

Most mammalian tissues contain resident macrophage populations. In addition to their role in host defense, macrophages may also provide important trophic signals necessary for homeostasis, and are critical effector cells in tissue repair, remodeling, and regeneration1. The adult mammalian myocardium contains a relatively small population of macrophages2; in adult mice, resident cardiac macrophages represent less than 10% of non-cardiomyocytes3. Macrophages may be inconspicuous in the healthy heart, but take center stage following myocardial injury. In the infarcted myocardium, induction of CC chemokines recruits abundant pro-inflammatory monocytes4,5 that differentiate into macrophages6 and exert phagocytotic actions. Clearance of the wound from dead cells and matrix debris triggers anti-inflammatory cascades, inhibiting leukocyte recruitment7; at this stage, local proliferation in response to growth factor stimulation contributes to renewal of the macrophage population in the healing infarct8,9. Infarct macrophages exhibit remarkable phenotypic and functional heterogeneity and may regulate cellular processes critical to cardiac repair. Depletion of macrophages following infarction in mice increased mortality, disrupted wound debridement, and impaired cardiac repair10,11. The protective effects of the macrophages in infarct healing may be due to their critical phagocytotic actions, but may also involve downmodulation of injurious pro-inflammatory signaling, secretion of cytoprotective mediators, activation of reparative fibroblasts and angiogenic cells, and (in some cases) stimulation of a regenerative program12,13,14.
Maturation of the infarct is associated with resolution of the leukocyte infiltrate and with formation of a collagen-based scar. As the scar matures, the ventricle remodels. After a large myocardial infarction, viable myocardial segments exhibit a range of molecular changes that lead to fibrosis, cardiomyocyte hypertrophy, and regional dysfunction, and contribute to the pathogenesis of chronic heart failure. Increased macrophage density has been observed in the remote remodeling myocardium following infarction15; however, the origin of these cells and their role in heart failure progression remain poorly understood.
In the current issue of the journal, Sager and co-workers16 provide the first systematic analysis on the origin, fate and function of macrophages in the remodeling failing myocardium. The authors demonstrated a marked expansion of the macrophage population in the remote non-infarcted myocardium that increased progressively 4–8 weeks after coronary occlusion. Fate mapping studies and parabiosis experiments suggested that expansion of local macrophages results from both new recruitment of monocytes and local proliferation of macrophages. Recruitment of monocytes was dependent on activation of the CCL2/CCR2 axis; in vitro experiments suggested that macrophage proliferation was activated by mechanical strain through mitogen activated protein kinase (MAPK)-dependent pathways. These macrophages exhibited a distinct phenotype, expressing high amounts of Interleukin (IL)-1β, Ym-1 and Vascular Endothelial Growth Factor (VEGF) mRNA, but low levels of matrix metalloproteinase (MMP)9, when compared with normal cardiac macrophages. Late silencing of five adhesion molecules implicated in leukocyte recruitment, in order to selectively inhibit monocyte infiltration in the non-infarcted segments, significantly attenuated adverse remodeling and dysfunction.
The study provides for the first time a systematic analysis of the origin and phenotype of macrophages in the failing infarcted myocardium, and suggests that macrophages in non-infarcted segments may play a critical role in heart failure progression following myocardial infarction. The findings not only contribute to our understanding of the role of macrophages in cardiac remodeling, but also open new directions by raising several important new questions.
What is the basis for the temporally distinct “waves” of macrophage expansion in infarcted and viable segments?
Although it is not surprising that early recruitment and activation of macrophages, triggered by cardiomyocyte necrosis17, primarily involves the infarcted region, the basis for the selective late expansion of the macrophage population in non-infarcted segments is unclear. 8 weeks after the acute event, and while the macrophage infiltrate has resolved in the mature scar, the viable non-infarcted myocardium harbors an active and expanding macrophage population (Figure 1). The authors attribute the proliferative activity of macrophages in viable segments to increased wall stress that activates a proliferative program in macrophages, in part through MAPK-dependent actions. However, left ventricular wall stress is inversely proportionate to wall thickness18 and would be expected to be higher in thinner infarcted segments. The spatial selectivity of the macrophage response may be explained by the microenvironmental differences between the structurally preserved non-infarcted myocardium and the collagen-rich mature scar. In the viable myocardium, the rich microvascular capillary network permits activation of endothelial adhesion cascades that trigger leukocyte extravasation. In contrast, the mature scar is characterized by a low capillary density and a relative abundance of mature neovessels coated with mural cells19, hampering leukocyte extravasation. Moreover, the cross-linked extracellular matrix in the mature scar may prevent monocyte migration and may deactivate macrophages, thus attenuating macrophage expansion. Due to its low cellular content, the mature scar may not be able to generate gradients of chemotactic mediators, necessary for monocyte recruitment.
Figure 1.
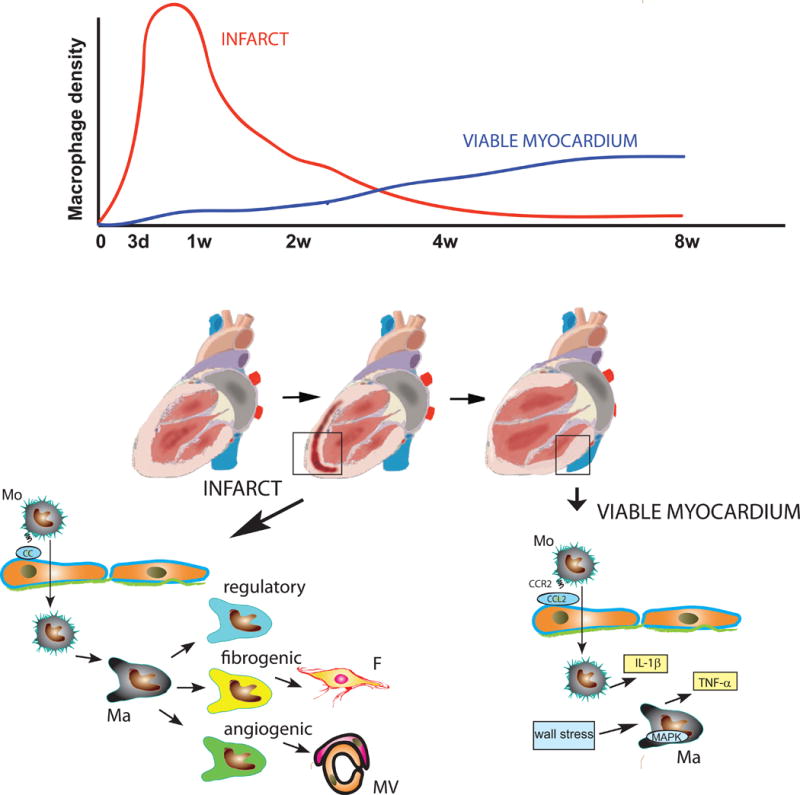
Distinct temporal patterns of macrophage expansion in infarcted and viable myocardial segments following myocardial infarction. The dramatic early increase in macrophage density in the infarcted region is triggered by chemokine-mediated recruitment of monocytes. At a later stage, as chemokine expression is reduced, renewal of macrophages in the infarct is dependent on proliferation. Infarct macrophages exhibit remarkable heterogeneity, acquiring regulatory, fibrogenic, or angiogenic phenotypes and regulating cellular responses critical to cardiac repair. In the viable failing myocardium, late expansion of the macrophage population is driven by CCL2/CCR2-mediated monocyte recruitment, and by stimulation of MAPK signaling in resident macrophages, triggered by increased wall stress. Activated macrophages in the viable non-infarcted myocardium may contribute to heart failure progression by secreting pro-inflammatory cytokines, such as IL-1β and TNF-α (Mo, monocyte; Ma, macrophage; F, fibroblast; MV, microvessel; d, days; w, weeks).
Do macrophages in non-infarcted segments contribute to progression of chronic heart failure?
In experimental studies, macrophage depletion, or genetic manipulations altering the composition or phenotype of infarct macrophages have profound effects on cardiac repair and on chronic remodeling of the heart following infarction10,11,4. Because these interventions cannot selectively target macrophages in infarcted vs. non-infarcted segments, dissection of the potential role for macrophage populations in viable remodeling myocardium is challenging. In order to investigate the effects of these cells, the authors attempted to selectively inhibit macrophage expansion in non-infarcted segments through late silencing of adhesion molecules, at a timepoint that would not be expected to affect monocyte recruitment in the infarcted area. Although the marked preservation of function and geometry in infarcted hearts after adhesion molecule knockdown is consistent with a potential role of macrophages in viable segments, alternative interpretations are possible. Adhesion molecule knockdown does not specifically reduce monocyte infiltration, but may affect recruitment of other leukocyte subpopulations. Moreover, endothelial adhesion molecules have been implicated in angiogenic responses and may have broader effects on endothelial cell function and gene expression in both infarcted and non-infarcted areas20.
Which macrophage-derived signals promote progression of heart failure in viable segments?
Although, in human patients, macrophage infiltration has been associated with segmental contractile dysfunction21, the basis for the deleterious effects of macrophages is unclear. Macrophages in remote remodeling myocardium synthesize large amounts of IL-1β and tumor necrosis factor (TNF)–α, pro-inflammatory cytokines that suppress myocardial contractile function22, and activate matrix-degrading proteases, thus contributing to adverse remodeling following myocardial infarction23. A systematic analysis of the gene expression profile of these cells may suggest additional candidate mediators.
Should we target macrophages in chronic heart failure?
Although the protective effects of attenuated monocyte recruitment in chronic heart failure reported in the current study are impressive, a word of caution is in order when suggesting translational implications. First, the model of infarctive heart failure used in the study does not recapitulate the pathophysiologic heterogeneity of the human disease. Second, we know very little about the phenotype and function of macrophages in healthy and diseased human hearts. Understanding the characteristics of human cardiac macrophages and the effects of myocardial diseases on their gene expression profile and functional properties is important in order to translate the growing body of evidence derived from mouse models into human pathologic conditions.
Acknowledgments
SOURCES OF FUNDING:
Dr Frangogiannis’ laboratory is supported by NIH grants R01 HL76246 and R01 HL85440.
Footnotes
DISCLOSURES:
None
References
- 1.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450–62. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118:400–9. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 5.Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J Immunol. 2013;191:4838–48. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–22. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–73. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frangogiannis NG, Mendoza LH, Ren G, Akrivakis S, Jackson PL, Michael LH, Smith CW, Entman ML. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H483–92. doi: 10.1152/ajpheart.01016.2002. [DOI] [PubMed] [Google Scholar]
- 9.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, Swirski FK, Weissleder R, Nahrendorf M. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res. 2014;115:284–95. doi: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frantz S, Hofmann U, Fraccarollo D, Schafer A, Kranepuhl S, Hagedorn I, Nieswandt B, Nahrendorf M, Wagner H, Bayer B, Pachel C, Schon MP, Kneitz S, Bobinger T, Weidemann F, Ertl G, Bauersachs J. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. Faseb J. 2013;27:871–81. doi: 10.1096/fj.12-214049. [DOI] [PubMed] [Google Scholar]
- 12.Frangogiannis NG. Emerging roles for macrophages in cardiac injury: cytoprotection, repair, and regeneration. J Clin Invest. 2015;125:2927–30. doi: 10.1172/JCI83191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382–92. doi: 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, Arditi M, Marban E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest. 2015 doi: 10.1172/JCI81321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee WW, Marinelli B, van der Laan AM, Sena BF, Gorbatov R, Leuschner F, Dutta P, Iwamoto Y, Ueno T, Begieneman MP, Niessen HW, Piek JJ, Vinegoni C, Pittet MJ, Swirski FK, Tawakol A, Di Carli M, Weissleder R, Nahrendorf M. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol. 2012;59:153–63. doi: 10.1016/j.jacc.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sager HB, Hulsmans M, Lavine KJ, Beltrami Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, Dahlman JE, Borodovsky A, Fitzgerald K, Anderson DG, Weissleder R, Libby P, Swirski FK, Nahrendorf M. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.116.309001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Elia N, D’Hooge J, Marwick TH. Association Between Myocardial Mechanics and Ischemic LV Remodeling. JACC Cardiovasc Imaging. 2015;8:1430–43. doi: 10.1016/j.jcmg.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–9. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 20.Kevil CG, Orr AW, Langston W, Mickett K, Murphy-Ullrich J, Patel RP, Kucik DF, Bullard DC. Intercellular adhesion molecule-1 (ICAM-1) regulates endothelial cell motility through a nitric oxide-dependent pathway. J Biol Chem. 2004;279:19230–8. doi: 10.1074/jbc.M312025200. [DOI] [PubMed] [Google Scholar]
- 21.Frangogiannis NG, Shimoni S, Chang SM, Ren G, Shan K, Aggeli C, Reardon MJ, Letsou GV, Espada R, Ramchandani M, Entman ML, Zoghbi WA. Evidence for an active inflammatory process in the hibernating human myocardium. Am J Pathol. 2002;160:1425–33. doi: 10.1016/S0002-9440(10)62568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hosenpud JD, Campbell SM, Mendelson DJ. Interleukin-1-induced myocardial depression in an isolated beating heart preparation. J Heart Transplant. 1989;8:460–4. [PubMed] [Google Scholar]
- 23.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]