetamide derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with isopentylamine (0.22 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.19 g, 46%). Mp 99 °C. 1H NMR (500 MHz, CDCl3) δ 6.12 (br s, 1H), 5.13 (br s, 1H), 4.11 (q, J=7.0 Hz, 2H), 3.21–3.29 (m, 4H), 2.22 (t, J=7.0 Hz, 2H), 1.83 (pentet, J=6.8 Hz, 2H), 1.62 (sextet, J=6.7, 13.39 Hz, 1H), 1.38–1.41 (q, 2H), 1.24 (t, J=7.1 Hz, 3H), 0.92 (d, J=6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 172.7, 157.4, 60.9, 40.4, 38.6, 38.1, 33.9, 26.4, 26.1, 22.6, 14.8. DART-HRMS (m/z) calculated for C12H25N2O3 [M+H]+ 245.1865, found 245.1870.
4.2.3.2. Ethyl (4-((2-((tert-butoxycarbonyl)amino)ethyl)amino)-4-oxobutyl)carbamate (24)
Converted to the N-Boc-acetamide derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with tert-butyl (2-aminoethyl)carbamate (0.3 g, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white solid (yield: 0.004 g, 8%). Mp 102–103 °C. 1H NMR (500 MHz, CDCl3) δ 6.54 (br s, 1H), 5.23 (br s, 1H), 5.03 (br s, 1H), 4.14 (q, J=7.0 Hz, 2H), 3.38 (q, J=5.5 Hz, 2H), 3.30 (t, 2H), 3.24 (t, 2H), 2.25 (t, J=6.9 Hz, 2H), 1.85 (pentet, J=6.7 Hz, 2H), 1.46 (s, 9H), 1.26 (t, J=7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.9, 157.1, 154.7, 79.6, 61.0, 40.7, 40.1, 37.7, 33.6, 28.6, 26.3, 14.8. DART-HRMS (m/z) calculated for C14H28N3O5 [M+H]+ 318.2029, found 318.2043.
4.2.3.3. Ethyl (4-oxo-4-(thiazol-2-ylamino)butyl)carbamate (25)
Converted to the N-aminothiozole derivative using the general procedure described and with the following specifications and alterations: 22 (0.30 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with 2-aminothiazole (0.19 g, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.32 g, 73%). Mp 173–174 °C. 1H NMR (500 MHz, CD3OD) δ 7.41 (d, J=2.9 Hz, 1H), 7.08 (d, J=2.9 Hz, 1H), 4.05 (q, J=3.0 Hz, 2H), 3.17 (t, 2H), 2.51 (t, 2H), 1.89 (pentet, J=6.7 Hz, 2H), 1.20 (t, 3H). 13C NMR (125 MHz, CD3OD) δ 170.3, 157.4, 156.7, 135.6, 111.7, 59.0, 38.3, 31.0, 23.9, 12.2. DART-HRMS (m/z) calculated for C10H16N3O3S [M+H]+ 258.0912, found 258.0906.
4.2.3.4. Ethyl (4-((4-(3-nitrophenyl)thiazol-2-yl)amino)-4-oxobutyl)carbamate (26)
Converted to the 3-nitrophenyl-thiazol-2-amine derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with 3-nitrophenyl-thiazol-2-amine (0.42 g, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.42 g, 65%). Mp 167–168 °C. 1H NMR (500 MHz, CDCl3) δ 10.38 (br s, 1H), 8.73 (s, 1H), 8.17 (d, J=7.9 Hz, 1H), 7.59 (t, 1H), 7.30 (t, 1H), 6.91 (s, 1H), 5.01 (br s, 1H), 4.34 (q, 2H), 3.85 (t, 2H), 2.60 (t, 2H), 2.07 (pentet, J=7.6 Hz, 2H), 1.37 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 170.6, 164.5, 158.3, 148.9, 147.6, 136.3, 131.8, 129.8, 122.6, 121.2, 109.8, 61.5, 40.0, 33.5, 26.7, 14.8. DART-HRMS (m/z) calculated for C16H19N4O5S [M+H]+ 379.1076, found 379.1043.
4.2.3.5. Ethyl (4-((4-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)amino)-4-oxobutyl)carbamate (27)
Converted to the toluene-OTBS derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with 4-(((tert-butyldimethylsilyl)oxy)methyl)aniline (0.45 g, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.089 g, 13%). Mp 102.7–103 °C. 1H NMR (500 MHz, CDCl3) δ 8.38 (br s, 1H), 7.57 (d, J=7.9 Hz, 2H), 7.29 (d, 2H), 4.99 (br s, 1H), 4.72 (s, 2H), 4.15 (q, J=7.1 Hz, 2H), 3.33 (q, J=5.8 Hz, 2H), 2.42 (t, J=6.7 Hz, 2H), 1.93 (pentet, J=6.5 Hz, 2H), 1.26 (t, J=7.1 Hz, 3H), 0.96 (s, 9H), 0.11 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 171.2, 157.9, 137.3, 137.2, 126.9, 119.8, 64.9, 61.3, 40.0, 34.8, 29.8, 27.1, 26.1, 14.8, −5.0. DART-HRMS (m/z) calculated for C20H35N2O4Si [M+H]+ 395.2366, found 395.2343.
4.2.3.6. Ethyl (4-((4-((tert-butyldimethylsilyl)oxy)phenyl)amino)-4-oxobutyl)carbamate (28)
Converted to the aniline-OTBS derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.1 mmol), phenethylamine replaced with 4-((tert-butyldimethylsilyl)oxy)aniline (0.42 g, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 mL, 1.89 mmol). Product was obtained as a light brown solid (yield: 0.32 g, 49%). Mp 112 °C. 1H NMR (500 MHz, CDCl3) δ 8.11 (br s, 1H), 7.36–7.38 (d, J=8.6 Hz, 2H), 6.72–6.74 (d, 2H), 4.86 (br s, 1H), 4.07 (q, J=7.4 Hz, 2H), 3.26 (q, J=6.2 Hz, 2H), 2.32 (t, 2H), 1.85 (pentet, J=6.5 Hz, 2H), 1.18 (t, J=7.1 Hz, 3H), 0.92 (s, 9H), 0.12 (s, 6H) 13C NMR (125 MHz, CDCl3) δ 172.9, 157.1, 154.7, 135.5, 129.0, 122.5, 61.1, 40.7, 33.6, 28.6, 26.3, 21.0, 14.8. DART-HRMS (m/z) calculated for C19H33N2O4Si [M+H]+ 381.2210, found 381.2197.
4.2.3.7. Ethyl (4-((4-chlorophenethyl)amino)-4-oxobutyl)carbamate (29)
Converted to the N-(para-chloro)-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 22 (0.30 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with para-chloro-phenethylamine (0.26 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.28 g, 51%). Mp 118–119 °C. 1H NMR (500 MHz, CDCl3) δ 7.27–7.29 (d, 2H), 7.14–7.15 (d, J=8.3 Hz, 2H), 6.19 (br s, 1H), 5.02 (br s, 1H), 4.11 (q, J=7.1 Hz, 2H), 3.51 (q, 2H), 3.19 (q, J=6.1 Hz, 2H), 2.81 (t, J=7.1 Hz, 2H), 2.19 (t, 2H), 1.80 (pentet, J=6.7 Hz, 2H), 1.25 (t, J=7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.8, 157.4, 137.6, 132.4, 130.3, 128.9, 61.0, 40.7, 40.2, 35.2, 33.8, 26.5, 14.8. DART-HRMS (m/z) calculated for C15H22N2ClO3 [M+H]+ 313.1319, found 313.1340.
4.2.3.8. Ethyl (4-((3-chlorophenethyl)amino)-4-oxobutyl)carbamate (30)
Converted to the N-(meta-chloro)-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with meta-chloro-phenethylamine (0.27 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.4 g, 74%). Mp 89–90 °C. 1H NMR (500 MHz, CDCl3) δ 7.29 (d, 2H), 7.15 (d, J=8.3 Hz, 2H), 6.19 (br s, 1H), 5.02 (br s, 1H), 4.11 (q, J=7.1 Hz, 2H), 3.51 (q, 2H), 3.19 (q, J=6.1 Hz, 2H), 2.81 (t, J=7.1 Hz, 2H), 2.19 (t, 2H), 1.80 (pentet, J=6.7 Hz, 2H), 1.25 (t, J=7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.8, 157.4, 137.6, 132.4, 130.3, 128.9, 61.0, 40.7, 40.2, 35.2, 33.8, 26.5, 14.8. DART-HRMS (m/z) calculated for C15H22N2ClO3 [M+H]+ 313.1319, found 313.1341.
4.2.3.9. Phenethyl 4-((ethoxycarbonyl)amino)butanoate (31)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), HOBt (0.26 g, 1.89 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with 2-phenylethan-1-ol (0.21 g, 1.71 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.32 g, 67%). Mp 122–123 °C. 1H NMR (500 MHz, CDCl3) δ 7.12–7.27 (m, 6H), 4.62 (br s, 1H), 4.23 (t, J=7.1 Hz, 2H), 4.03 (d, J=6.8 Hz, 2H), 3.09 (d, J=6.2 Hz, 2H), 2.87 (t, J=6.8 Hz, 2H), 2.26 (t, J=7.1 Hz, 2H), 1.72 (t, J=6.8 Hz, 2H), 1.16 (t, J=6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.0, 162.6, 160.7, 157.3, 134.6, 130.2, 115.4, 115.3, 60.8, 40.8, 40.1, 34.9, 33.6, 26.3, 14.6. DART-HRMS (m/z) calculated for C15H22NO4 [M+H]+ 280.1549, found 280.1553.
4.2.3.10. Methyl (4-((ethoxycarbonyl)amino)butanoyl)phenylalaninate (32)
Converted to the N-methyl-l-phenylalanine derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), methyl-l-phenylalanine (0.34 g, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.36 g, 63%). Mp 129–130 °C. 1H NMR (500 MHz, CDCl3) δ 7.26–7.33 (m, 3H), 7.13–7.15 (d, J=7.2 Hz, 2H), 6.35 (br s, 1H), 4.94 (br s, 1H), 4.88–4.92 (q, 1H), 4.13 (q, J=7.0 Hz, 2H), 3.75 (s, 3H), 3.17–3.21 (dd, J=5.7, 13.91 Hz, 2H), 3.07–3.11 (dd, 1H), 2.22–2.26 (dt, J=2.7, 7.1 Hz, 2H), 1.80 (pentet, J=6.8 Hz, 3H), 1.26 (t, J=7.1 Hz, 3H) 13C NMR (125 MHz, CDCl3) δ 172.4, 171.6, 157.2, 136.2, 129.4, 128.8, 127.3, 60.9, 53.4, 52.5, 40.1, 38.0, 33.5, 26.1, 14.8. DART-HRMS (m/z) calculated for C17H25N2O5 [M+H]+ 337.1763, found 337.1732.
4.2.3.11. Ethyl (4-((3-fluorophenethyl)amino)-4-oxobutyl)carbamate (33)
Converted to the N-(m-fluoro)-phenethalamine derivative using the general procedure described and with the following specifications and alterations: 22 (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine replaced with m-fluoro-phenethylamine (0.25 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.18 g, 28%). Mp 117–118 °C. 1H NMR (500 MHz, CDCl3) δ 7.18 (t, 2H), 7.01 (t, 2H), 6.09 (br s, 1H), 4.97 (br s, 1H), 4.12 (q, J=7.0 Hz, 2H), 3.52 (q, 2H), 3.20 (q, J=6.2 Hz, 2H), 2.82 (t, J=7.1 Hz, 2H), 2.20 (t, 2H), 1.81 (pentet, J=6.7 Hz, 2H), 1.25 (t, J=7.1 Hz, 3H) 13C NMR (125 MHz, CDCl3) δ 172.8, 162.8, 160.9, 134.7, 130.4, 130.3, 115.6, 115.5, 61.0, 40.9, 40.3, 35.0, 33.8, 26.4, 14.8. DART-HRMS (m/z) calculated for C15H22FN2O3 [M+H]+ 297.1614, found 297.1638.
4.2.4. Linker analogues
4.2.4.1. Ethyl (2-oxo-2-(phenethylamino)ethyl)carbamate (34)
Converted to the N-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 34a (0.22 g, 1.5 mmol), TEA (0.42 mL, 3.1 mmol), phenethylamine (0.21 mL, 1.71 mmol), DMAP (0.018 g, 0.15 mmol), EDC-HCl (0.32 g, 1.71 mmol). Product was obtained as a white powder (yield: 0.21 g, 56%). Mp 100–101 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.34 (m, 5H), 6.20 (br s, 1H), 5.37 (br s, 1H), 4.15 (q, J=7.1 Hz, 2H), 3.81 (d, J=5.8 Hz, 2H), 3.56 (q, J=6.9 Hz, 2H), 2.84 (t, J=7.0 Hz, 2H), 1.27 (t, J=7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 169.3, 157.0, 138.8, 128.9, 128.8, 126.8, 61.6, 44.8, 40.8, 35.8, 14.7. DART-HRMS (m/z) calculated for C13H19N2O3 [M+H]+ 251.1396, found 251.1383.
4.2.4.2. Ethyl (3-oxo-3-(phenethylamino)propyl)carbamate (35)
Converted to the N-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 35a (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine (0.24 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.21 g, 47%). Mp 105–106 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.35 (m, 5H), 5.63 (br s, 1H), 5.29 (br s, 1H), 4.12 (q, J=7.0 Hz, 2H), 3.56 (q, J=6.9 Hz, 2H), 3.46 (q, J=6.0 Hz, 2H), 2.84 (t, J=7.0 Hz, 2H), 2.37 (t, J=5.8 Hz, 2H), 1.26 (t, J=7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 171.4, 157.0, 138.9, 128.89, 128.87, 126.8, 60.9, 40.8, 37.2, 36.3, 35.8, 14.8. DART-HRMS (m/z) calculated for C14H21N2O3 [M+H]+ 265.1552, found 265.1538.
4.2.4.3. Ethyl (5-oxo-5-(phenethylamino)pentyl)carbamate (36)
Converted to the N-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 36a (0.32 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.36 g, 72%). Mp 102–103 °C. 1H NMR (500 MHz, CDCl3) δ 7.11–7.25 (m, 5H), 5.53 (br s, 1H), 4.71 (br s, 1H), 4.02 (q, J=6.7 Hz, 2H), 3.45 (q, J=6.7 Hz, 2H), 3.09 (q, J=6.2 Hz, 2H), 2.74 (t, J=7.0 Hz, 2H), 2.07 (t, J=7.3 Hz, 2H), 1.56 (pentet, J=7.5 Hz, 2H), 1.41 (pentet, J=7.1 Hz, 2H), 1.16 (t, J=7.0 Hz, 3H) 13C NMR (125 MHz, CDCl3) δ 172.9, 156.9, 139.0, 128.9, 128.8, 126.7, 60.9, 40.7, 40.4, 36.1, 35.9, 29.6, 22.7, 14.8. DART-HRMS (m/z) calculated for C16H25N2O3 [M+H]+ 293.1865, found 293.1866.
4.2.4.4. Ethyl (6-oxo-6-(phenethylamino)hexyl)carbamate (37)
Converted to the N-phenethylamine derivative using the general procedure described and with the following specifications and alterations: 37a (0.35 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenethylamine (0.24 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.35 g, 67%). Mp 99–100 °C. 1H NMR (500 MHz, CDCl3) δ 7.18–7.32 (m, 5H), 5.64 (br s, 1H), 4.78 (br s, 1H), 4.09 (q, J=6.8 Hz, 2H), 3.52 (q, J=6.8 Hz, 2H), 3.15 (q, J=6.2 Hz, 2H), 2.81 (t, J=7.0 Hz, 2H), 2.12 (t, J=7.5 Hz, 2H), 1.61 (pentet, J=7.6 Hz, 2H), 1.48 (pentet, J=7.3 Hz, 2H), 1.21–1.33 (m, 5H). 13C NMR (125 MHz, CDCl3) δ 173.0, 156.9, 139.1, 128.9, 128.8, 126.7, 60.8, 40.8, 40.7, 36.6, 35.9, 29.9, 26.4, 25.4, 14.8. DART-HRMS (m/z) calculated for C17H27N2O3 [M+H]+ 307.2022, found 307.2026.
4.2.4.5. Ethyl (5-(benzylamino)-5-oxopentyl)carbamate (38)
Converted to the N-phenmethylamine derivative using the general procedure described and with the following specifications and alterations: 36a (0.32 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), phenylmethanamine (0.21 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.35 g, 65%). Mp 102–103 °C. 1H NMR (500 MHz, CDCl3) δ 7.18–7.27 (m, 5H), 5.99 (br s, 1H), 4.76 (br s, 1H), 4.35 (d, J=5.7 Hz, 2H), 3.97 (q, J=6.6 Hz, 2H), 3.10 (q, J=6.1 Hz, 2H), 2.17 (t, J=7.5 Hz, 2H), 1.62 (pentet, J=7.5 Hz, 2H), 1.45 (pentet, J=7.1 Hz, 2H), 1.13 (t, J=7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.9, 157.0, 138.6, 128.9, 127.9, 127.7, 60.9, 43.8, 40.3, 36.0, 29.7, 22.8, 14.8. DART-HRMS (m/z) calculated for C15H23N2O3 [M+H]+ 279.1709, found 279.1696. Data matched that reported previously.46
4.2.4.6. Ethyl (6-oxo-6-(phenylamino)hexyl)carbamate (39)
Converted to the N-aniline derivative using the general procedure described and with the following specifications and alterations: 37a (0.35 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), aniline (0.17 mL, 1.89 mmol), DMAP (0.021 g, 0.171 mmol), EDC-HCl (0.36 g, 1.89 mmol). Product was obtained as a white powder (yield: 0.29 g, 60%). Mp 107–108 °C. 1H NMR (500 MHz, CDCl3) δ 7.53 (br s, 1H), 7.45–7.46 (d, J=8.0 Hz, 2H), 7.21–7.24 (t, J=7.8 Hz, 2H), 7.02 (t, J=7.3 Hz, 1H), 4.70 (br s, 1H), 4.03 (q, J=6.8 Hz, 2H), 3.09 (q, J=6.2 Hz, 2H), 2.28 (t, J=7.4 Hz, 2H), 1.67 (pentet, J=7.6 Hz, 2H), 1.46 (pentet, J=7.2 Hz, 2H), 1.32 (pentet, 2H), 1.15 (t, J=7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 171.5, 157.0, 138.3, 129.1, 124.3, 120.0, 60.9, 40.8, 37.6, 29.9, 26.4, 25.2, 14.8. DART-HRMS (m/z) calculated for C15H23N2O3 [M+H]+ 279.1709, found 279.1721. Data matched that reported previously.46
4.2.4.7. Ethyl (3-oxo-3-((4-phenylbutyl)amino)propyl)carbamate (40)
Converted to the N-phenylbutylamine derivative using the general procedure described and with the following specifications and alterations: 35a (0.3 g, 1.71 mmol), TEA (0.57 mL, 4.11 mmol), 4-phenylbutan-1-amine (0.3 mL, 1.89 mmol), DMAP (0.021 g, 0.17 mmol), EDC-HCl (0.36 g, 1.90 mmol). Product was obtained as a white powder (yield: 0.25 g, 49%). Mp 105–106 °C. 1H NMR (500 MHz, CDCl3) δ 7.07–7.22 (m, 5H), 5.58 (br s, 1H), 5.23 (br s, 1H), 4.02 (q, J=6.9 Hz, 2H), 3.38 (q, J=6.2 Hz, 2H), 3.20 (q, 2H), 2.56 (t, J=7.6 Hz, 2H), 2.30 (t, J=5.8 Hz, 2H), 1.58 (pentet, 2H), 1.46 (pentet, 2H), 1.15 (t, J=7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 171.3, 157.0, 142.2, 128.56, 128.54, 126.0, 60.9, 39.5, 37.2, 36.4, 35.6, 29.3, 28.8, 14.8. DART-HRMS (m/z) calculated for C16H25N2O3 [M+H]+ 293.1865, found 293.1893.
4.2.4.8. Ethyl (2-oxo-2-((4-phenylbutyl)amino)ethyl)carbamate (41)
Converted to the N-phenbutylamine derivative using the general procedure described and with the following specifications and alterations: 34a (0.22 g, 1.5 mmol), TEA (0.42 mL, 3.0 mmol), 4-phenylbutan-1-amine (0.26 mL, 1.65 mmol), DMAP (0.015g, 0.15 mmol), EDC-HCl (0.32 g, 1.65 mmol). Product was obtained as a white powder (yield: 0.039 g, 9%). Mp 101–102 °C. 1H NMR (500 MHz, CDCl3) δ 7.18–7.31 (m, 5H), 6.27 (br s, 1H), 5.47 (br s, 1H), 4.15 (q, J=7.1 Hz, 2H), 3.83 (d, J=5.4 Hz, 2H), 3.30 (q, J=6.6 Hz, 2H), 2.65 (t, J=7.5 Hz, 2H), 1.67 (pentet, J=7.6 Hz, 2H), 1.56 (pentet, 2H), 1.27 (t, J=7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 169.2, 157.1, 142.2, 128.56, 128.52, 126.0, 61.6, 44.8, 39.5, 35.6, 29.2, 28.7, 14.7. DART-HRMS (m/z) calculated for C15H23N2O3 [M+H]+ 279.1709, found 279.1713.
4.2.4.9. Ethyl phenethylcarbamate (42)
Converted to the N-phenylacetamide derivative using the general procedure described and with the following alterations: phenethylamine (0.31 mL, 2.5 mmol) dissolved in CH2Cl2 (10 mL) followed by the addition of TEA (0.69 mL, 5.0 mmol) which was cooled to 0 °C. Ethyl chloroformate (0.21 mL, 2.20 mmol) added dropwise at 0 °C, stirred for 24 h at rt, and concentrated. Resulting material dissolved in CH2Cl2 and washed sequentially with 15 mL of the following solutions: 1M HCl, H2O, sat. NaHCO3, H2O, and brine. Organic layer concentrated and dissolved in EtOAc. The solution was heated followed by the addition of cold hexane to result in precipitation. Solution cooled to 0 °C and precipitate collected through filtration. Product was obtained as a white powder (yield: 0.12 g, 29%). Mp 35–36 °C. 1H NMR (500 MHz, CDCl3) δ 7.20–7.36 (m, 5H), 4.71 (br s, 1H), 4.14 (q, J=7.0 Hz, 2H), 3.46 (t, 2H), 2.84 (t, J=6.9 Hz, 2H), 1.26 (t, 3H). 13C NMR (125 MHz, CDCl3) δ 156.8, 139.0, 128.9, 128.7, 126.7, 60.9, 42.3, 36.4, 14.8. DART-HRMS (m/z) calculated for C11H16NO2 [M+H]+ 194.1181, found 194.1209.
4.3. Biological evaluation
4.3.1. HDAC enzyme assay
All compounds were screened at 1 μM for inhibition of HDAC2 at Reaction Biology (Malvern, PA) using proprietary protocols. The substrate for HDAC2 was a fluorogenic peptide from p53 residues 379–382 (RHKKAC) at 50 μM. Reference compound trichostatin A (TSA) was tested in a 10-dose IC50 with 3-fold serial dilution starting at 10 μM. TSA exhibited an IC50 value of 25.8 nM.
Synthetic SCA and SCA/SAHA hybrid were also screened at 10 μM and 5 μM for inhibition against HDAC 1–11 at BPS Bioscience (San Diego, CA) using proprietary protocols. HDAC 1–3, 6, and 10 used 10 μM HDAC substrate 3. HDAC 4–5, 7–9, and 11 used 2 μM HDAC substrate Class 2a. Reference compound of either trichostatin A (TSA) at 100 μM or SAHA at 10 μM was tested against each HDAC isozyme.
4.3.2. Cell line maintenance
4.3.2.1. Solid Tumor
Human colon cancer cells (HCT116), breast cancer cells (MCF-7), and triple negative breast cancer cells (MDA-MB-231) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). HCT116 cells were cultured in McCoy’s 5A media (Gibco) supplemented with 10% FBS (Gibco) and 1× penicillin/streptomycin (Cellgro). MCF-7 cells were cultured in DMEM media (UConn Cell Culture Facility) supplemented with 10% FBS and 1× penicillin/streptomycin. MDA-MB-231 cells were cultured in the same supplemented media as MCF-7 with addition of 1× glucose. All cells were grown in Corning Cell Culture, canted neck T75 (Fisher Scientific) in an Autoflow IR water-jacketed CO2 incubator (37 °C, 5% CO2). Experiments performed in Celltreat Tissue Culture Treated 96 well flat bottom plates. DMSO was used to prepare all drug solutions and the final DMSO concentration was not greater than 1%.
4.3.2.2. Lymphocytic Lines
Human cutaneous T lymphocyte cells (HuT-78) and human T lymphoblast cells (Molt-4) were obtained from ATCC (Manassas, VA). HuT-78 and Molt-4 were cultured as described previously at 37 °C with 5% CO2.47 HuT-78 cells were grown in Hyclone RPMI-1640 medium supplemented with 10% FBS and the addition of 1× penicillin/streptomycin. Molt-4 cells were grown in Hyclone RPMI-1640 medium supplemented with 10% fetal clone III. Human peripheral blood mononuclear cells (hPBMCs) were purified by density centrifugation from human blood that was purchased from Research Blood Components (Brighton, MA) as described previously.48 Briefly, hPBMCs were frozen in freezing media (10% DMSO, 10% FBS, 80% media) in liquid nitrogen until needed. The cells were re-suspended prior to use at 1 million per mL in RPMI-1640 supplemented with 10% heat-inactivated FBS, 1× HEPES, pyruvate, non-essential amino acids, and basal medium eagle (BME) and added to 96 well plates.
4.3.3. Cell viability assay
4.3.3.1. Solid Tumor
Cytotoxicity of santacruzamate A analogues in the HCT116, MCF-7, and MDA-MB-231 cell lines were evaluated using a standard MTS-PMS assay. Briefly, a 96 well plate was seeded at 3,000 cells per well. Before the addition of test compounds, plates were incubated at 37 °C with 5% CO2 for 24 h. Then, 1 μL of test compound was added to the well and then incubated for another 72 h. All test compounds were screened at 50 μM in HCT116 and MCF-7 cell line and at 100 μM in MDA-MB-231. Positive controls for experiments were doxorubicin at 10 μM and 5 μM for HCT116 and MDA-MB-231, respectively. SAHA at 20 μM was used as a control for the MCF-7 cell line. MTS and PMS reagents were prepared according to the protocol below and the two reagents were mixed at a ratio of 20 (MTS): 1 (PMS). 20 μL of the MTS/PMS solution was added to each well. The plate was incubated for another 3 h and then the absorbance of the wells were measured using a Synergy H1 Hybrid Reader (Biotek) at 490 nm. All experiments were repeated at least two independent times performed in triplicate.
4.3.3.2. Preparation of MTS and PMS reagents
MTS reagent (Promega Celltiter 96) was dissolved in sterile PBS (UConn Cell Culture Facility) to make a 2 mg/mL solution. Phenazine methosulfate (PMS) reagent (Sigma) was dissolved in sterile PBS to make a 0.92 mg/mL solution. MTS and PMS reagents were stored at −20 °C until needed.
4.3.3.3. Lymphocytic Lines
Cytotoxicity of the santacruzamate A analogues in the HuT-78, Molt-4, and hPBMCs was evaluated by the Quanti-Blue (QB) Cell Viability Assay Kit from BioAssay Systems (Hayward, CA). For the QB Cell Viability Assay Kit, a 96 well plate was seeded with 100 μL of cells or a 384 well plate was seeded with 50 μL of cells at a concentration of 105 cells/mL for Molt-4 and HuT-78 cells and 106 cells/mL for hPBMCs. All test compounds were screened at 50 μM. After 70 h of incubation, 10 μL of the QB detection reagent was added into each well for the final 2 h of incubation. The fluorescence of the wells was measured using a Perkin-Elmer VICTOR X plate reader at λex 530 nm and λem 590 nm. In dose response experiments, a linear regression was used to obtain IC50 values. All experiments were repeated three independent times.
4.3.4. Western blot analysis
Molt-4 cells were re-suspended at 6×106 cells/4 mL in fresh media. Cells were treated for 24 h with the indicated concentrations of compounds. Cells were lysed with ice cold lysing buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) and total protein was quantified with a BCA assay (ThermoFisher) according to manufacturer’s protocol. Equivalent masses of proteins were resolved by SDS–PAGE on 16% gels, transferred to nitrocellulose, and blotted with antibodies to acetylated histones (Acetyl-Histone H3 (K9) #9649, Acetyl-Histone H3 (K23) #8848, or Acetyl-Histone H4 (K12) #2591) from Cell Signaling Technologies (Danvers, MA). The JLA20 actin antibody was obtained from the University of Iowa Developmental Studies Hybridoma Bank and used as a loading control.
4.3.5. Cytotoxic T-lymphocyte assay
The cytotoxic T-lymphocyte (CTL) assay was performed as previously described.39 Briefly, TALL-104 CTL human leukemia cells were prepared and maintained as previously described.49 Compounds were prepared at a stock concentration of 10 mM in DMSO for a final testing concentration of 100 μM. Due to the high-throughput nature of the assay, screening was performed once with dose response performed in duplicate for active compounds. Compounds (1 μL each) were added to wells followed by addition of cell solution (100 μL/well; 2.5 × 106 cells/mL). Plates were incubated at 37 °C for 30 min prior to addition of 10 μL stimulation solution, which was comprised of experimental saline (ES) and anti-LAMP antibody, supplemented with either anti-CD3 beads (experimental wells and positive control wells), 20 μM thapsigargin [TG] and 1 μM phorbol 12-myristate 13-acetate [PMA] to generate maximal exocytosis, or non-stimulating anti-CD8 beads to serve as negative controls). Plates were subject to a secondary incubation at room temperature for 50 min in the dark with constant rotation before being fixed with 1% paraformaldehyde (PFA) and subject to flow cytometric analysis, conducted on a two-laser BD FACSCalibur at the UConn Flow Cytometry Facility. To calculate effects on exocytosis, anti-LAMP staining fluorescence intensity was measured from cells determined based on forward and side scatter to have a single bead bound. Responses were normalized to control anti-CD3 levels using the equation:
where “test” indicates responses from a test sample, and “ctrl” indicates a control sample. Using this analysis, compounds that do not affect granule exocytosis give responses of ~ 100%, compounds that inhibit exocytosis give responses < 100% and compounds that augment exocytosis give responses > 100%.
Supplementary Material
Acknowledgments
Funds for this research were provided by a Fogarty International Center (FIC) K01 International Research Scientist Development Award (IRSDA) (K01 TW008002, M.J.B., P.I.) and by an FIC supplement to the International Cooperative Biodiversity Group (ICBG) grant based in Panama (U01 TW006634, W.H. Gerwick, P.I., M.J.B., P.I. for supplement). Support was also received from an American Association of Colleges of Pharmacy grant (AG140125, A.J.W., P.I.). We acknowledge M. K. Hadden for access to cell culture facilities and Georgina Appiah-Pippim, Brendan Stewart, and Anne Sung for assistance with compound management and organization. Molecular graphics and analyses were performed with the University of California, San Francisco Chimera package, developed by the UCSF Resource for Biocomputing, Visualization, and Informatics (supported by NIGMS P41-GM103311)]
Footnotes
Supplementary materials include individual models of SAHA and santacruzamate A binding without enzyme overlay, theoretical hydrogen bonding and shuttling of SAHA and HDAC2, as well as intermediate synthesis and characterization and spectra for final compounds. This material can be found online at http://dx.doi.org/10.1016/j.bmc.2016.XYZ (to be completed upon publication).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 2.Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Nat Rev Cancer. 2015;15:201–215. doi: 10.1038/nrc3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gnyszka A, Jastrzebski Z, Flis S. Anticancer Res. 2013;33:2989–2996. [PubMed] [Google Scholar]
- 4.Yuan Y, Tang AJ, Castoreno AB, Kuo SY, Wang Q, Kuballa P, Xavier R, Shamji AF, Schreiber SL, Wagner BK. Cell Death Dis. 2013;4:e690. doi: 10.1038/cddis.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gandhi AK, Shi T, Li M, Jungnelius U, Romano A, Tabernero J, Siena S, Schafer PH, Chopra R. PLoS One. 2013;8:e80437. doi: 10.1371/journal.pone.0080437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun S, Rao NL, Venable J, Thurmond R, Karlsson L. Inflamm Allergy Drug Targets. 2007;6:223–235. doi: 10.2174/187152807783334300. [DOI] [PubMed] [Google Scholar]
- 7.Iribarren K, Bloy N, Buque A, Cremer I, Eggermont A, Fridman WH, Fucikova J, Galon J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L. Oncoimmunology. 2016;5:e1088631. doi: 10.1080/2162402X.2015.1088631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams JL, Smothers J, Srinivasan R, Hoos A. Nat Rev Drug Discov. 2015;14:603–622. doi: 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- 9.Khan O, La Thangue NB. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Dymock BW. Expert Opin Ther Pat. 2009;19:1727–1757. doi: 10.1517/13543770903393789. [DOI] [PubMed] [Google Scholar]
- 11.Johnstone RW. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 12.Conley BA, Wright JJ, Kummar S. Cancer. 2006;107:832–840. doi: 10.1002/cncr.22064. [DOI] [PubMed] [Google Scholar]
- 13.Manal M, Chandrasekar MJ, Gomathi Priya J, Nanjan MJ. Bioorg Chem. 2016;67:18–42. doi: 10.1016/j.bioorg.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Choi SH, Heo K, Byun HM, An W, Lu W, Yang AS. Nucleic Acids Res. 2011;39:104–118. doi: 10.1093/nar/gkq774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostler KR, Davis EM, Payne SL, Gosalia BB, Exposito-Cespedes J, Le Beau MM, Godley LA. Oncogene. 2007;26:5553–5563. doi: 10.1038/sj.onc.1210351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ononye SN, van Heyst M, Falcone EM, Anderson AC, Wright DL. Pharm Pat Analyst. 2012;1:207–221. doi: 10.4155/ppa.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dougan M, Dranoff G. Immunotherapy of cancer. In: Wang R-F, editor. Innate Immune Regulation and Cancer Immunotherapy. Springer-Verlag; New York: 2012. pp. 391–414. [Google Scholar]
- 18.Pavlik CM, Wong CY, Ononye S, Lopez DD, Engene N, McPhail KL, Gerwick WH, Balunas MJ. J Nat Prod. 2013;76:2026–2033. doi: 10.1021/np400198r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 20.Flipo M, Charton J, Hocine A, Dassonneville S, Deprez B, Deprez-Poulain R. J Med Chem. 2009;52:6790–6802. doi: 10.1021/jm900648x. [DOI] [PubMed] [Google Scholar]
- 21.Maligres PE, Houpis I, Rossen K, Molina A, Sager J, Upadhyay V, Wells KM, Reamer RA, Lynch JE, Askin D, Volante RP, Reider PJ, Houghton P. Tetrahedron. 1997;53:10983–10992. [Google Scholar]
- 22.Jeffrey PD, McCombie SW. J Org Chem. 1982;47:587–590. [Google Scholar]
- 23.Narasimhan C, Lai CS, Joseph J. Bioorg Chem. 1996;24:50–58. [Google Scholar]
- 24.Buchini S, Buschiazzo A, Withers SG. Ang Chem Int Ed. 2008;47:2700–2703. doi: 10.1002/anie.200705435. [DOI] [PubMed] [Google Scholar]
- 25.Ying Y, Taori K, Kim H, Hong J, Luesch H. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 26.Taori K, Paul VJ, Luesch H. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 27.McKay AF, Tarlton EJ, Petri SI, Steyermark PR, Mosley MA. J Am Chem Soc. 1958;80:1510–1517. [Google Scholar]
- 28.Zhao XQ, Chang YL, Fowler FW, Lauher JW. J Am Chem Soc. 1990;112:6627–6634. [Google Scholar]
- 29.Fridman YD, Kebets NM, Nanaeva MT, Zurdinov AZ, Sabirova TS, Atarskaya LI. Pharm Chem J. 1989;23:879–883. [Google Scholar]
- 30.Hosseini MW, Comarmond J, Lehn JM. Helv Chim Acta. 1989;72:1066–1077. [Google Scholar]
- 31.Contino C, Maurizis JC, Ollier M, Rapp M, Lacombe JM, Pucci B. Eur J Med Chem. 1998;33:809–816. [Google Scholar]
- 32.Li A-H, Moro S, Forsyth N, Melman N, Ji X-d, Jacobson KA. J Med Chem. 1999;42:706–721. doi: 10.1021/jm980550w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dow GS, Chen Y, Andrews KT, Caridha D, Gerena L, Gettayacamin M, Johnson J, Li Q, Melendez V, Obaldia N, 3rd, Tran TN, Kozikowski AP. Antimicrob Agents Chemother. 2008;52:3467–3477. doi: 10.1128/AAC.00439-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng JH, Mori Y, Rogel O, Castle SL, McAtee JJ. J Am Chem Soc. 2000;122:7416–7417. [Google Scholar]
- 35.Bisswanger H. Perspect Sci. 2014;1:41–55. [Google Scholar]
- 36.Luesch H, Moore RE, Paul VJ, Mooberry SL, Corbett TH. J Nat Prod. 2001;64:907–910. doi: 10.1021/np010049y. [DOI] [PubMed] [Google Scholar]
- 37.Luesch H, Yoshida WY, Moore RE, Paul VJ, Mooberry SL, Corbett TH. J Nat Prod. 2002;65:16–20. doi: 10.1021/np010317s. [DOI] [PubMed] [Google Scholar]
- 38.Pettit GR, Xu J-p. 6,239,104 B1. US Patent. 2001 May 29;
- 39.Zhao Z, deMayo JA, West AM, Balunas MJ, Zweifach A. J Biomol Screen. 2016 doi: 10.1177/1087057116643260. [DOI] [PubMed] [Google Scholar]
- 40.Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE, Steiner P. J Biol Chem. 2013;288:26926–26943. doi: 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanommeslaeghe K, De Proft F, Loverix S, Tourwe D, Geerlings P. Bioorg Med Chem. 2005;13:3987–3992. doi: 10.1016/j.bmc.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 42.Chen K, Xu L, Wiest O. J Org Chem. 2013;78:5051–5055. doi: 10.1021/jo400406g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurard-Levin ZA, Kim J, Mrksich M. Chembiochem. 2009;10:2159–2161. doi: 10.1002/cbic.200900417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Liu Q, Lu WH, Ma MZ, Liao JW, Ganesan A, Hu YM, Wen SJ, Huang P. RSC Adv. 2015;5:1109–1112. [Google Scholar]
- 47.Oblak EZ, VanHeyst MD, Li J, Wiemer AJ, Wright DL. J Am Chem Soc. 2014;136:4309–4315. doi: 10.1021/ja413106t. [DOI] [PubMed] [Google Scholar]
- 48.Hsiao CH, Lin X, Barney RJ, Shippy RR, Li J, Vinogradova O, Wiemer DF, Wiemer AJ. Chem Biol. 2014;21:945–954. doi: 10.1016/j.chembiol.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Z, Haynes MK, Ursu O, Edwards BS, Sklar LA, Zweifach A. J Biomol Screen. 2015;20:359–371. doi: 10.1177/1087057114557620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.