

Abstract
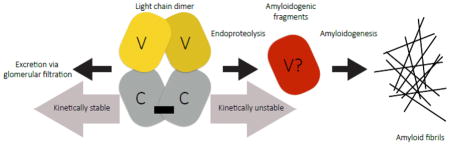
Light chain amyloidosis (AL amyloidosis) appears to be caused by the aggregation of an antibody light chain (LC) or fragment thereof, and is fatal if untreated. LCs are secreted from clonally expanded plasma cells as disulfide-linked dimers, with each monomer comprising one constant and one variable domain. The energetic contribution of each domain and the role of endoproteolysis in AL amyloidosis remain unclear. To investigate why only some LCs form amyloid and cause organ toxicity, we measured the aggregation propensity and kinetic stability of LC dimers and associated variable domains from AL amyloidosis patients and non-patients. All the variable domains studied readily form amyloid fibrils, whereas none of the full-length LC dimers, even those from AL amyloidosis patients, are amyloidogenic. Kinetic stability — that is, the free energy difference between the native state and the unfolding transition state — dictates the LC’s unfolding rate. Full-length LC dimers derived from AL amyloidosis patients unfold more rapidly than other full-length LC dimers and can be readily cleaved into their component domains by proteases, whereas non-amyloidogenic LC dimers are more kinetically stable and resistant to endoproteolysis. Our data suggest that amyloidogenic LC dimers are kinetically unstable (unfold faster) and thus are susceptible to endoproteolysis that results in the release amyloidogenic LC fragments, whereas other LCs are not as amenable to unfolding and endoproteolysis and are therefore aggregation resistant. Pharmacologic kinetic stabilization of the full-length LC dimer could be a useful strategy to treat AL amyloidosis.
Keywords: AL amyloidosis, systemic amyloidosis, amyloid fibrils, endoproteolysis, immunoglobulin
Graphical abstract
Introduction
There is strong genetic and pharmacologic evidence that the misfolding and/or aggregation of initially soluble proteins can cause dysfunction in post-mitotic tissue(s) in the systemic amyloidoses [1–3]. A relatively common systemic amyloid disease is immunoglobulin light chain amyloidosis (AL amyloidosis) [3], a fatal condition if untreated, that affects 10 patients per million per year [4]. AL amyloidosis is caused by the misfolding and misassembly of an immunoglobulin (Ig) light chain (LC) or fragment thereof, whose sequence is generally unique to each patient [5]. These LCs are secreted from neoplastic monoclonal plasma cells, often without an Ig heavy chain. Most LCs are efficiently removed from the blood by the kidneys [6, 7]. However, some LCs are amyloidogenic — they can aggregate after secretion from plasma cells to form a variety of structures including amyloid fibrils, causing proteotoxicity in organs, most commonly, the heart and kidneys. In AL amyloidosis, the population of clonal plasma cells is generally small and proteotoxicity occurs without fully-developed cancer symptoms, although amyloid deposition can also be a symptom of multiple myeloma, a more aggressive plasma cell cancer [8, 9]. The underlying cancer can often be treated with chemotherapy and/or stem cell replacement therapy, which removes the source of amyloidogenic LCs, stopping active aggregation and proteotoxicity [10]. However, many AL amyloidosis patients present with advanced cardiomyopathy, which makes it difficult for them to tolerate chemotherapy. Thus, there is an urgent need for both earlier diagnosis and therapeutic strategies that reduce LC cardiotoxicity [10, 11]. A better understanding of whether a given LC sequence will be amyloidogenic and why some are especially cardiotoxic could allow potentially dangerous LCs to be identified earlier [4].
The two classes of LCs, κ and λ, each consist of an N-terminal variable (V) Ig domain attached to a C-terminal constant (C) Ig domain; 22–25 kDa (Fig. 1a). Each Ig domain is stabilized by an intra-chain disulfide bond (Fig. 1a). The V-domains have diverse sequences used for antigen recognition, while the C-domains are much more conserved. LCs from the λ6a subclass are more commonly observed in amyloid deposits than in mature antibodies, so the presence of a λ6a clone seems to represent a risk factor for AL amyloidosis [12]. λ LCs are typically detected in the blood as disulfide-linked dimers (Fig. 1a) with a half-life of 3–6 h [13].
Fig. 1. Recombinant full-length light chains form disulfide-linked dimers.
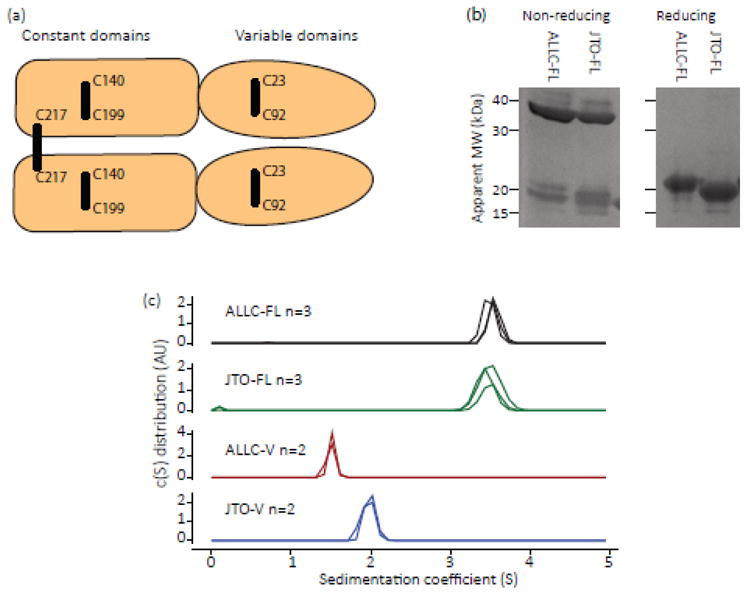
(a) Schematic diagram of a light chain homodimer, where each monomer has a variable and constant immunoglobulin domain. Each domain is stabilized by an internal disulfide bond and the two chains are linked by an inter-chain disulfide bond (shown by black lines). The cysteine residues forming each bond are indicated by their residue numbers in the germline sequence. (b) Reducing and non-reducing SDS-PAGE of ALLC-FL and JTO-FL, indicating the presence of the native inter-chain disulfide bond. Both ALLC-FL and JTO-FL have a higher apparent molecular weight (MW) when the inter-chain disulfide is reduced. Positions of MW markers are shown. (c) Sedimentation velocity analytical ultracentrifugation analysis of ALLC-FL and JTO-FL, and their respective V-domains at 20 °C, OD280 = 0.4 in PBS buffer, pH 7.4. Colors represent different LCs: ALLC-FL, black; ALLC-V, red; JTO-FL, green; JTO-V, blue. The sedimentation behavior of the full-length LCs is consistent with a dimeric quaternary structure. ALLC-V sediments more slowly than JTO-V, indicating that ALLC-V has a lower propensity to form dimers in solution.
Amyloid fibrils isolated from AL amyloidosis patients often contain LC fragments, which appear to result from endoproteolysis. Early reports identified V-domains as being the main constituent of fibrils [14], reviewed in [8, 15], but amyloid can also comprise C-domains and full-length, 2-domain LCs [8, 16–20]. It is not clear which fragment(s) play a role in the deposition or toxicity of LCs. Research to date on the mechanisms of LC amyloid formation has mainly focused on isolated V-domains [5, 21–26]. The propensity of V-domains to form amyloid generally correlates with their thermodynamic stability, suggesting that aggregation is driven by populating partially-folded states [5, 21–30]. The role of the C-domain and the amyloidogenic potential of full-length LCs have recently received more attention [28, 31–34], but to date there has been no systematic survey of how the structure and stability of full-length LCs relates to their amyloid propensity. Herein, we investigate the kinetic stability (unfolding rates) and amyloidogenicity of recombinant λ LCs. Under native-like conditions, all full-length λ LC dimers remained soluble and non-amyloidogenic, whereas both the V-domains studied formed amyloid fibrils. Full-length amyloidogenic LC dimers were less kinetically stable, and therefore more susceptible to endoproteolysis, than full-length non-amyloidogenic LC dimers. Thus, endoproteolysis of kinetically unstable LCs appears to release aggregation-prone V-domains.
Results
Full-length λ light chains do not readily form amyloid at 37°C
We recently showed [11] that the 2-domain, full-length amyloidogenic λ6a LC, ALLC, coming from an AL amyloidosis patient [35], was less stable than the analogous non-amyloidogenic λ6a LC protein JTO, originating from a multiple myeloma patient [25]. However, we did not observe amyloid formation from either full-length LC in vitro [11]. To investigate why ALLC forms amyloid fibrils in the patient, we expressed ALLC and JTO as full-length LCs (referred to as ALLC-FL and JTO-FL comprising the contiguous V- and C-domains, Fig. S1) and as isolated V-domains (referred to as ALLC-V and JTO-V). We measured the aggregation propensity of these LCs using the amyloid-binding dye thioflavin T (ThT) [36]. We used recombinant LCs, rather than urine-derived “Bence-Jones proteins”, to test the effects of specific mutations on kinetic stability in the absence of post-translational modifications other than disulfide bonds. All full-length constructs contained the λ3 constant domain encoded by the IGLC3*01 gene segment; Fig. S1.
Both full-length LCs (0.3 mg mL−1) formed disulfide-linked dimers in vitro as assessed by analytical ultracentrifugation (AUC) sedimentation velocity experiments and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), with and without disulfide reduction (Fig. 1b and 1c). Both V-domains sedimented more slowly than the full-length LCs, with ALLC-V sedimenting more slowly than JTO-V (Fig. 1c). This is consistent with JTO-V having a higher propensity than ALLC-V to form non-covalent dimers in solution, and agrees with the hypothesis that destabilization of the V-domain dimer interface is associated with amyloidogenesis [27, 30].
To investigate the aggregation of these LCs under native-like conditions, we monitored aggregation (5 μM monomer equivalent) in phosphate buffered saline (PBS, pH 7.4) at 37 °C with shaking. Both V-domains readily formed visible aggregates after 24 h at 1000 rpm in a plate shaker, which we detected by measuring turbidity at 350 nm (Fig. 2a). Notably, neither ALLC-FL nor JTO-FL showed an increase in turbidity under these conditions (Fig. 2a). To further scrutinize this apparent lack of aggregation, we quantified residual soluble LC starting material by size exclusion chromatography (SEC). After 48 h of shaking (1000 rpm) at 37 °C, insoluble material was pelleted in a microcentrifuge (16,000 g, 30 min) and the quantity and apparent molecular weight of the residual soluble LC was analyzed by analytical SEC. Both V-domains were completely depleted from solution under the aggregation conditions. In contrast, both full-length LCs remained soluble and dimeric (same retention time on the SEC column) at the starting concentration (Fig. 2b) indicating a lack of any kind of aggregation. The fluorescence spectrum of ALLC-FL after 48 h of incubation under aggregation conditions was indistinguishable from that of the starting material (Fig. 2c) after being subjected to centrifugation. The full-length LCs, therefore, remain in a native-like dimeric state under conditions where their V-domains aggregate.
Fig. 2. Full-length LCs remain soluble under conditions where V-domains aggregate.
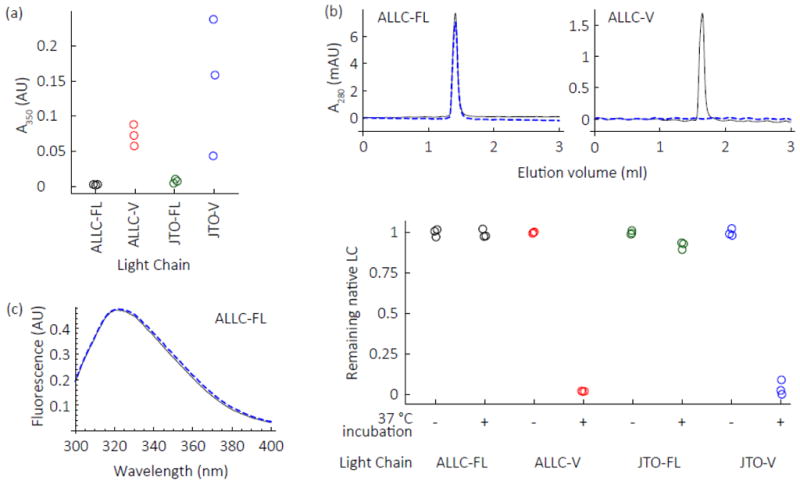
LCs (5 μM monomer equivalent) in PBS were shaken at 1000 rpm in a microwell plate at 37 C. Colors are as defined in the legend of Fig. 1. (a) After 24 h, V-domains aggregate, as determined by turbidity measurements at 350 nm, while full-length LCs do not aggregate. (b) Mass balance size exclusion chromatography (SEC) experiments (detected by absorbance at 280 nm) reveal no loss of native-like full-length ALLC or JTO dimers under aggregation conditions (48 h) that lead to variable domain aggregation. After each aggregation attempt, the sample was subjected to centrifugation at 16,000 g for 30 min, which typically precipitates aggregates. Representative chromatograms (upper panel) demonstrate that ALLC-FL retains its dimeric structure at the starting concentration after the aggregation reaction, unlike ALLC-V domains which are not detectable by SEC because they aggregate and thus are removed by the centrifugation step. Black lines represent starting concentration sample, blue dashed lines represent the sample after exposure to aggregation conditions and centrifugation. Quantitation of peak area is shown in the bottom panel. Values are normalized to the average peak area of the starting material. (c) Residual soluble ALLC-FL retains a native-like intrinsic tryptophan fluorescence spectrum (λex = 280 nm) after being subjected to aggregation conditions and centrifugation (blue dashed line), the spectrum is indistinguishable from that of the starting material (black line).
Since amyloid-like aggregates readily bind to the fluorogenic dye thioflavin T (ThT) [36, 37], we used ThT fluorescence to further characterize the differential aggregation of full-length LCs and V-domains. ALLC-V and JTO-V formed ThT-positive aggregates (λex = 440 nm, λem = 480 nm) after 24 h of shaking (250 rpm) at 37 °C, whereas neither full-length LC did so (Fig. 3a). Even after prolonged incubation (35 days in microwell plates at 1000 rpm), neither full-length LC aggregated under conditions where both V-domains formed ThT-positive aggregates in 1–2 days (Fig. 3b). The decrease in ThT fluorescence of the V-domain amyloid fibrils over the duration of the experiment is likely due to large aggregates adhering to the wells [38], increased light scattering, and/or structural annealing that alters ThT binding as the fibrils age [37]. Both V-domains aggregated similarly in PBS when monitored by ThT fluorescence in a cuvette at 37 °C with vigorous stirring, with kinetics characteristic of a nucleated polymerization (Fig. 3c) [37, 39]. The kinetics of ALLC-V could be fit to the Finke-Watzky mechanistic model [39], whereas the fits to the data for JTO-V are poor, due to the uneven baseline in the lag phase (Fig. 3c and 3d). Electron micrographs reveal that the aggregates formed by both V-domains at 37 °C exhibit a fibril morphology resembling amyloid (Fig. 4a and 4b). We were able to precipitate both ALLC-FL and JTO-FL at elevated temperature (55 °C) [11, 35], however these aggregates appear to be amorphous (Fig 4c and 4d) and do not bind to ThT [11]. Overall, ALLC-V and JTO-V aggregate in a similar manner, while ALLC-FL and JTO-FL remain similarly soluble. Therefore, it appears that some other physical chemical property may explain the differences in full-length LC pathology.
Fig. 3. Full-length light chains are resistant to aggregation in conditions where their variable domains form amyloid fibrils.
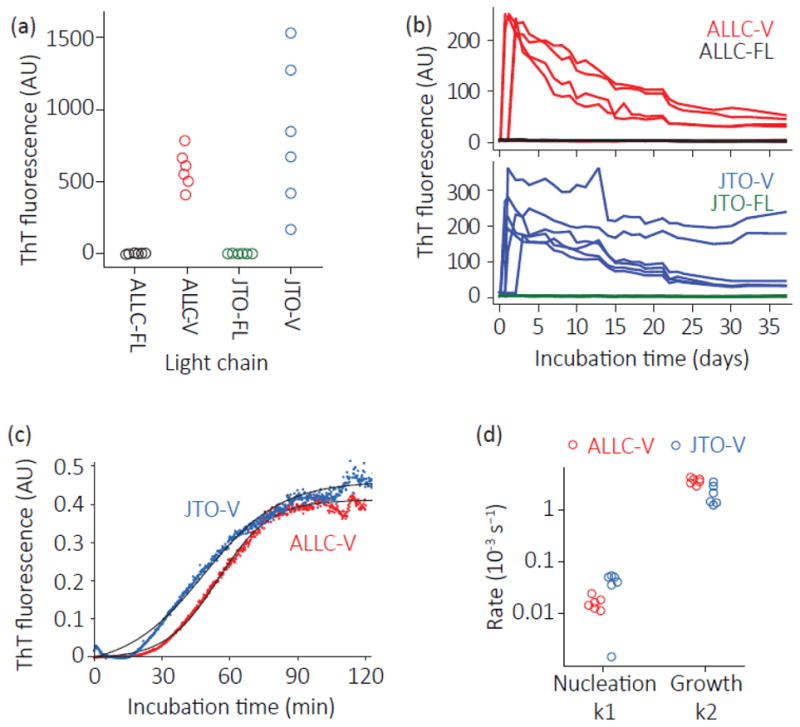
Aggregation of recombinant LCs was measured by thioflavin T (ThT) fluorescence. Colors as defined in the legend to Fig. 1. LCs (5 μM monomer equivalent) were incubated in PBS at 37 °C with agitation. (a) LCs were incubated for 24 h in microfuge tubes without ThT, after which samples were added to 10 μM ThT to detect amyloid-like aggregates (n=6). (b) Full-length and V-domains of ALLC (top) and JTO (bottom) were incubated in a microwell plate with 10 μM ThT (n=6). ThT fluorescence was measured daily. Note that only 4 of the 6 ALLC-V reactions formed amyloid. (c) Representative aggregation kinetics (n=6) of ALLC-V and JTO-V with 10 μM ThT. Reactions were vigorously stirred in a polystyrene cuvette in a fluorimeter. Black lines show fits to the Finke-Watzky model for nucleated polymerization [39]. (d) Fit parameters for ALLC-V and JTO-V aggregation (n=6), measured as in (c). ALLC-V and JTO-V aggregate with similar kinetics.
Fig. 4. LC variable domains form amyloid-like fibrils under native-like conditions, while full-length LCs form amorphous aggregates at high temperature.

Representative electron micrographs of aggregates formed by 20 μM ALLC-V (a) and 20 μM JTO-V (b) at 37 °C, 250 rpm in PBS, pH 7.4 for 48 h. Aggregates formed by ALLC-FL (c) and JTO-FL (d) at 55 °C in PBS, pH 7.4 for 96 h are amorphous. Scale bars = 0.2 μm. Both conditions result in the formation of amorphous aggregates, but only the V-domains additionally form amyloid fibrils.
Amyloidogenic LC dimers are less stable than non-amyloidogenic LC dimers
At equilibrium, proteins dynamically sample unfolded conformations, even in the absence of denaturant. Protein aggregation is driven by population of partially-folded intermediates [5, 24], access to which is limited by high-energy unfolding transition states. Less stable proteins generally have a higher equilibrium population of such intermediates (thermodynamic instability) or may access them more frequently (kinetic instability). Since thermodynamically less stable LC V-domains are known to aggregate more readily [5, 21–26], we measured the apparent thermodynamic stability of the LC V-domains and the full-length LC dimers by urea denaturation monitored by intrinsic tryptophan fluorescence (Fig. 5, λex = 280 nm, λem = 300–400 nm). Both LC V-domains unfolded and refolded reversibly from urea, as has been observed for other λ6a V-domains [21, 24, 25]. However, neither full-length, disulfide-bonded LC reversibly refolded after complete unfolding by urea. Data for ALLC-FL are shown in Figs. 5a and 5b. Similar results were observed for JTO-FL (data not shown). Both full-length LCs refolded from denaturant to a state with a non-native-like tryptophan fluorescence emission spectrum, with a red-shifted emission maximum that suggests greater solvent exposure of a tryptophan residue (Fig. 5a, dashed blue line). Urea titrations of ALLC-FL from either a folded state in 0 M urea or an unfolded state in 6 M urea show hysteresis in the plots of the average wavelength of the resulting emission spectrum (Fig. 5b). Similar irreversible folding from a heat denatured state has previously been observed for LCs [28, 31]. Therefore, for full-length LCs only an apparent equilibrium stability, indicated by the urea concentration at the unfolding midpoint, Cm, could be measured (Table 1). We previously showed that ALLC-FL is less stable than JTO-FL [11] (Fig. 5c). This difference is recapitulated in the V-domains: ALLC-V is less stable than JTO-V, although by a smaller margin (Fig. 5d and Table 1).
Fig. 5. Equilibrium urea denaturation of full-length ALLC and JTO LCs and their isolated V – domains.

(a) Intrinsic fluorescence spectra of ALLC-FL in 50 mM sodium phosphate buffer, pH 7, 25 °C. Both V- and C-domains have a tryptophan residue that packs against the intra-domain disulfide bond, quenching tryptophan fluorescence. When the each domain unfolds, the quenching is lost and the emission of the tryptophan residue increases. Folded ALLC-FL was diluted to a urea concentration of 0.6 M (black) or 6 M (grey dotted line) for the unfolding spectra. ALLC-FL unfolded in 6 M urea and equilibrated for one hour was diluted to a final urea concentration of 0.6 M and then incubated overnight for the refolded spectrum (blue dashed line). (b) Equilibrium denaturation curves for ALLC-FL starting from folded protein (black circles, unfolding) or protein unfolded in 6 M urea (blue squares, refolding). The average emission wavelength (λex = 280 nm, λem = 300-400 nm, see Materials and Methods) quantifies the emission spectra of the LCs. (c) Urea denaturation curves of ALLC-FL (black) and JTO-FL (green). Fraction folded was calculated from the average fluorescence emission wavelength (λem = 300-400 nm) measurements of the LCs, normalized to the values at 0 M and 6 M urea. Lines represent fits to a 2-state equilibrium denaturation model [59], from which the unfolding midpoint, Cm, was calculated. (d) Denaturation curves of ALLC-V (red) and JTO-V (blue), analyzed as for (c). Data for ALLC-FL and JTO-FL are from Cooley et al. [11]. Stability parameters are shown in Table 1.
Table 1. Stability parameters for all LCs analyzed at 25.
°C, pH 7. Errors indicate goodness-of-fit. Cm, urea unfolding midpoint from apparent equilibrium denaturation curves; kuH2O, the calculated unfolding rate in the absence of urea from stopped-flow unfolding measurements; mu, the urea dependence of the unfolding rate; t50, half-life of the folded state calculated from the unfolding rate. V-domain unfolding rates were calculated from only the unfolding rate data, for comparison with the full-length LCs.
| Light chain | Cm (M urea) | kuH2O (×10−3 s−1) | mu (kJ mol−1 M−1) | t50 (s) |
|---|---|---|---|---|
| ALLC-FL | 1.75 ± 0.2 | 5.44 ± 0.46 | 2.11 ± 0.036 | 184 |
| ALLC-V | 1.47 ± 0.19 | 32.7 ± 0.85 | 1.87 ± 0.01 | 30.6 |
| JTO-FL | 2.62 ± 0.15 | 0.079 ± 0.01 | 3.12 ± 0.05 | 12600 |
| JTO-V | 1.82 ± 0.13 | 26.0 ± 0.30 | 1.43 ± 0.005 | 38.4 |
| 6aJL2-FL | 2.67 ± 0.2 | 0.174 ± 0.02 | 2.85 ± 0.047 | 5740 |
| 6aJL2-FL R25G | 2.59 ± 0.14 | 0.687 ± 0.06 | 2.58 ± 0.04 | 1460 |
| WIL-FL | 1.93 ± 0.16 | 1.45 ± 0.08 | 2.18 ± 0.02 | 689 |
| WIL-FL H101Δ | 1.68 ± 0.17 | 1.51 ± 0.07 | 2.25 ± 0.02 | 661 |
| 6aJL7AL-FL | 2.4 ± 0.33 | 0.65 ± 0.05 | 2.81 ± 0.03 | 1540 |
| ALLC-FL C217S | 2.09 ± 0.37 | 22.3 ± 0.51 | 1.4 ± 0.01 | 44.9 |
| JTO-FL C217S | 2.46 ± 0.4 | 12.1 ± 0.28 | 1.54 ± 0.01 | 82.9 |
| 6aJL2-FL C217S | 2.84 ± 0.2 | 2.45 ± 0.10 | 1.88 ± 0.02 | 408 |
The free energy difference between the native state and the unfolding transition state, which we define as a protein’s kinetic stability, can be estimated in the absence of denaturant by extrapolation from the unfolding rate in the presence of denaturant. Unlike equilibrium denaturant titrations, measurement of unfolding rates does not require reversible folding [40]. Kinetic stability can be thought of as the average lifespan of a folded molecule between transient excursions to an unfolded state, described as its half-life or t50. LCs are folded in the endoplasmic reticulum of plasma cells before secretion into the blood [11], so kinetic stability may provide a useful estimate of LC stability in blood [40]. The unfolding rates of full-length LCs and their respective V-domains were measured as a function of urea concentration by stopped-flow fluorescence (Fig. 6, λex = 280 nm, λem = 350 nm). Both full-length LC and V-domain unfolding was cooperative; thus, the unfolding of the two domains in full-length LCs appears to be kinetically linked (Fig. 6). At low urea concentrations the unfolding of JTO showed a minor lag phase (Fig. 6, 5 M urea transient) which may indicate a more complex unfolding pathway, but we have not been able to characterize this further. The unfolding kinetics of other LCs fit well to a single exponential equation (Fig. 6) and the denaturant dependence of the unfolding rates is linear, which suggests that unfolding is a 2-state process under these conditions. We analyzed the LC unfolding data under the assumption of a 2-state process, although we note that the unfolding pathway may be more complex in the absence of urea. Both full length LCs unfolded more slowly than their isolated V-domains, and ALLC-FL unfolded more rapidly than JTO-FL, in agreement with their apparent thermodynamic stabilities (Fig. 6 and Table 1). Unfolding of LC V-domains is reversible [21, 24, 25, 29], so we also measured folding rates of ALLC-V and JTO-V as a function of urea concentration (Fig. 6b, λex = 280 nm, λem = 350 nm). The folding rates of ALLC-V and JTO-V were similar (Fig. 6b and Table 2), despite the 16 residues that differ between these proteins (Fig. S1), implying that stability differences between them are driven by differences in their unfolding rates, as has been observed for other single-domain proteins [41]. The overall similarity of the folding, unfolding and aggregation rates of ALLC-V and JTO-V suggests that they have similar folding free energy landscapes. The clearest measurable difference is between the kinetic stability of the full-length native dimers.
Fig. 6. Folding and unfolding rates of ALLC and JTO.

(a) Unfolding rates of ALLC-FL (black) and JTO-FL (green) as a function of urea concentration. The change in fluorescence emission at 350 nm over time was fitted to an exponential equation and the observed rate extracted. Representative transients (gray) and fits (colored as for Fig. 1) are shown on the right. In the case of JTO-FL the large amplitude rate from a double-exponential fit was used to account for the minor lag phase present at low urea concentrations. The logarithm of the rates versus urea concentration was fitted to a linear equation [59]; for JTO-FL the rate associated with the large amplitude change was used. The unfolding rate in the absence of urea, kuH2O, is the y-intercept of the fit, and the denaturant dependence of the unfolding rate, mu, is its slope. (b) Folding and unfolding kinetics of ALLCV (red) and JTO-V (blue), analyzed as for (a) except that the data were fitted to a 2-state kinetic folding model [59]. Representative transients and fits are shown on the right. Parameters are shown in Tables 1 and 2.
Table 2. Folding and unfolding rates of LC V-domains (.
Fig. 4b).kfH2O and kuH2O are the calculated folding and unfolding rates, respectively, in the absence of urea from stopped flow measurements; mf and mu, the urea dependence of the folding and unfolding rates. Errors indicate goodness-of-fit.
| Light chain | kfH2O (×10−3 s−1) | mf (kJ mol−1 M−1) | kuH2O (×10−3 s−1) | mu (kJ mol−1 M−1) |
|---|---|---|---|---|
| ALLC-V | 4060 ± 89 | −1.60 ± 0.019 | 22.5 ± 0.66 | 0.607 ± 0.0047 |
| JTO-V | 5100 ± 130 | −1.83 ± 0.024 | 25.2 ± 0.60 | 0.789 ± 0.0040 |
The >50-fold faster unfolding rate of the amyloidogenic ALLC-FL relative to the non-amyloidogenic JTO-FL suggests that unfolding rate differences (kinetic stability) could explain why only certain LCs form amyloid in vivo — those which unfold more readily. To ask whether this is a general trend, we measured the unfolding midpoints (apparent equilibrium stability) and rates of an additional five full-length LCs (Figs. 7, S1 and S2), wherein different V-domains were fused to identical C-domains. We used the λ6a germline 6aJL2 sequence, which was previously shown to have a more stable V-domain than that of JTO [42]. We made the destabilizing R25G mutation in 6aJL2, which is a germline gene polymorphism found in approximately 25% of λ6 LCs from AL amyloidosis patients [42]. This mutation was shown to reduce the thermodynamic stability of the 6aJL2 V-domain by 5.9 kJ mol−1 and increase its amyloidogenicity [42]. We also used two LC sequences from AL amyloidosis patients: WIL, which was previously compared to JTO [25], and the Genbank sequence AF490967 [12], which we refer to as 6aJL7AL since it originates from the λ 6a and JL7 gene segments. Our WIL sequence contained a histidine residue at position 101 that was not present in the original protein studied [25], which we refer to as WIL 101H. As with ALLC and JTO, we use the notation -FL for the full-length LCs. Stability data is shown in Figs. 7 and S2, and Table 1. The germline λ6a sequence, 6aJL2-FL, was substantially more kinetically stable than the amyloidogenic full-length LCs but less kinetically stable than JTO-FL (Fig. 7). The R25G mutation reduced 6aJL2-FL’s kinetic stability, consistent with its association with amyloidogenic λ6a sequences [42] (Fig. 7). Both 6aJL2-FL variants exhibited similar apparent thermodynamic stabilities relative to JTO-FL, based on Cm values (Fig. S2). The two patient-derived full-length amyloidogenic sequences (WIL-FL and 6aJL7AL-FL) had lower Cm values, and unfolded faster than JTO-FL, although neither was as kinetically unstable as ALLC-FL (Fig. 7). WIL-FL 101H had a slightly increased Cm value compared to WIL-FL (Fig. S2), but its unfolding rate was indistinguishable from that of WIL-FL (Fig. 7).
Fig. 7. Amyloidogenic full-length LCs are less stable than non-amyloidogenic full-length LCs but do not readily form amyloid fibrils.

(a) Time constants (t50, 1/kuH2O) for the unfolding of full-length LCs determined by extrapolation from unfolding rates in the presence of urea as for Fig. 5. Such t50 values represent the average lifetime of the folded state at equilibrium. Amyloidogenic LCs are shown in red, non-amyloidogenic LCs are shown in blue. Error bars represent goodness-of-fit. Data are shown in Table 1. (b) Denaturation midpoint (Cm urea) versus unfolding rate for all LCs studied. Full-length, wildtype proteins are indicated by triangles, V-domains by black circles, C217S variants by green squares and variants of 6aJL2-FL and WIL-FL by crosses. Amyloidogenic full-length LCs are colored red and non-amyloidogenic full-length LCs are colored blue. Data and fits are shown in Figs. 5, 6, S2 and S3; parameters are summarized in Table 1. (c) Light chain unfolding rates at 37 °C correlate with unfolding rates at 25 °C. The unfolding rates of LCs were measured at 37 °C in 50 mM sodium phosphate buffer, pH 7, and the unfolding rates in the absence of urea calculated. kuH2O is the calculated unfolding rates in the absence of urea from stopped flow measurements. Errors indicate goodness-of-fit, shown by the length of the bars on the plot. Fit parameters are shown in Table 3. (d) ThT fluorescence of full-length light chains incubated at 37 °C in PBS in a microplate for 30 days with shaking. Colors correspond to those in (b). None of the full-length proteins forms ThT-binding aggregates (n=6). The data for ALLC-FL, ALLC-V, JTO-FL and JTO-V are taken from the time courses shown in Fig. 3b, and the aggregation of the other light chains was measured in parallel.
LC dimer inter-chain disulfide elimination reduces kinetic stability
Since the disulfide-mediated dimerization of full-length LCs seems to be important in preventing amyloidosis, perhaps by enhancing their kinetic stability (Figs. 6 and 7), we reasoned that disrupting the dimer interface might reduce the kinetic stability of full-length LCs enough to cause them to aggregate. We therefore mutated the conserved cysteine residue in the C-domain near the C-terminus (residue 217 in the germline sequence, which forms an inter-chain disulfide bond, Fig. 1a) to serine in ALLC-FL, JTO-FL and 6aJL2-FL. Sedimentation velocity AUC analysis showed that ALLC-FL C217S sedimented more slowly than ALLC-FL, and had a broader distribution of sedimentation coefficients, consistent with it populating a monomer-dimer equilibrium (Fig. S3). JTO-FL C217S exhibited similar sedimentation properties to JTO-FL, those consistent with a prominent dimer quaternary structure (Fig. S3). These results are analogous to the data for the variable domains (Fig. 1c), suggesting that the dimer interface is weaker in ALLC-FL than in JTO-FL, which may be related to their relative stabilities. The apparent thermodynamic stabilities of the LCs were only slightly altered by this mutation (Table 1 and Figs. 7b and S4). However, all three proteins unfolded faster than their C217 variants (Table 1 and Figs. 7b and S4). The denaturant dependences of the rates and equilibrium transitions are lower for all three C217S LCs than their C217 variants (Fig. S4). This may reflect increased solvent exposure in the native state, consistent with reduced dimer interface stability.
The stability data for all of the LCs investigated is summarized in Fig. 7b. The non-amyloidogenic LCs JTO-FL and 6aJL2-FL are more stable than the LCs associated with amyloidosis. We verified that LC unfolding rates at 25°C and 37°C correlated with each other, as expected for folded proteins (Table 3 and Fig. 7c). Notably however, none of the 2-domain LCs formed amyloid after incubation for 30 days at 37 °C with shaking (Fig. 7d), indicating that even when the dimer interface is disrupted, the C-domain exhibits a striking protective effect against aggregation in the context of the full-length LC.
Table 3. Stability parameters for all LCs analyzed at 37.
°C, pH 7. Errors indicate goodness-of-fit. kuH2O, the calculated unfolding rate in the absence of urea from stopped-flow unfolding measurements; mu, the urea dependence of the unfolding rate; t50, half-life of the folded state calculated from the unfolding rate.
| Light chain | kuH2O (×10−3 s−1) | mu (kJ mol−1 M−1) | t50 (s) |
|---|---|---|---|
| ALLC-FL | 49.4 ± 2.46 | 0.582 ± 0.0085 | 20.2 |
| JTO-FL | 0.375 ± 0.02 | 1.15 ± 0.0083 | 82.4 |
| 6aJL2-FL | 2.26 ± 0.14 | 0.931 ± 0.010 | 442 |
| 6aJL2-FL R25G | 12.1 ± 0.74 | 0.781 ± 0.010 | 91.6 |
| WIL-FL 101H | 9.81 ± 0.4 | 0.744 ± 0.0067 | 2670 |
| Wil-FL | 16.1 ± 0.73 | 0.700 ± 0.0073 | 62.1 |
| 6aJL7AL-FL | 10.9 ± 0.76 | 0.788 ± 0.011 | 102 |
Kinetically unstable full-length amyloidogenic LCs are susceptible to proteolysis — a critical step in triggering the amyloidogenesis cascade?
Amyloid deposits isolated from patients often contain LC fragments, as well as full-length LCs [8, 16–20]. It is not clear whether the LCs are proteolyzed before or after amyloid formation, although amyloid formation from full-length LCs was first demonstrated after proteolysis of LCs with pepsin in vitro [43]. Since proteases generally prefer unstructured polypeptides as substrates, a protein’s rate of proteolysis is often controlled by its global or local unfolding rate [44, 45]. Our observation that full-length amyloidogenic LCs are less kinetically stable than full-length non-amyloidogenic LCs (Fig. 7), but do not form amyloid (Figs. 2, 3 and 7d), suggests that limited endoproteolysis may initiate aggregation by releasing an amyloidogenic V-domain from its protective C-domain.
We assessed the endoproteolysis of full-length LCs by measuring proteinase K sensitivity at 37 °C in PBS buffer. The extent of proteolysis after 2 h (Fig. 8a) correlated with the logarithm of the LC unfolding rates (Fig. 8b, R² = 0.93, P < 0.005, Student’s t-test), in line with the model that proteinase K can endoproteolyze LCs whenever they visit an extended unfolded conformation [44]. Strikingly, the amyloidogenic 2-domain LCs (ALLC-FL, WIL-FL, 6aJL7AL-FL and 6aJL2-FL R25G; Fig. 7 and 8a; red data points) were cleaved to a greater extent than full-length LC sequences not capable of amyloid formation in vivo (6aJL2-FL and JTO-FL; Figs. 7 and 8a; blue data points). The products visible on the SDS-PAGE gels in Fig. 8a were identified by mass spectrometry as the C-domains (Fig. S5). This is consistent with rapid digestion of the V-domain by proteinase K, either directly from the full-length LC or following cleavage of the unstructured linker between the C- and V-domains. Similarly, both ALLC-V and JTO-V were rapidly proteolyzed by proteinase K, consistent with their kinetic and thermodynamic instability (Fig. 8c).
Fig. 8. Kinetically unstable amyloidogenic LC dimers are susceptible to endoproteolysis.

LCs were incubated with proteinase K at 37 °C, pH 7.4, then quenched with phenylmethyl sulfonyl fluoride (PMSF, 5 mM). (a) SDS-PAGE gels of purified, recombinant full-length LCs (10 μM monomer equivalent) incubated with proteinase K (100 nM) for 2 h at 37 °C in PBS buffer. Quantitation is shown below (n=3). Patient-derived amyloidogenic LC sequences (ALLC-FL, WIL-FL and 6aJL7AL-FL; red) are cleaved more readily than the non-amyloidogenic JTO-FL and 6aJL2-FL (blue), while the R25G mutation in 6aJL2-FL (red) increases its proteolytic susceptibility. LCs are initially cleaved into fragments the size of an isolated Ig domain; ALLC-V is shown in the right-most lane for reference (arrow). The product was identified as the C-domain by mass spectrometry (Fig. S5). (b) Correlation between unfolding rate and extent of proteolysis at 37 °C. The mean of the measurements in (a) is shown. (c) ALLC-V and JTO-V are completely proteolyzed by 30 min incubation with proteinase K (n=3).
In accordance with their reduced kinetic stability, the C217S variants of ALLC-FL and JTO-FL were cleaved much more rapidly than the full-length disulfide-bonded LCs (Fig. 9a). ALLC-FL was more sensitive than JTO-FL to digestion by trypsin in PBS at 37 °C (Fig. 9b), consistent with the hypothesis that the relative endoproteolysis rate is limited by the unfolding rate, rather than protease activity [44, 45]. Endoproteolysis by pepsin at pH 3, however, as historically used to generate amyloid fibrils from full-length LCs [43], digested full-length LCs to similar extents (Fig. 9c). Even the highly stable JTO-FL (Fig. 6) was digested (Fig. 9c), suggesting that acidic conditions disrupt the tertiary and quaternary structure of LC dimers. These results also seem to explain the finding that pepsin treatment of LCs does not always result in amyloidogenesis [46], which we propose may be due to overproteolysis.
Fig. 9. Proteolysis of LCs depends on solution conditions.

LCs were incubated with either proteinase K, trypsin, or pepsin at 37 °C at the pH shown, quenched with PMSF (5 mM) or by increasing the pH, and analyzed by SDS-PAGE. Representative gels (n=3) are shown. Quantitation of the remaining intact LCs is shown. The lines show the averaged time courses, and each point represents a single replicate. Black, ALLC-FL; orange, ALLC-FL C217S; green, JTO-FL; purple, JTO-FL C217S. (a) SDS-PAGE gels of full-length LCs (10 μM) incubated with 100 nM proteinase K as a function of time at 37 °C in PBS buffer (pH 7.4). (b) In PBS buffer, ALLC-FL is proteolyzed by trypsin to a greater extent than JTO-FL after 4 h. (c) At pH 3, JTO-FL is proteolyzed by pepsin more rapidly than ALLC-FL, indicating disruption of the native state under these conditions.
Incubation of ALLC-FL after endoproteolysis, under conditions where ALLC-V aggregates, did not result in amyloidogenesis — suggesting that cleavage in the right place in the full length sequence is critical (Fig. 10a). Furthermore, overproteolysis may be a factor as ALLC-V is completely proteolyzed by a 30-min incubation with proteinase K (Fig. 8c). Incubation of LC proteinase K fragments at pH 4, which can accelerate aggregation [24, 31, 46], did not lead to fibril formation either (Fig. 10a). To further explore the hypothesis that kinetic instability and cleavage of full-length LCs at the proper sequence location are both needed for amyloidogenesis, we engineered a thrombin cleavage site between the two domains of ALLC-FL and JTO-FL, to make proteins referred to as ALLC-Th-FL and JTO-Th-FL. The λ LC hexapeptide subsequence, 113GQPKAA118, which forms the junction between the linker region and the C-domain (Fig. S1), is similar to the thrombin consensus sequence, LVPRGS. Insertion of the thrombin cleavage site in ALLC-FL and JTO-FL seems to destabilize both LCs since its incorporation increases the susceptibility of both LCs to proteinase K endoproteolysis (Fig. 10b), although ALLC-Th-FL is less stable than JTO-Th-FL. Incubation of ALLC-Th-FL and JTO-Th-FL with thrombin resulted in cleavage of ALLC-Th-FL to smaller species, while less cleavage of JTO-Th-FL was observed (Fig. 10c). This suggests that endoproteolysis of ALLC-Th-FL and JTO-Th-FL depends on the kinetic stability of the LC. Mass spectrometry analysis of the products of thrombin digestion of ALLC-Th-FL showed fragments with masses consistent with both the predicted V-domain and C-domain products (Fig. S5). Incubation of ALLC-FL and JTO-FL with thrombin for 16 h at 37 °C did not result in endoproteolysis (Fig. 10c). Incubation of ALLC-Th-FL with thrombin at pH 7.4, 37 °C with agitation did not result in amyloid formation. However, we were able to induce ALLC-Th-FL to form amyloid by digestion with thrombin, followed by incubation at pH 4, 37 °C with shaking for 24 h (Fig. 10d). Notably, the same treatment did not cause JTO-Th-FL, ALLC-FL or JTO-FL to aggregate after digestion with thrombin (Fig. 10d).
Fig. 10. LC fragments released by proteolysis can form amyloid fibrils under acidic conditions.

(a) ALLC-FL and ALLC-V (10 μM monomer equivalent each) were incubated with (red) or without (black) proteinase K (100 nM) for 2 h, before the proteolysis reaction was quenched with PMSF (1 mM) and the resulting polypeptides incubated at pH 4 or pH 7.4 for 24 h at 37 °C with agitation. Samples were added to ThT (10 μM) for fluorescence measurements. (b) Insertion of a thrombin consensus sequence, LVPRGS, into the LC constant domain makes both ALLC-FL and JTO-FL more susceptible to cleavage by proteinase K. Light chains (10 μM) were incubated with proteinase K (100 nM) at 37 °C for 30 min in PBS (pH 7.4), and the digestion products visualized by SDS-PAGE. (c) SDS-PAGE gel of ALLC and JTO variants (10 μM) incubated with thrombin (100 nM) for 16 h at 37 °C in PBS buffer. ALLC-Th-FL is more readily cleaved by thrombin than JTO-Th-FL. (d) 10 μM ALLC-FL, JTO-FL (n=3), ALLC-Th-FL and JTO-Th-FL (n=9) were incubated with (red) or without (black) thrombin (100 nM) for 16 h. The proteolysis reaction was quenched with PMSF (1 mM) and the resulting polypeptides diluted 2-fold with sodium phosphate/citric acid buffer at pH 4 then incubated for 24 h at 37 °C with agitation. Samples were added to ThT (10 μM) for fluorescence measurements. Fragments of ALLC-Th-FL form amyloid fibrils following such treatment, while fragments of JTO-Th-FL do not.
Discussion
The results presented above are consistent with the hypothesis that the propensity of full length LC dimers to form amyloid is influenced by at least two factors: the kinetic stability of the LC dimer and endoproteolytic release of fragments from the dimer that are themselves amyloidogenic.
Although full-length LCs aggregate at high temperature [11, 33, 35, 47], they do not readily do so at physiological temperature and pH, conditions under which V-domains will aggregate (Figs. 2, 3 and 7d). Contrary to our initial expectations, full-length LCs do not readily form either amyloid-like (Fig. 3) or amorphous (Fig. 2) aggregates under the native-like aggregation conditions explored herein. Slow aggregation kinetics, often requiring native state destabilization, have been observed in other full-length LCs [31, 35, 37]. Full-length amyloidogenic LCs unfold faster than non-amyloidogenic LCs (Fig. 6), but this instability is not by itself sufficient to cause amyloid formation (Figs. 2 and 6d), even when a LC is destabilized by a Cys-to-Ser mutation. We have been unable to find native-like conditions where full-length LCs aggregate. This suggests that the C-terminal domains of LCs prevent aggregation of their attached V-domains, through a mechanism that remains to be determined.
In addition to LC dimer unfolding, endoproteolysis by the appropriate protease, one that affords an amyloidogenic sequence, appears to be required to trigger LC amyloidogenesis (Fig. 10). The kinetic stability of a LC appears to define its resistance to endoproteolysis under native-like conditions in vitro (Figs. 8 and 9). The diversity of light chain fragments found in patients [8] suggests that several proteases may contribute to endoproteolysis-triggered LC amyloidogenesis, and indeed there are many candidate proteases in the secretory pathway and the extracellular space. In fact, different LC sequences may be susceptible to endoproteolysis by different proteases when the LC dimer is sufficiently kinetically unstable, producing distinct peptide sequences with varying amyloidogenicity. This may include C-domains or fragments thereof, which have been found as the main constituent of some amyloid deposits [16, 19, 34]. Although cleavage by proteinase K does not yield amyloidogenic fragments, we suggest that proteolysis by other proteases could do so, a process that is rate limited by the LC unfolding rate [44]. Insertion of a thrombin cleavage site into LCs demonstrates the principle that impaired kinetic stability dependent endoproteolysis can generate amyloidogenic fragments, although we do not understand why these fragments are less amyloidogenic than recombinant V-domains. Recent work by Nokwe and coworkers suggests that a C-terminal arginine residue (as results from thrombin cleavage of ALLC-Th-FL) stabilizes a LC V-domain and reduces its amyloidogenicity [48], highlighting the sensitivity of amyloidogenesis to the precise sequence of the aggregating LC, as well as its kinetic stability. Post-translational modifications could also play a role. For example, glycosylation of κ light chains is commonly observed in amyloid deposits, but rarely in mature antibodies [16, 49], perhaps rendering full-length LCs less kinetically stable and more prone to endoproteolysis. LC dimers do not efficiently refold from a chaotrope-denatured state; hence, the refolded state could be susceptible to endoproteolysis, although we did not experimentally test this hypothesis herein. It is established that extracellular matrix components hasten the aggregation of amyloidogenic proteins [50, 51], a variable that merits further exploration, although we have not observed such an effect in our experiments to date (data not shown). It is also possible that amyloid fibrils composed of LC fragments can seed or accelerate the aggregation of full-length LCs, although we have not observed this in vitro with these LCs (data not shown).
The finding that full-length amyloidogenic LC dimers are kinetically unstable relative to non-amyloidogenic LC dimers has implications for treating AL amyloidosis. Since destabilizing C-domain dimerization increases the unfolding rate of the LC and reduces the resistance of the whole protein to endoproteolysis, it is possible that stabilizing this interface with an antibody or pharmacological chaperone could make full-length LCs less prone to endoproteolysis, and hence less amyloidogenic. A similar kinetic stabilization approach is currently being used to treat the transthyretin amyloidoses [2, 52].
Materials and Methods
Protein sequences
All gene sequences were synthesized by IDT (Coralville, IA) with the exception of the V-domain of WIL, which was generously provided by Dr. Steve Bourgault of Université du Québec à Montréal. We previously measured the stabilities of ALLC-FL and JTO-FL [11]. The V-domains of JTO, WIL, 6aJL2 and 6aJL2 R25G have been studied previously [25, 42]. The sequence of 6aJL7AL was chosen from a study of sequence determinants of amyloidogenicity [12], and the gene was synthesized from the Genbank entry AF490967. Full-length light chain genes included the human CL3 C-domain sequence, and were cloned into a construct derived from pDEST-14 (Thermo Fisher, Carlsbad, CA), under the control of a T7 promoter. V-domains were cloned into pET-22 (EMD Millipore, Billerica, MA), fused to the E. coli pelB leader sequence. Mutagenesis was carried out using either QuikChange (Agilent, Santa Clara, CA) or Q5 Site-Directed Mutagenesis (NEB, Ipswich, MA) according to the manufacturers’ instructions.
Protein expression and purification
Full-length LCs were expressed as inclusion bodies in E. coli, as described previously [11, 32]. BL21 (DE3) E. coli were grown in ZYP-5052 autoinduction media [53] containing glucose, lactose and ampicillin for 24 h at 37 °C, 250 rpm. Cells were harvested by centrifugation and frozen until purification, typically overnight. Pellets were thawed, resuspended in phosphate buffered saline (PBS: 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) containing 1 mM phenylmethyl sulfuryl fluoride (PMSF) and lysed by sonication on ice. DNase I (Roche, Indianapolis, IN) was added to the lysate, and inclusion bodies were collected by centrifugation. Inclusion bodies were resuspended and sonicated three times in 1% (v/v) Triton-X in PBS, then once in PBS. The proteins were dissolved in 4 M guanidine hydrochloride containing 5 mM dithiothreitol and incubated at 4°C for 2 h. Insoluble material was removed by centrifugation, then LCs were refolded by dropwise dilution into 50 mM Tris-Cl (pH 8.5 on ice) containing 5 mM reduced glutathione and 0.5 mM oxidized glutathione. LCs were stirred overnight on ice, then (NH4)2SO4 was added to 25% saturation and allowed to equilibrate. Insoluble material was removed by centrifugation. Further (NH4)2SO4 was added to reach 75% saturation, which caused the LCs to precipitate. Insoluble material was collected by centrifugation and resuspended in 25 mM Tris-Cl pH 8.5 at 4 °C and dialyzed overnight against the same buffer. Any precipitate was removed by centrifugation, and the solution was filtered before being loaded onto a Source 15Q column (GE, Pittsburgh, PA) at pH 8.5, eluting with a 0–0.5 M NaCl gradient. Fractions containing LCs were pooled, concentrated, and further purified by size exclusion chromatography on a Superdex 75 column (GE) in PBS at 4°C. All LCs eluted at a volume consistent with a dimer of around 45 kDa. LC purity was assessed by SDS-PAGE. LC solutions were filter sterilized and stored at 4°C, and dialyzed into appropriate buffers for further analysis. Concentrations were measured by absorbance at 280 nm, using extinction coefficients calculated by the ProtParam website [54].
LC V-domains were expressed as fusions with the pelB leader sequence, which directs proteins to the E. coli periplasm. Transformed E. coli BL21 (DE3) cells were grown to an OD600 of 1 in ZYP-glucose media [53] containing ampicillin at 37°C, 250 rpm, then cooled to 20 °C and induced for 4 h at 20°C, 100 rpm by addition of 0.1 mM isopropyl c-D-1-thiogalactopyranoside. Cells were harvested by centrifugation. Periplasmic extracts were prepared by resuspending cells on ice in 0.2 M Tris pH 8.5, 0.5 M sucrose and 1 mM EDTA, shaking at 100 rpm on ice for 30 min, then diluting 3-fold with ice-cold water [55]. Cell debris was removed by centrifugation, and the LC V-domains purified by 25–75% (NH4)2SO4 precipitations, ion exchange and size exclusion chromatography as described for the full-length proteins.
Analytical ultracentrifugation
Sedimentation velocity measurements were carried out on a Beckman Optima XL-I ultracentrifuge using an AN 60 Ti rotor. LCs were diluted to an OD280 of 0.4 in PBS (approximately 13 μM monomer equivalent for full-length LCs, and 30 μM for V-domains), and PBS was used as a buffer blank. Samples were equilibrated to 20°C at 3,000 rpm before acceleration to 50,000 rpm. 500 absorbance scans at 280 nm were recorded for each sample over the 40-hour run. Data were processed using Sedfit software [56]. Absorbance traces were fitted to a continuous distribution c(S) model to produce the sedimentation coefficient distributions shown.
Aggregation reactions
All amyloid formation assays were carried out in PBS at 37 °C, with 5 or 20 μM (monomer equivalent) LC. Thioflavin T (ThT, Sigma, St. Louis, MO) was made up in ethanol to a stock concentration of 5 mM, then diluted in PBS to the desired concentration and filtered before use. Final ethanol concentrations were 0.2–0.4% (v/v). Fluorescence was measured in an Aviv ATF 105 spectrofluorimeter (Lakewood, NJ) in disposable polystyrene cuvettes, or a Spectramax Gemini EM plate reader (Molecular Devices, Sunnyvale, CA) using clear-bottom, black-wall, 96-well microwell plates (Corning, Tewksbury, MA, item #3631). Excitation and emission wavelengths were 440 nm and 480 nm, respectively. For endpoint reactions, 100 μl of protein was shaken at 250 rpm in a V-profile 2 mL microcentrifuge tube (Axygen, Union City, CA, item # 311-10-081), and samples were added to 10 μM ThT solutions in PBS for measurement. After subtraction of buffer fluorescence, LCs that had formed amyloid typically showed an increase in sample fluorescence between 5-fold and 100-fold compared to the native protein, although the precise relationship between the amount of fibrils in a sample and the resulting ThT fluorescence is not clear. The presence of non-amyloid aggregates was assessed by measuring turbidity at 350 nm in a spectrophotometer (Nanodrop 2000c using a quartz cuvette) after 24 h incubation at 37 °C in PBS. Amorphous LC aggregates were formed by incubating LCs at 55 °C in PBS for 96 h [35].
For V-domain aggregation kinetics, 5 μM protein was stirred in PBS containing 10 μM ThT in a polystyrene cuvette. Fluorescence readings were taken every 5 s. Data were normalized and fitted to the Finke-Watzky model [39]:
Where for the reaction A → B, A0 is the initial concentration of A, k1 is the initiation rate and k2 is the elongation rate.
Long-term aggregation reactions were carried out in microwell plates sealed with Crystal Clear tape (Hampton). LCs (5 μM) were incubated with 10 μM ThT on a plate shaker at 37 °C, 1000 rpm. Fluorescence was measured daily. Aggregation at pH 4 was carried out in 20 mM sodium phosphate/citric acid buffer.
Mass balance measurements
After aggregation, samples were centrifuged at 16,000 g in a microfuge for 30 min. 5 μL of supernatant was injected onto a BEH200 SEC column (Waters, Milford, MA) on a Waters Acquity H-Class Bio-UPLC (ultra performance liquid chromatography) instrument. The column was equilibrated and run at 0.2 mL min−1 in 10 mM sodium phosphate buffer, pH 7.6, containing 100mM KCl and 1mM EDTA. Protein detection was by absorbance at 280 nm.
Electron microscopy
Electron micrographs were recorded by the Core Microscopy Facility at The Scripps Research Institute. Copper grids (carbon coated 400 mesh (Electron Microscopy Sciences, Hatfield, PA) were glow discharged and inverted on a 7 μL aliquot of sample for 2 min. Excess sample was removed and the grids immediately placed on a droplet of 2% uranyl acetate solution or phosphotungstic acid for 2 min. Excess stain was removed and the grid allowed to dry thoroughly. Grids were then examined on a Philips CM100 electron microscope (FEI, Hillsbrough OR) at 80kv and images collected using a Megaview G2 ccd camera (ResAlta Research Technologies, Golden, Co).
Equilibrium unfolding experiments
LCs were incubated overnight at 25°C in 50 mM sodium phosphate buffer, pH 7, containing varying urea concentrations. Protein concentration was around 0.1 mg mL−1. Intrinsic tryptophan fluorescence was measured on an Aviv ATF 105 spectrofluorimeter at 25°C, using an excitation wavelength of 280 nm and recording emission spectra between 300 and 400 nm. To assess spectral changes as a function of urea concentration, we calculated average wavelength, <λ> [57]:
where λi and Ii are the wavelength and intensity at i, respectively. To compare light chains, we calculated the folded fraction of each protein:
where <λ>x is the average wavelength at urea concentration x. The data was fit to a 2-state equilibrium unfolding equation [58] from which Cm values were calculated:
where x is the urea concentration (M), G is the free energy of unfolding (kJ mol−1), m is the denaturant dependence of the unfolding reaction (kJ mol−1 M−1), R is the ideal gas constant, T is the temperature in K, a and c are the fluorescence of the folded and unfolded states, and b and d are the denaturant dependence of the fluorescence of the folded and unfolded states, respectively. Cm, the midpoint concentration, is:
Kinetic unfolding and folding experiments
LCs in 50 mM sodium phosphate buffer, pH 7 at 25 °C were rapidly mixed with varying concentrations of urea in the same buffer using an Aviv ATF 105 spectrofluorimeter fitted with a stopped flow mixing system. To measure folding rates, V-domains were equilibrated in 6 M urea overnight at 25 °C before experiments. Stock urea concentrations in the instrument’s reservoirs were calculated from their measured diffractive indices. The final urea concentrations were controlled by varying the protein-to-diluent ratio injected into the stopped flow mixer. The instrument’s dead time was around 100 ms. Fluorescence excitation and emission wavelengths were 280 nm and 350 nm, respectively. Kinetic transients were fitted to an exponential function with a correction for photobleaching. JTO-FL showed low-amplitude decrease in fluorescence at the lower range of urea concentrations which preceded the larger-amplitude increase in fluorescence corresponding to Ig domain unfolding. Data for JTO-FL were therefore fitted to an exponential function with two phases, and only the rate which corresponded to the major phase (which typically had an amplitude 10-fold greater than that of the minor phase) was analyzed further. The natural logarithm of the observed rate as a function of urea concentration was fitted to a linear model:
Where ku is the unfolding rate, kuH2O is the unfolding rate in the absence of urea, mu is the denaturant dependence of the unfolding rate, R is the ideal gas constant and T is the temperature in K. The errors on the fit parameters are reported in Table 1. To generate the “chevron plots” shown in Fig. 6b, folding kinetic data measured for the V-domains were fit to a 2-state folding model [59]:
Where kobs is the observed folding or unfolding rate, ku is the unfolding rate, kuH2O is the unfolding rate in the absence of urea, mu is the denaturant dependence of the unfolding rate, kf is the folding rate, kfH20 is the folding rate in the absence of urea and mf is the denaturant dependence of the folding rate.
Endoproteolysis
LCs (10 μM monomer equivalent) were incubated with 100 nM proteinase K (Fermentas), trypsin (Sigma) or thrombin (EMD Millipore) at 37°C in PBS. Reactions were quenched by addition of 5 mM PMSF and cooling on ice. For pepsin proteolysis, 10 μM LCs were incubated with 100 nM pepsin (Protea Biosciences, Morgantown, WV) in 20 mM sodium phosphate/citric acid buffer at pH 3, 37°C. Reactions were quenched by adding 10% (v/v) 1 M Tris-Cl pH 8. The extent of proteolysis was measured by electrophoresing the proteins on 15% SDS PAGE gels and quantifying the intact LC by reaction with 2,2,2-trichloroethanol to give a fluorescent product [60], or by Coomassie staining. Gels were visualized on a Bio-Rad Chemidoc MP. Band intensity was quantified using ImageJ [61] or Bio-Rad Image Lab software. Apparent molecular weights are indicated based on the positions of Invitrogen SeeBlue Plus2 (Thermo, Carlsbad, CA) or Precision Plus (BioRad, Hercules, CA) molecular weight standards.
Mass Spectrometry
The products of thrombin digestion (Fig. 10c) were identified by liquid chromatography (LC) and mass spectrometry (MS) following unfolding in 6 M urea and reduction in 5 mM DTT. The peaks identified correspond to adducts of the peptide fragments expected from a single cleavage event between residues 114 and 115. Similar treatment of the proteinase K proteolysis products (Fig. 8a) showed a complex mixture of species that we were unable to unambiguously identify. The products were therefore separated from the intact protein by SDS-PAGE and excised from the gel. The gel band was destained, reduced with 10 mM DTT, alkylated with 55 mM iodoacetamide and digested overnight with trypsin. The resulting peptides were analyzed by nano-LC-MS/MS and compared to the sequence of ALLC-FL. Mass spectrometry was carried out by the Center for Metabolomics and Mass Spectrometry at the Scripps Research Institute, La Jolla, CA, USA.
Aggregation after thrombin proteolysis
LC (10 μM) was incubated with thrombin in PBS at 37 °C for 16 h, then diluted 2-fold with 200 mM sodium phosphate/citric acid buffer at pH 4. Samples were shaken for 24 h at 37 °C, 250 rpm in 2 mL microcentrifuge tubes, and ThT fluorescence measured in microwell plates as above.
Supplementary Material
Highlights.
Aggregation of antibody light chains (LCs) is associated with AL amyloidosis
Full-length LCs are much less amyloidogenic than their variable domains
LCs from AL amyloidosis patients are less kinetically stable than other LCs
Amyloidogenesis may be initiated by endoproteolysis of kinetically unstable LCs
Proteolytic fragments of LCs can form amyloid fibrils
Acknowledgments
This work was supported by NIH grant DK46335 (JWK), the Skaggs Institute for Chemical Biology and the Lita Annenberg Hazen Foundation. Steve Bourgault of the Université du Québec à Montréal generously provided the WIL expression plasmid. We thank Joel Buxbaum, Luke Wiseman, Zina Polonskaya, Colleen Fearns and members of the Kelly laboratory for helpful discussions and critical reading of the manuscript.
Abbreviations
- AUC
analytical ultracentrifugation
- Ig
immunoglobulin
- LC
light chain
- PBS
phosphate buffered saline
- PMSF
phenylmethyl sulfonyl fluoride
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
- ThT
thioflavin T
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blancas-Mejia LM, Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem. 2013;82:745–74. doi: 10.1146/annurev-biochem-072611-130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21:221–4. doi: 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- 4.Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121:5124–30. doi: 10.1182/blood-2013-01-453001. [DOI] [PubMed] [Google Scholar]
- 5.Ramirez-Alvarado M. Amyloid formation in light chain amyloidosis. Curr Top Med Chem. 2012;12:2523–33. doi: 10.2174/1568026611212220007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzmann JA, Clark RJ, Abraham RS, Bryant S, Lymp JF, Bradwell AR, et al. Serum reference intervals and diagnostic ranges for free kappa and free lambda immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem. 2002;48:1437–44. [PubMed] [Google Scholar]
- 7.Solomon A, Waldmann TA, Fahey JL, McFarlane AS. Metabolism of Bence Jones Proteins. J Clin Invest. 1964;43:103–17. doi: 10.1172/JCI104884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buxbaum J. Mechanisms of disease: monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease. Hematol Oncol Clin North Am. 1992;6:323–46. [PubMed] [Google Scholar]
- 9.Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346:564–9. doi: 10.1056/NEJMoa01133202. [DOI] [PubMed] [Google Scholar]
- 10.Chaulagain CP, Comenzo RL. New insights and modern treatment of AL amyloidosis. Curr Hematol Malig Rep. 2013;8:291–8. doi: 10.1007/s11899-013-0175-0. [DOI] [PubMed] [Google Scholar]
- 11.Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, et al. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci USA. 2014;111:13046–51. doi: 10.1073/pnas.1406050111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abraham RS, Geyer SM, Price-Troska TL, Allmer C, Kyle RA, Gertz MA, et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL) Blood. 2003;101:3801–8. doi: 10.1182/blood-2002-09-2707. [DOI] [PubMed] [Google Scholar]
- 13.Waldmann TA, Strober W, Mogielnicki RP. The renal handling of low molecular weight proteins. II. Disorders of serum protein catabolism in patients with tubular proteinuria, the nephrotic syndrome, or uremia. J Clin Invest. 1972;51:2162–74. doi: 10.1172/JCI107023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glenner GG, Harbaugh J, Ohma JI, Harada M, Cuatrecasas P. An amyloid protein: the amino-terminal variable fragment of an immunoglobulin light chain. Biochem Biophys Res Commun. 1970;41:1287–9. doi: 10.1016/0006-291x(70)90227-5. [DOI] [PubMed] [Google Scholar]
- 15.Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts) N Engl J Med. 1980;302:1283–92. doi: 10.1056/NEJM198006053022305. [DOI] [PubMed] [Google Scholar]
- 16.Engvig JP, Olsen KE, Gislefoss RE, Sletten K, Wahlstrom O, Westermark P. Constant region of a kappa III immunoglobulin light chain as a major AL-amyloid protein. Scand J Immunol. 1998;48:92–8. doi: 10.1046/j.1365-3083.1998.00352.x. [DOI] [PubMed] [Google Scholar]
- 17.Lavatelli F, Perlman DH, Spencer B, Prokaeva T, McComb ME, Theberge R, et al. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. Mol Cell Proteomics. 2008;7:1570–83. doi: 10.1074/mcp.M700545-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen KE, Sletten K, Westermark P. Fragments of the constant region of immunoglobulin light chains are constituents of AL-amyloid proteins. Biochem Biophys Res Commun. 1998;251:642–7. doi: 10.1006/bbrc.1998.9508. [DOI] [PubMed] [Google Scholar]
- 19.Solomon A, Weiss DT, Murphy CL, Hrncic R, Wall JS, Schell M. Light chain-associated amyloid deposits comprised of a novel kappa constant domain. Proc Natl Acad Sci USA. 1998;95:9547–51. doi: 10.1073/pnas.95.16.9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–9. doi: 10.1182/blood-2009-07-230722. [DOI] [PubMed] [Google Scholar]
- 21.Blancas-Mejia LM, Tischer A, Thompson JR, Tai J, Wang L, Auton M, et al. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J Mol Biol. 2014;426:347–61. doi: 10.1016/j.jmb.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feige MJ, Groscurth S, Marcinowski M, Yew ZT, Truffault V, Paci E, et al. The structure of a folding intermediate provides insight into differences in immunoglobulin amyloidogenicity. Proc Natl Acad Sci USA. 2008;105:13373–8. doi: 10.1073/pnas.0802809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helms LR, Wetzel R. Specificity of abnormal assembly in immunoglobulin light chain deposition disease and amyloidosis. J Mol Biol. 1996;257:77–86. doi: 10.1006/jmbi.1996.0148. [DOI] [PubMed] [Google Scholar]
- 24.Hurle MR, Helms LR, Li L, Chan W, Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc Natl Acad Sci USA. 1994;91:5446–50. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, Solomon A. Thermodynamic instability of human λ6 light chains: correlation with fibrillogenicity. Biochemistry. 1999;38:14101–8. doi: 10.1021/bi991131j. [DOI] [PubMed] [Google Scholar]
- 26.Wall JS, Gupta V, Wilkerson M, Schell M, Loris R, Adams P, et al. Structural basis of light chain amyloidogenicity: comparison of the thermodynamic properties, fibrillogenic potential and tertiary structural features of four Vlambda6 proteins. J Mol Recognit. 2004;17:323–31. doi: 10.1002/jmr.681. [DOI] [PubMed] [Google Scholar]
- 27.Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, Thompson JR. Altered dimer interface decreases stability in an amyloidogenic protein. J Biol Chem. 2008;283:15853–60. doi: 10.1074/jbc.M705347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klimtchuk ES, Gursky O, Patel RS, Laporte KL, Connors LH, Skinner M, et al. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry. 2010;49:9848–57. doi: 10.1021/bi101351c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blancas-Mejia LM, Tellez LA, del Pozo-Yauner L, Becerril B, Sanchez-Ruiz JM, Fernandez-Velasco DA. Thermodynamic and kinetic characterization of a germ line human lambda6 light-chain protein: the relation between unfolding and fibrillogenesis. J Mol Biol. 2009;386:1153–66. doi: 10.1016/j.jmb.2008.12.069. [DOI] [PubMed] [Google Scholar]
- 30.Wolwertz ML, Nguyen PT, Quittot N, Bourgault S. Probing the role of lambda6 immunoglobulin light chain dimerization in amyloid formation. Biochim Biophys Acta. 2016;1864:409–18. doi: 10.1016/j.bbapap.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Blancas-Mejia LM, Horn TJ, Marin-Argany M, Auton M, Tischer A, Ramirez-Alvarado M. Thermodynamic and fibril formation studies of full length immunoglobulin light chain AL-09 and its germline protein using scan rate dependent thermal unfolding. Biophys Chem. 2015;207:13–20. doi: 10.1016/j.bpc.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rognoni P, Lavatelli F, Casarini S, Palladini G, Verga L, Pedrazzoli P, et al. A strategy for synthesis of pathogenic human immunoglobulin free light chains in E. coli. PLoS One. 2013;8:e76022. doi: 10.1371/journal.pone.0076022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sikkink LA, Ramirez-Alvarado M. Biochemical and aggregation analysis of Bence Jones proteins from different light chain diseases. Amyloid. 2008;15:29–39. doi: 10.1080/13506120701815324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto K, Yagi H, Lee YH, Kardos J, Hagihara Y, Naiki H, et al. The amyloid fibrils of the constant domain of immunoglobulin light chain. FEBS Lett. 2010;584:3348–53. doi: 10.1016/j.febslet.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 35.Arendt BK, Ramirez-Alvarado M, Sikkink LA, Keats JJ, Ahmann GJ, Dispenzieri A, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC–2. Blood. 2008;112:1931–41. doi: 10.1182/blood-2008-03-143040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LeVine H., 3rd Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–84. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 37.Wall J, Murphy CL, Solomon A. In vitro immunoglobulin light chain fibrillogenesis. Methods Enzymol. 1999;309:204–17. doi: 10.1016/s0076-6879(99)09016-3. [DOI] [PubMed] [Google Scholar]
- 38.Murray AN, Palhano FL, Bieschke J, Kelly JW. Surface adsorption considerations when working with amyloid fibrils in multiwell plates and Eppendorf tubes. Protein Sci. 2013;22:1531–41. doi: 10.1002/pro.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris AM, Watzky MA, Agar JN, Finke RG. Fitting neurological protein aggregation kinetic data via a 2-step, minimal/”Ockham’s razor” model: the Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry. 2008;47:2413–27. doi: 10.1021/bi701899y. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez-Ruiz JM. Protein kinetic stability. Biophys Chem. 2010;148:1–15. doi: 10.1016/j.bpc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence C, Kuge J, Ahmad K, Plaxco KW. Investigation of an anomalously accelerating substitution in the folding of a prototypical two-state protein. J Mol Biol. 2010;403:446–58. doi: 10.1016/j.jmb.2010.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.del Pozo Yauner L, Ortiz E, Sanchez R, Sanchez-Lopez R, Guereca L, Murphy CL, et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins. 2008;72:684–92. doi: 10.1002/prot.21934. [DOI] [PubMed] [Google Scholar]
- 43.Glenner GG, Ein D, Eanes ED, Bladen HA, Terry W, Page DL. Creation of “amyloid” fibrils from Bence Jones proteins in vitro. Science. 1971;174:712–4. doi: 10.1126/science.174.4010.712. [DOI] [PubMed] [Google Scholar]
- 44.Imoto T, Yamada H, Ueda T. Unfolding rates of globular proteins determined by kinetics of proteolysis. J Mol Biol. 1986;190:647–9. doi: 10.1016/0022-2836(86)90250-0. [DOI] [PubMed] [Google Scholar]
- 45.Park C, Marqusee S. Probing the high energy states in proteins by proteolysis. J Mol Biol. 2004;343:1467–76. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 46.Linke RP, Tischendorf FW, Zucker-Franklin D, Franklin EC. The formation of amyloid-like fibrils in vitro from Bence Jones Proteins of the VλI subclass. J Immunol. 1973;111:24–6. [PubMed] [Google Scholar]
- 47.Neet KE, Putnam FW. Characterization of the thermal denaturation of Bence-Jones proteins by ultracentrifugation at elevated temperatures. J Biol Chem. 1966;241:2320–5. [PubMed] [Google Scholar]
- 48.Nokwe CN, Hora M, Zacharias M, Yagi H, John C, Reif B, et al. The Antibody Light-Chain Linker Is Important for Domain Stability and Amyloid Formation. J Mol Biol. 2015;427:3572–86. doi: 10.1016/j.jmb.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 49.Omtvedt LA, Bailey D, Renouf DV, Davies MJ, Paramonov NA, Haavik S, et al. Glycosylation of immunoglobulin light chains associated with amyloidosis. Amyloid. 2000;7:227–44. doi: 10.3109/13506120009146437. [DOI] [PubMed] [Google Scholar]
- 50.McLaughlin RW, De Stigter JK, Sikkink LA, Baden EM, Ramirez-Alvarado M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci. 2006;15:1710–22. doi: 10.1110/ps.051997606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren R, Hong Z, Gong H, Laporte K, Skinner M, Seldin DC, et al. Role of glycosaminoglycan sulfation in the formation of immunoglobulin light chain amyloid oligomers and fibrils. J Biol Chem. 2010;285:37672–82. doi: 10.1074/jbc.M110.149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109:9629–34. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 54.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools in ExPASy server. In: Walker J, editor. Proteomics Protoc Handb. Totowa, NJ: Humana Press; 2005. pp. 571–608. [Google Scholar]
- 55.Quan S, Hiniker A, Collet JF, Bardwell JC. Isolation of bacteria envelope proteins. Methods Mol Biol. 2013;966:359–66. doi: 10.1007/978-1-62703-245-2_22. [DOI] [PubMed] [Google Scholar]
- 56.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–19. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Royer CA, Mann CJ, Matthews CR. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 1993;2:1844–52. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maxwell KL, Wildes D, Zarrine-Afsar A, De Los Rios MA, Brown AG, Friel CT, et al. Protein folding: defining a “standard” set of experimental conditions and a preliminary kinetic data set of two-state proteins. Protein Sci. 2005;14:602–16. doi: 10.1110/ps.041205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30:10428–35. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 60.Ladner CL, Yang J, Turner RJ, Edwards RA. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal Biochem. 2004;326:13–20. doi: 10.1016/j.ab.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 61.Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with Image. J Biophotonics Int. 2004;11:36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.